
Abstract
Cs exchanged heteropolyacid catalysts functionalized with various Sn contents were prepared by wet impregnation method. These catalysts were characterized by X-ray diffraction, FT-IR, Raman spectroscopy, temperature programmed desorption of ammonia and BET surface area measurements. The catalytic properties of Sn–CsPW catalysts were evaluated for the synthesis of glycerol carbonate and they exhibit an unprecedented activity for the higher glycerol conversion and selectivity towards glycerol carbonate under vacuum conditions. Sn-functionalized Cs exchanged heteropolyacid catalysts (CsPW) play a significant role in the enhancement of acidity, catalytic activity and stability. The glycerol conversion and the selectivity of carbonate formation mainly depend on the Sn content and acidity of the catalysts. Different reaction parameters such as Sn molar ratio, glycerol to urea molar ratio, reaction temperature were investigated and also optimum conditions were established. The catalyst containing molar ratio of 3:1 Sn–CsPW has shown highest conversion and glycerol carbonate selectivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There has been an increasing demand in recent years on the biomass-derived fuels as an alternative energy sources for petroleum crudes. Biodiesel has attracted one of the choices in the recent past as an alternative fuel because of its environmental benefits as it can be produced from renewable resources including vegetable oils or animal fats [1]. However, huge amounts of glycerol produced during biodiesel production as a by-product at the rate of 10 % by weight are challenging task for its conversion into commodity chemicals. Therefore, the conversion of renewable biomass-derived glycerol is a promising alternative for the sustainable production of chemical intermediates and fuels [2–4]. Thus, glycerol can be used as platform chemical for the production of more valuable commodity chemicals [5, 6]. The effective utilization of the by-product glycerol derived from the bio-diesel synthesis is a key solution for bio-diesel commercialization. Thus, many researchers are presently attempting the conversion of glycerol into value-added chemicals by various catalytic methods, such as dehydration [7], hydrogenolysis [8] and carbonylation [9]. The use of glycerol as raw material for the preparation of highly valued bio-based building blocks such as glycerol carbonate could be one of the potential routes. This process is a more attractive method utilizing glycerol as a renewable and cheap raw material for the preparation of glycerol carbonate.
Glycerol carbonate (GC) is a valuable and interesting material in the fine chemical industry. It has been investigated as a novel component in gas separation membranes, polyurethane foams, a component in coatings, paints and detergents [10, 11]. In particular, inexpensive GC can be used to synthesize the high-value glycidol, a source of new polymeric material for the production of a number of polymers. Glycerol carbonate has a wide application due to its dual functionality of carbonate ring and pendant hydroxyl group. Its oxo group and dioxolane group can react with alcohols, amines, carboxylic acids, ketones and isocyanates, while its hydroxy group enables esterification, urethane formation with isocyanates and hydroxy activation through tosylation. Furthermore, its utilization as an intermediate has received several industrial applications, because of its high boiling point, high flash point and low volatility [12].
The glycerol carbonate can be prepared by the reaction of glycerol with CO2 or carbon monoxide and oxygen in the presence of Cu catalysts and also by sodium metal impregnated on Y-zeolite [13]. The reaction of glycerol with carbon dioxide was carried out in a super critical CO2 medium in the presence of zeolite and ethylene carbonate as a co-source of carbonate groups [14]. In all these processes, glycerol carbonate has to be purified prior to the reaction at relatively high temperature (150 °C). The preparation of glycerol carbonate using urea as a reactant is favorable due to its cheaper price and easy handling.
Glycerol carbonate prepared using glycerol and a gaseous mixture of CO and CO2 over tin complexes have shown very low yield 6 % [15]. It can be also prepared by glycerol with dimethyl carbonate over different catalysts, such as lipase catalyst [16], CaO, MgO [17] and calcined La2O3 [18]. Recently, glycerol carbonate preparation was reported using KF/gamma Al2O3 catalyst [19]. Glycerol carbonate can also be synthesized with urea over Au-supported MgO [20] and Zinc solid catalysts [21]. However, these methods for the synthesis of GC using urea over novel catalytic materials require high molar ratio of glycerol to urea. Recently, the synthesis of glycerol carbonate from glycerol and alkyl carbonates over Mg–Al hydrotalcite catalysts was reported under vapor phase conditions with low yields [22].
