
Abstract
Delayed vasospasm that develops 3–7 days after aneurysmal subarachnoid hemorrhage (SAH) has traditionally been considered the most important determinant of delayed ischemic injury and poor outcome. Consequently, most therapies against delayed ischemic injury are directed towards reducing the incidence of vasospasm. The clinical trials based on this strategy, however, have so far claimed limited success; the incidence of vasospasm is reduced without reduction in delayed ischemic injury or improvement in the long-term outcome. This fact has shifted research interest to the early brain injury (first 72 h) evoked by SAH. In recent years, several pathological mechanisms that activate within minutes after the initial bleed and lead to early brain injury are identified. In addition, it is found that many of these mechanisms evolve with time and participate in the pathogenesis of delayed ischemic injury and poor outcome. Therefore, a therapy or therapies focused on these early mechanisms may not only prevent the early brain injury but may also help reduce the intensity of later developing neurological complications. This manuscript reviews the pathological mechanisms of early brain injury after SAH and summarizes the status of current therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) accounts for 5% of all stroke cases and affects up to 30,000 North Americans yearly [1]. Early brain injury that occurs at the time of bleed is the leading cause of mortality (30–70%) after SAH [1, 2]. SAH survivors are at risk of developing delayed cerebral vasospasm, delayed cerebral ischemia, or delayed ischemic neurological deficits during the hospital course [2]. Delayed vasospasm develops in approximately 70% of patients between 3 and 14 days after SAH [1, 2]. For decades, delayed vasospasm has been considered the single and the most important cause of delayed cerebral ischemia and poor outcome [3]. The basic and clinical research has been focused on finding strategies to prevent and/or treat delayed vasospasm. However, lack of prevention of delayed cerebral ischemia and improved outcome in a recent clinical trial (CONSCIOUS-1) that successfully prevented the development of delayed vasospasm has raised doubts on the importance of vasospasm in delayed ischemic injury and the outcome after SAH [4]. Recent reviews of the experimental and clinical literature indicate that the presence of delayed vasospasm is not a pre-requisite for delayed ischemic injury and poor outcome after SAH [2, 5]. In fact, 21% of SAH survivors, who do not develop vasospasm, develop delayed ischemic injury, and only 20–30% of those, who do develop delayed vasospasm actually, suffer from delayed ischemic injury [2]. It maybe that the pathological mechanisms that activate within minutes after SAH and lead to early brain injury play an important role in the pathogenesis of delayed ischemic injury and poor outcome [6]. This manuscript summarizes the animal and human literature addressing the mechanisms of early brain injury after SAH and the importance of its early treatment.
Early Brain Injury by SAH
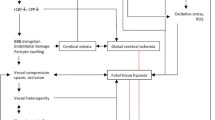
Early brain injury is the product of pathological mechanisms triggered in the brain during the first 72 h after SAH (Fig. 1). These mechanisms are activated at aneurysm rupture and evolve with time affecting the course and the outcome of SAH (Table 1) [7–9]. Below, we discuss the pathological mechanisms most pertinent to early brain injury after SAH.

Mechanisms of early brain injury after SAH: A number of changes in cerebral environment and function occur during the first 72 h after SAH. Some of the major changes are listed. See text for explanation. ICP intracranial pressure, CPP cerebral perfusion pressure, CBF cerebral blood flow, NO nitric oxide, NOS nitric oxide synthase, ET-1 endothelin-1
Mechanical Trauma
The first injury to the brain after the aneurysm rupture is mechanical. This trauma evokes constriction of the artery harboring the ruptured aneurysm and its compression by blood filling in the subarachnoid cisterns [10, 11]. Sudden rise in intracranial pressure that may reach as high as 2,000 mm H2O (161.8 mmHg) [12] stops further bleed and compresses cerebral arteries and tissue. Depending upon the amount released, blood not only stretches the subarachnoid space, but also flows into the branching channels and envelops branches of the conducting artery [13]. The stretching of the subarachnoid space by blood is mechanically transferred to the vessels near the aneurysm leading to spasm of surrounding arteries [14]. Over the course of its presence, the subarachnoid blood clot evokes the early brain injury [15] and the delayed spasm [16].
