
Abstract
The physiological functions of endogenous amyloid-β (Aβ), which plays important role in the pathology of Alzheimer's disease (AD), have not been paid enough attention. Here, we review the multiple physiological effects of Aβ, particularly in regulating synaptic transmission, and the possible mechanisms, in order to decipher the real characters of Aβ under both physiological and pathological conditions. Some worthy studies have shown that the deprivation of endogenous Aβ gives rise to synaptic dysfunction and cognitive deficiency, while the moderate elevation of this peptide enhances long term potentiation and leads to neuronal hyperexcitability. In this review, we provide a new view for understanding the role of Aβ in AD pathophysiology from the perspective of physiological meaning.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer's disease (AD) is an irreversible neurodegenerative disorder and the most common cause of dementia [1, 2], which clinically manifests as progressive cognitive impairment and is pathologically characterized by extracellular amyloid-β (Aβ) plaques and intraneuronal neurofibrillary tangles [3]. Indisputable human genetic evidence and abundant data from biochemistry, histology, and animal models have established that Aβ is a key player in the pathogenesis of AD. However, along with a series of failures in clinical trials for the treatment and prevention of AD targeting Aβ [4, 5], there is a growing debate about its critical role in the pathogenesis of the disease.
More than three decades have passed since Aβ was first identified in 1984 when Aβ was recognized as an endogenous neuropeptide that is physiologically metabolized in the central nervous system [6]. The Aβ sequence can be dated to ~ 500 million years ago, and the sequence homology in mammals exceeds 95% [7]. The conservation in evolution means that Aβ is critical to providing a selective advantage in the survival of species. Recently, accumulating studies have implied that Aβ plays roles in cognitive functions, synaptic functions, angiogenesis, antimicrobial response, tumor suppression, recovery from injury, and neurogenesis [8]. Especially, the roles at the synapse and antimicrobial role of Aβ [9,10,11], potentially explain the lack of efficacy and adverse effects in the clinical trials targeting Aβ production (Fig. 1).

Schematic representation of the suggested physiological and pathological roles of Aβ in the synapse. Created with https://biorender.com/.
The synapse is widely regarded as the basic biological structure of memory. As early as 1991, it was recognized that synaptic loss is a factor correlated with the cognitive deficit in AD [12] and an important cytopathological feature of cognitive decline [13]. It has been reported that Aβ regulates synaptic function in early AD [14, 15]. Given the pivotal role of the synapse in the mechanisms of learning and memory, elucidating how Aβ influences synaptic activity may benefit the understanding of AD pathology.
Here, we concentrate on evidence from research on the functions of Aβ in synaptic terminals. To begin with, several key points concerned with physiological conditions, which are usually omitted, will be elucidated. Next, the potential necessary and sufficient role of Aβ in synaptic function will be expanded into two parts (Table 1). The necessary role will be drawn from laboratory data in which Aβ itself was ablated or the generation pathway was blocked, mainly referring to amyloid precursor protein (APP) and BACE1 (β-site APP-cleaving enzyme 1). In contrast, the sufficient role will be discussed by underscoring the effect of moderately increased Aβ, but not toxic levels, on synaptic plasticity and neural excitability. Further, the underlying mechanism and several contradictions in these evidence will be listed. Last, we considered the physiological role of Aβ at the synapse in AD therapeutics and research on its pathology.
Keys Concerns with Physiological Aβ
Biogenesis and Metabolism
APP is encoded by 19 exons on the long arm of chromosome 21, of which exons 16 and 17 are responsible for encoding Aβ. APP family proteins are type I single-pass transmembrane proteins; the other two isoforms, amyloid precursor-like proteins 1 and 2 (APLP1/2) cannot produce Aβ peptide. According to the splicing sequence, APP695, APP751, and APP770 have been described most often, and APP695 is the main isoform in the human brain. See the biosynthesis and metabolic fate of Aβ in Figure 2.

Schematic representation of the biogenesis and metabolism of Aβ. (1) Canonical APP processing. APP inserted on the cellular membrane is cleaved by α-secretase in an amyloidogenic manner, and internalized APP is proteolyzed by β-secretase in subcellular compartments to produce Aβ; (2) Transporting. Aβ along with CTF is packaged into vesicles or is secreted into extracellular space, and Aβ can be transported intracellularly in both anterograde and retrograde directions; (3) Functioning at synapse. Aβ performs the function in the intra- and extra-cellular space, and the presynaptic nicotinic acetylcholine receptor (nAChR) mediates Aβ reuptake at the synaptic terminal; (4) Degrading. Aβ is transported by lipoprotein receptor-related protein (LRP) and receptor for advanced glycation end products (RAGE). In cells, Aβ can be degraded by insulin-degrading enzymes and neprilysin or be bound by peripheral substances. Created with https://biorender.com/.
Although Aβ is generated from APP in a complex manner, canonical processing by α/β/γ-secretase is dominant, including an amyloidogenic and a non-amyloidogenic pathway. The former pathway happens in subcellular compartments like endoplasmic reticulum/intermediate compartment, and Golgi apparatus/trans-Golgi network [16,17,18], where internalized APP is proteolyzed by β-secretase on the 671–672 amino-acid sequence [19], exposing the N-terminus of Aβ, and then γ-secretase works to generate the C-terminus, forming a chain with 37–49 amino-acids named Aβ. Generally, Aβ40 (~ 90% of total Aβ) and Aβ42 (~ 5%–10% of total Aβ) are predominant [20, 21], and Aβ42 is more prone to deposition than Aβ40 due to the strong hydrophobicity of the C-terminal amino-acid residue. The unhydrolyzed APP is located at the cell surface and is processed by the latter means, in which α-secretase cleaves at amino-acids 16–17 on the Aβ sequence to generate a soluble fragment αAPPs and α C‑terminal fragments, which are further catalyzed by γ‑secretase to generate p3 [22, 23].
The mature Aβ along with the C-terminal fragment (CTF) is packaged into vesicles or is secreted into extracellular space. Intracellularly, Aβ can be transported in both anterograde and retrograde directions. APP [24] as well as somatic Aβ [25] are transported in the fast anterograde component, while retrograde transport to cell bodies occurs when Aβ is absorbed by synaptic reuptake or is produced by APP internalized from distal axon terminals [26]. Besides, transport of the compartment containing BACE and PS1 requires APP, which may function as a kinesin-I membrane receptor [27]. After performing its function in the intra- and extra-cellular space (see below), Aβ under physiological conditions maintains a balance that relies on a clearance mechanism. On the one hand, central Aβ can be transported through the blood-brain barrier mediated by lipoprotein receptor-related protein and receptor for advanced glycation end products. On the other hand, Aβ can be degraded by insulin-degrading enzymes and neprilysin, or be bound by peripheral substances [28]. In addition, Aβ reuptake into neurons occurs in the presynaptic compartment [29].
Distribution and Localization
Central Aβ is mainly produced in the brain. The cerebral cortex and hippocampus are believed to be regions that are enriched in Aβ and start their propagation. In AD brains, the Aβ deposits first appear in the neocortex, followed by allocortical regions, diencephalic nuclei, the striatum, and the cholinergic nuclei of the basal forebrain [60]; the entorhinal cortex is one of the most vulnerable regions [61]. Similarly, in normal brains, the cortex and hippocampus strongly express APP, suggesting the regions where Aβ abounds [62, 63].
The distribution of Aβ in subtypes of neural cells can be revealed by evidence of APP, and β- and γ-secretase. Although APP is widely expressed in a variety of tissues and cells, previous studies have shown that Aβ is more readily metabolized in neurons where BACE1 protein is abundant [38, 64], whereas other cell types mainly express BACE2, which is not involved in amyloidogenesis [65]. Similarly, neuronal APP has been identified as predominantly APP695 [66]. Besides neurons, glial cells, endothelial cells [67], and meninges [68] also express APP. Early studies showed that APPs in microglia and astrocyte were expressed in internal membranous vesicles [69] as isoforms containing Kunitz-type protease inhibitors [68, 70,71,72,73] rather than APP695. It was believed that the main source of Aβ was not glia cells but neurons [74], except for type I (GFAP+ A2B5–) astrocytes [75] or in a morbid environment [76]. Although this evidence is still in vitro, the contribution to the physiological Aβ biogenesis of astrocytes should be stressed since high levels of Aβ have been detected in human iPSC-derived astrocytes [77].
However, the specific neuron type that generates Aβ is still controversial. An immunocytochemical analysis has shown that APP is more frequently associated with glutamatergic rather than GABAergic or cholinergic terminals, indicating that endogenous Aβ is predominantly derived from excitatory neurons [78]. It has been reported that reducing neuronal activity using GABA-A receptor enhancers or increasing it with GABA-A channel blockers significantly reduces or increases Aβ levels (both Aβ40 and Aβ42) [6], stressing the contribution of GABAergic neurons to Aβ production. Given the high expression of APP in a heterogeneous subset of GABAergic interneurons, it has been reported that these interneurons take part in ~ 17% of the soluble Aβ and ~ 30% of the total hippocampal plaque burden, and interneurons are also located in the CA1 region, where plaques are most prevalent (accounting for ~ 75%) [79].
Within neurons, Aβ is located in neurites [80, 81]: biochemical, immunostaining, and electron microscopic studies have found APP and its fragments [66] in dendrites and axon terminals [24, 82]. Further, Aβ is supposed to be primarily released by synapses [83, 84]. It has been reported that Aβ levels in the brain interstitial fluid are considerably regulated by synaptic activity and synaptic vesicle exocytosis, implicating a mechanism on the presynaptic side of the synaptic cleft [51]. Notably, neuronal activity-dependent endocytosis of APP is involved in ~ 70% of the regulatory mechanisms in synaptic Aβ release [52].