Heteropolyacids (HPAs) are well-known typical strong Brønsted acids and employed in a wide variety of acid catalyzed reactions in both homogeneous and heterogeneous conditions. The disadvantage of HPAs is their relatively low thermal stability and low surface area (<10 m2/g). To overcome these disadvantages, high surface area and thermally stable supports such as SBA-15 and MCM-41 are employed to disperse the active phase [23, 24]. Recently, metal-doped CsPW catalysts have attracted much attention due to their high surface area and tunable porosity which enable them for the use in glycerol dehydration and glycerol hydrogenolysis [25–27].
In the present work, we report for the first time Sn-doped CsPW catalyst for glycerol carbonolysis reaction. A systematic study has been made on the different molar ratios of Sn and CsPW during preparation of the catalysts, surface area and acidic functionalities. The catalytic properties were correlated with the acidity and textural properties.
Experimental
Catalyst preparation
Cesium salt was prepared by the dropwise addition of a stoichiometric quantity of aqueous Cs2CO3 (0.47 M) to an aqueous solution of H3PW12O40 (0.75 M) at 40 °C with continuous stirring [28]. The precipitate obtained was dried for 48 h in an oven at 100 °C and calcined at 500 °C for 5 h. A series of catalysts are prepared by changing the molar ratio from 0 to 5 of Sn by impregnation method. A known amount of Sn precursor (nitrate) was dissolved in distilled water and the Cs salt was also kept constant. The resulting mixture was dried in hot oven at 100 °C. All the prepared catalysts were calcined at 500 °C for 4 h.
Catalyst characterization
X-ray diffraction studies
X-ray powder diffraction patterns of the samples were obtained with a model: D8 Diffractometer (Advance, Bruker, Germany), using Cu Kα radiation (1.5406 Å) at 40 kV and 30 mA. The measurements were recorded in steps of 0.045° with a count time of 0.5 s in the range of 2–80°.
Raman spectroscopy
The Raman spectra of the catalyst samples were collected with a Horiba-Jobin-Yvon LabRam-HR spectrometer equipped with a confocal microscope, 2400/900 grooves/mm gratings, and a notch filter. The visible laser excitation at 532 nm (visible/green) was supplied by a Yag doubled diode pumped laser (20 mW). The scattered photons were dried and focused on to a single-stage monochromator and measured with a UV-sensitive LN2-cooled CCD detector (Horiba-Jobin-Yvon CCD-3000 V).
FT-IR spectroscopy
FT-IR spectra of the catalysts were recorded on an IR (model: GC-FT-IR Nicolet 670) spectrometer by KBr disc method at room temperature [29].
Temperature programmed desorption
Temperature programmed desorption (TPD) studies of NH3 were conducted on Auto Chem 2910 (Micromeritics, USA) instrument. In a typical experiment, 100 mg of calcined sample was taken in a U-shaped quartz cell. The catalyst sample was packed in one arm of the sample tube on a quartz wool bed. Prior to TPD studies, the catalyst sample was pre-treated by passing high purity helium (50 mL/min) at 200 °C for 1 h. After pre-treatment, the sample was saturated by passing (50 mL/min) high purity anhydrous ammonia at 80 °C for 1 h and subsequently flushed with He flow (50 mL/min) at 150 °C for 1 h to remove the physisorbed ammonia. TPD analysis was carried out from ambient temperature to 600 °C at a heating rate of 10 °C/min. The ammonia concentration in the effluent stream was monitored with the thermal conductivity detector and the area under the peak was integrated using the software GRAMS/32 to determine the amount of desorbed ammonia.
BET studies
The surface area of catalysts was estimated using N2 adsorption isotherms at −196 °C by the multipoint BET method taking 0.162 nm2 as its cross-sectional area.