Altered Cerebral Physiology
Intracranial Pressure
Intracranial pressure (ICP) rises when blood released at the time of aneurysm rupture fills up the subarachnoid cisterns displacing the cerebrospinal fluid (CSF). Most awake patients describe this moment as the onset of “the worst headache of my life” [12]. Two patterns of ICP rise are recognized and compress brain tissue by different mechanisms. In the first pattern, observed in most patients, ICP peaks to a value near diastolic blood pressure and then falls and settles near but slightly above the baseline [17]. In this pattern, the volume of bleed is small but cerebral edema is present [12]. In the second pattern, observed in some patients, ICP remains elevated, due to the mass effect from enlarging hematoma or due to the development of acute hydrocephalus [12, 18]. The second pattern of ICP rise is associated with high mortality [12, 19]. The terms “ischemic-edematous lesion” and “hemorrhagic-compressive lesion” have been used to differentiate the brain compressing forces associated with the two ICP patterns [12, 19]. The intensity of ICP rise is linked to hemorrhage volume, obstruction of the CSF outflow, diffuse vasoparalysis, and distal cerebral arteriolar vasodilation. The severity of ICP is associated with changes in cerebral metabolism, inflammation, fall in cerebral blood flow (CBF), and development of early and delayed cerebral ischemia [20]. Hence, the extent of ICP rise is often used to predict outcome of SAH [15, 21] (Fig. 2).

The timeline of physiological alterations during the first 72 h after SAH. Within seconds after SAH: ICP rises, CBF and CPP fall, and BP increases [17]. Five minutes after SAH: ICP declines towards basal value and CPP and BP recover to the basal value, CBF remains decreased, and CBF autoregulation is impaired [17, 37]. Sixty minutes after SAH: ICP stabilizes to a new plateau that is above the basal value, CPP and BP have recovered, CBF is still decreased, and CBF autoregulation still impaired [17, 37]. Twenty-four hours after SAH: ICP is still at the 60-min value, CPP and BP are recovered, CBF is decreased, and CBF autoregulation is still impaired [140]. Seventy-two hours after SAH: ICP, CPP, and BP are at baseline [140], CBF is recovered or decreased [140], and CBF autoregulation is still impaired [141]. Insert: In animals, the higher the ICP rise at SAH and the lower the 60-min CBF recovery, the smaller the changes of 24 h survival [142]
Cerebral Perfusion Pressure
CPP falls profoundly during and immediately after SAH. Animal studies indicate that CPP fall is not sufficient to cause perfusion arrest [17]. Similarly, in SAH patients, CPP reductions are not clearly associated with poor neurological outcome. Consequently, decreased CPP may contribute to early ischemic brain injury but is not solely responsible for it [17, 22].
Cerebral Blood Flow
CBF falls after SAH and may or may not recover depending upon the severity of the bleed [17]. In animals, CBF reduction after SAH is accompanied by constriction of large cerebral blood vessels [23]. By contrast, in humans, little arteriographic evidence of acute arterial spasm is found [24, 25]. Therefore, initial fall in CBF in humans is often attributed to the brief period of no-reflow, due to elevated ICP and decreased CPP [26].
CBF Autoregulation
Autoregulatory mechanisms of CBF are frequently impaired after SAH leading to inadequate CBF response to a change in systemic blood pressure (pressure autoregulation) or to a change in partial pressure of carbon dioxide (chemoregulation) [22, 27]. In animals, a severe disturbance in autoregulation occurs within 2–3 h and continues for months after SAH [22]. In patients, this impairment is most pronounced during the first 72 h and correlates well with the SAH severity [27].
