
Abstract
Generalized pustular psoriasis (GPP) is a rare disease that has only recently benefited from a consistent definition and clinical coding standard. A lack of disease awareness combined with clinical similarities to other types of psoriasis have historically complicated the diagnosis of GPP. It is now clear that GPP requires a differential diagnosis from psoriasis vulgaris (plaque psoriasis), and better understanding of the genetic characteristics underlying GPP may improve the accuracy of diagnoses in the future. GPP can present at any age but is most common in the fifth decade of life. There appears to be a female preponderance in GPP, although there is notable variability in prevalence by geographical region and between ethnicities. GPP is potentially life-threatening, associated with several serious complications, and may require emergency treatment, particularly for complications arising from systemic inflammation. As with many rare diseases, there are inherent challenges to understanding the epidemiology of GPP. In addition to small patient numbers, estimating the prevalence of rare diseases is further complicated by studies that use non-standardized methodologies and that are conducted in different populations. These complications in data gathering have led to marked variability in GPP case estimates by geographical region and between ethnicities. There is ongoing research into disease characteristics, and insights into regional measures of prevalence are essential to increasing our understanding of GPP.
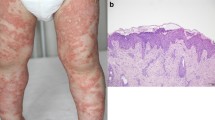
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.16823503. |
Generalized pustular psoriasis (GPP) is a rare and potentially life-threatening disease. While it shares some signs and symptoms with other, less serious, forms of psoriasis, GPP is a separate condition and requires an accurate diagnosis, which should lead to distinct treatment and management approaches. |
Estimating the number of individuals who are affected by GPP is challenging because rare diseases are often misdiagnosed and patient numbers are small. Several claims database analyses have been conducted that might increase our understanding of the prevalence of GPP. |
Medical claims database analyses can provide up-to-date insights into various aspects of GPP, including disease characteristics and estimates of disease prevalence in defined cohorts. |
1 Introduction
Over the past two decades, knowledge of skin pathologies, particularly psoriasis, has increased markedly [1]. Psoriasis is categorized into several distinct subtypes: plaque (or psoriasis vulgaris, which accounts for around 90% of cases), guttate, inverse, erythrodermic, and pustular psoriasis. Pustular psoriasis is subclassified into generalized pustular psoriasis (GPP) and localized pustular psoriasis (palmoplantar pustulosis and acrodermatitis continua of Hallopeau). Erythrodermic psoriasis can develop alongside any other psoriasis subtype, presenting as erythematous inflamed skin that covers over 90% of the total body surface area [1].
GPP was first described by Leopold von Zumbusch in 1910. However, since its discovery, the disease characteristics have not been consistently defined and several descriptions and diagnostic criteria have been reported in the literature [2]. Following recent international consensus by the European Rare and Severe Psoriasis Expert Network (ERASPEN), GPP is defined as primary, sterile, visible pustules on non-acral skin (excluding cases in which pustulation is restricted to psoriatic plaques). GPP can occur with or without systemic inflammation and with or without psoriasis vulgaris, and can be relapsing (more than one episode) or persistent (> 3 months) [3].
Despite the heterogeneity of rare diseases, there are common barriers to achieving accurate disease prevalence estimates. A lack of widespread awareness and accurate diagnoses, combined with research approaches that use very different data sources and non-standardized methodologies, mean that gathering insights into disease prevalence at a regional, national, or global level is challenging [4]. Recognition of GPP as a rare disease is increasing globally; however, describing the epidemiology of GPP is complicated by the challenges associated with studying rare diseases.
In this first chapter of the supplement titled 'Understanding Generalized Pustular Psoriasis to Improve Patient Outcomes', the characteristics of GPP and key differentiators between GPP and plaque psoriasis are considered, alongside current epidemiology estimates.
2 Disease Characteristics of GPP
2.1 Describing GPP
The clinical course of GPP is heterogeneous; it can be considered a relapsing disease with recurrent flares, or a persistent disease with intermittent flares [3, 5]. Disease flares manifest as the sudden appearance of large areas of painful, non-infectious pustules, which may co-present with fever, chills, and loss of appetite [6, 7]. Symptom severity may vary with each flare for a given individual, and flares may occur several times per year or with long dormant periods between each episode [6, 8].