Catalytic activity
The catalytic reaction was performed in a 25 mL two neck round-bottom (RB) flask under reduced pressures. In a typical experiment, glycerol (2 g), urea (1.3 g) and catalysts (0.2 g) were taken in the round-bottom flask and heated in an oil bath at 140 °C with constant stirring under reduced pressure. One neck of the RB flask was connected to vacuum line. The reaction was carried out under a reduced pressure to remove the formation of ammonia during the reaction. The reaction was monitored in different time intervals. After completion of the reaction, methanol was added and the catalyst was separated by filtration. The products were analyzed by a gas chromatograph (Shimadzu, 2010) equipped with flame ionization detector using innowax capillary column (diameter 0.25 mm, length 30 m). The obtained products were analyzed by gas chromatograph–mass spectrometry (Shimadzu GC-2010) equipped with flame ionization detector using a DB-wax 123-7033 capillary column and methanol as the internal standard.
Results and discussion
XRD studies
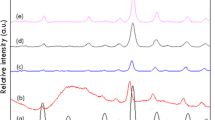
The XRD patterns of pure CsPW and the samples containing Sn/Cs exchanged tungstophosphoric acid (PTA) with different molar ratios are shown in the Fig. 1. It can be seen from Fig. 1 that the diffraction peaks observed at 2θ = 10.9°, 27.5° and 33.5° suggest that the Keggin ion structure of PTA was clearly observed even after the exchange of cesium and Sn. The XRD patterns did not show any reflections due to SnO2 in the samples of Sn-doped CsPW [30–32]. The absence of SnO2 peaks is probably due to the active phase which is highly dispersed on CsPW. Furthermore, the intensity of XRD peaks due to Keggin ion gradually decreased with increase in the molar ratio of Sn and CsPW.

X-ray diffraction patterns of various molar ratios of Sn–CsPW catalysts. a 5:1, b 4:1, c 3:1, d 2:1, e 1:1 and f pure CsPW
Fourier transform infrared spectroscopy
The FT-IR spectra of pure CsPW and the spectra for various molar ratios of Sn/CsPW catalysts are shown in Fig. 2. The spectra exhibit typically four characteristic IR bands located at 1080, 985, 890 and 839 cm−1 which are attributed to the absorption modes of Keggin structure of tungstophosphoric acid. The bands at 1080, 985, 890 and 839 cm−1 are exclusively assigned to the stretching modes of oxygen atom bond to tungsten and phosphorous, P–O, W=O, W–Ob–W in corner-shared octahedral and W–Oc–W in edge-shared octahedral, respectively [33, 34]. It can be seen from Fig. 2 that as Sn loading increases the intensity of characteristic bands of Keggin ion was decreased. The above results suggest that the active phase Sn/CsPW was well dispersed on the primary structure of Keggin ion and intensity of Keggin ion bands decreased. These results are in well agreement with the above XRD findings.

FT-IR spectra of various molar ratios of Sn–CsPW catalysts. a Pure CsPW, b 1:1, c 2:1, d 3:1, e 4:1 and f 5:1
Temperature programmed desorption of ammonia
NH3-TPD profiles of various catalysts of Sn and CsPW with molar ratio 1:1–5:1 are shown in Fig. 3 and the amount of NH3 desorbed during TPD is reported in Table 1. It can be seen from Fig. 3 that the desorption peak observed below 150 °C is assigned to weak acidic sites and the peak noticed between 150 and 300 °C was attributed to moderate acidic sites. The TPD peak observed between 400 and 600 °C is attributed to strong acidic sites of the catalyst. TPD profiles of samples containing Sn on the CsPW showed three desorption peaks indicating the presence of three types of acidic sites (weak, moderate and strong). With the influence of Sn content on CsPW, the total acidity of catalyst increases gradually for samples with molar ratio from 1:1 to 3:1 and thereafter it decreased. Whereas the strong acidic sites increase with increase in Sn loading up to 3:1 molar ratio and it decrease further at higher Sn loadings. Ammonia uptake values of different molar ratio of Sn on CsPW are summarized in Table 1. It is interesting to see that the total acidity of catalyst increases up to 3:1 and decreases at higher molar ratio of Sn loading. The decrease in acidity at higher molar ratio was attributed to the formation of bulk Sn metal oxide which leads to a decrease in the acidity. Therefore, the findings suggest that a large amount of acidic sites are observed in the sample containing 3:1 molar ratio of Sn and CsPW than the other samples.

