
Abstract
Generalized pustular psoriasis (GPP) is a rare, severe form of pustular psoriasis characterized by widespread, recurrent episodes of neutrophil-rich pustule formation in the epidermis, which can be accompanied by fever and systemic inflammation. Recent clinical, histologic, and genetic evidence indicates that GPP is a distinct entity from plaque psoriasis, with different cytokine pathways predominant in the manifestation of each disease. The interleukin-36 (IL-36) signaling cascade plays a key role in regulating the innate immune system, and its dysregulation appears central to the pathogenesis of GPP. The altered expression of various IL-36 pathway constituents has been shown to cause a positive feedback loop of uncontrolled signaling and excess production of inflammatory cytokines, which in turn leads to chemokine induction and neutrophil recruitment in the epidermis. Given the potentially life-threatening nature of GPP episodes, drug interventions that rapidly achieve disease resolution are required. Early phase data indicate that treatments targeting various components of the IL-36 inflammatory cascade represent promising areas of research. However, there are currently no therapeutic agents specifically approved for GPP in the USA or Europe. Understanding the inflammatory pathways, associated risk factors, and role of neutrophils in the manifestation and perpetuation of GPP flares remains a key goal in developing effective therapeutics. In this article, we summarize the current understanding of GPP, describe novel therapeutic opportunities, and detail how the unique pathophysiology of the disease may inform future treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.16823503. |
Generalized pustular psoriasis (GPP) is a rare, neutrophilic skin disease characterized by sudden episodes of widespread rash and sterile pustules. |
While GPP can present with pre-existing plaque psoriasis, it is now recognized as a separate clinical entity with clear distinctions in genetic and immunologic determinants and response to treatment. |
Identifying a key role for the interleukin-36 immune signaling pathway in the pathogenesis of GPP has paved the way for the development of new therapies. |
1 Introduction
There are several subtypes of psoriasis, the most common of which is plaque psoriasis, which is characterized by scaly, sharply demarcated plaques affecting the extensor surfaces [1]. Pustular psoriasis, a rarer form of psoriasis, represents approximately 1% of all clinical cases; it is an immune-mediated systemic skin disorder, distinguished by sterile, neutrophil-rich pustules with a variety of distribution patterns [2]. Pustular psoriasis can be further divided into specific subtypes based on the clinical presentation and location of the pustules; it can be classified as either localized, comprising palmoplantar pustulosis (PPP) and acrodermatitis continua of Hallopeau, or generalized, comprising generalized pustular psoriasis (GPP), pustular psoriasis of pregnancy, and infantile/juvenile pustular psoriasis [1].
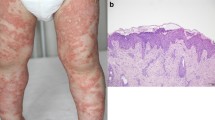
The pathogenesis of GPP appears to be driven by a combination of select genetic loci, predominantly inflammatory cytokine signaling constituents, and environmental risk factors, including viral infections, medications, and discontinuation of corticosteroids [3]. Histologic examination of GPP lesions reveals parakeratosis, substantial mononuclear and neutrophilic infiltration into the epidermis, and epidermal edema and hyperplasia [4, 5]. Spongiform pustules of Kogoj, hyperplasia of the suprapapillary capillaries, and Munro’s microabscesses typical of plaque psoriasis are also seen. Presentation of GPP may occur in parallel with plaque psoriasis, which can impede accurate clinical identification [4,5,6].
Greater understanding of the pathophysiology of GPP is required to develop effective, targeted, disease-specific therapies. In this article, we summarize current understanding of GPP and describe how its unique pathophysiology may inform future treatment strategies.
2 Immunopathology of GPP
GPP is a rare, neutrophilic skin disease characterized by sudden episodes of widespread rash and sterile pustules that can occur with or without systemic inflammation, as denoted by fever, leukocytosis, and elevated C-reactive protein levels [7]. A key characteristic of GPP is repeated episodes of generalized sterile pustule formation caused by extensive neutrophilic and mononuclear inflammatory infiltrates in the epidermis [5, 8]. Severe flares typically require intensive hospital treatment [9, 10].