GPP has been subcategorized into four forms based on the onset of flares and the morphology of lesions [9]. GPP (also called GPP of von Zumbusch) presents with rapid onset (7 days or fewer) of a generalized pustular flare and may be experienced by up to 90% of patients with GPP [9, 10]; it is recognized as the most severe presentation of the disease [11]. Annular GPP presents with a generalized pustular flare that develops between 7 days and 3 months, and is often associated with relatively mild symptoms [2, 12]. Chronic acral GPP presents with the onset of a generalized pustular flare over more than 3 months, with lesions that begin with an acral distribution in the extremities and then gradually spread into a generalized pustular flare [9]. The ‘mixed GPP’ category captures patients who have features that are associated with more than one subtype, with localized pustular psoriasis that develops into a generalized pustular flare over a range of onset periods [9].
GPP can manifest with or without concomitant plaque psoriasis [7]. It is important to recognize that GPP is a distinct clinical entity from plaque psoriasis and that a differential diagnosis is critical [6, 11, 13]. A diagnosis of GPP should be considered in patients with sudden onset of erythema and pustulosis, and emergency care may be required in patients with fever or severe pain, or with signs of systemic inflammation that may lead to respiratory, cardiac, or renal failure [2, 11]. GPP should be distinguished from the transient, primary pustule formation that may occur at the periphery of plaques in patients with psoriasis vulgaris [2, 3]. See the article on Diagnosis of Generalized Pustular Psoriasis (https://doi.org/10.1007/s40257-021-00652-1).
2.2 Common Disease Characteristics
Several characteristics of GPP have become apparent from published case reports. It has been suggested that there may be a female preponderance in GPP; however, the extent of this varies by geographical region and between ethnicities. A study in 102 patients from Malaysia reported a 2:1 ratio of GPP in females versus males; a study of 74 patients in the US reported a 1.03:1 ratio; and in a study of over 700 patients in Japan, 51.5% were female [9, 10, 14]. Findings from a recent inpatient database of 1516 people with GPP in Japan reported that 56% were male, and a registry in Western Japan observed that 52% of 102 GPP cases were male [15, 16]. GPP can present at any age, including as juvenile GPP [8]; however, the majority of cases present in the fifth decade and the mean age at diagnosis has been reported as 45.6–50.0 years [5, 9, 10]. It has been suggested that females may be diagnosed earlier (mean age 39.4 years) than males [17]. The prognosis of GPP in older patients may be poorer than in younger patients due to the systemic complications of the disease, including cardiorespiratory failure and risk of infection and sepsis [2, 6].
2.3 Genetic Markers in GPP
The etiology of GPP is not fully understood; however, the interleukin (IL)-36 pathway appears to be pivotal in the pathogenesis of the disease. The potential role of IL-36 has been supported by the identification of loss-of-function mutations in the IL-36 receptor antagonist gene (IL36RN) and the overexpression of IL-36 cytokines in GPP skin lesions [18]. The IL36RN gene is responsible for suppressing proinflammatory responses triggered by IL-36α, IL-36β, and IL-36γ via the encoding of a functional IL-36Ra protein [6]. In addition, variants of CARD14, which encodes a keratinocyte adaptor protein, and AP1S3, which encodes a subunit of the adaptor protein 1 complex, have also been implicated in GPP [2, 19]. CARD14 variants are relatively rare, with some regional variability. A recent review of published evidence on the genetic basis of GPP noted that CARD14 variations rarely present in patients with GPP alone and are more commonly found in cases of GPP with concomitant psoriasis vulgaris; however, CARD14 variations specific to either GPP or psoriasis vulgaris have not yet been characterized [20]. In a recent variant-screening study conducted in 863 patients with pustular psoriasis, no CARD14 substitutions were observed among European patients with GPP, but were present in Chinese patients [19]. A CARD14 variant was identified in Japanese patients with both GPP and psoriasis vulgaris, but was not associated with psoriasis vulgaris alone, and was not reported in patients with GPP alone, in this population [21].