Laser Raman spectra of various molar ratios of Sn–CsPW catalysts. a 5:1, b 4:1, c 3:1, d 2:1 and e 1:1
Laser Raman spectroscopy
The Raman spectroscopy of the samples with various molar ratios of Sn and CsPW catalysts is shown in Fig. 4. Pure tungstophosphoric acid exhibits four Raman bands in the region of 1100–200 cm−1. The Raman bands at 1011 and 990 cm−1 correspond to W=Ot symmetric and asymmetric stretching modes, respectively. The weaker Raman bands at 890 and 530 cm−1 are assigned to characteristic bands of asymmetric stretching vibration of bridging W–Ob–W and symmetric stretching of bridging W–Oc–W, respectively [35]. These findings show that all the catalysts exhibit four Raman bands at 1011, 990, 890 and 530 cm−1 which are attributed to characteristic bands of Keggin ion structure of tungstophosphoric acid. It can be seen from Fig. 4 that with Sn loading increases the intensity of characteristic bands of Keggin ion was slightly declined. The above results suggest that Sn metal oxide peaks were not observed and it indicates that the active phase of Sn is well dispersed and decreases the intensity of Keggin ion bands. These results are well agreement with the above XRD, FT-IR findings.

Temperature programmed desorption peaks of various molar ratios of Sn–CsPW catalysts. a 1:1, b 2:1, c 3:1, d 4:1 and e 5:1
BET surface area
The surface area of Sn–CsPW catalysts with different molar ratio of Sn is summarized in Table 1. The BET surface area of pure Cs exchanged tungstophosphoric acid is 117 m2/g. It was observed that modification in CsPW support lowers the surface area 103–91 m2/g for catalysts loaded with 1:1 and 2:1 molar ratio of Sn, respectively. Further increase of Sn mole ratio on the support resulted in the reduction of surface area of the catalysts which indicate that the pores of CsPW are partially filled with Sn metal particles.
Catalytic activity
The catalytic properties of various Sn/CsPW catalysts during liquid phase carbonolysis of glycerol are reported in Table 2. The catalysts containing various molar ratios of Sn/CsPW and without Sn are tested for carbonolysis of glycerol at 140 °C under vacuum condition by taking 200 mg of the catalyst. The product distribution during glycerol carbonate synthesis using glycerol and urea is reported along with the other side products obtained such as urethane and glycidol. Even though the reaction was carried out under vacuum condition, a small amount of urethane is also noticed. This might be due to the faster reaction of liberated ammonia gas with glycerol leading to the formation of glycerol urethane [36]. The formation of Glycidol is due to the release of CO2 from glycerol carbonate which occurs generally at higher temperature or by employing more acidic catalysts during the reaction [37]. To investigate the effect of metal component on the carbonolysis of glycerol, the catalyst samples with various molar ratio of Sn were tested along with pure CsPW and the results are reported in Table 2. To optimize the conditions to achieve better yield of glycerol carbonate, the effect of reaction temperature was investigated and the results are represented in Table 3. The catalyst which has shown the best catalytic activity was subjected to different calcination temperatures and the results are shown in Table 4. The reaction was conducted using various molar ratios of glycerol and urea to find optimum mole ratio suitable for this reaction and the results are summarized in Table 5. A comparison of carbonolysis reaction using the present catalysts (PTA) was made using Sn-functionalized phosphomolybdic acid (PMA) silicotungsticacid (STA) and the results are shown in Table 6. The reaction was also conducted in the absence of catalyst and noticed that there was no considerable amount of glycerol carbonate formed.