In synapses, Aβ is predominantly distributed in the presynaptic membrane [78, 85]. Consistent with this, a meta-analysis of AD synaptic pathology showed that presynaptic markers are affected more than postsynaptic markers [86]. In normal or 5XFAD mice, BACE1 is localized to vesicles (possibly endosomes) at the ends of hippocampal mossy fibers, and in some cases, BACE1-positive vesicles are located near the synaptic active zone, suggesting Aβ production in the presynaptic membrane [87]. And it has been found that APP and BACE1 interact in biosynthesis and endocytosis, particularly along circulating microdomains such as dendritic spines and presynaptic boutons [88]. However, it has also been shown that in cultured murine neurons, γ-secretase is located both presynaptically and postsynaptically [89]. Furthermore, a recent super-resolution microscopy study found that co-labeling with APP is stronger postsynaptically than presynaptically [90]. Therefore, more evidence is needed to clarify the distribution of Aβ at the synapse.
Dosage Effect
Under physiological conditions, the level of Aβ in the human brain and cerebrospinal fluid lies in the picomolar range [91, 92]. As the studied concentrations of Aβ42 ranged from femtomolar to millimolar, covering over twelve orders of magnitude [93], concentration matters for the physiological function of Aβ. According to the existing results, both too high and too low Aβ has negative effect on synaptic function, but only positive regulation has been reported in the physiological concentration range.
First, the detrimental effect of high levels of Aβ has been analyzed in the brains of AD and AD animal models. In these brains, Aβ concentrations tend to be in the nanomolar-to-micromolar range [94], which is much higher than the physiological level, thereby impairing synaptic function [95,96,97]. For example, the senile plaque requires a concentration of 100 nmol/L to aggregate Aβ, while Aβ42 is considered to gather when the concentration is up to 90 nmol/L [98]. Consequently, high concentrations of Aβ oligomers can cause the collapse of dendritic spines [84, 99] and disruption of LTP [100]. As the toxicity of pathologically overloaded Aβ is beyond the scope of this paper, further summary can be seen in reviews [14, 101]. Interestingly, inhibition of endogenous Aβ does not protect synaptic transmission as verso of a phenomenon in AD. The genetic knockout (KO) and pharmacological inhibition of Aβ production also have adverse effects on synapses. Varying degrees of cognitive deficits and synaptic damage are induced by knocking out the APP or BACE1 gene, interfering with siRNA, or applying an inhibitor to wild-type mice, and interestingly, some evidence suggested this damage can be rescued by moderate amounts of Aβ (Table1).
Second, however, positive effects on synaptic regulation have gradually been discovered. Several studies have shown (Table1) that low concentrations (picomolar) of Aβ can enhance LTP [31], increase dendritic spine density [59], and promote docking vesicles [36]. The dose-dependence was demonstrated in an electrophysiological study at different concentrations (100, 200, and 300 pmol/L) of Aβ [49]. In another study, the full recovery of potentiation was at 300 pmol/L Aβ42, the threshold required for normal synaptic plasticity may be ~ 380 pmol/L [31]. Notably, the APP mutant A673T reduced Aβ by 40%–50% [102, 103] in a laboratory study and by ~ 28% in human plasma [104], which is thought to be protective against AD. Compared with mutations accelerating AD, the A673T mutation seems to reveal Aβ maintains a delicate balance to be a friend or foe in a dose-dependent manner.
Although the precise concentrations of Aβ to execute different acts in synaptic function are controversial, according to the available evidence, a “hormetic effect” seems to exist in Aβ roles: that is, a positive effect in the optimal dose range and a negative effect either above or below the range. The hormesis hypothesis may be a suitable explanation for the etiology of sporadic AD.
Species Differences
Although it was recognized as early as the mid-1980s that Aβ is an endogenously-produced peptide, significant deposition of Aβ is often achieved by chimeras in animals with humanized mutations, so as to partially mimic the pathology of anthropic AD. However, the animal sequences of Aβ are distinct from humanized fragments to some extent. Blockade of endogenous Aβ with specific antibodies or ablation of APP expression impairs LTP and memory function [31]. Conversely, neurotransmitter release and recycling of synaptic vesicles are enhanced by increased endogenous Aβ1-40 or Aβ1-42 via interfering with clearance, or by applying picomolar amounts of synthetic fragments [53, 55, 105]. Therefore, the species differences in Aβ sequences matter as they function in synaptic regulation, although this seems to be complex.
On the one hand, 96.6% consistency has been identified between human and mouse APP, and only three amino-acid residues differ in the Aβ sequence [106]. However, endogenous picomolar Aβ does not induce Ca2+ homeostasis and synaptic integrity in neurons in mice, while high concentrations of Aβ from Tg2576 primary cortical neurons cause Ca2+ overload and synaptic damage [99]. This may be due to the sequence difference itself, or changes that occurred during biogenesis [107]. On the other hand, the impaired LTP, contextual fear memory, and reference memory induced by anti-rodent Aβ antibodies and siRNA against murine APP can be rescued by human Aβ42 [31]. Likewise, deletion of the Drosophila APP-like protein (Appl) is not lethal but has subtle behavioral defects that are partially rescued by expressing human APP [108]. Interestingly, the function of humanized APP varies with different mutations. Knockout of APP results in a significantly shorter body length and a short, curly tail in zebrafish. Wild-type human APP, rather than Swedish mutant APP, a mutation associated with familial AD, prevented these phenotypes [109]. In summary, the evidence suggests subtle relationships among Aβ sequences in various species, which needs more studies to clarify how much its functions are distinct or overlap.
Isoforms and Aggregation
More than 20 forms of Aβ can be produced by enzymatic reactions and modification. Physiologically, Aβ40 is in the majority while Aβ37, Aβ38, Aβ39, and Aβ42 are in the minority, and peptides such as Aβ34, Aβ36, Aβ41, and Aβ43 are detectable in some instances [91, 110, 111]. Aβ segments are highly ordered, with 95% sequence identity between Aβ42 and Aβ40, except for a C-terminus of increased rigidity at Aβ42, which makes Aβ42 more prone to aggregation than Aβ40 [112]. As Aβ varies among monomers, oligomers, fibrils, and mature plaques, it remains difficult to identify the roles of endogenous pathological Aβ in AD patients. A widely held view is that Aβ oligomers, rather than fibrils or monomers, are the neurotoxic forms [100]. In the late 20th century and early 2000s, several studies showed that the soluble form of Aβ causes the loss of dendritic spines in cultured neurons, whereas fibrils and monomers are relatively inert [113,114,115]. Even at physiological concentrations, Aβ dimers, trimers, but not monomers, are deemed to cause synaptic dysfunction and loss [116]. However, the latest research on PS1 and PS2 conditional double-KO mice has shown that a reduced Aβ42 level is harmful to cognitive function, and the cognitive decline can be alleviated by giving exogenous soluble Aβ1-42 monomers [46].
In addition to the aggregated form, Aβ monomers themselves are also thought to have different functions. The hydrophobic C-terminal domains associated with oligomer formation are closely associated with neurotoxicity, especially at high levels (μmol/L) of Aβ. However, the hydrophilic N-terminal domain may mediate the protective action of Aβ at physiological levels (pmol/L–nmol/L). It has been found that the N-terminal Aβ fragment and shorter Aβ core (Aβ10–15) protect against or even reverse the effects of Aβ-induced neurotoxicity, memory deficits, and apoptosis [57]. Moreover, the rescue effect also occurs in 5XFAD mice and APP/PS1 mice, transgenic models of AD with significant Aβ deposition, especially at the level of synaptic plasticity [56]. Furthermore, a recent study has shown that fragments containing Aβ1–16 but not Aβ17–42 increase the size of the recycling pool of synaptic vesicles [54]. Therefore, it is critical to clarify the specific length of segments and aggregative form of Aβ in vivo when we explore its function.
Potential Physiological Roles of Aβ in Regulating Synaptic Function
Reduced Endogenous Aβ Impairs Synaptic Function
Blocking Aβ by Antibodies
The absence of Aβ appears to be detrimental to synapses. After antagonizing endogenous Aβ42 in rodents using JRF/rAb2 [31] or 4G8 [30], animals displayed cognitive deficits in behavior tests and impairment of LTP in electrophysiology. Moreover, injection of human Aβ42 rescued the above phenotypes, suggesting that endogenous Aβ plays a crucial role in normal LTP and memory. Abramov et al. further revealed a mechanism indicating that Aβ may positively regulate basal synaptic transmission in a presynaptic and history-dependent manner, particularly in excitatory neurons [55]. It was reported that a reduction in presynaptic strength by 53% ± 6% and inhibited exocytosis of synaptic vesicles occurred after using the monoclonal antibody HJ5.1 against murine Aβ, and this was reversible after a 30-min washout. In fact, the facilitation of vesicle release is diminished by both increasing and decreasing the endogenous extracellular Aβ concentrations. Similarly, short-term facilitation, which is believed to be closely related to memory formation, is not only impaired when Aβ excessively increases but when it dramatically decreases (> 60%), further suggesting that the action of Aβ exhibits dose-dependent [55].
Notably, Aβ is also thought to be involved in memory consolidation [30] and the forgetting mechanism by preventing subsequent modifications to provide adaptive physiological functions [45]. For instance, intracerebroventricular injection of 4G8 or knockdown of Fcgr2b, a receptor for soluble Aβ, regulates memory maintenance and forgetting in a novel object recognition test [117].
