
Abstract
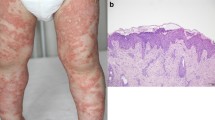
Generalized pustular psoriasis (GPP) is a rare, chronic, and severe inflammatory skin disorder characterized by sudden eruption of sterile pustules, often accompanied by systemic inflammation. GPP flares can be life-threatening if untreated, owing to potential serious complications such as sepsis and cardiovascular failure. Diagnosis and clinical measurement of disease severity in GPP are often difficult. Lack of standardized criteria in the international guidelines and the heterogeneity of cutaneous and extracutaneous symptoms make the diagnosis of GPP difficult. Clinical criteria for description and diagnosis of pustular conditions, including GPP, are variable and there is no specific agreement on commonly sustained concepts. Differentiation of GPP from other similar conditions/diseases is important and requires careful assessments. The evidence that supports current topical or systemic therapies is largely based on case reports and small studies. Some biologic agents that target key cytokines involved in the activation of inflammatory pathways have been used as treatments for GPP. Recently, spesolimab, an IL-36R antagonist, has been approved in the USA and Japan for the treatment of GPP flares in adults, but there are no currently approved treatments for GPP in Europe. The IL-36 pathway has recently emerged as a central axis driving the pathogenic inflammatory mechanisms of GPP. Biologic agents that inhibit the IL-36 pathway have shown efficacy and safety in patients with GPP, addressing a generally considered unmet medical need.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Generalized pustular psoriasis (GPP) is a rare, chronic, and severe inflammatory skin disorder characterized by sudden eruption of sterile pustules, often accompanied by systemic inflammation. |
Lack of standardized criteria in the international guidelines and the heterogeneity of cutaneous and extracutaneous symptoms make the diagnosis of GPP difficult. Differential diagnosis with entities like AGEP and Sneddon–Wilkinson disease, among others, is key. |
The IL-36 pathway has recently emerged as a central axis driving the pathogenic inflammatory mechanisms of GPP. |
Spesolimab has recently been approved in the USA and Japan for the treatment of GPP flares in adults. However, there are neither guidelines nor specific treatments approved in Europe for GPP. Biologic agents that inhibit the IL-36 pathway have shown efficacy and safety in patients with GPP, addressing a generally considered as unmet medical need. |
Limited disease awareness, combined with inaccurate diagnosis and similarity to other variants of psoriasis, has classically complicated the patient journey. |
Introduction
Psoriasis is a systemic immune-mediated disorder that affects an estimated 2–4% of the Western population [1,2,3]. Skin manifestations of psoriasis include five forms or phenotypes: plaque psoriasis or psoriasis vulgaris, inverse, pustular, guttate, and erythrodermic psoriasis. Pustular psoriasis, which often occurs with concurrent plaque psoriasis, is a rare entity with sterile pustules that can be localized or generalized [4, 5]. Classically recognized subtypes of pustular psoriasis include generalized pustular psoriasis (GPP), palmoplantar pustular psoriasis (PPP), and acrodermatitis continua of Hallopeau (ACH) [4,5,6,7,8]. This article presents an updated review on the clinical characteristics and pathogenesis of GPP, also revising current concepts on diagnosis, evaluation, and new therapeutic options.
Methods
The objective of the present paper is to conduct a semi-structured literature review on epidemiology, clinical characteristics, diagnosis, and treatment of GPP. The MEDLINE database was searched via PubMed to retrieve relevant articles on generalized pustular psoriasis (GPP), using appropriate controlled vocabulary. Variants, subtypes, and synonyms were included (e.g., impetigo herpetiformis, extensive pustular psoriasis, and Von Zumbusch psoriasis), and MESH (Medical Subject Headings) terms were preferred. A first search retrieved 524 articles. After applying a language filter (only English and Spanish languages were allowed) and a 5-year time limit, we identified 215 articles.
Selected articles included clinical trials, clinical series, systematic reviews, observational studies, cohort studies, cross-sectional studies, real-world evidence studies, consensus, surveys, registry reports, and guidelines for diagnosis or treatment. Other types of articles (e.g., letters, comments) were excluded. Then, a set of 75 articles was chosen for the evaluation of titles and abstracts. From these, 40 main papers were selected using different keywords and the full text of these articles was assessed to form the core of the review. Other relevant articles were subsequently added based on the personal knowledge and experience of the authors. Individual assessment of selected articles was used as the basis for the review.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Results
Clinical Characteristics and Diagnosis
The condition currently known as GPP was first described by Leopold von Zumbusch in 1910. Since its first description, clinical characteristics have not been consistently defined and several descriptions and different diagnostic criteria have been used [4,5,6,7,8,9,10,11,12,13,14].
Although there is no international consensus, according to the consensus by the European Rare and Severe Psoriasis Expert Network (ERASPEN), GPP is defined as macroscopically visible primary sterile pustules occurring on non-acral skin and not restricted within psoriasis plaques [5].
Sterile pustules are considered primary lesions. The pustules that dry up can form scale heaps or brownish scabs that slowly cast off. These scabs may be considered evidence of pustulation in cases where fresh pustules are not detected. The influence of trigger factors, albeit clinically recognized, is not considered diagnostic criterion.
GPP is subclassified according to the presence or absence of associated features such as systemic inflammation, lesions of plaque psoriasis, and the clinical course (relapsing or persistent). Criteria for systemic inflammation, besides high erythrocyte sedimentation rate (ESR) and serum C-reactive protein levels (CRP), include fever higher than 38 °C and leukocytosis defined by a white blood cell (WBC) count greater than 12 × 109/l. The condition should only be diagnosed when it has relapsed at least once (relapsing type) or when it persists for more than 3 months (persistent type) [5].