Effect of molar ratio of Sn/CsPW
The glycerol conversion and product selectivity as a function of Sn content on Cs exchanged heteropolyacid were investigated for the liquid phase carbonolysis of glycerol with urea and the product distribution results are given in Table 2. The catalytic results show that glycerol conversion increased with an increase in Sn loading and attains a maximum conversion with 3:1 SnCsPW. The increase in conversion and selectivity at 3:1 molar ratio of Sn and CsPW is mainly due to the presence of strong acidic sites which decrease at higher ratio. These results are well in correlation with the acidic functionalities of the catalysts determined by NH3-TPD method. The increase in the selectivity towards glycerol carbonate as a function of Sn loading (3:1 molar ratio) suggests that a conversion of 91 % is achieved with 83 % selectivity of glycerol carbonate. Therefore, the above results suggest that 3:1 molar ratio of Sn–CsPW is the optimum for the conversion of glycerol carbonate from glycerol and urea.
Effect of reaction temperature
The change in the reaction temperature plays a pronounced change in the glycerol conversion and the selectivity of the desired product glycerol carbonate. The effect of reaction temperature on the product distribution during carbonolysis reaction is shown in Table 3. The increase in reaction temperature from 140 to 160 °C yields increases in the selectivity of glycerol carbonate from 64 to 83 % with substantial increase in glycerol conversion from 56 to 91 %, respectively. The raise of reaction temperature further by 20 °C led to a decline in the selectivity of the glycerol carbonate to 46 % at glycerol conversion of 67 %. Furthermore, the selectivity to glycidol is also increased to 41 % at the expenses of glycerol carbonate. This may be due to decarboxylation of glycerol carbonate at higher temperature (160 °C). The above results clearly suggest that moderate reaction temperatures are suitable to achieve best catalytic properties during carbonolysis of glycerol.
Effect of mole ratio glycerol to urea
The effect of glycerol to urea mole ratio was investigated on the catalytic performance using the sample containing 3:1 molar ratio of Sn/CsPW catalyst without altering other reaction parameters. Glycerol conversion is found to be much lower while using 1:0.5 mol ratio of glycerol to urea in the reactant mixture. The lower activity could be due to the less number of moles of urea present in the reactant mixture. As the number of moles of urea increases in the reactant mixture (1:1), glycerol conversion increased drastically and high selectivity towards glycerol carbonate was achieved. Whereas, in the case of 1:1.5 mol ratio the conversion did not change appreciably but the selectivity towards the desired product was decreased with the formation of considerable amount of urethanes. This is mainly because the liberation of more amount of ammonia from urea favors the formation of urethane. The above results show that 1:1 mol ratio of glycerol to urea was found to be the optimum mole ratio in the reaction mixture for the glycerol carbonate synthesis.
Effect of calcination temperature
To understand the structural changes and surface acidity of catalyst, the sample containing 3:1 molar ratio of Sn and CsPW was calcined at different temperatures in the range 400–600 °C. It is known that the acidity of the catalysts changes with a change in calcination temperature. As the calcination temperature increases, the acidity of the catalyst increased up to 500 °C and decreased later. This is due to crystallite CsPW the catalysts and further leads to decrease in the surface acidity [38]. The catalytic results over various calcination temperatures are presented in Table 5. It can be seen from the table that the catalytic activity and selectivity increase with the increase in calcination temperature from 400 to 500 °C and thereafter decreased in the catalytic performance. The glycerol conversion increased from 81 to 91 % and selectivity towards glycerol carbonate also increased from 72 to 83 % with increase in the calcination temperature of the catalyst. Thus, it clearly indicates that the catalytic performance depends on the calcination temperature of the catalysts. It is found that the samples calcined at 500 °C exhibit higher catalytic activity than other calcination temperatures.