Deficiency or inhibition of APP
In early studies, APP-null mutant mice showed weight loss, abnormalities in locomotion, astrocyte gliosis at 14 weeks [32], and age-dependent cognitive deficits [33], with the cognition, altered weakly [34]. Recently, a reduction of LTP was reported when APP was knocked out or siRNA [31] interference was applied. In primary hippocampal neurons from APP-KO mice, there was synapse loss, restricted neurite growth, and reduced branching [35]. Interestingly, APP released by astrocytes was able to partially rescue this defect [118]. Furthermore, the absence of APP was shown to increase neuronal excitability. Although the two homologous analogues of APP, APLP1, and APLA2, do not produce Aβ, hippocampal neurons exhibit hyperexcitability when all three APP family genes are knocked out simultaneously in excitatory neurons [37]. Similarly, genetic loss of APP selectively impairs GABA-B receptor-mediated presynaptic inhibition and reduces axonal GABA-B receptor expression [119], indicating that this is a potential mechanism by which APP can regulate synaptic activity. The above studies that directly target APP somewhat of a contribution of Aβ to synaptic structural development and functions, but the role of APP itself should not be ignored.
Deficiency or Inhibition of BACE1
As a rate-limiting enzyme in Aβ processing, using BACE1 inhibitors seems to be a viable approach to attenuating Aβ and then benefiting AD. However, although significantly reducing Aβ production and amyloid deposition in the brain, BACE1 inhibitors can not improve the cognitive or functional decline in subjects with mild-to-moderate AD [120,121,122]. Compared to placebo, individuals who received a BACE1 inhibitor showed a dose-dependent cognitive deterioration and treatment-related adverse events, such as neuropsychiatric deficits and hippocampal volume loss in phase II and III clinical trials [123, 124], leading to the early termination of clinical trials. Interestingly, cognition returned to baseline levels after cessation of treatment [124]. This clinical evidence suggests that, at least in AD, remarkably reducing Aβ with a BACE1 inhibitor needs to be approached with prudence.
While clinical data are always limited to AD patients, pharmacological inhibition or genetic modification of BACE1 in wild-type mice can partly reveal the physiological roles of Aβ. In animal experiments, gavage of the blood-brain-barrier-permeable BACE1 inhibitors Verubecestat (MK-8931) and Lanabecestat (AZD3293) to mice resulted in a dose-dependent decrease in LTP [125]. Oral administration of the BACE1 inhibitors SCH1682496 or LY2811376 also caused a dose-dependent decrease in Aβ40 levels, but prolonged treatment suppressed dendritic spine formation in layer V pyramidal neurons, which recovered after drug discontinuation [44]. Consistent with this, BACE1-deficient mice exhibit impaired synaptic transmission and plasticity, evidenced by reduced LTP in Schaffer collateral branch-to-CA1 synapses and mossy fiber-to-CA3 synapses [41, 43]. In another study, deficits in paired-pulse facilitation and de-depression implicated in presynaptic release and synaptic plasticity were recorded in BACE1(-/-) mice, and the poor performance on tests of cognition was prevented by APP/PS1 transgenic mice [38]. Moreover, it has been suggested that inhibition or deficiency of BACE1 leads to reduced docking of synaptic vesicles to the active zone and the ensuing glutamate release [125]. To avoid the developmentally-relevant phenotypes in germline mutant mice, researchers have turned to conditional KO of exon 2 of BACE1 in adult mice, in which impairment of synaptic and axonal function also occurs [39, 126]. For example, in BACE1fl/fl; R26CreERT2-TAM mice, BACE1 is reduced by 90%–95%. and Aβ is inhibited by ~ 60%–90%, followed by axonal dysfunction [126]. Collectively, the failure of BACE1 inhibitors, which cause a strong reduction in Aβ deposition, may largely be due to their role in synaptic function.
Given the harmful effects associated with synaptic damage, it appears that complete or significant inhibition of BACE1 neutralizes or even overwhelms the anticipated therapeutic effect against an Aβ burden. Both clinical [123, 124] and laboratory [125] results have confirmed that this impairment is dose-dependent; in addition, this can be partly explained by the fact that germline heterozygous BACE1-KO mice with 50% of normal BACE1 levels do not differ significantly from wild-type mice [38, 127, 128]. Therefore, the dosage is vital. Admittedly, however, seizures [129, 130], axon guidance [131], impaired peripheral nerve myelination [132, 133], and low anxiety or depressive tendencies [38] have been sequentially reported in BACE1-null mice. All these side-effects are consistent with adverse events in clinical trials with BACE1 inhibitors [123, 124], although some of the phenotypes remain controversial [42]. This indicates that the functions of BACE1 itself should be taken into account.
Collectively, Aβ is necessary for maintaining the normal synaptic function, reduced endogenous Aβ by genetic or pharmaceutical inhibition of Aβ or its biogenic necessities, APP and BACE1, disturb synaptic morphology, synaptic vesicle transmission, synaptic plasticity, and even cognitive function. Although several studies have revealed a rescue effect [30, 31, 47] and implied a dosage effect, more detailed and persuasive results are needed to draw firmer conclusions.
Interplay: Aβ and Neural Hyperexcitability
Moderately Increased AΒ Enhances LTP and Neuronal Excitability
As previously noted, the body produces Aβ endogenously at picomolar concentrations, and either too low or too high Aβ may impair synaptic function. However, a number of early and recent studies have demonstrated that a modest increase of Aβ can enhance synaptic transmission and neuronal excitability, providing further evidence for its physiological function. Puzzo et al. have worked long on the role of Aβ in physiological states, particularly in synaptic regulation and cognitive function. They initially administered intrahippocampal injections or delivered picomolar levels of Aβ42 to mouse brain slices, and found that Aβ42 enhanced LTP and behavior performance [50]. Given the toxic effects of excess Aβ42 in AD, the team investigated the LTP variation with different concentrations of Aβ42 in order to clarify the dose-effect relationship, which finally took on a bell-shaped curve [49]. Besides, the exposure time also matters [48]. Furthermore, by inhibiting thiorphan, an enzyme degrading Aβ in the synaptic cleft, the acute effects of endogenously-released Aβ were investigated at single presynaptic terminals and synaptic connections [53, 55]. These studies demonstrated that Aβ mediates presynaptic enhancement and synaptic transmission by increasing miniature synaptic vesicle release and mEPSC frequency, which depends on the history of activation. As deprivation of endogenous Aβ reduces presynaptic activity, it has been speculated that Aβ maintains basal presynaptic activity and spontaneous activity [55]. In recent years, more studies have focused on the aggregated forms and effective sites of Aβ. So, several studies have shown that it is the N-terminal Aβ, particularly the 1–16 fragment, that exerts excitatory effects and promotes vesicular recycling [54, 58], even reversing the Aβ toxicity. Besides, Aβ42 oligomers, commonly regarded as toxic, have been reported to enhance synaptic plasticity at picomolar concentrations [36, 46, 47].
Since the direct application of soluble Aβ in wild-type mice increases neuronal activation [134], what is the situation in early AD or AD model mice with a mild to moderate increase in Aβ?
In vivo Ca2+ imaging of somatic, dendritic, and axonal activity patterns in cortical neurons has shown that both healthy ageing and AD-related mutations have neuronal hyperactivity [135]. In the hippocampus of young AD model mice, hyperexcitable neurons are selectively increased prior to plaque formation. In these animal models, acute treatment with the γ-secretase inhibitor LY-411575 reduces soluble Aβ levels and rescues the neuronal dysfunction, while administration of soluble Aβ oligomers re-establishes the excitatory state [134]. Here, soluble forms rather than aggregates matter. However, in the AD mouse model, two-photon data displayed that not all neuronal activity is reduced or increased, and it is in the vicinity of plaques where part of the neurons with hyperactivity are exclusively found [136, 137]. The mechanism is attributed to the fact that low levels of Aβ enhance glutamate release and regulate Ca2+ homeostasis, particularly in the early stages of AD [99].
Aβ is altered 20–25 years prior to the onset of AD [138, 139]. Individuals at risk for AD always manifest hyperactivation in memory-related brain regions in functional magnetic resonance imaging (fMRI). APOE (apolipoprotein E) ε4 carriers at 25–35 years old present increased co-activation of the default mode network and a more activated hippocampus during encoding tasks compared to non-carriers in fMRI studies [140]. Another study not only found that cognitively normal APOE ε4 allele carriers have a greater magnitude and greater extent of brain activation than APOE ε3 allele carriers during a memory activation task but also showed that the extent of baseline brain activation correlated with the degree of memory decline after a 2-year longitudinal assessment [141]. In addition, young subjects with normal cognition who carry the familial AD gene E280A PS1 mutation, have hippocampal activation before the onset of symptoms at ~ 45 years old [142]. The same phenomenon has been demonstrated in patients with amnestic mild cognitive impairment (aMCI) [143,144,145,146]. Therefore, increased Aβ at an early stage may take part in the regulation of cognitive function, possibly by inducing synaptic dysfunction, but the underlying mechanisms remain to be solved.
Neural Hyperexcitability Promotes Aβ Production
Endogenous Aβ increases neuronal excitability [147], while neural activity also regulates Aβ production. Laboratory studies have shown that neuronal and synaptic activity dynamically regulates soluble extracellular Aβ concentrations [6, 31, 148]. The rapid effects (a timescale of minutes to hours) of synaptic activity on Aβ were investigated by microdialysis combined with field potential recordings, in which it was demonstrated that synaptic activity dynamically and directly regulated Aβ in the brain interstitial fluid (ISF) [51]. Further, ISF Aβ levels were elevated by enhancing synaptic transmission and were prevented by inhibiting endocytosis mediated by clathrin. The above evidence suggests that Aβ release depends on synaptic activity mediated by endocytosis [52].