AP1S3 variants have been reported consistently across pustular psoriasis subtypes. Over 95% of patients with variations in this gene were female, indicating that sex-specific factors may alter the penetrance of AP1S3 variants [19].
Recently, loss-of-function variants of myeloperoxidase gene (MPO) that are associated with increased neutrophil accumulation and activity have been reported in some patients with GPP [22]. A rare loss-of-function variant in SERPINA3, which encodes serine protease inhibitor A3 (serpin A3), has also been identified in patients with GPP [23].
Loss-of-function variants of IL36RN have been identified in 23–37% of familial and sporadic GPP cases [18, 24]. The frequency of IL36RN variants shows regional differences, from 28.8% in Malaysian patients to 34.7% in European patients with GPP, and 46.8–60.5% in Han Chinese patients; one study noted that IL36RN may be the major gene associated with the pathogenesis of GPP in the Han population in China [5, 25]. Further analysis in the Chinese population suggested that the mechanisms of IL-36 overexpression differ between patients with GPP and those with psoriasis vulgaris, which may explain previous observations of a lack of association between IL36RN variation and the development of psoriasis vulgaris [13]. In patients with homozygous IL36RN variants, the age of onset of GPP can be earlier than usual, and IL36RN disease alleles have a dose-dependent effect on age of onset in all pustular psoriasis subtypes [19].
While IL36RN and CARD14 variations are implicated in the pathogenesis of GPP, clinical experience of patients presenting with both mutations simultaneously is limited. In 2019, the first published case described a patient with heterozygous IL36RN variation (compared with typical homozygous or combined heterozygous variation in GPP), with the authors suggesting that coexisting mutations in IL36RN and CARD14 may also predispose individuals to GPP [26].
Screening of IL36RN, CARD14, and AP1S3 is not routinely indicated; however, IL36RN status is becoming increasingly recognized as a useful tool in the diagnosis of GPP [2].
2.4 GPP in Pregnancy
Impetigo herpetiformis, or GPP during pregnancy, has a typical onset in the last trimester, and while the etiology is unclear, associations with hypocalcemia and hypoparathyroidism have been suggested in some cases [27]. Pregnancy has been identified as a precipitating factor for flares in patients with GPP [10], and GPP in pregnancy is associated with severe outcomes, including placental insufficiency leading to an increased risk of stillbirth, neonatal death, and fetal abnormalities [28].
3 GPP and Plaque Psoriasis: Contrasting Conditions with Key Differentiators
GPP is clinically, phenotypically, and genetically different from plaque psoriasis and requires a differential diagnosis [5,6,7]. This is important because GPP flares are potentially life-threatening and may require immediate treatment [2]. In practice, distinguishing the two diseases is challenging because of the relatively low familiarity with GPP among physicians and the apparent similarities between GPP and plaque psoriasis, especially when pustules develop at plaque sites. Since GPP presents in over two-thirds of individuals with history of or existing plaque psoriasis, it is often incorrectly categorized as a form of plaque psoriasis [6]. Previous studies have suggested that around 1% of people with psoriasis have GPP; however, these estimates were based on unclear definitions of GPP.
While there may be morphological similarities at presentation, several key factors exist to differentiate GPP from plaque psoriasis. As a first principle, it might be considered that GPP is an autoinflammatory disease [6] and that plaque psoriasis is an autoimmune disease [29]. Genetic studies suggest that GPP and plaque psoriasis have immunologic pathways that both overlap and are independent from one another [30]. A key difference is that IL36RN variants that drive proinflammatory responses are not as common in plaque psoriasis (36.8–37.8%) as they are in GPP (70.6–79.2%) [13, 25]. This important distinction could play a key role in GPP differential diagnosis and screening.
Edema is notably greater in GPP than in plaque psoriasis, in which it is relatively rare, and inflammatory cell infiltrate is also more prominent in GPP [2, 8]. Plaque psoriasis characteristically presents as acanthosis, parakeratosis, and dermal inflammatory infiltrates. In contrast, in GPP, acanthotic changes are accompanied by epidermal neutrophilic infiltrates and neutrophil accumulation beneath the stratum corneum, which cause pustule formation [6, 8]. Such Kogoj’s spongiform pustules may not be present in plaque psoriasis lesions, although they may be observed histologically [8].