Altered Ionic Homeostasis
A rapid alteration in ionic homeostasis occurs after SAH and affects especially sodium, potassium, calcium, and magnesium ions. The alteration evokes immediate effects such as vasoconstriction, an electrical activity disturbance, and slowly developing effects, such as activation and expression of proteins, that develop in a delayed fashion but last for a long time. Both of these effects can be detrimental to the injured brain. The timeline of SAH-derived alteration in ionic homeostasis is presented in Table 2.
Cortical Spreading Depolarization
Cortical Spreading Depolarization (CSD) describes a wave of mass neuronal depolarization associated with net influx of cations and water [28] and is an effect of breakdown of ion homeostasis in the cerebral cortex. CSD is associated with massive neuronal influx of sodium and calcium. Elevated intracellular calcium is possibly the predominant mediator of neuronal death from ischemia [29]. Animal and human studies indicate that CSDs occur early and late after SAH. In human SAH, CSDs can occur as clusters or as isolated events [28]. The cooperative study on brain injury and depolarizations (COSBID group) found that clustered CSDs occurred in spatial and temporal correlation to the development of early and delayed brain damage. Electrocortical and regional cerebral blood flow recordings provided evidence of three different neurovascular responses to CSD in SAH patients, similar to the findings in animals: (1) spreading hyperemia, (2) spreading ischemia, and (3) neurovascular uncoupling [28]. Experimental evidence suggests that subarachnoid oxyhemoglobin, elevated extracellular potassium, decline in NO availability, glutamate, and endothelin-1 are involved in the development of CSD and spreading ischemia after SAH [30, 31].
Decreased Serum Magnesium
Approximately 38% of the patients admitted within 48 h after SAH have abnormally low serum magnesium [32]. In animals, decrease in serum and CSF magnesium occurs within minutes and in humans within hours after SAH [33]. Because magnesium is a physiological antagonist of calcium and controls the NMDA receptor-derived calcium influx, its decrease after SAH contributes to the rise in cellular calcium. In addition, magnesium dilates blood vessels, inhibits aggregation of platelet, inhibits release of excitatory amino-acids, and inhibits synthesis of endothelin-1 (ET-1) [34]. Therefore, decrease in magnesium after SAH exacerbates early brain injury and promotes mechanisms of delayed brain damage.
Mechanical and Biochemical Alterations
The timeline of SAH-derived mechanical and biochemical alterations is presented in Fig. 3. These alterations are described in the next three paragraphs.

The timeline of biochemical alterations during the first 72 h after SAH. Sixty minutes after SAH: Glutamate concentration in cerebral interstitial fluid is increased [23], ventricles swell and brain water content increases [35], and the status of plasma electrolytes at this time is not established. Twenty-four hours after SAH: Glutamate concentration in cerebral interstitial fluid is still increased [143], hydrocephalus [35, 96] and hyponatremia [40, 96] have set in. Seventy-two hours after SAH: Glutamate concentration cerebral interstitial fluid is still increased [143], hydrocephalus [35] and hyponatremia [41] are still present. Insert: SAH patients with large bleeds and poor clinical status at admission are more likely to develop acute hydrocephalus and have poor outcomes [38]
Hydrocephalus
Approximately 20% to 30% patients develop acute hydrocephalus within the first 3 days after SAH [35]. In most cases, these patients have larger hemorrhages, poor cerebral perfusion, and reduced CBF at admission [36]. In animals, symptoms of hydrocephalus are present within an hour after initial bleed and are associated with intensity of CBF reduction and cerebral ischemia [37]. The mechanisms of acute hydrocephalus include sudden obstruction of cerebrospinal fluid circulation [35], presence of blood in the ventricles, hemorrhage from a posterior circulation aneurysm, diffused spread of subarachnoid blood, rebleeding, hypertension, and increased sympathetic activity [38].