Furthermore, clinical phenomena abound suggesting that alterations of brain activity are accompanied by changes in Aβ level. The regions active in the default state in young adults have a higher propensity for Aβ deposition in the old with AD [149]. In addition, evidence from epilepsy and post-traumatic states provide a good illustration. First, patients with epilepsy, particularly late-onset epilepsy of unknown etiology, are at higher risk of developing dementia. Simultaneously, seizures have been detected in the early stages of AD [150]. As previously noted, because patients with aMCI exhibit elevated hippocampal activation in the dentate gyrus or CA3 region, Bakker et al. [151] reduced hippocampal hyperactivity in aMCI with the antiepileptic drug levetiracetam, and, as expected, cognitive function was improved. Second, the increased ISF Aβ in 18 patients with acute brain injury showed a strong positive correlation between Aβ level in the ISF and neurological status [152]. The fact that ISF Aβ varies along with neuronal function further implies that the extracellular Aβ level is regulated by neuronal activity.
Underlying Mechanisms in the Regulation of Aβ at Synapses
nAChR
In the central nervous system, the nicotinic acetylcholine receptors (nAChRs) are located at synapses in most neuron populations [153] as well as being expressed in non-neuronal cells [154,155,156]. nAChRs are ligand-gated ion channels. Depolarization of the membrane and excitatory effects are caused by the application of nAChR agonists followed by opening ion channels, and consequently, increasing permeability to Na+/K+/Ca2+. The α7 nAChR has the highest Ca2+ permeability among nAChR isoforms, so its relative permeability is comparable to that of the N-methyl-D-aspartate receptor (NMDAR) [157]. Overall, α7 nAChRs are involved in a variety of biological processes, including neuronal excitability, neurotransmitter release, signal transduction, synaptic plasticity, and neurogenesis [158,159,160].
In the brains of AD patients, the reduction of nAChRs is correlated with disease progression [161, 162], and cholinesterase inhibitors are widely used in the treatment of mild to moderate AD. α7 nAChRs have been found to co-localize with intracellular Aβ42-positive neurons in the post-mortem brain tissue of AD patients [163]. Similarly, an increase of Aβ/nAChR-like complexes has been found in carriers of APOE ε4 [164], a strong risk factor for AD [165]. In fact, nAChRs interact with Aβ under physiological conditions, particularly the α7 isoform, which has a high affinity for Aβ [166, 167]. Furthermore, 12-month-old α7 KO mice exhibit an AD-like pathology, in which elevated Aβ is thought to be a compensatory response to the deletion of nAChRs [168]. It has been reported that low levels of Aβ (picomolar the low nanomolar range) activate α7 nAChR channels [50, 169] possibly via the nitric oxide/cGMP/protein kinase G pathway [36]. In contrast, higher levels (nanomolar the low micromolar range) reduce the duration of ACh-induced activation [170], leading to dysregulation of electrical activity at synapses [171]. However, controversially, nicotine, another ligand of nAChRs, is reported to improve cognition and protect neurons from Aβ damage by agonizing nAChRs [172, 173]. This paradox has been explained by the suggestion that different cellular pathways and downstream mechanisms are initiated. That is, nicotine acts through PI3K–AKT, JAK–2/STAT-3, and other mechanisms to exert protective effects, whereas Aβ is thought to initiate intracellular signaling cascades like the MAPK kinase pathway and leads to cell death [174].
In addition, nAChR subtypes other than α7 participate the synaptic mechanism of Aβ. For instance, α7β2, a variant of α7, is more sensitive to pathological concentrations of Aβ [175]; mice with β2nAChR deletion display neurodegeneration [176] despite the amelioration of spatial reference memory in APP/PS1 mice by β2 deficiency [177]; and α4β2 nAChR is particularly associated with episodic memory and working memory [178], while selective co-activation of α7 and α4β2 nAChRs is also sufficient to reverse Aβ-induced AMPA receptor dysfunction and LTP alterations. Due to the structural differences of nAChRs [179], Aβ might interact with specific subtypes to varying degrees. Aβ and nAChRs form complexes through multiple sites [180] to mediate the physiological effects of Aβ or toxicity to cholinergic neurons. For example, when cell lines expressing α4β2 nAChRs are exposed to nanomolar Aβ 42, the expression of genes related to Ca2+ signaling and axonal vesicle transport is upregulated while genes related to metabolic, apoptotic, or DNA repair pathways are downregulated [181]. Notably, the results did not mimic physiological stations because the high concentration of Aβ and overexpressed nAChR receptors were used in this research.
The complexity of relationships between Aβ and nAChRs is evident, but the dose of Aβ applied and the aggregation state still need to be considered [56, 182]. Although nAChRs are weakly expressed in AD, they maintain normal or even increased mRNA (for review see [174]). Besides, the extreme susceptibility of nAChRs to desensitization may partially explain the paradox between nicotine and Aβ, or even the variation of the Aβ dosage effect.
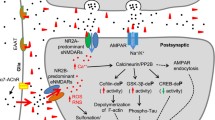
The N-methyl-D-aspartate Receptor
The NMDAR belongs to the ionotropic glutamate receptor family, and it enhances synaptic transmission and plasticity [183] mediated by Ca2+/calmodulin-dependent protein kinase II [184, 185], which triggers a signaling cascade. The NMDAR has been found to be critical for neurons [186, 187]. Antagonism of NMDARs gives rise to apoptosis and degeneration, while moderate activation of this receptor benefits neuron survival; however, the excessive activation of NMDARs causes Ca2+ overload, resulting in excitotoxicity. Therefore, both inactivation and overactivation are potentially harmful [188, 189]. In addition, it has recently been suggested that synaptic NMDARs and extrasynaptic NMDARs play very different roles [190, 191], where the former is thought to be beneficial and the latter to mediate toxic effects [183, 192,193,194,195].
Accumulation of Aβ oligomers has been observed in the synapses of glutamatergic neurons in AD brains [196, 197]. Although plenty of studies have demonstrated that Aβ mediates neurotoxicity by directly or indirectly regulating NMDARs [6, 198,199,200,201,202], and NMDAR antagonists can rescue Aβ-induced damage [116, 203], interestingly, genetic deletion of the NMDAR subunit GluN3A results in neuropathological changes like AD, including psychological/cognitive deficits and amyloid-β/tau pathology [204]. Moreover, blocking NMDARs may reduce neurodegeneration [205]. Therefore, as a non-competitive, specific, low-affinity NMDAR antagonist with a fast closing rate, memantine is used to treat moderate to severe AD [206] since it can reduce excitotoxicity while preserving normal NMDAR activity at the same time. Notably, it has been shown that memantine preferentially targets the extrasynaptic NMDAR [193] which is regarded as a detrimental characteristic of AD.
The multiple possibilities for NMDARs in terms of dose, subunit type, and subcellular localization make research on the relationship between Aβ and NMDARs difficult. Complete inhibition, low to mild activation, and over-activation have dramatically distinct effects. Besides, different subunits vary: for example, GluR2A and GluR2B each interact with Aβ to cause opposite results [207]. Moreover, careful investigation is needed, for example, on the roles of D-serine and glycine, co-agonists of synaptic NMDARs and extrasynaptic NMDARs, respectively [208]; which downstream pathways are activated by NMDARs with different subcellular localizations; whether or not NMDARs are translocated on the cell membrane.
Vesicular Circulation
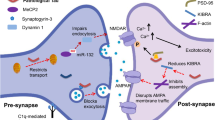
The synaptic vesicle cycle (SVC), comprising vesicle trafficking, docking, fusion, transmitter release, and regeneration of fresh vesicles [29], plays a crucial role in the biology of synaptic terminals by way of recurrent exocytosis and endocytosis [209]. Due to the strong positive correlation between cognitive decline and synaptic loss [13, 210], research on synapses exposed to Aβ is increasing [14, 211]. A convergence of results points out a reciprocal relationship between Aβ and the SVC. For one thing, Aβ regulates the SVC via dosage effect, sites of action (pre- and post-synaptic), and pattern of action (local autocrine or paracrine), for another, the SVC also affects the production of Aβ [29].
Studies so far suggest that the SVC can be regulated by Aβ. The absence of Aβ impairs vesicular docking in active zones [125]. Picomolar or low levels [28] of Aβ have been shown to enhance synaptic transmission by upregulating the presynaptic neurotransmitter release probability (Pr) [55]. Furthermore, Lazarevic et al. systematically studied the concentration effect in Aβ regulation at the synapse. There, they found that the SVC decreases when Aβ is depleted by modulating production while the SVC increases by using an endogenous Aβ degradation inhibitor [53, 54].
On the contrary, a high level of Aβ inhibits Pr [96]. Either natural or synthetic Aβ oligomers but not monomers [116] at high doses inhibit synaptic transmission and plasticity [212,213,214]. Intracellular administration of nanomolar Aβ42 significantly cuts down LTP, reduces mEPSC amplitude, and decreases the number of intrasynaptic vesicles and/or Pr [215]. Moreover, direct injection of Aβ42 oligomers into presynaptic axon terminals results in a blockade of synaptic transmission [216], and even acute exposure to Aβ oligomers reduces postsynaptic current frequency by ~ 50% [116]. A series of studies have proposed that Aβ is involved in many steps of the SVC. First, Aβ perturbs the formation of fusion complexes, as reported in postmortem AD brains, where the SNARE complex, which is essential in driving synaptic vesicle fusion in the presynaptic active zone, is significantly reduced [217]. Second, the interaction of SNARE protein vesicle-associated protein 2 (VAMP2) with synaptophysin is necessary and sufficient to recruit VAMP2 to synaptic contacts, and it is disrupted by internalized Aβ42 [218]. Further, the ability of clathrin-dependent endocytosis is a critical step in the SVC, and a wealth of evidence, including genomics and proteomics, shows that such endocytosis is severely disturbed in AD [219,220,221,222]. In Aβ oligomer-treated neurons, only 50% of the released vesicles are recycled back in time, leading to a considerable delay in readily-releasable pool recovery [223]. In particular, atypical cyclin-dependent kinase 5 (CDK5) [224] plays a major role in regulating the size of the synaptic vesicle pool by targeting synaptic vesicle endocytosis [225], and consistently, CDK5 is significantly higher in postmortem AD brains [226, 227]. Although substantial studies have been devoted to the mechanisms of Aβ toxicity, from another perspective, some of these results also imply that Aβ is a potent target for presynaptic regulation both in physiology and pathology. Together, endogenously released Aβ peptides are crucial for maintaining a normal SVC in the functional range.