GPP does not feature the transient primary pustule formation at the periphery of plaques as often seen during exacerbation or irritation of plaque psoriasis [2]. In addition, flare recurrence is a hallmark of GPP, which may not be seen as consistently in plaque psoriasis [2, 7]. There are also several common laboratory anomalies that may differentiate GPP from plaque psoriasis, including leukocytosis with neutrophilia and elevated erythrocyte sedimentation rate, as well as increased alkaline phosphatase, transaminase, and bilirubin levels [2, 7, 8]. The Japanese guidelines for the management and treatment of GPP note that fever, C-reactive protein level, hypoalbuminemia, and white blood cell count are important in the diagnosis, and the grading of severity, of GPP [7]. Concurrent increased alkaline phosphatase, transaminase, and bilirubin levels are linked to neutrophilic cholangitis associated with the flare’s skin eruption [31]; neutrophilic cholangitis is not a common feature of plaque psoriasis, and thus may support a diagnosis of GPP.
The female preponderance in GPP is not apparent in plaque psoriasis, in which equal numbers of males and females are affected.
4 Challenges in Determining the Epidemiology of GPP
Since GPP is a rare disease, clinical awareness is limited and inaccurate diagnosis presents challenges to estimating disease prevalence. Considering the available evidence, GPP is understood to affect all populations, but prevalence varies by geographical region and between ethnicities.
There is a large degree of global and regional variability in the prevalence of GPP in the published literature. Interpreting the available data is challenging because of the different methodologies, populations, and definitions of GPP used across studies, as well as the inclusion, or lack of differentiation from, palmoplantar pustulosis. In many cases, data from hospital audits or claims database analyses have not been extrapolated to reflect national population-level prevalence estimates. This means caution must be used in drawing wide-ranging or comparative conclusions of prevalence estimates from the available studies. Over the past two decades, several initiatives that provide national population-level insights into the epidemiology of GPP have been published (Table 1) [32,33,34].
In France, a study conducted in 2004 using a questionnaire-based assessment in 121 hospital dermatology wards showed 99 cases of GPP across 46 wards, leading to an estimated disease occurrence of 1.76 cases per 1,000,000 persons [32]. The authors noted this estimate was similar to earlier findings in 1998 and 2001 from the Caisse Nationale d'Assurance Maladie des Travailleurs Salariés (CNAMTS) French health insurance database [32].
Prevalence data in another study were obtained using questionnaires sent to 575 community center hospitals throughout Japan that captured details of visiting patients with GPP from 1983–1989. The study included 541 patients with GPP and reported a GPP case estimate of 7.46 patients per million, with 2.87 patients per million presenting with acute GPP [33]. A recent analysis of the Japanese national database of health insurance claims suggests this figure could be higher. The database covers approximately 90% of Japanese residents and recorded 4636 codings of pustular psoriasis, which would indicate over 30 cases per million people [35].
A review of a national insurance claims database in the Republic of Korea conducted between 2011 and 2015 estimated a prevalence of 1.2 cases per 10,000 persons. In patients with psoriasis (range 219,429–233,909 across study years), 2.0–2.7% were categorized as having GPP. The authors equated prevalence in the database to national prevalence because of compulsory enrollment in the insurance database for all residents of the Republic of Korea [34].
A US insurance claims review was conducted between 2016 and 2019 using the IQVIA PharMetrics® Plus database. Patients were required to be continuously enrolled in the medical and pharmacy benefits for 6 months before and at least 2 months after the index date (date of diagnosis). This database analysis identified 990 patients with GPP over the study period [36].
It should be noted that the large variations reported across the published studies do not necessarily transcribe an epidemiological reality, and that available data should perhaps be considered as indicative of prevalence only. The variability is likely driven by challenges related to the study of a rare and relatively unknown disease, as well as differences in the distribution of genetic variants and cofactors in separate populations, and reported estimates represent a range that must be interpreted while considering methodological differences.