GPP may present with pre-existing plaque psoriasis, and closely interlinked immunologic pathways appear to underpin the pathogenesis of both conditions [11, 12]. However, GPP has also been shown to present independently and is now recognized as a clinical entity separate from plaque psoriasis, with clear distinctions in genetic and immunologic determinants as well as in response to treatment [13, 14]. Understanding these distinctions is key to the development of effective, targeted treatments for this disease. GPP is predominantly characterized by innate immune inflammation and is considered an autoinflammatory pustular neutrophilic disease. In this respect, GPP is considered representative of autoinflammatory keratinization diseases, which are characterized by inflammation in the epidermis, hyperkeratosis, and primary genetic causative factors associated with the hyperactivation of innate immunity (autoinflammation) [15]. By contrast, plaque psoriasis has both innate and adaptive immunopathogenic responses and is considered an autoimmune condition [16, 17]. Studies have identified different cytokine pathways that are predominant in the manifestation of each disease. For example, while the interleukin (IL)-23/IL-17 axis appears to drive plaque psoriasis [18, 19], a growing body of evidence implicates the IL-36 pathway as central to the development of GPP [16, 20].
IL-36 cytokines belong to the IL-1 family and are expressed by and act upon various cell types, including keratinocytes, epithelial cells, and immune cells, in an autocrine or paracrine manner [21]. With a key role in regulating the innate immune system, the uncontrolled expression and activation of IL-36 cytokines can lead to self-perpetuating inflammatory cascades [22, 23]. IL-36 signaling occurs via a complex of IL-36 receptor (IL-36R) and IL-1R accessory protein, propagating inflammatory responses in the epithelium. Dysregulation of this signaling pathway appears central to the immunopathogenesis of GPP [23, 24] (Fig. 1). Overexpression of IL-36 agonists (IL-36α, β, or γ) or expression of a dysfunctional IL-36R antagonist (IL-36RA), encoded by IL36RN, can lead to a positive feedback loop of uncontrolled signaling and excess production of inflammatory cytokines [20, 25]. This in turn leads to the induction of chemokines such as CXCL1 and CXCL8 (IL-8), producing a chemokine gradient that attracts a high number of neutrophils into the epidermis [20]. Here, spongiform pustules of Kogoj and sub-corneal accumulation of neutrophils manifest as “lakes of pus,” which are characteristic of patients with GPP [3, 7, 16] (Fig. 1).

IL-36 autocrine and autoinflammatory circuits. Keratinocytes are the major source of IL-36 in the skin. IL-36 cytokines are secreted from keratinocytes as precursors that require processing by neutrophil-derived proteases. Upon protease cleavage, mature IL-36 agonists have > 500-fold biological activity and bind to IL-36R on the surface of keratinocytes, inducing an inflammatory cascade that promotes IL-36 expression. IL-36 cytokines also induce the expression of numerous cytokines (IL-1β, IL-17A, IL-23, and TNF-α), neutrophilic chemokines (CXCL1, CXCL2, and CXCL8), and lymphokines, which further propagate this pro-inflammatory cycle [23, 24]. AP-1 activating protein-1, CARD caspase recruitment domain, CCL chemokine (C–C motif) ligand, CXCL chemokine (C–X–C motif) ligand, IL interleukin, MAPK mitogen-activated protein kinase, mRNA messenger RNA, NET neutrophil extracellular trap, NFκB nuclear factor kappa-light-chain-enhancer of activated B cells, R receptor RA receptor antagonist, RAcP receptor accessory protein, TNF tumor necrosis factor
The video (available online) gives an overview of IL-36 autocrine and autoinflammatory circuits (MP4 29712 KB)
.