Vice versa, the SVC takes part in Aβ production [22, 23]. Indeed, non-amyloid cleavage of APP occurs on the cell membrane, while amyloid cleavage of APP by β- and γ-secretase is facilitated in vesicles, leading to Aβ production and release [19]. This process has been shown to be upregulated by increased neuronal activity and clathrin-dependent endocytosis [51, 52], thereby promoting Aβ production and even affecting the ratio of Aβ42 to Aβ40 [55].
Other Mechanisms
Other than the above mechanisms, glial cells, energy metabolism, and other factors participate in the role of Aβ in synapses through direct or indirect regulation. (1) Microglia. On the one hand, microglia can be activated by Aβ and then mediate synapse pruning and elimination [228]; on the other hand, the activated immune system influences the aggregation of Aβ to cause diverse effects [229] (e.g. microglia secrete galectin3 to promote the oligomerization of Aβ [230]). (2) Astrocytes. Astrocytes participate in the processes of synapse engulfment [231], besides which, they secrets APOE, a risk factor of sporadic AD, to result in synaptic degeneration by enhancing the abnormal aggregation of Aβ at synapses [232, 233]. (3) Energy metabolism. The synapse is vulnerable to energy deficiency as a highly energy-consuming structure, especially in vesicle cycling [12, 234]. It has been shown that subthreshold amyloid deposition or the distribution of Aβ is correlated with increased aerobic glycolysis in early adulthood [235,236,237], whereas aerobic glycolysis decreases in the normal aging brain [238].
Perspectives
The underlying role of Aβ in regulating synaptic functions seems to have being revealed gradually. However, several key limitations need to be noted. (1) Animal models. Although diverse animal models have been developed to study AD and Aβ, most of them are genetically-manipulated mice carrying mutations of human familial AD [106, 239,240,241]. Species differences should be taken into account when they poorly mimic the pathological process of human AD. In fact, human-derived Aβ fragments are more likely to be deposited, and hAPP transgenic mice without expression of endogenous murine APP display more plaques and faster Aβ deposition [230, 242]. (2) The role of APP or BACE1. APP performs its functions concurrently with its various products including Aβ, all of which also join in the regulation of synapses. For example, sAPP contributes to synaptic function [243, 244], partly as a ligand to regulate synaptic transmission [245]. Possibly, Aβ does not act alone [246]. Similarly for BACE1, in models with BACE1 deletion, impaired axonal guidance is associated with reduced hydrolysis of CHL1 (cell adhesion molecule L1-like) [131], and synaptic damage is associated with seizure protein 6 [247], both of which are substrates of BACE1. (3) The complexity of Aβ itself. All the following factors matter in Aβ functions: length of fragments [248], concentration, intracellular or extracellular localization, and aggregation state [47]. For instance, nanomolar concentrations of intra-axonal oligomeric Aβ42 (o Aβ42), but not oAβ40 or extracellular oAβ42, acutely inhibit synaptic transmission in squid [6]. Besides, the effect of Aβ varies with time under both physiological [48] and pathological conditions [249]. Population studies have shown that Aβ deposition does not increase all the time, while CSF Aβ42 is significantly negatively correlated with disease progression [139]. All the above reveals that Aβ plays different roles through a dynamic balance of time and state. Generally, limited by the complicated biophysical characteristics of Aβ aggregation [250], the state of Aβ in the laboratory is not always comparable with that in vivo. Therefore, research on the physiological mechanisms of Aβ still needs a more rigorous and unified paradigm.
In conclusion, although the history of research on the mechanism of Aβ is long, the role of Aβ itself under physiological conditions is still poorly understood. Successive failure in clinical trials has brought investigators back to the original and intrinsic question: what is the physiological role of Aβ ? Undeniable evidence has established that Aβ plays a key role in AD, and this appears to imply an equally important role in physiological memory regulation. As an essential structural base of memory formation, the synapse is a promising target for research. However, research is difficult because of the above challenges. In the future, we should design effective approaches to imitate the physiological Aβ environment as much as possible, and more animal models with increased homologous Aβ should be developed, to reveal the physiology and understand the pathology in AD.
References
Chan KY, Wang W, Wu JJ, Liu L, Theodoratou E, Car J. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: A systematic review and analysis. Lancet 2013, 381: 2016–2023.
Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers 2021, 7: 33.
2020 Alzheimer's disease facts and figures. Alzheimers Dement. https://doi.org/10.1002/alz.12068.
Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol 2019, 15: 73–88.
Musiek ES, Gomez-Isla T, Holtzman DM. Aducanumab for Alzheimer disease: The amyloid hypothesis moves from bench to bedside. J Clin Invest 2021, 131: e154889.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron 2003, 37: 925–937.
Tharp WG, Sarkar IN. Origins of amyloid-Β. BMC Genomics 2013, 14: 290.
Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol 2020, 140: 417–447.
Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 2018, 14: 1602–1614.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated β-amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron 2018, 99: 56-63.e3.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 2016, 8: 340ra72.
Li S, Sheng ZH. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat Rev Neurosci 2022, 23: 4–22.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991, 30: 572–580.
Palop JJ, Mucke L. Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 2010, 13: 812–818.
Canter RG, Penney J, Tsai LH. The Road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539: 187–196.
Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med 1998, 4: 730–734.
Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, et al. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci U S A 1997, 94: 3748–3752.
Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, et al. Alzheimer’s Aβ(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 1997, 3: 1021–1023.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286: 735–741.
Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, et al. Gamma-Cleavage is dependent on zeta-cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem 2005, 280: 37689–37697.
Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem 1992, 267: 17082–17086.
Müller UC, Deller T, Korte M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat Rev Neurosci 2017, 18: 281–298.
Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem 2008, 283: 29615–29619.
Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, et al. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 1990, 87: 1561–1565.
Brahic M, Bousset L, Bieri G, Melki R, Gitler AD. Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol 2016, 131: 539–548.
Yamazaki T, Selkoe DJ, Koo EH. Trafficking of cell surface beta-amyloid precursor protein: Retrograde and transcytotic transport in cultured neurons. J Cell Biol 1995, 129: 431–442.
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 2001, 414: 643–648.
Wang YJ, Zhou HD, Zhou XF. Clearance of amyloid-beta in Alzheimer’s disease: Progress, problems and perspectives. Drug Discov Today 2006, 11: 931–938.
Ovsepian SV, O’Leary VB, Zaborszky L, Ntziachristos V, Dolly JO. Synaptic vesicle cycle and amyloid β: Biting the hand that feeds. Alzheimers Dement 2018, 14: 502–513.
Garcia-Osta A, Alberini CM. Amyloid beta mediates memory formation. Learn Mem 2009, 16: 267–272.
Puzzo D, Privitera L, Fa’ M, Staniszewski A, Hashimoto G, Aziz F, et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 2011, 69: 819–830.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJS, Hopkins R, Smith DW, et al. β-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995, 81: 525–531.
Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O’Dowd G, et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the β-amyloid precursor protein. Neuroscience 1999, 90: 1–13.
Dierich M, Hartmann S, Dietrich N, Moeser P, Brede F, Johnson Chacko L, et al. β-secretase BACE1 is required for normal cochlear function. J Neurosci 2019, 39: 9013–9027.
Southam KA, Stennard F, Pavez C, Small DH. Knockout of amyloid β protein precursor (APP) expression alters synaptogenesis, neurite branching and axonal morphology of hippocampal neurons. Neurochem Res 2019, 44: 1346–1355.
Gulisano W, Melone M, Ripoli C, Tropea MR, Li Puma DD, Giunta S, et al. Neuromodulatory action of picomolar extracellular Aβ42 oligomers on presynaptic and postsynaptic mechanisms underlying synaptic function and memory. J Neurosci 2019, 39: 5986–6000.
Lee SH, Kang J, Ho A, Watanabe H, Bolshakov VY, Shen J. APP family regulates neuronal excitability and synaptic plasticity but not neuronal survival. Neuron 2020, 108: 676-690.e8.
Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 2005, 25: 11693–11709.
Lombardo S, Chiacchiaretta M, Tarr A, Kim W, Cao T, Sigal G, et al. BACE1 partial deletion induces synaptic plasticity deficit in adult mice. Sci Rep 2019, 9: 19877.
Wang H, Megill A, Wong PC, Kirkwood A, Lee HK. Postsynaptic target specific synaptic dysfunctions in the CA3 area of BACE1 knockout mice. PLoS One 2014, 9: e92279.
Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci 2008, 28: 8677–8681.
Hu X, Das B, Hou H, He W, Yan R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med 2018, 215: 927–940.
Wang H, Song L, Lee A, Laird F, Wong PC, Lee HK. Mossy fiber long-term potentiation deficits in BACE1 knock-outs can be rescued by activation of alpha7 nicotinic acetylcholine receptors. J Neurosci 2010, 30: 13808–13813.