According to the Japanese guidelines issued by the Japanese Dermatological Association (JDA), GPP patients must present systemic symptoms such as fever and fatigue, systemic or extensive flush (sic) accompanied by multiple sterile pustules that sometimes merge to form lakes of pus, and neutrophilic subcorneal pustules corresponding to Kogoj’s spongiform pustules. The aforementioned clinical and histological features recur repeatedly [6,7,8]. A comparison between Japanese and European guidelines can be seen in Table 1 [5,6,7,8].
Epidemiology
GPP is a rare disease, which implies challenges in collecting epidemiologic data. Population-based estimates of GPP prevalence and incidence are scarce. In addition, clinical awareness is limited and the difficulty reaching a diagnosis challenges the estimation of disease prevalence. A high variability in prevalence between different ethnicities and geographical regions has been reported [9,10,11,12,13,14,15].
GPP can present at any age, from childhood to old age, but the median reported age at diagnosis is around 50 years [9,10,11,12,13,14,15]. In general, female preponderance has been observed in GPP patients. Early onset of GPP is more likely in patients with the IL36RN mutation [16,17,18,19].
Globally, prevalence of GPP can be estimated as 1–7 cases per million persons [8,9,10,11,12,13,14,15]. However, data are variable, and estimations range from 1.76 cases per million in France to 7.46 cases per million in Japan, 180 cases per million in Italy, and 88–124 cases per million in Korea [8,9,10,11,12,13,14,15].
Recently, Löfvendahl et al. conducted an observational retrospective study to estimate the prevalence of GPP in the Swedish general population [15]. They used data from physician-diagnosed GPP in a population-based healthcare registry over a 12-year observational period and found a prevalence of 9.1 per 100,000. The prevalence of GPP in women was 1.6 times higher than in men (11.2 vs. 7.0 per 100,000), and 43% of GPP patients also had a plaque psoriasis code in the registry [15].
Clinical Course
The clinical course of GPP is highly variable. GPP can appear as a relapsing disease with recurrent flares and no pustulation between flares, or it can be a persistent disease with permanent pustulation in which flares of greater severity can also occur [4,5,6,7,8,9].
Four forms are recognized according to the onset of flares and the morphology of lesions. Von Zumbusch GPP is the most severe presentation, which may be life-threatening. It presents with rapid onset (7 days or fewer) of a generalized pustular flare that may be experienced by up to 90% of patients with GPP. Annular GPP (Lapière-Millian) presents with a generalized flare of circinate or annular lesions with peripheral pustules that develops in between 7 days and 3 months and is often associated with relatively mild symptoms. Chronic acral GPP is characterized by lesions that begin in acral areas and spread gradually to form a generalized pustular flare. “Mixed GPP” has features associated with more than one subtype [9].
Choon et al. summarized the clinical characteristics and outcomes from main GPP studies [4]. Clinical symptoms are heterogenous, as are their reported frequencies. High fever has been recorded in 24–96% of patients during a GPP flare and approximately 30–70% of patients had leukocytosis with neutrophilia. GPP tend to be associated with trigger factors (reported in 41–85% of cases), previous plaque psoriasis (31–78%), arthritis (31–34%), and malaise/fatigue (100%) [4].
Flares of GPP vary in frequency, severity, and duration between patients and also between episodes in the same patient. Severe flares can be life-threatening and are usually accompanied by systemic symptoms, such as high fever, general malaise, and fatigue. The most frequently reported cutaneous signs and symptoms of GPP include pustules, scaling, dryness, swelling, erythema, skin pain, itching, and burning sensation.
Extracutaneous manifestations, including arthritis, uveitis, and neutrophilic cholangitis, can also appear. Laboratory abnormalities, including neutrophilia, elevated CRP levels, abnormal liver function tests, hypocalcemia, and hypoalbuminemia, may be associated with flares [4,5,6, 9,10,11,12,13].
Patients may have numerous flares per year or a flare every few years. Although the course of flares is highly variable, most flares last between 2 and 5 weeks but may persist longer than 3 months and approximately 50% may require hospitalization. Potentially life-threatening extracutaneous complications, including sepsis and renal, hepatic, respiratory, and heart failure, can occur and the reported mortality rates range from 2 to 16% [4, 9, 13].
Triggers or Precipitating Factors
Flares may appear without an evident cause or may be provoked by certain triggers or precipitating factors such as rapid withdrawal of systemic corticosteroids, infections, pregnancy, menstruation, and stress. Other potential triggers that have been implicated (mostly based on small series or anecdotal evidence) include hypocalcemia and vaccination. The use of medications has also been reported as a precipitating factor. The most commonly reported triggers are detailed in Table 2 [3, 4, 9,10,11,12,13].
Differential Diagnosis
In general, the diagnosis of GPP is mainly clinical. Laboratory tests are helpful for diagnostic confirmation and to assess the level of systemic inflammation and possible systemic complications that may lead to fatal outcomes. Potassium hydroxide direct examinations can also be performed to exclude generalized tinea corporis and disseminated candidiasis, which may cause scaling and pustulosis. Biopsies do not always confirm the diagnosis of GPP (e.g., versus acute generalized exanthematous pustulosis [AGEP]). However, the diagnosis is clinico-pathological and skin biopsies may reinforce the clinical diagnosis of GPP [7]
At least 29 different primary pustuloses have been described over the years with a wide range of possible associated clinical features [5]. The rarity of these conditions does not allow collecting enough cases to establish diagnostic and differential criteria. The clinical criteria for description and diagnosis of pustular conditions, including GPP, are variable, and it is difficult to reach a commonly accepted agreement. Proposed definitions of pustuloses show several discrepancies on subtypes, localizations, extension, presence of arthritis, pain, systemic inflammation, and other characteristics [4,5,6,7,8]. Because of that, differential diagnosis in GPP is of paramount importance.