Further evidence on the differences in immunopathologic determinants driving GPP and plaque psoriasis pathogenesis comes from gene expression analyses. Compared with healthy controls, levels of tumor necrosis factor-α, IL-1, IL-17A, and IL-36 are increased in skin biopsy samples from patients with GPP or plaque psoriasis; however, GPP lesions were shown to have higher levels of IL-1 and IL-36 and lower levels of IL-17A and interferon-γ compared with plaque psoriasis lesions [16]. Immunohistochemical analysis also revealed that IL-36 expression localizes to keratinocytes that surround the neutrophilic pustules in GPP [16]. Furthermore, GPP lesions contain higher levels of neutrophilic chemokines and neutrophil and monocyte transcripts compared with plaque psoriasis lesions [16]. It is important to note that IL-36 and IL-23 cytokine pathways closely interact and may crosstalk extensively, and dysregulation of either pathway is capable of perpetuating an inflammatory response [19].
In addition to histologic analyses, the central role of the IL-36 pathway in GPP is supported by the identification of associated mutations in patients with the disease. To date, IL36RN mutations appear to be the main determinant of pathology in individuals whose disease has a recognized genetic component [26]. These genetic mutations induce a response pathway whereby IL-36R-activating ligands (IL-36α, β, and γ) are not regulated by IL-36RA, leading to self-amplifying IL-36 production, notably in highly differentiated epidermal keratinocytes [27] (Fig. 1). IL36RN mutations leading to an aberrant IL-36RA with decreased affinity for its receptor were discovered in a study of Tunisian families with a severe form of GPP (with some phenotypical variation even in members of one given family) known as deficiency of IL-36R antagonist (DITRA) [25]. In an international cohort, 7.7% of patients with GPP were heterozygous and 21% had biallelic mutations in IL36RN [28], whereas in a study of Chinese patients, IL36RN mutations were identified in 75% of patients with GPP [29, 30]. Therefore, the occurrence of IL36RN mutations may differ by ethnicity, indicating the likelihood of genetic diversity in the pathophysiology of GPP. GPP may often present with existing or prior plaque psoriasis but can also arise in patients with no history of plaque psoriasis [7]. Evidence that GPP alone is a distinct subtype of psoriasis, with its own etiology, comes from a study in Japanese patients in which IL36RN mutations were observed in a much higher proportion of patients with GPP alone compared with those presenting with GPP and plaque psoriasis [30]. IL36RN mutations are also associated with an earlier age of onset and more severe GPP [26]; furthermore, the onset of GPP has been found to be substantially delayed in individuals with monoallelic compared with biallelic IL36RN mutations, suggesting a gene dosage effect [28].
Despite the strong correlation between IL36RN mutations and GPP, not all patients with GPP have evidence of mutations in this gene [31]. The pathogenesis of GPP also appears to be mediated by transcripts of alternative genetic mutations associated with the IL-36-mediated inflammatory cascade [20]. Gain-of-function mutations in CARD14, which facilitates activation of nuclear factor-κB (NF-κB) in keratinocytes, are associated with GPP in Japanese populations [32]. CARD14 mutations have also been reported in patients with GPP and pre-existing plaque psoriasis, but are rarely reported in patients with GPP alone [33]. Furthermore, loss-of-function mutations in AP1S3 are associated with pustular psoriasis in individuals of European origin, but are rarely seen in individuals from East Asia [34]. AP1S3 encodes a protein implicated in autophagosome formation and AP1S3-deficient cells demonstrate NF-κB activation, upregulated IL-1 signaling, and overexpression of IL-36α [35].
In addition, overproduction of IL-36 agonists is observed in patients with GPP with mutations in SERPINA3, which encodes alpha-1-antichymotrypsin, an inhibitor of cathepsin G, a protease secreted by neutrophils that cleaves (and activates) IL-36 precursors [36]. Several proteases secreted by neutrophils (cathepsin G, proteinase 3, elastase), and even keratinocytes (cathepsin S), have been found to activate IL-36, which may further contribute to positive inflammatory feedback [37]. Mutations in myeloperoxidase, a neutrophil-associated lysosomal hemoprotein, have also been identified in patients with GPP [38], although not recognized as the initial trigger of the disease. Overall, while the evidence indicates that the IL-36 pathway is central to the development of GPP, a greater understanding of the associated immunologic components, as well as the pathogenic contribution of genetic and ethnic variations, is required.