Filser S, Ovsepian SV, Masana M, Blazquez-Llorca L, Brandt Elvang A, Volbracht C, et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry 2015, 77: 729–739.
Finnie PSB, Nader K. Amyloid beta secreted during consolidation prevents memory malleability. Curr Biol 2020, 30: 1934-1940.e4.
Duan Y, Lv J, Zhang Z, Chen Z, Wu H, Chen J, et al. Exogenous Aβ1-42 monomers improve synaptic and cognitive function in Alzheimer’s disease model mice. Neuropharmacology 2022, 209: 109002.
Gulisano W, Melone M, Li Puma DD, Tropea MR, Palmeri A, Arancio O, et al. The effect of amyloid-β peptide on synaptic plasticity and memory is influenced by different isoforms, concentrations, and aggregation status. Neurobiol Aging 2018, 71: 51–60.
Koppensteiner P, Trinchese F, Fà M, Puzzo D, Gulisano W, Yan S, et al. Time-dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Aβ42: An early index of Alzheimer’s disease. Sci Rep 2016, 6: 32553.
Puzzo D, Privitera L, Palmeri A. Hormetic effect of amyloid-β peptide in synaptic plasticity and memory. Neurobiol Aging 2012, 33(1484): e15-1484.e24.
Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, et al. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 2008, 28: 14537–14545.
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 2005, 48: 913–922.
Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 2008, 58: 42–51.
Lazarevic V, Fieńko S, Andres-Alonso M, Anni D, Ivanova D, Montenegro-Venegas C, et al. Physiological concentrations of amyloid beta regulate recycling of synaptic vesicles via Alpha7 acetylcholine receptor and CDK5/calcineurin signaling. Front Mol Neurosci 2017, 10: 221.
Anni D, Weiss EM, Guhathakurta D, Akdas YE, Klueva J, Zeitler S, et al. Aβ1-16 controls synaptic vesicle pools at excitatory synapses via cholinergic modulation of synapsin phosphorylation. Cell Mol Life Sci 2021, 78: 4973–4992.
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 2009, 12: 1567–1576.
Forest KH, Taketa R, Arora K, Todorovic C, Nichols RA. The neuroprotective beta amyloid hexapeptide core reverses deficits in synaptic plasticity in the 5xFAD APP/PS1 mouse model. Front Mol Neurosci 2021, 14: 576038.
Forest KH, Alfulaij N, Arora K, Taketa R, Sherrin T, Todorovic C, et al. Protection against β-amyloid neurotoxicity by a non-toxic endogenous N-terminal β-amyloid fragment and its active hexapeptide core sequence. J Neurochem 2018, 144: 201–217.
Lawrence JLM, Tong M, Alfulaij N, Sherrin T, Contarino M, White MM, et al. Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal β-amyloid fragment. J Neurosci 2014, 34: 14210–14218.
Ortiz-Sanz C, Gaminde-Blasco A, Valero J, Bakota L, Brandt R, Zugaza JL, et al. Early effects of Aβ oligomers on dendritic spine dynamics and arborization in hippocampal neurons. Front Synaptic Neurosci 2020, 12: 2.
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58: 1791–1800.
Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, et al. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 2010, 68: 428–441.
Hick M, Herrmann U, Weyer SW, Mallm JP, Tschäpe JA, Borgers M, et al. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol 2015, 129: 21–37.
Wang B, Wang Z, Sun L, Yang L, Li H, Cole AL, et al. The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J Neurosci 2014, 34: 13314–13325.
Zhao J, Paganini L, Mucke L, Gordon M, Refolo L, Carman M, et al. Beta-secretase processing of the beta-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J Biol Chem 1996, 271: 31407–31411.
Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2012, 2: a006270.
DeBoer SR, Dolios G, Wang R, Sisodia SS. Differential release of β-amyloid from dendrite- versus axon-targeted APP. J Neurosci 2014, 34: 12313–12327.
Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: Modulation by interleukin-1. Mol Brain Res 1992, 16: 128–134.
LeBlanc AC, Chen HY, Autilio-Gambetti L, Gambetti P. Differential APP gene expression in rat cerebral cortex, meninges, and primary astroglial, microglial and neuronal cultures. FEBS Lett 1991, 292: 171–178.
Haass C, Hung AY, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci 1991, 11: 3783–3793.
De Rohan Silva HA, Jen A, Wickenden C, Jen LS, Wilkinson SL, Patel AJ. Cell-specific expression of β-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Mol Brain Res 1997, 47: 147–156.
Young MJ, Lee RKK, Jhaveri S, Wurtman RJ. Intracellular and cell-surface distribution of amyloid precursor protein in cortical astrocytes. Brain Res Bull 1999, 50: 27–32.
Gaul G, Dutly F, Frei K, Foguet M, Lübbert H, Paul G. APP RNA splicing is not affected by differentiation of neurons and glia in culture. FEBS Lett 1992, 307: 329–332.
Ohyagi Y, Takahashi K, Kamegai M, Tabira T. Developmental and differential expression of beta amyloid protein precursor mRNAs in mouse brain. Biochem Biophys Res Commun 1990, 167: 54–60.
LeBlanc AC, Xue R, Gambetti P. Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. J Neurochem 1996, 66: 2300–2310.
Berkenbosch F, Refolo LM, Friedrich VL Jr, Casper D, Blum M, Robakis NK. The Alzheimer’s amyloid precursor protein is produced by type I astrocytes in primary cultures of rat neuroglia. J Neurosci Res 1990, 25: 431–440.
Liang Y, Raven F, Ward JF, Zhen S, Zhang S, Sun H, et al. Upregulation of Alzheimer’s disease amyloid-β protein precursor in astrocytes both in vitro and in vivo. J Alzheimers Dis 2020, 76: 1071–1082.
Liao MC, Muratore CR, Gierahn TM, Sullivan SE, Srikanth P, De Jager PL, et al. Single-cell detection of secreted Aβ and sAPPα from human IPSC-derived neurons and astrocytes. J Neurosci 2016, 36: 1730–1746.
Rodrigues DI, Gutierres J, Pliássova A, Oliveira CR, Cunha RA, Agostinho P. Synaptic and sub-synaptic localization of amyloid-β protein precursor in the rat hippocampus. J Alzheimers Dis 2014, 40: 981–992.
Rice HC, Marcassa G, Chrysidou I, Horré K, Young-Pearse TL, Müller UC, et al. Contribution of GABAergic interneurons to amyloid-β plaque pathology in an APP knock-in mouse model. Mol Neurodegener 2020, 15: 3.
Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol 2010, 119: 523–541.
Padmanabhan P, Kneynsberg A, Götz J. Super-resolution microscopy: A closer look at synaptic dysfunction in Alzheimer disease. Nat Rev Neurosci 2021, 22: 723–740.
Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, et al. Alzheimer amyloid protein precursor in the rat hippocampus: Transport and processing through the perforant path. J Neurosci 1998, 18: 9629–9637.
Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci 2002, 22: 9785–9793.
Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 2010, 13: 190–196.
Yu Y, Jans DC, Winblad B, Tjernberg LO, Schedin-Weiss S. Neuronal Aβ42 is enriched in small vesicles at the presynaptic side of synapses. Life Sci Alliance 2018, 1: e201800028.
de Wilde MC, Overk CR, Sijben JW, Masliah E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement 2016, 12: 633–644.
Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol 2013, 126: 329–352.
Das U, Wang L, Ganguly A, Saikia JM, Wagner SL, Koo EH, et al. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 2016, 19: 55–64.
Schedin-Weiss S, Caesar I, Winblad B, Blom H, Tjernberg LO. Super-resolution microscopy reveals γ-secretase at both sides of the neuronal synapse. Acta Neuropathol Commun 2016, 4: 29.
Kedia S, Ramakrishna P, Netrakanti PR, Jose M, Sibarita JB, Nadkarni S, et al. Real-time nanoscale organization of amyloid precursor protein. Nanoscale 2020, 12: 8200–8215.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 1992, 359: 325–327.
Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement 2009, 5: 18–29.
Raskatov JA. What is the relevant amyloid β42 concentration? Chembiochem 2019, 20: 1725–1726.
Mehta PD, Pirttilä T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol 2000, 57: 100–105.
Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. β-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem 2010, 285: 2506–2514.
He Y, Wei M, Wu Y, Qin H, Li W, Ma X, et al. Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4, 5-bisphosphate. Nat Commun 2019, 10: 1193.
Park D, Na M, Kim JA, Lee U, Cho E, Jang M, et al. Activation of CaMKIV by soluble amyloid-β1-42 impedes trafficking of axonal vesicles and impairs activity-dependent synaptogenesis. Sci Signal 2017, 10: 8661.
Novo M, Freire S, Al-Soufi W. Critical aggregation concentration for the formation of early Amyloid-β (1–42) oligomers. Sci Rep 2018, 8: 1783.
Arbel-Ornath M, Hudry E, Boivin JR, Hashimoto T, Takeda S, Kuchibhotla KV, et al. Soluble oligomeric amyloid-β induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener 2017, 12: 27.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416: 535–539.
Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener 2014, 9: 48.
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488: 96–99.
Kokawa A, Ishihara S, Fujiwara H, Nobuhara M, Iwata M, Ihara Y, et al. The A673T mutation in the amyloid precursor protein reduces the production of β-amyloid protein from its β-carboxyl terminal fragment in cells. Acta Neuropathol Commun 2015, 3: 66.
Martiskainen H, Herukka SK, Stančáková A, Paananen J, Soininen H, Kuusisto J, et al. Decreased plasma β-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann Neurol 2017, 82: 128–132.
Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, et al. APP homodimers transduce an amyloid-β-mediated increase in release probability at excitatory synapses. Cell Rep 2014, 7: 1560–1576.
Götz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci 2018, 19: 583–598.
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 2014, 17: 661–663.
Luo LQ, Martin-Morris LE, White K. Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. J Neurosci 1990, 10: 3849–3861.
Joshi P, Liang JO, DiMonte K, Sullivan J, Pimplikar SW. Amyloid precursor protein is required for convergent-extension movements during Zebrafish development. Dev Biol 2009, 335: 1–11.
Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J Biol Chem 1996, 271: 31894–31902.
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ-secretase inhibitors and modulators. Biochim Biophys Acta BBA Biomembr 2013, 1828: 2898–2907.
Yan Y, Wang C. Abeta42 is more rigid than Abeta40 at the C terminus: Implications for Abeta aggregation and toxicity. J Mol Biol 2006, 364: 853–862.
Klein WL. Synaptic targeting by Aβ oligomers (ADDLS) as a basis for memory loss in early Alzheimer’s disease. Alzheimers Dement 2006, 2: 43–55.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998, 95: 6448–6453.
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 2009, 106: 4012–4017.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 2007, 27: 2866–2875.
Lee S, Kim J, Choi S. Endogenous amyloid-β mediates memory forgetting in the normal brain. Biochem Biophys Res Commun 2018, 506: 492–497.
Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J Neurosci 1997, 17: 9407–9414.
Dinamarca MC, Raveh A, Schneider A, Fritzius T, Früh S, Rem PD, et al. Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat Commun 2019, 10: 1331.
Lo AC, Evans CD, Mancini M, Wang H, Shcherbinin S, Lu M, et al. Phase II (NAVIGATE-AD study) results of LY3202626 effects on patients with mild Alzheimer’s disease dementia. J Alzheimers Dis Rep 2021, 5: 321–336.
Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 2018, 378: 1691–1703.
Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, et al. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med 2019, 380: 1408–1420.
Hrabinova M, Pejchal J, Kucera T, Jun D, Schmidt M, Soukup O. Is it the twilight of BACE1 inhibitors? Curr Neuropharmacol 2021, 19: 61–77.
Sperling R, Henley D, Aisen PS, Raman R, Donohue MC, Ernstrom K, et al. Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical alzheimer disease: A truncated randomized phase 2b/3 clinical trial. JAMA Neurol 2021, 78: 293–301.
Das B, Singh N, Yao AY, Zhou J, He W, Hu X, et al. BACE1 controls synaptic function through modulating release of synaptic vesicles. Mol Psychiatry 2021, 26: 6394–6410.
Ou-Yang MH, Kurz JE, Nomura T, Popovic J, Rajapaksha TW, Dong H, et al. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med 2018, 10: eaa5620.
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci 2001, 4: 231–232.
Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: Implications for Alzheimer’s disease therapeutics. Hum Mol Genet 2001, 10: 1317–1324.
Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, et al. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 2010, 30: 8819–8829.
Hitt BD, Jaramillo TC, Chetkovich DM, Vassar R. BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener 2010, 5: 31.
Vassar R. Editorial: Implications for BACE1 inhibitor clinical trials: Adult conditional BACE1 knockout mice exhibit axonal organization defects in the hippocampus. J Prev Alzheimers Dis 2019, 6: 78–84.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, et al. Control of peripheral nerve myelination by the beta-secretase BACE1. Science 2006, 314: 664–666.
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 2006, 9: 1520–1525.
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, et al. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 2012, 109: 8740–8745.
Lerdkrai C, Asavapanumas N, Brawek B, Kovalchuk Y, Mojtahedi N, Olmedillas Del Moral M, et al. Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 2018, 115: E1279–E1288.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321: 1686–1689.
Korzhova V, Marinković P, Njavro JR, Goltstein PM, Sun F, Tahirovic S, et al. Long-term dynamics of aberrant neuronal activity in awake Alzheimer’s disease transgenic mice. Commun Biol 2021, 4: 1368.
Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84: 608–622.
Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012, 367: 795–804.
Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A 2009, 106: 7209–7214.
Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 2000, 343: 450–456.
Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, et al. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann Neurol 2010, 68: 865–875.
Dickerson BC, Salat DH, Bates JF, Atiya M, Killiany RJ, Greve DN, et al. Medial temporal lobe function and structure in mild cognitive impairment. Ann Neurol 2004, 56: 27–35.
Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005, 65: 404–411.
Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: An independent component analysis. J Neurosci 2006, 26: 10222–10231.
Hämäläinen A, Pihlajamäki M, Tanila H, Hänninen T, Niskanen E, Tervo S, et al. Increased fMRI responses during encoding in mild cognitive impairment. Neurobiol Aging 2007, 28: 1889–1903.
Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, et al. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365: 559–565.
Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, et al. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci 2009, 29: 9704–9713.
Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005, 25: 7709–7717.
Romoli M, Sen A, Parnetti L, Calabresi P, Costa C. Amyloid-β: A potential link between epilepsy and cognitive decline. Nat Rev Neurol 2021, 17: 469–485.
Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012, 74: 467–474.
Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 2008, 321: 1221–1224.
Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol 2000, 527: 515–528.
Papouin T, Dunphy JM, Tolman M, Dineley KT, Haydon PG. Septal cholinergic neuromodulation tunes the astrocyte-dependent gating of hippocampal NMDA receptors to wakefulness. Neuron 2017, 94: 840-854.e7.
Suzuki T, Hide I, Matsubara A, Hama C, Harada K, Miyano K, et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J Neurosci Res 2006, 83: 1461–1470.
Báez-Pagán CA, Delgado-Vélez M, Lasalde-Dominicci JA. Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol 2015, 10: 468–476.
Castro NG, Albuquerque EX. Alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J 1995, 68: 516–524.
Letsinger AC, Gu Z, Yakel JL. α7 nicotinic acetylcholine receptors in the hippocampal circuit: Taming complexity. Trends Neurosci 2022, 45: 145–157.
Gomez-Varela D, Berg DK. Lateral mobility of presynaptic α7-containing nicotinic receptors and its relevance for glutamate release. J Neurosci 2013, 33: 17062–17071.
Townsend M, Whyment A, Walczak JS, Jeggo R, van den Top M, Flood DG, et al. α7-nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J Neurophysiol 2016, 116: 2663–2675.
Burghaus L, Schütz U, Krempel U, de Vos RAI, Jansen Steur ENH, Wevers A, et al. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Mol Brain Res 2000, 76: 385–388.
Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 1981, 10: 122–126.
Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110: 199–211.
Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer’s disease: Progress to date and the path forward. Neuron 2019, 101: 820–838.
Wang HY, Trocmé-Thibierge C, Stucky A, Shah SM, Kvasic J, Khan A, et al. Increased Aβ42-α7-like nicotinic acetylcholine receptor complex level in lymphocytes is associated with apolipoprotein E4-driven Alzheimer’s disease pathogenesis. Alzheimers Res Ther 2017, 9: 54.
Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Abeta(1–42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem 2000, 75: 1155–1161.
Cecon E, Dam J, Luka M, Gautier C, Chollet AM, Delagrange P, et al. Quantitative assessment of oligomeric amyloid β peptide binding to α7 nicotinic receptor. Br J Pharmacol 2019, 176: 3475–3488.
Tropea MR, Li Puma DD, Melone M, Gulisano W, Arancio O, Grassi C, et al. Genetic deletion of α7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Prog Neurobiol 2021, 206: 102154.
Dougherty JJ, Wu J, Nichols RA. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 2003, 23: 6740–6747.
Lasala M, Fabiani C, Corradi J, Antollini S, Bouzat C. Molecular modulation of human α7 nicotinic receptor by amyloid-β peptides. Front Cell Neurosci 2019, 13: 37.
Stoiljkovic M, Kelley C, Hajós GP, Nagy D, Koenig G, Leventhal L, et al. Hippocampal network dynamics in response to α7 nACh receptors activation in amyloid-β overproducing transgenic mice. Neurobiol Aging 2016, 45: 161–168.
Inestrosa NC, Godoy JA, Vargas JY, Arrazola MS, Rios JA, Carvajal FJ, et al. Nicotine prevents synaptic impairment induced by amyloid-β oligomers through α7-nicotinic acetylcholine receptor activation. Neuromol Med 2013, 15: 549–569.
Kihara T, Shimohama S, Urushitani M, Sawada H, Kimura J, Kume T, et al. Stimulation of α4β2 nicotinic acetylcholine receptors inhibits β-amyloid toxicity. Brain Res 1998, 792: 331–334.
Buckingham SD, Jones AK, Brown LA, Sattelle DB. Nicotinic acetylcholine receptor signalling: Roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol Rev 2009, 61: 39–61.
Wu J, Liu Q, Tang P, Mikkelsen JD, Shen J, Whiteaker P, et al. Heteromeric α7β2 nicotinic acetylcholine receptors in the brain. Trends Pharmacol Sci 2016, 37: 562–574.
Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP. Increased neurodegeneration during ageing in mice lacking high-affinity nicotine receptors. EMBO J 1999, 18: 1235–1244.
George AA, Vieira JM, Xavier-Jackson C, Gee MT, Cirrito JR, Bimonte-Nelson HA, et al. Implications of oligomeric amyloid-beta (oAβ42) signaling through α7β2-nicotinic acetylcholine receptors (nAChRs) on basal forebrain cholinergic neuronal intrinsic excitability and cognitive decline. J Neurosci 2021, 41: 555–575.
Sabri O, Meyer PM, Gräf S, Hesse S, Wilke S, Becker GA, et al. Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain 2018, 141: 1840–1854.