Increase in Extracellular Glutamate
Cerebral glutamate level increases within minutes after SAH and peaks at approximately 40 min [23]. This biochemical change is associated with the intensity of initial insult [39]. An increased interstitial glutamate concentration after SAH is linked to cellular leakage, altered synaptic transmission, blood–brain barrier opening, and inhibited glutamate uptake [39].
Hyponatremia
Hyponatremia is either present in 10% to 30% of SAH patients at admission or develops within 1–2 days from the initial bleed [40]. Cerebral salt-wasting syndrome and inappropriate secretion of anti-diuretic hormone are implicated in its development. SAH-related hyponatremia is difficult to treat and is associated with the risk of developing cerebral ischemia and infarctions [41].
Vascular Pathology
Cerebral vasculature constricts in response to SAH [42–45]. In animals, constriction of large and small (≤100 μm) parenchymal vessels is visible within minutes after the initial bleed (Table 3) [23, 46]. In humans, vascular imaging is mostly restricted to large vessels; these imaging studies reveal that the large vessels constrict with a delay of 3–7 days after the SAH. However, more recently, Uhl et al. confirmed constriction of small vessels in patients undergoing surgery within the first 72 h after SAH [45]. Thus, it appears that the response of small vessels to SAH in humans (constriction) is the same as in animal models.
The morphology and function of small vessels is assessed in animals and in autopsy samples from patients who died within the first 72 h after SAH. These studies show corrugation, disruption, and detachment of the endothelium from the basement membrane [7, 43]. Therefore, it is not surprising that therapeutic agents that require a functional endothelium to elicit response are ineffective during the early hours after SAH [47, 48]. Another morphological alteration found in small vessels after SAH is destruction of the basement membrane [49–51]. Although at present destruction of basement membrane is established in experimental studies only, its pathological consequence—increased vessel permeability—is established in animals and in humans [50, 52, 53]. In most cases, an increase in vascular permeability precedes and correlates well with the development of delayed cerebral ischemia and poor clinical outcome [50, 52].
Cell Death
Necrosis, apoptosis, and autophagy cell death pathways activate early in the brain after SAH (Table 3). Cerebral targets of these cell death pathways include brain cells (neurons and glia) and cerebral vasculature (smooth muscle and endothelium) [6, 51, 54]. It appears that more than one cell death pathway is active at any given time after SAH. For example; Dreier and colleagues found necrotic and apoptotic cell death and cerebral infarction in animals 24 h after SAH [55]. Similarly, Lee et al. report neuronal apoptosis in the superficial layers of the fronto-basal cortex and autophagy in deep cortical structures of animals 24 h after SAH [56]. For how long, after SAH, do these cell death pathways remain active is not clear at present. However, animal and human autopsy studies indicate that apoptotic cell death of neurons increases during the first 7 days and then decreases by 11 days after SAH [9, 57]. In SAH animals, the early cell death is associated with neurological deficits [54, 58].
Molecular Alterations
The timeline of SAH-derived molecular alterations is presented in Fig. 4. These molecular alterations are described below.