3 Therapeutic Perspectives
There is no globally accepted therapeutic guidance for GPP, and treatment often reflects the recommendations for plaque psoriasis [9]. Therefore, there is an urgent need for well-designed trials to provide clinical evidence on the safety and efficacy of therapies specifically for GPP. Given the potentially life-threatening nature of GPP flares, ideal therapeutic agents should have a rapid onset of action, a rapid time to disease clearance, the ability to prevent flares, and a favorable safety profile [7]. However, there is a lack of robust therapeutic data available, which is an inevitable consequence of the rarity of the disease, and the sudden, self-limiting, episodic nature of pustular flares limits the availability of suitable patients with active disease to power randomized controlled trials [39].
To date, biologics have only been approved for the treatment of GPP in a limited number of countries, with no GPP-specific therapeutic agents approved in the USA or Europe [7]. The IL-17/IL-17R inhibitors secukinumab and ixekizumab [40,41,42,43,44] and the IL-23 inhibitors risankizumab and guselkumab [45, 46] are approved for use in Japan, and the IL-17 antagonist brodalumab is approved for use in Japan, Taiwan, and Thailand [8, 47]. Approval of these IL-17/IL-17R and IL-23 inhibitors was based on data from prospective but small-scale, open-label, single-arm, phase III studies [41, 45, 47]. Furthermore approval of the tumor necrosis factor inhibitors adalimumab [48, 49], infliximab [50, 51], and certolizumab pegol for the treatment of GPP in Japan was based largely on case studies [52].
Phase II and III clinical trials investigating the effects of IL-36R-blocking monoclonal antibodies in patients with GPP are ongoing. Interim results from a phase II study of imsidolimab (ANB019, NCT03619902) suggest a favorable safety profile [53]; phase III trails were due to begin in the third quarter of 2021. Spesolimab (BI 655130) has demonstrated efficacy in a phase I, open-label, proof-of-concept study of seven biologic-naive adult patients presenting with a moderate or severe GPP flare [54], with phase II trials currently underway (NCT04399837) [55]. Both imsidolimab and spesolimab are also under investigation for other putative indications, including PPP (AB019, NCT03633396; BI 655130, NCT03633396) [56, 57]. In addition, phase I development of an antibody targeting the IL-36R for the treatment of PPP is underway (REGN6490).
Regarding the treatment of other psoriatic conditions, IL-36γ blockade with a small IL-36γ-binding molecule has been shown to inhibit inflammation in plaque psoriasis by preventing interactions with IL-36R [58]. In addition, the IL-36 cytokine pathway has been identified as an important factor in the pathogenesis of psoriatic arthritis, and therefore may be a suitable therapeutic target [59]. Further studies of additional components of the IL-36-related pathways have been initiated, with the IL-1β inhibitor gevokizumab having shown promise on treatment of two patients with GPP [60].
4 Conclusions
GPP places a considerable burden on patients, and while efforts have been made to better understand the disease and its management, progress is slowed by the small number of cases. As a result, there are many important knowledge gaps and unmet treatment needs. Identification of the key role of the IL-36 pathway in the immunopathology of GPP has paved the way for new therapies targeting disease-associated genetic and molecular pathways, and these agents have shown promise in patients presenting with GPP flares. Further elucidation of the unique pathogenesis of GPP may identify additional genetic variants that could provide effective therapeutic targets. Accurate knowledge of the pathophysiology of GPP and its genetic determinants will help physicians to personalize treatment and more effectively manage patients with this rare disease.
References
Benjegerdes KE, Hyde K, Kivelevitch D, Mansouri B. Pustular psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:131–44.