Roberts JP, Stokoe SA, Sathler MF, Nichols RA, Kim S. Selective coactivation of α7- and α4β2-nicotinic acetylcholine receptors reverses beta-amyloid-induced synaptic dysfunction. J Biol Chem 2021, 296: 100402.
Barykin EP, Garifulina AI, Tolstova AP, Anashkina AA, Adzhubei AA, Mezentsev YV, et al. Tetrapeptide Ac-HAEE-NH2 protects α4β2 nAChR from inhibition by Aβ. Int J Mol Sci 2020, 21: 6272.
Arora K, Belcaid M, Lantz MJ, Taketa R, Nichols RA. Transcriptome profile of nicotinic receptor-linked sensitization of beta amyloid neurotoxicity. Sci Rep 2020, 10: 5696.
Mehta TK, Dougherty JJ, Wu J, Choi CH, Khan GM, Nichols RA. Defining pre-synaptic nicotinic receptors regulated by beta amyloid in mouse cortex and hippocampus with receptor null mutants. J Neurochem 2009, 109: 1452–1458.
Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis 2017, 57: 1041–1048.
Cook SG, Goodell DJ, Restrepo S, Arnold DB, Bayer KU. Simultaneous live imaging of multiple endogenous proteins reveals a mechanism for Alzheimer’s-related plasticity impairment. Cell Rep 2019, 27: 658-665.e4.
Opazo P, da Viana Silva S, Carta M, Breillat C, Coultrap SJ, Grillo-Bosch D, et al. CaMKII metaplasticity drives Aβ oligomer-mediated synaptotoxicity. Cell Rep 2018, 23: 3137–3145.
Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem 2006, 6: 787–799.
Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans 2006, 34: 936–938.
Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci 2003, 26: 81–89.
Lipton SA, Nakanishi N. Shakespeare in love—With NMDA receptors? Nat Med 1999, 5: 270–271.
Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat Rev Neurosci 2010, 11: 682–696.
Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82: 279–293.
Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci 2000, 20: 22–33.
Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, et al. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 2008, 22: 4258–4271.
Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A 2009, 106: 9854–9859.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 2002, 5: 405–414.
Kokubo H, Kayed R, Glabe CG, Yamaguchi H. Soluble Aβ oligomers ultrastructurally localize to cell processes and might be related to synaptic dysfunction in Alzheimer’s disease brain. Brain Res 2005, 1031: 222–228.
Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, et al. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol Dis 2012, 45: 381–387.
Kawamoto EM, Lepsch LB, Boaventura MFC, Munhoz CD, Lima LS, Yshii LM, et al. Amyloid beta-peptide activates nuclear factor-kappaB through an N-methyl-D-aspartate signaling pathway in cultured cerebellar cells. J Neurosci Res 2008, 86: 845–860.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A 2013, 110: E2518–E2527.
Alberdi E, Sánchez-Gómez MV, Cavaliere F, Pérez-Samartín A, Zugaza JL, Trullas R, et al. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47: 264–272.
Texidó L, Martín-Satué M, Alberdi E, Solsona C, Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49: 184–190.
Dore K, Carrico Z, Alfonso S, Marino M, Koymans K, Kessels HW, et al. PSD-95 protects synapses from β-amyloid. Cell Rep 2021, 35: 109194.
de Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, et al. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the alzheimer drug memantine*. J Biol Chem 2007, 282: 11590–11601.
Zhong W, Wu A, Berglund K, Gu X, Jiang MQ, Talati J, et al. Pathogenesis of sporadic Alzheimer’s disease by deficiency of NMDA receptor subunit GluN3A. Alzheimers Dement 2022, 18: 222–239.
Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci U S A 2000, 97: 12885–12890.
Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat Rev Drug Discov 2006, 5: 160–170.
Domingues A, Almeida S, Da Cruz E, Silva EF, Oliveira CR, Rego AC. Toxicity of β-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D. Neurochem Int 2007, 50: 872–880.
Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150: 633–646.
Südhof TC. The synaptic vesicle cycle: A cascade of protein–protein interactions. Nature 1995, 375: 645–653.
DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 1990, 27: 457–464.
Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251: 51–65.
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 2003, 100: 10417–10422.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62: 788–801.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008, 14: 837–842.
Ripoli C, Cocco S, Li Puma DD, Piacentini R, Mastrodonato A, Scala F, et al. Intracellular accumulation of amyloid-β (Aβ) protein plays a major role in Aβ-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci 2014, 34: 12893–12903.
Moreno H, Yu E, Pigino G, Hernandez AI, Kim N, Moreira JE, et al. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc Natl Acad Sci U S A 2009, 106: 5901–5906.
Sharma M, Burré J, Südhof TC. Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci Transl Med 2012, 4: 147113.
Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P. Amyloid-β acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One 2012, 7: e43201.
Wu F, Yao PJ. Clathrin-mediated endocytosis and Alzheimer’s disease: An update. Ageing Res Rev 2009, 8: 147–149.
Yao PJ. Synaptic frailty and clathrin-mediated synaptic vesicle trafficking in Alzheimer’s disease. Trends Neurosci 2004, 27: 24–29.
Yao PJ, Morsch R, Callahan LM, Coleman PD. Changes in synaptic expression of clathrin assembly protein AP180 in Alzheimer’s disease analysed by immunohistochemistry. Neuroscience 1999, 94: 389–394.
Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, et al. Defects in expression of genes related to synaptic vesicle traffickingin frontal cortex of Alzheimer’s disease. Neurobiol Dis 2003, 12: 97–109.
Park J, Jang M, Chang S. Deleterious effects of soluble amyloid-β oligomers on multiple steps of synaptic vesicle trafficking. Neurobiol Dis 2013, 55: 129–139.
Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron 2010, 67: 797–809.
Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 2003, 5: 701–710.
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405: 360–364.
Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A 2011, 108: 14330–14335.
Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367: 688–694.
Spitzer P, Walter M, Göth C, Oberstein TJ, Linning P, Knölker HJ, et al. Pharmacological inhibition of amyloidogenic APP processing and knock-down of APP in primary human macrophages impairs the secretion of cytokines. Front Immunol 1967, 11: 1967.
Tao CC, Cheng KM, Ma YL, Hsu WL, Chen YC, Fuh JL, et al. Galectin-3 promotes Aβ oligomerization and Aβ toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ 2020, 27: 192–209.
Luo T, Gao TM. A novel phagocytic role of astrocytes in activity-dependent elimination of mature excitatory synapses. Neurosci Bull 2021, 37: 1256–1259.
Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 2012, 135: 2155–2168.
Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, et al. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. Am J Pathol 2013, 182: 1750–1768.
Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156: 825–835.
Goyal MS, Gordon BA, Couture LE, Flores S, Xiong C, Morris JC, et al. Spatiotemporal relationship between subthreshold amyloid accumulation and aerobic glycolysis in the human brain. Neurobiol Aging 2020, 96: 165–175.
Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, et al. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc Natl Acad Sci U S A 2010, 107: 17763–17767.
Pascoal TA, Mathotaarachchi S, Kang MS, Mohaddes S, Shin M, Park AY, et al. Aβ-induced vulnerability propagates via the brain’s default mode network. Nat Commun 2019, 10: 2353.
Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab 2017, 26: 353-360.e3.
Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer’s disease: Genetic studies and transgenic models. Annu Rev Genet 1998, 32: 461–493.
Drummond E, Wisniewski T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol 2017, 133: 155–175.
Benitez DP, Jiang S, Wood J, Wang R, Hall CM, Peerboom C, et al. Knock-in models related to Alzheimer’s disease: Synaptic transmission, plaques and the role of microglia. Mol Neurodegener 2021, 16: 47.
Steffen J, Krohn M, Schwitlick C, Brüning T, Paarmann K, Pietrzik CU, et al. Expression of endogenous mouse APP modulates β-amyloid deposition in hAPP-transgenic mice. Acta Neuropathol Commun 2017, 5: 49.
Richter MC, Ludewig S, Winschel A, Abel T, Bold C, Salzburger LR, et al. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. EMBO J 2018, 37: e98335.
Livingstone RW, Elder MK, Barrett MC, Westlake CM, Peppercorn K, Tate WP, et al. Secreted amyloid precursor protein-alpha promotes arc protein synthesis in hippocampal neurons. Front Mol Neurosci 2019, 12: 198.
Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, et al. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 2019, 363: eaa4827.
Wang Z, Jackson RJ, Hong W, Taylor WM, Corbett GT, Moreno A, et al. Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J Neurosci 2017, 37: 11947–11966.
Zhu K, Xiang X, Filser S, Marinković P, Dorostkar MM, Crux S, et al. Beta-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol Psychiatry 2018, 83: 428–437.
Borgstedt L, Blobner M, Musiol M, Bratke S, Syryca F, Rammes G, et al. Neurotoxicity of different amyloid beta subspecies in mice and their interaction with isoflurane anaesthesia. PLoS One 2020, 15: e0242989.
Sebastian Monasor L, Müller SA, Colombo AV, Tanrioever G, König J, Roth S, et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 2020, 9: e54083.
Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem 2009, 284: 4749–4753.
Acknowledgements
This review was supported by grants from the Shanghai Municipal Science and Technology Major Project, the National Key Research and Development Program Foundation of China (2016YFC1306403), the National Natural Science Foundation of China (81870822, 91332201, 81901081, 81600930, 82171408, and 82171411), and the Natural Science Foundation of Fujian Province (2020CXB049).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, W., Li, L., Sang, S. et al. Physiological Roles of β-amyloid in Regulating Synaptic Function: Implications for AD Pathophysiology. Neurosci. Bull. 39, 1289–1308 (2023). https://doi.org/10.1007/s12264-022-00985-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-022-00985-9