Effect of Cs exchanged heteropolyacid
A comparison of the catalytic performance of Sn-functionalized CsPW catalysts is made with two other heteropolyacids, such as Cs exchanged phosphomolybdic acid (CsPMA) and Cs exchanged silicotungsticacid (CsSTA) under similar experimental conditions. The samples were prepared with 3:1 molar ratio of Sn and (CsSTA or CsPMA). The product distribution of these three catalysts during glycerol carbonate synthesis is represented in Table 6. The results suggest that the glycerol conversion and selectivity were found to be higher in 3:1 molar ratio of Sn and CsPW catalyst compared to other heteropolyacid catalysts. The better catalytic properties exhibited by Sn-functionalized CsPW are due to the presence of more strong acidic sites than the Sn–CsPMA or Sn–CsSTA catalysts [39]. The above results strongly suggest that the more number of acidic sites favors higher glycerol conversion and also the formation of glycerol carbonate.
Energy dispersive spectroscopy (EDS)
Energy dispersive spectroscopy is employed to confirm the presence of Sn in the best catalyst containing 3:1 molar ratio of Sn–CsPW and the results are shown in Fig. 5. It can be seen from Fig. 5 that Sn was successfully functionalised with Keggin ion of phosphotungstic acid catalyst. The results also reveal that the Sn–CsPW molar ratio is maintained at 3.1 ratios after the preparation of the catalyst (Fig. 5). Therefore, the EDS analysis clearly represents the presence of Sn in the synthesis of catalyst.

EDS results of 3:1 mol ratio Sn–CsPW catalyst
Reaction mechanism
It is recognized that acidic and basic properties of the catalysts influence the formation of glycerol carbonate from glycerol and urea. Furthermore, solid Lewis acid catalysts exhibit high yields of glycerol carbonate [9]. Based on the above results, the possible reaction mechanism for glycerol carbonylation with urea over Sn-functionalised CsPW catalysts has been proposed and shown in Scheme 1. It is necessary that Lewis acidic sites (Sn) activated the carbonyl group of urea and the weak conjugated framework oxygen from Keggin ion activates the hydroxyl group of glycerol. It is concluded that the presence of these Lewis acidic sites in Sn–CsPW catalyst shows high activity towards glycerol carbonate synthesis.

Reaction scheme: carbonolysis of glycerol to glycerol carbonate
Conclusions
Sn–CsPW catalysts exhibited excellent catalytic performance during glycerol carbonate synthesis. The XRD results show that the active phase of Sn was highly dispersed on Cs exchanged phosphotungstic acid. The FT-IR and Raman results reveal that the characteristic peaks of Keggin ion structure intact even after the formation of Cs salt of heteropolyacid. TPD of ammonia results shows that acidity increased up to 3:1 molar ratio of Sn/CsPW catalyst. It was found that glycerol conversion and selectivity of glycerol carbonate remarkably increased with the increase in Sn loading. The calcination of samples containing 3:1 molar ratio of Sn/CsPW at 500 °C exhibits higher conversion and selectivity. The catalytic results are well correlated with the acidity of the catalysts.