Shah M, Al Aboud DM, Crane JS, Kumar S. Pustular psoriasis. Treasure Island (FL): StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC; 2021.
Boehner A, Navarini AA, Eyerich K. Generalized pustular psoriasis - a model disease for specific targeted immunotherapy, systematic review. Exp Dermatol. 2018;27(10):1067–77.
Ly K, Beck KM, Smith MP, Thibodeaux Q, Bhutani T. Diagnosis and screening of patients with generalized pustular psoriasis. Psoriasis (Auckl). 2019;9:37–42.
Kardaun SH, Kuiper H, Fidler V, Jonkman MF. The histopathological spectrum of acute generalized exanthematous pustulosis (AGEP) and its differentiation from generalized pustular psoriasis. J Cutan Pathol. 2010;37(12):1220–9.
Navarini AA, Burden AD, Capon F, Mrowietz U, Puig L, Köks S, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11):1792–9.
Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–19.
Fujita H, Terui T, Hayama K, Akiyama M, Ikeda S, Mabuchi T, et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11):1235–70.
Robinson A, Van Voorhees AS, Hsu S, Korman NJ, Lebwohl MG, Bebo BF Jr, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67(2):279–88.
Kharawala S, Golembesky AK, Bohn RL, Esser D. The clinical, humanistic, and economic burden of generalized pustular psoriasis: a structured review. Expert Rev Clin Immunol. 2020;16(3):239–52.
Choon SE, Lai NM, Mohammad NA, Nanu NM, Tey KE, Chew SF. Clinical profile, morbidity, and outcome of adult-onset generalized pustular psoriasis: analysis of 102 cases seen in a tertiary hospital in Johor, Malaysia. Int J Dermatol. 2014;53(6):676–84.
Rendon A, Schäkel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. 2019;20(6):1475.
Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, et al. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol. 2013;133(5):1366–9.
Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89(3):432–7.
Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. J Allergy Clin Immunol. 2017;140(6):1545–7.
Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140(1):109–20.
Liang Y, Sarkar MK, Tsoi LC, Gudjonsson JE. Psoriasis: a mixed autoimmune and autoinflammatory disease. Curr Opin Immunol. 2017;49:1–8.
Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. 2018;201(6):1605–13.
Furue K, Yamamura K, Tsuji G, Mitoma C, Uchi H, Nakahara T, et al. Highlighting interleukin-36 signalling in plaque psoriasis and pustular psoriasis. Acta Derm Venereol. 2018;98(1):5–13.
Zhou J, Luo Q, Cheng Y, Wen X, Liu J. An update on genetic basis of generalized pustular psoriasis (review). Int J Mol Med. 2021;47(6):118.
Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281(1):169–78.
Boutet MA, Nerviani A, Pitzalis C. IL-36, IL-37, and IL-38 cytokines in skin and joint inflammation: a comprehensive review of their therapeutic potential. Int J Mol Sci. 2019;20(6):1257.
Iznardo H, Puig L. Exploring the role of IL-36 cytokines as a new target in psoriatic disease. Int J Mol Sci. 2021;22(9):4344.
Uppala R, Tsoi LC, Harms PW, Wang B, Billi AC, Maverakis E, et al. “Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell Mol Immunol. 2021;18(2):307–17.
Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365(7):620–8.
Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol. 2019;143(3):1021–6.
Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20(13):3318.
Hussain S, Berki DM, Choon SE, Burden AD, Allen MH, Arostegui JI, et al. IL36RN mutations define a severe autoinflammatory phenotype of generalized pustular psoriasis. J Allergy Clin Immunol. 2015;135(4):1067-70.e9.
Wang TS, Chiu HY, Hong JB, Chan CC, Lin SJ, Tsai TF. Correlation of IL36RN mutation with different clinical features of pustular psoriasis in Chinese patients. Arch Dermatol Res. 2016;308(1):55–63.