The differential diagnosis of GPP flares vs AGEP represents a clinical challenge and requires a detailed clinical, histopathological, and laboratory evaluation.
According to Fujita et al., the subcorneal pustules in AGEP are spongiform and less outstanding than GPP pustules [7]. The localization and spongiform characteristic of the pustules can help to differentiate AGEP from GPP in a skin biopsy specimen. Necrotic keratinocytes are more commonly found in AGEP pustules. Generally, pustules in GPP do not contain eosinophils, have more apoptotic keratinocytes, and are localized at a higher epidermal level than those of AGEP [7].
Mutations of the gene encoding interleukin-36 receptor antagonist (IL-36RN) have been found in some cases of AGEP, suggesting a close relationship or overlap with GPP. However, a drug reaction such as AGEP should be actively ruled out to confirm the diagnosis of GPP [5].
Due to the lack of published evidence for the differential diagnosis of GPP, the authors have developed a detailed list of clinical features and associated variables, as shown in the “Discussion” section.
Another differential diagnosis of GPP is Sneddon–Wilkinson disease or subcorneal pustular dermatosis, a rare neutrophilic dermatosis in which recurrent sterile pustules appear in the most superficial (subcorneal) layers of the skin [7]. Sneddon–Wilkinson disease has been reported most commonly in adult or elderly females and its etiology is unknown. It presents with many flaccid pustules, several millimeters in diameter, on normal or mildly erythematous skin. The pustules are usually distributed in annular or serpiginous patterns and are most commonly located on flexural surfaces and on intertriginous skin. A gravity-induced demarcation in some vesiculopustules, with clear fluid superiorly and pus inferiorly, is characteristic of this disease. It is a chronic and benign condition; unlike GPP, subcorneal pustular dermatosis does not appear to be associated with systemic symptoms [7], but associated disorders should be considered, especially gammopathies [20].
Etiology and Pathophysiology
Current histologic, genetic, and pathophysiologic data demonstrate that GPP is a distinct entity from psoriasis vulgaris, and different cytokine pathways are involved in the development of each condition [6, 7, 16,17,18,19, 21, 22].
GPP may appear with pre-existing plaque psoriasis, and common interlinked immunologic pathways appear to contribute to the pathogenetic basis of both entities. However, unlike plaque psoriasis, where the adaptive immune system and the interleukin (IL)-23/IL-17 axis play a central pathogenic role, GPP appears to be driven by an inflammatory response resulting from hyperactivation of innate immunity, with predominant participation of the IL-36 axis.
Johnston et al. reported that gene expression studies on skin biopsy samples from patients with GPP or plaque psoriasis showed that GPP lesions have higher levels of IL-1 and IL-36 and lower levels of IL-17A and interferon-γ compared with plaque psoriasis lesions [23]. In addition, immunohistochemical analysis revealed that GPP lesions contain higher levels of neutrophilic chemokines compared with plaque psoriasis lesions [23].
The IL-36 signaling cascade plays a role in regulating the innate immune system, and its dysregulation seems to be key to the pathogenesis of GPP. The dysregulate expression of different IL-36 pathway components provokes a feedback loop of altered signaling and excess production of inflammatory cytokines, which in turn enhance chemokine induction and recruitment of neutrophils in the epidermis [16,17,18,19, 21, 22, 24].
The IL-36 Inflammatory Pathway
The interleukin (IL)-1 family of cytokines is composed of 11 members, including the proinflammatory cytokines IL-36α, IL-36β, and IL-36γ. Similar to IL-1, IL-36 cytokines can act as initiators and amplifiers of inflammation [24]. The IL-36 cytokine agonists, i.e., IL-36α, IL-36β, and IL-36γ, bind to a common receptor to stimulate inflammatory responses (IL-36R). According to Bassoy et al., the IL-36 cytokines are secreted predominantly by epithelial cells and act on several cell types, including immune cells, epithelial cells, fibroblasts, and keratinocytes [25]. IL-36 cytokines are released as precursors that require N-terminal truncation for full pro-inflammatory activity. Their processing and activation are produced by elastase, cathepsin G, proteinase 3 (neutrophil-derived), and cathepsin S (derived from keratinocytes and fibroblasts). However, protease inhibitors like alpha-1 antitrypsin (codified by SERPINA1) and alpha-1 antichymotrypsin (codified by SERPINA3) inhibit IL-36 cytokines processing performed by elastase and cathepsin G proteases, respectively [26]. The role of IL-36 has been demonstrated in the skin, where it can act on keratinocytes and immune cells to induce an intense inflammatory response. A role of this family of cytokines in joint inflammation and in pulmonary and intestinal pathophysiology has also been shown [24, 25].
Uncontrolled expression and activation of IL-36 cytokines can lead to self-perpetuating inflammatory cascades [26]. IL-36 signaling involves a complex of IL-36 receptor (IL-36R) and IL-1R accessory protein (IL-1RAcP), disseminating inflammatory responses in the epithelium [26]. Therapeutic responses to IL-1 receptor inhibition in GPP tend to be incomplete, which suggests that IL-1 does not play a central role in GPP but acts in a positive feedback loop inducing and being induced by IL-36 [23, 26].
According to Zhou et al., GPP can occur through overexpression of IL-36 agonists or by the expression of a dysfunctional IL-36R antagonist (IL-36RA), encoded by mutations in the IL-36RN gene that lead to a stimulating feedback loop of uncontrolled signaling and excess production of proinflammatory cytokines [17]. This induces some chemokines to attract high numbers of neutrophils into the epidermis, where the accumulation of neutrophils manifests as pustules, a hallmark of GPP (Fig. 1) [26].