References
Zhou CH, Beltramini JN, Fan YX, Lu GQ (2008) Chemo selective catalytic conversion of glycerol as a bio renewable source to valuable commodity chemicals. Chem Soc Rev 37:527–549
Carpenter D, Westover TL, Czernika S, Jablonskia W (2014) Biomass feedstocks for renewable fuel production: a review of the impacts of feedstock and pre-treatment on the yield and product distribution of fast pyrolysis, bio-oils and vapours. Green Chem 16:384–406
Ma Fangru, Hanna Milford A (1999) Biodiesel production: a review. Bio Resour Technol 70:1–15
Padhi SK, Panda K, Singh RK (2013) A brief introduction on renewable glycerol. Int J Eng Res Technol 8:1119–1169
Viswanadham B, Srikanth A, Chary KVR (2014) Characterization and reactivity of 11-molybdo-1-vanadophosphoric acid catalyst supported on zirconium for dehydration of glycerol to acrolein. J Chem Sci 126:445–454
Kumar VP, Harikrishna Y, Nagaraju N, Chary KVR (2014) Characterization and reactivity of TiO2 supported nano ruthenium catalysts for vapour phase hydrogenolysis of glycerol. Indian J Chem A 53:516–523
Viswanadham B, Srikanth A, Kumar VP, Chary KVR (2014) Vapour phase dehydration of glycerol to acrolein over SBA-15 supported vanadium substituted phosphomolybdic acid catalyst. JNN 14:1–14
Li J, Wang T (2010) Coupling reaction and azeotropic distillation for the synthesis of glycerol carbonate from glycerol and dimethyl carbonate. Chem Eng Process Process Intensif 5:530–535
Climent MJ, Corma A, De Frutos P, Iborra S, Noy M, Velty A, Concepción P (2010) Chemicals from biomass: synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts: the role of acid-base pairs. J Catal 269:140–149
Brooker AT (2011) A solid detergent composition comprising glycerol carbonate. EP Patent 2,380,958
Kargar M, Hekmatshoar R, Gandhi M, Mostashari A (2013) Use of glycerol carbonate in an efficient one pot and solvent free synthesis of 1, 3-sn-diglycerides. J Am Oil Chem Soc 90:259–264
Sonnati MO, Amigoni S, Givenchy Taffin EP, Darmanin T, Choulet O, Guittard F (2013) Glycerol carbonate as a versatile building block for tomorrow: synthesis, reactivity, properties and applications. Green Chem 15:283–306
Vieville C, Yoo JW, Pelet S, Mouloungui Z (1998) Synthesis of glycerol carbonate by direct carbonation of glycerol in supercritical CO2 in the presence of zeolites and ion exchange resins. Catal Lett 56:245
Teles JH, Rieber N, Harder W (1994) Preparation of glycerol carbonate. US Patent 5,359,094
Aresta M, Dibenedetto A, Nocito F, Pastore C (2006) A study on the carboxylation of glycerol to glycerol carbonate with carbon dioxide: the role of the catalyst, solvent and reaction conditions. J Mol Catal A 2579:149–153
Kimb SC, Kima YH, Lee H, Yoon DY, Song BK (2007) Lipase catalyzed synthesis of glycerol carbonate from renewable glycerol and dimethyl carbonate through transesterification. J Mol Catal B 49:75–78
Gomez JRO, Aberasturi OGJ, Madurga BM, Rodriguez AP, Lopez CR, Ibarreta LL, Soria JT, Velasco MCV (2009) Synthesis of glycerol carbonate from glycerol and dimethyl carbonate by transesterification: catalyst screening and reaction optimization. Appl Catal A Gen 366:315–324
Wang L, Ma Y, Wang Y, Liu S, Deng Y (2011) Efficient synthesis of glycerol carbonate from glycerol using lanthanum oxide as a base catalyst. Catal Commun 12:1458–1462
Liu Z, Wang J, Kang M, Yin N, Wang X, Tanand Y, Zhu Y (2014) Synthesis of glycerol carbonate by transesterification of glycerol and dimethyl carbonate over KF/γ-Al2O3 catalyst. J Braz Chem Soc 25:152–160
Hammond C, Sanchez JL, Rahim MH, Dimitratos N, Jenkins RL, Carley AF, He Q, Kiely CJ, Knight DW, Hutchings GJ (2011) Synthesis of glycerol carbonate from glycerol and urea with gold based catalysts. Dalton Trans 40:3927–3937
Fujita SI, Yamanishi Y, Arai M (2013) Synthesis of glycerol carbonate from glycerol and urea using zinc containing solid catalysts: a homogeneous reaction. J Catal 297:137–141