Sugiura K, Takemoto A, Yamaguchi M, Takahashi H, Shoda Y, Mitsuma T, et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J Invest Dermatol. 2013;133(11):2514–21.
Mössner R, Wilsmann-Theis D, Oji V, Gkogkolou P, Löhr S, Schulz P, et al. The genetic basis for most patients with pustular skin disease remains elusive. Br J Dermatol. 2018;178(3):740–8.
Sugiura K, Muto M, Akiyama M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J Invest Dermatol. 2014;134(6):1755–7.
Li L, You J, Fu X, Wang Z, Sun Y, Liu H, et al. Variants of CARD14 are predisposing factors for generalized pustular psoriasis (GPP) with psoriasis vulgaris but not for GPP alone in a Chinese population. Br J Dermatol. 2019;180(2):425–6.
Setta-Kaffetzi N, Simpson MA, Navarini AA, Patel VM, Lu H-C, Allen MH, et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am J Hum Genet. 2014;94(5):790–7.
Mahil SK, Twelves S, Farkas K, Setta-Kaffetzi N, Burden AD, Gach JE, et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J Invest Dermatol. 2016;136(11):2251–9.
Frey S, Sticht H, Wilsmann-Theis D, Gerschütz A, Wolf K, Löhr S, et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol. 2020;140(7):1451-5.e13.
Ainscough JS, Macleod T, McGonagle D, Brakefield R, Baron JM, Alase A, et al. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc Natl Acad Sci U S A. 2017;114(13):E2748–57.
Vergnano M, Mockenhaupt M, Benzian-Olsson N, Paulmann M, Grys K, Mahil SK, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet. 2020;107(3):539–43.
Choon SE, Lebwohl MG, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Study protocol of the global EffisayilTM 1 Phase II, multicentre, randomised, double-blind, placebo-controlled trial of spesolimab in patients with generalized pustular psoriasis presenting with an acute flare. BMJ Open. 2021;11(3):e043666.
Wilsmann-Theis D, Schnell LM, Ralser-Isselstein V, Bieber T, Schön MP, Hüffmeier U, et al. Successful treatment with interleukin-17A antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J Dermatol. 2018;45(7):850–4.
Imafuku S, Honma M, Okubo Y, Komine M, Ohtsuki M, Morita A, et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: a 52-week analysis from phase III open-label multicenter Japanese study. J Dermatol. 2016;43(9):1011–7.
Kromer C, Loewe E, Schaarschmidt ML, Pinter A, Gerdes S, Herr R, et al. Drug survival in the treatment of generalized pustular psoriasis: a retrospective multicenter study. Dermatol Ther. 2021;34(2):e14814.
Saeki H, Nakagawa H, Ishii T, Morisaki Y, Aoki T, Berclaz PY, et al. Efficacy and safety of open-label ixekizumab treatment in Japanese patients with moderate-to-severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis. J Eur Acad Dermatol Venereol. 2015;29(6):1148–55.
Taguchi R, Takamura S, Teraki Y. Combination therapy with biologic and colchicine for generalized pustular psoriasis. Int J Dermatol. 2020;59(11):e400–2.
Sano S, Kubo H, Morishima H, Goto R, Zheng R, Nakagawa H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J Dermatol. 2018;45(5):529–39.
Suleiman AA, Khatri A, Oberoi RK, Othman AA. Exposure-response relationships for the efficacy and safety of risankizumab in Japanese subjects with psoriasis. Clin Pharmacokinet. 2020;59(5):575–89.
Yamasaki KN, Kubo H, Ootaki Y. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: results from a 52-week, open-label study. Br J Dermatol. 2017;176(3):741–51.
Kawakami H, Matsumoto Y, Abe N, Katori Y, Takahashi K, Tsuboi R, et al. Perioperative management of tumor necrosis factor-alpha blocker-treated psoriatic patients: case reports and review. J Dermatol. 2016;43(2):190–3.