Adapted from Iznardo et al. [26]
Schematic representation of the signal transduction pathway activated by cytokines and genes involved in IL-36 autocrine and autoinflammatory circuits. The pathogenesis of GPP is related to mutations in multiple genes, such as the human IL-1Ra gene (IL1RN), IL-36Ra (IL36RN), caspase recruitment domain-containing protein 14 (CARD14), adapter protein complex 1 subunit sigma 3 (AP1S3), TNFAIP3-interacting protein 1 (TNIP1), and the gene coding for alpha-1 antichymotrypsin, also known as serine protease inhibitor gene serpin family A member 3 (SERPINA3). IL-1, TNF, and IL-17A promote the expression of IL-36 by keratinocytes. IL-36 cytokines are released as precursors requiring enzymatical cleavage by neutrophil-derived proteases (elastase, cathepsin G, or protease 3) and keratinocyte-derived cathepsin S. Mature IL-36 cytokines have 500-fold greater biological activity than their precursors and bind to IL-36R on the keratinocyte cell surface, acting in an autocrine manner to further induce IL-36 expression. In addition, they induce the production and secretion of neutrophil chemokines CXCL1, CXCL2, CXCL6, and CXCL8 (IL-8), increasing the attraction of neutrophils to the skin. Serine protease inhibitors such as alpha-1 antitrypsin or alpha-1 antichymotrypsin (encoded by SERPINA1 and SERPINA3, respectively) can inhibit neutrophil proteases.
GPP Genetics
In recent years, allelic variations and mutations of IL36RN, CARD14, AP1S3, and MPO genes have been found to be associated with GPP. Among those genes, IL36RN mutations are the most frequently encountered genetic abnormality. These disease-related genes play a role in common signaling pathways, especially in the IL-1/IL-36 axis, the pathogenic inflammatory pathway typically activated in GPP [17].
In as much as the correlation between IL36RN mutations and GPP has been robustly evidenced, not all patients with GPP harbor mutations in this gene [27]. IL 36RN mutation are present in 34.7% of European GPP patients, and in 28.8% of Asian patients [28]. Overproduction of IL-36 agonists is observed in GPP patients with mutations in SERPINA3, the gene encoding alpha-1 antichymotrypsin, an inhibitor of cathepsin G, a protease secreted by neutrophils that activates IL-36 precursors [29] (Fig. 1).
GPP pathogenesis also appears to be mediated by transcripts of other mutated genes. In Asian patients, mutations in CARD14 have been described as associated with GPP; the transcript of CARD14 facilitates activation of nuclear factor-κB (NF-κB) in keratinocytes and other cell types [18, 19, 21, 22, 24]. Mutations in MPO leading to a deficit of myeloperoxidase—a neutrophil-associated lysosomal hemoprotein that regulates protease activity, neutrophil extracellular trap formation, apoptosis of neutrophils and their clearance by monocytes—have also been associated with GPP [16,17,18,19, 21, 22, 30,31,32] (Fig. 1).
Therefore, while genetic and pathophysiologic data indicate that the IL-36 pathway is key to the development of GPP, a better understanding of the associated complex immunologic components is still needed.
Severity/Outcome Measures and Follow-Up of Patients
The uncommonness of GPP and its heterogeneous cutaneous and extracutaneous symptoms represent a challenge to the adoption of comprehensive and accurate disease measures for the routine clinical assessment and follow-up of patients. Plaque psoriasis disease scores are commonly used measures for evaluating patients with GPP. However, typical psoriasis measures such as PGA (Physician Global Assessment) or PASI (Psoriasis Area and Severity Index) do not assess pustules, a key feature of GPP. Therefore, specific scales to assess patients with GPP are required, and some have recently been developed (Table 3) [33].
Modified psoriasis clinical disease measures, such as the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) and Generalized Pustular Psoriasis Area and Severity Index (GPPASI), have been developed to specifically assess GPP by replacing the induration component with a pustular component.
For GPPASI, the score for each body region is calculated. The product of the sum of severity scores and its corresponding BSA (body surface area) score for erythema, scaling, and pustulation, is multiplied by a weighting factor for each body region. Then, the total GPPASI score is determined by the sum of the individual scores from all body regions (Fig. 2) [33].

Reproduced with permission from Burden et al. [33]
Generalized Pustular Psoriasis Area and Severity Index (GPPASI).
The total GPPGA score is based on averaging the individual scores for erythema, scaling, and pustulation. The GPPGA score calculation is detailed in Fig. 3 [34]. As pustules are the main characteristic of GPP, a variation of GPPGA total score is the GPPGA pustulation subscore, which ranges from 0 to 4 points, where 0 means no visible pustules and 4 represents severe pustulation.

Adapted from Choon et al., AAD-VMX 2021 [34]. GGPGA, Generalized Pustular Psoriasis Physician Global Assessment. AAD-VMX American Academy of Dermatology—Virtual Meeting Experience
Components and methodology of the GPPGA score for evaluation and follow-up of GPP patients.
GPP Treatment
Up to date, spesolimab has been recently approved in the USA and Japan for the treatment of GPP flares in adults. There are no specific treatments approved for GPP in Europe [33, 35,36,37,38]. The evidence that supports the efficacy and safety of treatments for GPP is mainly based on case reports, case series, and non-randomized studies. In general, the quality of evidence according to GRADE methodology is low [4,5,6,7,8,9, 18, 33, 35,36,37,38]. There is also a lack of GPP treatment guidelines. Treatment guidelines from Japan (JDA 2018) and the USA (NPF 2012) have not been recently updated [5, 6]. The Psoriasis Group of the Spanish Academy of Dermatology and Venereology (GPs) classifies GPP as a severe variant of psoriasis but does not provide any treatment recommendations [36].