5 Adding Evidence to the Understanding of GPP Disease Characteristics and Prevalence
To date, GPP has been described by a range of clinical definitions, which may have hindered accurate collection of prevalence data. These include the classification of GPP as a variant of plaque psoriasis, given the frequent comorbidity of this type of psoriasis in patients with GPP [6]. The introduction of a specific GPP diagnostic code (code L40.1) into the International Classification of Diseases, 10th Revision (ICD-10) in 2015 may aid accurate recording of cases and allow more precise estimates of disease prevalence. It is hoped that the increased awareness of GPP and better differential diagnosis, coupled with the availability of specific pustular psoriasis subtype codes, will drive more accurate prevalence estimates in the future.
As described, the considerable interstudy variation in methodologies and populations means that physicians have an incomplete view of the epidemiology of GPP. It is likely that determining national population-level prevalence estimates will remain challenging for some time. However, database assessments can provide specific insights into certain aspects of GPP, including regional variations, and there is value to the pustular psoriasis community in continuing to generate insights into aspects of GPP epidemiology. While database studies cannot act as proxies for accurate estimates of disease prevalence in the wider population, they can contribute to disease understanding.
In Japan, data were collected for 718 patients with GPP and 27,773 patients with plaque psoriasis between January 2015 and December 2019 using the Japanese Medical Data Vision database. There was a slight female preponderance in patients with GPP (51.5%) compared with those with plaque psoriasis, in which a minority of patients were female (38.7%). Patients with GPP were more likely to have comorbidities than patients with plaque psoriasis; these comorbidities included hypertension, peptic ulcer disease, osteoporosis, type 2 diabetes, gout, psoriatic arthritis, chronic obstructive pulmonary disease, obesity, asthma, insomnia, interstitial pneumonia, thyroid disorders, and other forms of psoriasis [14]. These findings suggested that patients with GPP in Japan had a higher disease burden than those with psoriasis vulgaris [37]. However, it is important to consider that some comorbidities may be related to lifestyle in any given country and will not necessarily be specific to GPP or be generalizable to other geographical regions or ethnicities [10].
6 Conclusions
Since GPP is a rare disease with limited clinical awareness, it is associated with several challenges, including consistent descriptions of the disease characteristics and accurate prevalence estimates. The characteristics of GPP, previously considered to be idiopathic, are gradually being elucidated, and this is aided by increased understanding of genetic variants that contribute to the development of pustular psoriasis, particularly GPP. Although GPP can present with plaque psoriasis, the two diseases are clinically and genetically distinct and require a differential diagnosis and distinct management approaches. Key morphological, disposition, and histological differences between GPP and plaque psoriasis are emerging, as well as genetic markers that could aid disease screening in the future.
As with many rare diseases, forming a clear picture of the clinical characteristics and disease prevalence is challenging. Key barriers include limited disease awareness and the different approaches to assessing prevalence, which are often performed using database studies or small hospital audits. In addition, the data gained from these initiatives are not commonly extrapolated to a national population-level prevalence. Despite the inherent limitations of medical database analyses, these studies can contribute up-to-date insights into various aspects of GPP, including disease characteristics and prevalence in defined cohorts.
References
Rendon A, Schakel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. 2019;20(6):1475.
Ly K, Beck KM, Smith MP, Thibodeaux Q, Bhutani T. Diagnosis and screening of patients with generalized pustular psoriasis. Psoriasis (Auckl). 2019;9:37–42.
Navarini AA, Burden AD, Capon F, Mrowietz U, Puig L, Koks S, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11):1792–9.
Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28(2):165–73.
Kharawala S, Golembesky AK, Bohn RL, Esser D. The clinical, humanistic, and economic burden of generalized pustular psoriasis: a structured review. Expert Rev Clin Immunol. 2020;16(3):239–52.
Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–19.
Fujita H, Terui T, Hayama K, Akiyama M, Ikeda S, Mabuchi T, et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11):1235–70.
Umezawa Y, Ozawa A, Kawasima T, Shimizu H, Terui T, Tagami H, et al. Therapeutic guidelines for the treatment of generalized pustular psoriasis (GPP) based on a proposed classification of disease severity. Arch Dermatol Res. 2003;295(Suppl 1):S43–54.