The timeline of molecular alterations during the first 72h after SAH. Ten minutes after SAH: Cerebral NO level is decreased [59] and platelet aggregates are present in parenchymal vessels [62]. Sixty minutes after SAH: cerebral NO level remains decreased [59], platelet aggregates persist in parenchymal vessels [62], plasma ET-1 level increases [74], oxidative stress is in progress [77, 78], and inflammatory cytokines are expressed [144]. Three hours after SAH: Cerebral NO level is increasing towards recovery [59], platelet aggregates are still present in the cerebral vessels [62], oxidative stress persists [77, 78], and inflammatory cytokines are expressed. Twenty-four hours after SAH: Cerebral NO level increases above basal value [60, 61], platelet aggregation in parenchymal vessels continues [62], plasma ET-1 level remains increased [73], oxidative stress persists [145], expression of inflammatory cytokine persists [144], and their markers appear in serum and CSF [88]. Seventy-four hours after SAH: CSF level of NO [61] and of ET-1 is increased [73], oxidative stress persists [145], CSF inflammatory cytokine level remains increased [88], and blood platelet count remains decreased indicating activation, sequestration/aggregation in the brain [146]. Insert: Antioxidant system activity is decreased and lipid peroxidation products accumulate within 72 h after SAH and correlate well with poor clinical conditions and outcome [79, 145]
Nitric Oxide/Nitric Oxide Synthase Pathway
A time-dependent alteration in nitric oxide (NO)/nitric oxide synthase (NOS) pathway occurs during the first 24 h after SAH. In animals, three phases of alteration in cerebral NO are recognized: a decrease within 10 min, return to basal value at 3 h [59], and an increase above basal value at 24 h after SAH [60]. In humans, due to early timing, the first two phases of cerebral NO alteration are not illustrated; the third phase, presenting an increase in NO 24 h after SAH, however, is established [61].
The NO/NOS pathway plays a major role in regulating the cerebral hemodynamic. Therefore, any alteration in this pathway can have pathological consequences. For example, NO regulates CBF and blood pressure by dilating blood vessels and by inhibiting platelet aggregation and leukocyte adherence to the vascular endothelium. After SAH, as cerebral NO level falls, CBF falls, cerebral vessels constrict, platelets aggregate, and neutrophils adhere to the vascular endothelium [59, 62, 63]. Similarly, the pathological rise in cerebral NO, 24 h after SAH, can exacerbate the brain injury. For example, NO as a free radical itself and in the form of peroxynitrite (powerful oxidant) can attack cell membrane; cause damage to the mitochondria, vascular endothelium, and smooth muscle cells [64]l; and activate cell death [65]. The initial fall and later rise in cerebral NO are linked to pathogenesis of delayed vasospasm and poor clinical outcome after SAH [61, 66, 67]. Consequently, alterations in NO occurring during the first 24 h after SAH carry acute, delayed, and prolonged consequences.
Endothelin-1
ET-1 is a potent vasoconstrictor released by astrocytes and leukocytes in response to inflammation and early ischemia after SAH [68, 69]. ET-1 is implicated in early brain injury and in the pathogenesis of delayed vasospasm and delayed ischemic deficits after SAH [70–72]. A number of observations support this concept: (1) ET-1 level increases in serum and plasma within minutes after SAH and expression of its receptors increases 24 to 48 h later [73–75]; (2) ET-1 has the capacity to produce long-lasting constriction [73]; (3) ET-1 level is increased at the time that cerebral NO is reduced (see above) and thus has a perfect unopposed opportunity to elicit a sustained contraction in cerebral vessels after SAH; and (4) ET-1 creates the degenerative morphological changes in the vascular wall similar to those that occur after SAH [76]. Therefore, ET-1 provides yet another mechanism that activates minutes after SAH and has early, delayed, and prolonged consequences.
Oxidative and Nitrosative Stress
Animal and human studies indicate that oxygen-free radicals (ROS) are generated early after SAH and consume enzymatic and non-enzymatic antioxidant defense systems [77–79]. ROS are mostly generated during lipid peroxidation and hemoglobin auto oxidation and induce oxidative stress that contributes to rapidly developing early and more slowly developing delayed ischemic injury after SAH [77, 78]. The mechanisms of ROS-induced brain injury after SAH include: (1) damage of vascular smooth muscle and endothelium, (2) disruption of blood–brain barrier, (3) production of strong spasmogens, and (4) induction of pro-apoptosis enzymes [6, 77]. Accordingly, mice that overexpress superoxide dismutase exhibit ameliorated delayed vasospasm and significantly reduced 24-h mortality [80, 81].