Takagaki A, Iwatani K, Nishimura S, Ebitani K (2010) Synthesis of glycerol carbonate from glycerol and dialkyl carbonates using hydrotalcite as a reusable heterogeneous base catalyst. Green Chem 12:578–581
Cardoso LAM, Alves W, Gonzaga ARE, Aguiar LMG, Andrade HMC (2004) Friedel–Crafts acylation of anisole with acetic anhydride over silica-supported heteropolyphosphotungstic acid (HPW/SiO2). J Mol Catal A 209:189–197
Gagea BC, Lorgouilloux Y, Altintas Y, Jacobs PA, Martens JA (2009) Bifunctional conversion of n-decane over HPW heteropoly acid incorporated into SBA-15 during synthesis. J Catal 265:99–108
Rana S, Mallick S, Rath D, Parida KM (2012) Characterization of novel Cs and K substituted phosphotungstic acid modified MCM-41 catalyst and its catalytic activity towards acetylation of aromatic alcohols. J Chem Sci 124:117–1125
Dias JA, Caliman E, Dias SCL (2004) Effects of cesium ion exchange on acidity of 12 tungstophosphoric acid. Microporous Mesoporous Mater 76:221–232
Izumi Y, Ogawa M, Urabe K (1995) Alkali metal salts and ammonium salts of Keggin-type heteropoly acids as solid acid catalysts for liquid-phase Friedal–Crafts reactions. Appl Catal A Gen 132:127–140
Soled S, Miseo S, McVicker G, Gates WE, Gutierrez A, Paes J (1997) Preparation of bulk and supported heteropoly acid salts. Catal Today 36:441–450
Kaba MS, Barteau MA, Lee WY, Song IK (2000) Nanoscale characterization of acid properties of heteropoly acids by scanning tunnelling microscopy and tunnelling microscopy. Appl Catal A Gen 194:1292–1298
Wan Q, Duan L, He K, Li J (2011) Removal of gaseous elemental mercury over a CeO2-WO3/TiO2 nanocomposite in simulated coal-fired flue gas. J Chem Eng 170:512–517
Yang H, Shi R, Zhang K, Hu Y, Tang A, Li X (2005) Synthesis of WO3/TiO2 nanocomposites via sol-gel method. J Alloy Compd 398:200–202
Akurati KK, Vital A, Dellemann JP, Michalow K, Graule T, Ferri D, Baiker A (2008) Flame made WO3/TiO2 nanoparticles: relation between surface acidity, structure and photocatalytic activity. Appl Catal B 79:53–62
Chimienti ME, Pizzio LR, Cáceres CV, Blanco MN (2001) Tungstophosphoric and tungstosilicic acids on carbon as acidic catalysts. Appl Catal A Gen 208:7–19
Kulkarni MG, Gopinath R, Meher LC, Dalai AK (2006) Solid acid catalyzed biodiesel production by simultaneous esterification and transesterification. Green Chem 8:1056–1062
Kaba MS, Barteau MA, Lee WY, Song IK (2000) Nano scale characterization of acid properties of heteropoly acids by scanning tunnelling microscopy and tunnelling. Appl Catal A Gen 194:129–136
Turney TW, Patti A, Gates W, Shaheena U, Kulasegaram S (2013) Formation of glycerol carbonate from glycerol and urea by metal monoglycerolates. Green Chem 15:1925–1931
Diaz CLB, Casilda VC, Marcos FR, Fernadez JF, Banares MA (2013) New concepts for process intensification in the conversion of glycerol carbonate to glycidol. Appl Catal B 129:575–579
Viswanadham B, Kumar VP, Chary KVR (2014) Vapor phase dehydration of glycerol to acrolein over phosphotungstic acid catalysts supported on niobia. Catal Lett 4:744–755
Bardin BB, Bordawekar SV, Neurock M, Davis RJ (1998) Acidity of Keggin-type heteropoly compounds evaluated by catalytic probe reactions, sorption microcalorimetry and density functional quantum chemical calculations. J Phys Chem B 102:10817–10825
Acknowledgments
ASR thanks Director, CSIR-IICT for project Assistant fellowship and BV and VPK thank CSIR-New Delhi for awarded Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Srikanth, A., Viswanadham, B., Kumar, V.P. et al. Synthesis and characterization of Cs-exchanged heteropolyacid catalysts functionalized with Sn for carbonolysis of glycerol to glycerol carbonate. Appl Petrochem Res 6, 145–153 (2016). https://doi.org/10.1007/s13203-015-0140-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-015-0140-z