In general, for psoriasis, the treatment is topical, phototherapy, systemic non-biological or systemic biological. Topical treatments are not recommended for GPP flares. In certain cases, topical treatments such as steroids, calcipotriene (calcipotriol), and tacrolimus should be considered as maintenance or adjuvant therapy [6, 35]. However, its use should be monitored carefully because of the potential induction of pustules after discontinuation of topical steroids and also few cases of induction of GPP have been reported with the use of topical calcipotriol [6]), maybe because calcipotriol ointment is a known irritant. In irritative contact dermatitis, keratinocytes deliver various cytokines, including IL-1, IL-6, TNF-α, and IFN-γ. If the area of irritation dermatitis is considerably large, it is possible that the amount of delivered cytokines may be sufficient to precipitate GPP [39]. The current classification for GPP therapies classifies treatment options as biological and non-biological systemic agents.
Non-biologic systemic therapies, including corticosteroids, acitretin, cyclosporine, and methotrexate have been typically used as first-line options for GPP although the evidence in support of this is limited. Other non-biologic agents that have been used for the treatment of GPP, also with limited evidence, include mycophenolate mofetil, hydroxyurea, apremilast, and colchicine. Considering the acute life-threatening characteristics of GPP flares, cyclosporine sometimes is preferred due to its rapid onset of action [7, 8, 33, 35,36,37,38, 40].
In Japan and other Asian countries, several biologics are approved for treatment of the disease, including TNF inhibitors (infliximab, adalimumab, and certolizumab pegol), IL-17/IL-17R inhibitors (secukinumab, brodalumab, and ixekizumab), and IL-23 inhibitors (risankizumab and guselkumab) [7, 8, 33, 35,36,37,38, 40]. Although these therapies are approved for GPP in Japan, all of them present uncertainties in terms of safety and efficacy in GPP treatment [36]. Their effectiveness in treating acute flares is unknown and clinical evidence supporting their use is scarce, coming typically from open-label trials or case series in GPP patients. Therefore, treatment of GPP flares and long-term management of GPP patients are still unmet medical needs [33, 35,36,37,38, 40]. Spesolimab has recently been approved in Japan for the treatment of GPP flares in adults.
According to a survey that included 29 dermatologists from the USA and Canada participating in the Corrona Psoriasis Registry, 72% of respondents indicated that treatments were too slow to control flares and 67% of them indicated that treatment does not help in preventing new flares [38].
IL-36 Targeted Therapies
Advances in the intricate details of genetic mutations and immunological mechanisms have led to a better understanding of GPP pathogenesis and brought up considerable implications on diagnosis and treatment. The identification of the central role of the IL-36 signaling pathway in the pathogenesis of GPP gave rise to potential new therapies. Given the life-threatening nature of GPP episodes, drug interventions that rapidly achieve disease resolution are required. Recent data indicate that IL-36 pathway inhibitors (spesolimab and imsidolimab) represent novel potential therapeutic options for GPP patients [34, 41,42,43,44].
In a phase II, multicenter, double-blind, placebo-controlled trial, (Effisayil 1 study), Bachelez et al. randomized patients with a GPP flare in a 2:1 ratio to receive intravenous spesolimab (n = 35) or placebo (n = 18) [43]. The primary endpoint was a Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at the end of week 1. At baseline, 46% of the patients in the spesolimab arm and 39% in the placebo arm had a GPPGA pustulation subscore of 3, while 37 and 33%, respectively, had a pustulation subscore of 4. At the end of week 1, a total of 19/35 patients (54%) in the spesolimab group had a pustulation subscore of 0 (complete pustular clearance), as compared with 1/18 patients (6%) in the placebo group. In addition, a total of 15/35 patients (43%) achieved a GPPGA total score of 0 or 1 (complete or almost complete clearance of skin lesions), as compared with 2/18 patients (11%) in the placebo group, showing a significant difference (P = 0.02) between treatment groups [43]. Related to safety, similar rates of adverse events were reported in the spesolimab and placebo arms (66 vs. 56% of the patients, respectively) at week 1. Spesolimab demonstrated an acceptable safety profile compared to placebo. Finally, according to Morita et al., the efficacy and safety of spesolimab in Asian patients was comparable to that in the overall population, supporting its use in the treatment of GPP flares [18].
Clinical trials on imsidolimab are ongoing but their results have not yet been published [26, 44].
Special Populations
There are two special populations that can be affected by GPP, pediatric patients and pregnant women [4, 10, 45,46,47,48].
GPP can occur as a rare form of childhood psoriasis, often requiring systemic therapy. In pediatric GPP, the circinate annular form is considered apart. A specific case of childhood GPP is the DITRA syndrome. DITRA (deficiency of interleukin-36 receptor antagonist) is a life-threatening autoinflammatory disease caused by autosomal-recessive mutations in the interleukin-36 receptor. DITRA patients have a higher risk of recurrent episodes of GPP with systemic inflammation and fever [47]. Treatment of pediatric or juvenile GPP is challenging because there is a paucity of randomized controlled trials and standardized guidelines. Biologic agents have been used in pediatric psoriasis but the evidence of their efficacy in refractory cases of pediatric GPP has been scarce and slow in gathering. Many case series have reported the successful use of biologic agents in pediatric GPP, but a better understanding of the efficacy and safety profile of biologic agents in this population remains an unmet need [10, 19, 45, 47, 48].
Pregnancy-associated GPP, also known as impetigo herpetiformis, is most frequently observed in the last trimester of pregnancy. It is now believed to be the same disease as acute GPP because most patients have other triggers in addition to pregnancy [4, 18]. Multiple sterile pustules are observed with annular configuration on an erythematous base, usually starting at flexural areas with subsequent spreading over the body. Systemic symptoms, such as fever, chills, fatigue, nausea, diarrhea, polyarthralgia, and laboratory abnormalities, such as hypocalcemia, can be observed. Impetigo herpetiformis, especially if severe and long-lasting, may lead to poor neonatal outcomes, and even to maternal death. Most patients experience prompt remission in the post-partum period, and recurrences with subsequent pregnancies are frequent, sometimes with earlier onset and greater severity. For some researchers, it is still controversial whether this entity should be considered different from GPP [4, 18, 47].