Zelickson BD, Muller SA. Generalized pustular psoriasis. A review of 63 cases. Arch Dermatol. 1991;127(9):1339–45.
Choon SE, Lai NM, Mohammad NA, Nanu NM, Tey KE, Chew SF. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: analysis of 102 cases seen in a tertiary hospital in Johor. Malaysia Int J Dermatol. 2014;53(6):676–84.
Crowley JJ, Pariser DM, Yamauchi PS. A brief guide to pustular psoriasis for primary care providers. Postgrad Med. 2021;133(3):330–44.
Naik HB, Cowen EW. Autoinflammatory pustular neutrophilic diseases. Dermatol Clin. 2013;31(3):405–25.
Li X, Chen M, Fu X, Zhang Q, Wang Z, Yu G, et al. Mutation analysis of the IL36RN gene in Chinese patients with generalized pustular psoriasis with/without psoriasis vulgaris. J Dermatol Sci. 2014;76(2):132–8.
Morita A, Kotowsky N, Gao R, Shimizu R, Okubo Y. Patient characteristics and burden of disease in Japanese patients with generalized pustular psoriasis: results from the Medical Data Vision claims database. J Dermatol. 2021;48(10):1463–73.
Miyachi H, Konishi T, Kumazawa R, Matsui H, Shimizu S, Fushimi K, et al. Treatments and outcomes of generalized pustular psoriasis: a cohort of 1516 patients in a nationwide inpatient database in Japan. J Am Acad Dermatol. 2021. https://doi.org/10.1016/j.jaad.2021.06.008 (Epub 8 Jun 2021).
Ohata C, Tsuruta N, Yonekura K, Higashi Y, Saito K, Katayama E, et al. Clinical characteristics of Japanese pustular psoriasis: a multicenter observational study. J Dermatol. 2021. https://doi.org/10.1111/1346-8138.16217 (Epub 1 Nov 2021).
Benjegerdes KE, Hyde K, Kivelevitch D, Mansouri B. Pustular psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:131–44.
Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365(7):620–8.
Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. 2019;143(3):1021–6.
Zhou J, Luo Q, Cheng Y, Wen X, Liu J. An update on genetic basis of generalized pustular psoriasis (Review). Int J Mol Med. 2021;47(6):118.
Sugiura K, Muto M, Akiyama M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J Invest Dermatol. 2014;134(6):1755–7.
Vergnano M, Mockenhaupt M, Benzian-Olsson N, Paulmann M, Grys K, Mahil SK, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet. 2020;107(3):539–43.
Frey S, Sticht H, Wilsmann-Theis D, Gerschutz A, Wolf K, Lohr S, et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol. 2020;140(7):1451–5.e13.
Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89(3):432–7.
Li Z, Yang Q, Wang S. Genetic polymorphism of IL36RN in Han patients with generalized pustular psoriasis in Sichuan region of China: a case-control study. Medicine (Baltimore). 2018;97(31):e11741.
Sawabe Y, Hayashi K, Kamata M, Nagata M, Fukaya S, Fukuyasu A, et al. Case of generalized pustular psoriasis with coexisting mutations in IL36RN and CARD14. J Dermatol. 2019;46(10):e368–70.
Kondo RN, Araujo FM, Pereira AM, Lopes VC, Martins LM. Pustular psoriasis of pregnancy (impetigo herpetiformis)—case report. An Bras Dermatol. 2013;88(6 Suppl 1):186–9.
Jones S, Black M. Obstetric and Gynecologic Dermatology (Third Edition): Chapter 10 – Effect of Pregnancy on Other Skin Disorders; 2008.
Takeichi T, Akiyama M. Generalized pustular psoriasis: clinical management and update on autoinflammatory aspects. Am J Clin Dermatol. 2020;21(2):227–36.
Boehner A, Navarini AA, Eyerich K. Generalized pustular psoriasis—a model disease for specific targeted immunotherapy, systematic review. Exp Dermatol. 2018;27(10):1067–77.