Matsumoto A, Komine M, Karakawa M, Kishimoto M, Ohtsuki M. Adalimumab administration after infliximab therapy is a successful treatment strategy for generalized pustular psoriasis. J Dermatol. 2017;44(2):202–4.
Adachi A, Komine M, Hirano T, Tsuda H, Karakawa M, Murata S, et al. Case of generalized pustular psoriasis exacerbated during pregnancy, successfully treated with infliximab. J Dermatol. 2016;43(12):1439–40.
Sugiura K, Endo K, Akasaka T, Akiyama M. Successful treatment with infliximab of sibling cases with generalized pustular psoriasis caused by deficiency of interleukin-36 receptor antagonist. J Eur Acad Dermatol Venereol. 2015;29(10):2054–6.
Viguier M, Aubin F, Delaporte E, Pagès C, Paul C, Beylot-Barry M, et al. Efficacy and safety of tumor necrosis factor inhibitors in acute generalized pustular psoriasis. Arch Dermatol. 2012;148(12):1423–5.
A study to evaluate the efficacy and safety of anb019 in subjects with generalized pustular psoriasis. https://ClinicalTrials.gov/show/NCT03619902.
Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380(10):981–3.
A study to test whether BI 655130 (spesolimab) prevents flare-ups in patients with generalized pustular psoriasis. https://ClinicalTrials.gov/show/NCT04399837.
A study to evaluate the efficacy and safety of ANB019 in subjects with palmoplantar pustulosis. https://ClinicalTrials.gov/show/NCT03633396.
A study to test long-term treatment with spesolimab in people with palmoplantar pustulosis (PPP) who took part in previous studies with spesolimab. https://ClinicalTrials.gov/show/NCT04493424.
Todorović V, Su Z, Putman CB, Kakavas SJ, Salte KM, McDonald HA, et al. Small molecule IL-36γ antagonist as a novel therapeutic approach for plaque psoriasis. Sci Rep. 2019;9(1):9089.
Boutet MA, Nerviani A, Lliso-Ribera G, Lucchesi D, Prediletto E, Ghirardi GM, et al. Interleukin-36 family dysregulation drives joint inflammation and therapy response in psoriatic arthritis. Rheumatology (Oxford). 2020;59(4):828–38.
Mansouri B, Richards L, Menter A. Treatment of two patients with generalized pustular psoriasis with the interleukin-1β inhibitor gevokizumab. Br J Dermatol. 2015;173(1):239–41.
Acknowledgements
All authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE) and made the decision to submit the manuscript for publication. The authors did not receive payment related to the development of the manuscript. Agreements between Boehringer Ingelheim and the authors included the confidentiality of the study data. In the preparation of this manuscript, James Parkinson, PhD from OPEN Health Communications (London, UK), provided medical writing, editorial support, and/or formatting support, which was contracted and funded by Boehringer Ingelheim. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure statement
This article has been published as part of a journal supplement wholly funded by Boehringer Ingelheim.
Funding
Medical writing support was funded by Boehringer Ingelheim.
Conflicts of interest
Lluis Puig has received honoraria or fees for serving on advisory boards, as a speaker, and as a consultant, as well as grants as an investigator from AbbVie, Almirall, Amgen, Baxalta, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Fresenius-Kabi, Janssen, JS BIOCAD, LEO Pharma, Lilly, Mylan, Novartis, Pfizer, Regeneron, Roche, Sandoz, Samsung-Bioepis, Sanofi, and UCB. Slaheddine Marrakchi has received consulting fees from Boehringer Ingelheim.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
The sponsor of the supplement and all authors identified and discussed the concept for each chapter and the supplement as a whole; further development of each review article was at the direction of the authors who provided guidance to OPEN Health on the content of the article, critically revised the work, and approved the content for publication.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Marrakchi, S., Puig, L. Pathophysiology of Generalized Pustular Psoriasis. Am J Clin Dermatol 23 (Suppl 1), 13–19 (2022). https://doi.org/10.1007/s40257-021-00655-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-021-00655-y