Discussion
GPP is a rare, chronic, and potentially life-threatening disease that poses multiple diagnostic and management challenges to dermatologists. The rarity of GPP and their variable cutaneous and extracutaneous manifestations represent a challenge to the development of disease measures for the clinical assessment and follow-up of disease and its response to treatment [34]. The authors recommend using GPPGA or GPPASI for outcome assessment and patient follow-up. Its implementation in routine clinical practice will depend on its incorporation into hospital protocols or future guidelines.
The unusual nature of GPP can make diagnosis problematic, and the existence of numerous different pustular diseases further complicates it. For example, the similarity between GPP and AGEP, a severe cutaneous drug reaction, poses diagnostic and therapeutic problems [3,4,5,6,7, 49, 50].
Since differential diagnosis is key to correctly diagnose GPP and there is a lack of evidence in the literature, the authors propose a table with the characteristics of the most similar diseases that need to be ruled out before diagnosing GPP (Table 4) [51].
Particular interest has been generated in this area by some recent reports of different types of pustular psoriasis, including de novo or exacerbated GPP, related to COVID-19 or following COVID-19 vaccination [52,53,54,55].
The only Spanish evidence of GPP patient characteristics comes from a multicentric study presented by Aragon et al. including 24 GPP patients [46]. Regarding comorbidities, 58.3% of patients had dyslipidemia, 45.8% had hypertension, and 41.7% had psoriatic arthritis. Twenty-three out of 24 patients required systemic treatment during a median of 18 months for the management of the flares, and one-third of these patients were treated previously with biologic agents. Regarding current therapy, almost all patients (23/24) were treating with any systemic therapy: 41.7% (10/24) with biologic therapy, 9/24 (37.5%) with conventional systemic (methotrexate, acitretin phototherapy) and 4/24 (16.7%) with apremilast [46].
According to the results of a recent systematic literature review (SLR), there is a scarcity of high-quality evidence for treatments of GPP that can rapidly and completely resolve symptoms with an acceptable safety profile. The most commonly used oral systemic options, according to the SLR conducted by Puig et al. that included 114 studies, were cyclosporine, methotrexate, and acitretin. However, the overall grade of evidence regarding their efficacy and safety was low. The evidence for biologic agents, although generally coming from uncontrolled clinical studies and case series, shows favorable efficacy, faster time to clearance of GPP pustules, and a good safety profile [45].
Treatment goals in GPP are not well defined due to a lack of consistent treatment guidelines. Therefore, immediate treatment of GPP flares and long-term management of patients with GPP are riddled with uncertainty [4, 35]. Therapeutic goals can be classified as immediate/short term and long term. Immediate therapeutic goals during a GPP flare include improving skin manifestations (stopping and preventing the appearance of pustules) and reducing the burden of systemic symptoms. Rapid control of skin symptoms, within a week of treatment, is now a feasible treatment goal based on the results of clinical trials with IL-36 inhibitors [42]. Short-term treatment goals should focus on maintaining a low and stable GPPGA score (between 0 and 1 points), without systemic inflammation. Regarding long-term management, treatment goals should focus on the prevention of new GPP episodes or disease worsening, with effective periodic monitoring strategies (e.g., GPPGA assessments) and clinical controls to detect or limit recurrence of flares [4, 7, 18, 35]. However, efficacy measurements (e.g., PASI) often used to evaluate effectiveness are not designed to measure effectiveness in GPP. In addition, there is also a lack of evaluation of early timepoints, e.g., during the first week of treatment, which is crucial for GPP.
Conclusions
In conclusion, GPP is a rare, chronic, and systemic disease that has recently benefited from advances in our understanding of its genetic and molecular basis. Limited disease awareness, combined with inaccurate diagnosis and similarity to other variants of psoriasis, has classically complicated the patient journey. It is common for the GPP patient to be seen at emergency rooms by non-expert physicians, and inappropriate diagnosis and therapeutic interventions may worsen the prognosis. Therefore, medical education programs are required to increase clinical awareness and improve the patient journey pathway.
The role of the IL-36 pathway as a key inflammatory axis in the pathogenic mechanism of GPP is driving the development of new treatments for this condition and possibly other rare inflammatory cutaneous disorders which have traditionally been considered to represent unmet medical needs.
References
Papp KA, Gniadecki R, Beecker J, et al. Psoriasis prevalence and severity by expert elicitation. Dermatol Ther (Heidelb). 2021;11(3):1053–64.
World Health Organization. Global report on Psoriasis. Geneva: World Health Organization; 2016.
Boehncke WH, Schön MP. Psoriasis. Lancet. 2015;386(9997):983–94.
Choon SE, Navarini AA, Pinter A. Clinical course and characteristics of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):21–9.
Navarini AA, Burden AD, Capon F, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11):1792–9.
Fujita H, Terui T, Hayama K, et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11):1235–70.
Fujita H, Gooderham M, Romiti R. Diagnosis of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):31–8.
Ly K, Beck KM, Smith MP, Thibodeaux Q, Bhutani T. Diagnosis and screening of patients with generalized pustular psoriasis. Psoriasis (Auckl). 2019;9:37–42.
Zheng M, Jullien D, Eyerich K. The prevalence and disease characteristics of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):5–12.
Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–19.
Reynolds KA, Pithadia DJ, Lee EB, Clarey D, Liao W, Wu JJ. Generalized pustular psoriasis: a review of the pathophysiology, clinical manifestations, diagnosis, and treatment. Cutis. 2022;110(2 Suppl):19–25.