Viguier M, Allez M, Zagdanski AM, Bertheau P, de Kerviler E, Rybojad M, et al. High frequency of cholestasis in generalized pustular psoriasis: evidence for neutrophilic involvement of the biliary tract. Hepatology. 2004;40(2):452–8.
Augey F, Renaudier P, Nicolas JF. Generalized pustular psoriasis (Zumbusch): a French epidemiological survey. Eur J Dermatol. 2006;16(6):669–73.
Ohkawara A, Yasuda H, Kobayashi H, Inaba Y, Ogawa H, Hashimoto I, et al. Generalized pustular psoriasis in Japan: two distinct groups formed by differences in symptoms and genetic background. Acta Derm Venereol. 1996;76(1):68–71.
Lee JY, Kang S, Park JS, Jo SJ. Prevalence of psoriasis in Korea: a population-based epidemiological study using the Korean National Health Insurance Database. Ann Dermatol. 2017;29(6):761–7.
Kubota K, Kamijima Y, Sato T, Ooba N, Koide D, Iizuka H, et al. Epidemiology of psoriasis and palmoplantar pustulosis: a nationwide study using the Japanese national claims database. BMJ Open. 2015;5(1):e006450.
Hanna ML, Singer D, Valdecantos WC. Economic burden of generalized pustular psoriasis and palmoplantar pustulosis in the United States. Curr Med Res Opin. 2021;37(5):735–42.
Okubo Y, Kotowsky N, Gao R, Saito K, Morita A. Clinical characteristics and health-care resource utilization in patients with generalized pustular psoriasis using real-world evidence from the Japanese Medical Data Center database. J Dermatol. 2021;48(11):1675–87.
Acknowledgements
All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE) and made the decision to submit the manuscript for publication. The authors did not receive payment related to the development of the manuscript. Agreements between Boehringer Ingelheim and the authors included the confidentiality of the study data. Colin Griffin, PhD, of OPEN Health Communications (London, UK) provided medical writing, editorial and/or formatting support, which was contracted and funded by Boehringer Ingelheim. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure statement
This article has been published as part of a journal supplement wholly funded by Boehringer Ingelheim.
Funding
Medical writing support was funded by Boehringer Ingelheim.
Conflict of interest
Kilian Eyerich has received consulting fees from AbbVie, Almirall, Bristol Myers Squibb, LEO Pharma, Janssen, Lilly, Novartis, UCB, Pfizer, and Boehringer Ingelheim; grants and/or contracts from AbbVie, LEO Pharma, UCB, and Lilly; and speaking fees from AbbVie, LEO Pharma, Janssen, and Lilly. Kilian Eyerich also declares stock or stock options with Dermagnostix and Dermagnostix R&D. Min Zheng has received consulting fees from AbbVie, Boehringer Ingelheim, Janssen-Cilag, LEO Pharma China, Novartis, Pfizer, and Xian-Janssen. Denis Jullien has received consulting fees from AbbVie, Almirall, Amgen, Biogen, Boehringer Ingelheim, Celgene, Fresenius Kabi, Janssen-Cilag, LEO Pharma, Lilly, MSD, MEDAC, Novartis, Pfizer, UCB, and Sanofi; payment for speakers’ bureaus from AbbVie, Celgene, Janssen-Cilag, LEO Pharma, Lilly, MEDAC, Novartis, and Pfizer; and travel/accommodation expenses reimbursed by AbbVie, Amgen, Biogen, Celgene, Fresenius Kabi, Janssen-Cilag, LEO Pharma, Lilly, MSD, MEDAC, Novartis, Pfizer, UCB, and Sanofi.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
The sponsor of the supplement and all authors identified and discussed the concept for each chapter and the supplement as a whole; further development of each review article was at the direction of the authors, who provided guidance to OPEN Health Communications on the content of the article, critically revised the work, and approved the content for publication.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zheng, M., Jullien, D. & Eyerich, K. The Prevalence and Disease Characteristics of Generalized Pustular Psoriasis. Am J Clin Dermatol 23 (Suppl 1), 5–12 (2022). https://doi.org/10.1007/s40257-021-00664-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-021-00664-x