Bachelez H, Barker J, Burden AD, Navarini AA, Krueger JG. Generalized pustular psoriasis is a disease distinct from psoriasis vulgaris: evidence and expert opinion. Expert Rev Clin Immunol. 2022;18(10):1033–47.
Kharawala S, Golembesky AK, Bohn RL, Esser D. The clinical, humanistic, and economic burden of generalized pustular psoriasis: a structured review. Expert Rev Clin Immunol. 2020;16:239–52.
Lee JY, Kang S, Park JS, et al. Prevalence of psoriasis in Korea: a population-based epidemiological study using the Korean National Health Insurance Database. Ann Dermatol. 2017;29:761–7.
Löfvendahl S, Norlin JM, Schmitt-Egenolf M. Prevalence and incidence of generalised pustular psoriasis in Sweden - a population-based register study. Br J Dermatol. 2022.
Marrakchi S, Puig L. Pathophysiology of generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):13–9.
Zhou J, Luo Q, Cheng Y, Wen X, Liu J. An update on genetic basis of generalized pustular psoriasis (Review). Int J Mol Med. 2021;47(6):118.
Morita A, Tsai TF, Yee EYW, et al. Efficacy and safety of spesolimab in Asian patients with a generalized pustular psoriasis flare: Results from the randomized, double-blind, placebo-controlled Effisayil™ 1 study. J Dermatol. 2022. https://doi.org/10.1111/1346-8138.16609.
Liu ZJ, Tian YT, Shi BY, Zhou Y, Jia XS. Association between mutation of interleukin 36 receptor antagonist and generalized pustular psoriasis: A PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore). 2020;99(45): e23068.
von dem Borne PA, Jonkman MF, van Doorn R. Complete remission of skin lesions in a patient with subcorneal pustular dermatosis (Sneddon–Wilkinson disease) treated with antimyeloma therapy: association with disappearance of M-protein. Br J Dermatol. 2017;176:1341.
Wang H, Jin H. Update on the aetiology and mechanisms of generalized pustular psoriasis. Eur J Dermatol. 2021;31(5):602–8.
Sugiura K. Role of interleukin 36 in generalised pustular psoriasis and beyond. Dermatol Ther (Heidelb). 2022;12(2):315–28.
Johnston A, Xing X, Wolterink L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140(1):109–20.
Boutet MA, Nerviani A, Pitzalis C. IL-36, IL-37, and IL-38 cytokines in skin and joint inflammation: a comprehensive review of their therapeutic potential. Int J Mol Sci. 2019;20(6):1257.
Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281(1):169–78.
Iznardo H, Puig L. The interleukin-1 family cytokines in psoriasis: pathogenetic role and therapeutic perspectives. Expert Rev Clin Immunol. 2021;17(2):187–99.
Mossner R, Wilsmann-Theis D, Oji V, Gkogkolou P, Lohr S, Schulz P, et al. The genetic basis for most patients with pustular skin disease remains elusive. Br J Dermatol. 2018;178(3):740–8.
Romiti R, Hirayama ALDS, Arnone M, Magalhães RF. Generalized pustular psoriasis (von Zumbusch). An Bras Dermatol. 2022;97(1):63–74.
Frey S, Sticht H, Wilsmann-Theis D, et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol. 2020;140(7):1451-5.e13.
Haskamp S, Bruns H, Hahn M, et al. Myeloperoxidase modulates inflammation in generalized pustular psoriasis and additional rare pustular skin diseases. Am J Hum Genet. 2020;107:527–38.
Vergnano M, Mockenhaupt M, Benzian-Olsson N, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease [published correction in Am J Hum Genet 2021;108:757]. Am J Hum Genet. 2020;107:539–43.
Haskamp S, Frey B, Becker I, et al. Transcriptomes of MPO-deficient patients with generalized pustular psoriasis reveals expansion of CD4+ cytotoxic T cells and an involvement of the complement system. J Invest Dermatol. 2022;142(8):2149-2158.e10.
Burden AD, Choon SE, Gottlieb AB, Navarini AA, Warren RB. Clinical disease measures in generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):39–50.
Choon SE, et al. AAD VMX; April 23–25, 2021; Virtual Meeting. https://www.psoriasiscouncil.org/docs/2021_esdr_congress_report.pdf. Accessed 15 May 2022.
Krueger J, Puig L, Thaçi D. Treatment options and goals for patients with generalized pustular psoriasis. Am J Clin Dermatol. 2022;23(Suppl 1):51–64.
Carrascosa JM, Puig L, Belinchón Romero I, et al. Practical update of the recommendations published by the Psoriasis Group of the Spanish Academy of Dermatology and Venereology (GPs) on the treatment of psoriasis with biologic therapy. Actas Dermo-Sifiliográficas. 2022; 113(3).
Kearns DG, Chat VS, Zang PD, Han G, Wu JJ. Review of treatments for generalized pustular psoriasis. J Dermatol Treat. 2021;32(5):492–4.
Strober B, Kotowsky N, Medeiros R, et al. Unmet medical needs in the treatment and management of generalized pustular psoriasis flares: evidence from a survey of Corrona Registry Dermatologists. Dermatol Ther (Heidelb). 2021;11(2):529–41.
Tamiya H, Fukai K, Moriwaki K, Ishii M. Generalized pustular psoriasis precipitated by topical calcipotriol ointment. Int J Dermatol. 2005;44:791–2.
van de Kerkhof PC. From empirical to pathogenesis-based treatments for psoriasis. J Invest Dermatol. 2022:S0022-202X(22)00082–3.
Baum P, Visvanathan S, Garcet S, et al. Pustular psoriasis: molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol. 2021;S0091–6749(21):01554–62.
Baum P, Visvanathan S, Garcet S, et al. Pustular psoriasis: molecular pathways and effects of spesolimab in generalized pustular psoriasis. J Allergy Clin Immunol. 2022;149(4):1402–12.
Bachelez H, Choon SE, Marrakchi S, et al. Trial of spesolimab for generalized pustular psoriasis. N Engl J Med. 2021;385(26):2431–40.
ClinicalTrials.gov Identifier: NCT03619902. A study to evaluate the efficacy and safety of imsidolimab (ANB019) in adults with generalized pustular psoriasis (GPP). https://clinicaltrials.gov/ct2/show/NCT03619902. Accessed 17 May 2002.
Puig L, Fujita H, Thaçi D, et al. 30th European Academy of Dermatology and Venereology Congress (29 September – 2 October 2021, Virtual). EADV 2021, p. 1055.
Aragón Miguel R, SahuquilloTorralba A, Ara Martin M, et al. Psoriasis pustulosas generalizadas y localizadas: estudio multicéntrico con 119 pacientes. 7º Congreso de Psoriasis, Presencial+Virtual, Madrid, Jan 21–22, 2022.
Hospach T, Glowatzki F, Blankenburg F, et al. Scoping review of biological treatment of deficiency of interleukin-36 receptor antagonist (DITRA) in children and adolescents. Pediatr Rheumatol Online J. 2019;17(1):37.
Miao C, Chen Y, Wang Z, Xiang X, Liu Y, Xu Z. Real-world data on the use of secukinumab and acitretin in pediatric generalized pustular psoriasis. J Dermatol. 2022. https://doi.org/10.1111/1346-8138.16551.
Sussman M, Napodano A, Huang S, Are A, Hsu S, Motaparthi K. Pustular psoriasis and acute generalized exanthematous pustulosis. Medicina (Kaunas). 2021;57(10):1004.
Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73(5):843–8.
Shadi Kourosh A. Subcorneal pustular dermatosis. https://www.uptodate.com/contents/subcorneal-pustular-dermatosis.
Miladi R, Janbakhsh A, Babazadeh A, et al. Pustular psoriasis flare-up in a patient with COVID-19. J Cosmet Dermatol. 2021;20(11):3364–8.
Perna D, Jones J, Schadt CR. Acute generalized pustular psoriasis exacerbated by the COVID-19 vaccine. JAAD Case Rep. 2021;17:1–3.
Elamin S, Hinds F, Tolland J. De novo generalized pustular psoriasis following Oxford-AstraZeneca COVID-19 vaccine. Clin Exp Dermatol. 2022;47(1):153–5.
Shahidi Dadras M, Diab R, Ahadi M, Abdollahimajd F. Generalized pustular psoriasis following COVID-19. Dermatol Ther. 2021;34(1): e14595.
Acknowledgements
Funding
Boehringer Ingelheim supported the publication of this article, including the journal’s Rapid Service Fee.
Medical Writing/Editorial Assistance
Writing and editorial assistance was provided by Content Ed Net (Madrid, Spain) with funding from Boehringer Ingelheim.
Author Contributions
Raquel Rivera-Díaz, Esteban Daudén, José Manuel Carrascosa, Pablo de la Cueva, and Luis Puig have contributed to the conception, design or acquisition of data, or analysis and interpretation of data. All authors have participated in in drafting, reviewing, and/or revising the manuscript and have approved its submission. The authors meet the authorship criteria recommended by the International Committee of Medical Journal Editors (ICMJE) and did not receive payment related to the development of this article.
Disclosures
R. Rivera-Díaz has the following conflict of interests: Advisory Board member, consultant, grants, research support, participation in clinical trials, honorarium for speaking, research support, with the following pharmaceutical companies: AbbVie/Abbott, Almirall, Amgen, Boehringer-Ingelheim, Celgene, Janssen-Cilag, Leo Pharma, Lilly, MSD-Schering-Plough, Novartis, Pfizer, and UCB. E. Daudén has the following conflict of interests: Advisory Board member, consultant, grants, research support, participation in clinical trials, honorarium for speaking, research support, with the following pharmaceutical companies: AbbVie/Abbott, Almirall, Amgen-Celgene, Janssen-Cilag, Leo-Pharma, Novartis, Pfizer, MSD-Schering-Plough, Lilly, UCB, Bristol-Myers and Boehringer-Ingelheim. J.M. Carrascosa has received honoraria or fees as investigator, speaker, and/or advisor from Pfizer, Sanofi, Lilly, AbbVie, Leo-Pharma, Boehringer-Ingelheim and UCB. P. de la Cueva has the following conflict of interests: Consultant, advisory board member, honorary for speaking and participation in clinical trials with the following pharmaceutical companies: AbbVie, Almirall, Astellas, Biogen, Boehringer-Ingelheim, Celgene, Janssen, LEO Pharma, Lilly, MSD, Novartis, Pfizer, Roche, Sanofi and UCB. L. Puig has received consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by AbbVie, Almirall, Amgen, Baxalta, Biogen, Boehringer- Ingelheim, Celgene, Gebro, Janssen, JSC BIOCAD, Leo-Pharma, Lilly, Merck-Serono, MSD, Mylan, Novartis, Pfizer, Regeneron, Roche, Sandoz, Samsung-Bioepis, Sanofi and UCB. Boehringer Ingelheim had the opportunity to review the content of the manuscript for medical and scientific accuracy, as well as intellectual property considerations.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rivera-Díaz, R., Daudén, E., Carrascosa, J.M. et al. Generalized Pustular Psoriasis: A Review on Clinical Characteristics, Diagnosis, and Treatment. Dermatol Ther (Heidelb) 13, 673–688 (2023). https://doi.org/10.1007/s13555-022-00881-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00881-0