Inflammation
Substantial amount of data supports early activation of inflammatory cascade after SAH. Components especially important in post-SAH inflammation and injury include adhesion molecules, cytokines, leukocytes, and complement. Adhesion molecules (such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin) are required for leukocyte migration and represent inflammation. In animals, endothelial expression of adhesion molecules and their serum levels increase within 24 h after SAH and their selective inhibition improves outcome [82, 83]. Similarly, in SAH patients, serum ICAM-1 and VCAM-1 concentration increases at the day of hemorrhage, remains high for 6–8 days, and associates with delayed ischemic injury [84–86].
An early increase in pro-inflammatory cytokines (such as interlukin-6, interleukin-1 receptor antagonist, and tumor necrosis factor-alpha) is also noted in the serum and CSF of SAH patients and relates to early and delayed ischemia and poor outcome [87, 88]. Similarly, systemic complement, another promoter of inflammation, activates within the first 48 h in SAH patients and associates with delayed neurological complications [89]. In animals, early inhibition of complement prevents pathogenesis of delayed vasospasm [90].
Platelets
Platelets activate within minutes after SAH. A reduction in venous jugular platelet count and shape change indicating sequestration and activation is observed 5 min after SAH in animals and 48 h after ictus in SAH patients [91, 92]. Furthermore, platelet aggregates in the lumen of small cerebral vessels are found within 10 min after SAH in animals [62] and within 2 days after SAH in human autopsy studies [8].
The presence of platelets in the small arteries leads to “no-reflow” phenomenon: the absence of vascular filling after a period of global cerebral ischemia [93]. In addition, luminal platelet aggregates activate and promote mechanisms that cause structural injury and functional deficits in small vessels and devastate the already compromised brain. For example, they (1) mechanically obstruct and biochemically constrict (via releasing platelet-derived serotonin, ADP and PDGF) the vessel lumen to promote hypoperfusion [53, 94], (2) injure the vascular endothelium to promote further aggregation [7, 95], and (3) digest the major protein, collagen IV of the vascular basement membrane (via releasing collagenases such as matrix metalloproteinases-2 and 9) to increase vascular permeability and gain access to the brain parenchyma [7]. In brain parenchyma, platelets may activate additional inflammatory mechanisms to further aggravate brain injury after SAH.
Therapeutic Options
Animal studies demonstrate that treatment of early brain injury improves outcome after SAH. Human data supporting these finding, however, are lacking as delayed vasospasm and delayed ischemic injury remain the focus of treatment in clinics and clinical trials. The success of these clinical trials unfortunately has been limited. It is time that a new strategy for treating SAH, aimed at reducing the progression of early activated injurious mechanisms, identified in the section “Early Brain Injury by SAH”, is considered. As explained earlier, these mechanisms activate within minutes after aneurysm rupture and may evolve with time and contribute to poor outcome.
Treatment under this new strategy will begin soon after SAH patient is stabilized and will use pharmacological agents that decrease the progression of the mechanisms identified in the section “Early Brain Injury by SAH”. Pharmacological agents may include a vasodilator (such as NO donors, calcium channel blockers, magnesium or ET-1 antagonists) to prevent further constriction, improve CBF, and reduce the intensity of spreading cerebral ischemia and progressing brain injury. Similarly, an antioxidant, anti-inflammatory, or antiplatelet agent may also be used to reduce ongoing inflammation and oxidation stress. In some cases, inhibition of a single mechanism may not provide substantial protection, and an agent or a combination of agents that inhibit multiple injurious mechanisms may be needed. In such a scenario, nitric oxide donors may be of special interest; they dilate cerebral arteries, recover CBF, and inhibit platelet aggregation. Care, off course, will need to be taken not to exacerbate the brain injury. Consequently, stabilization of the patient and continuous monitoring of vital signs, including ICP, CBF, BP, and heart rate, will be of crucial importance for an early treatment to begin and continue. Nevertheless, given the failure of current therapeutic focus in improving outcome, it is clear that the new strategy aimed at prevention of early brain injury to improve SAH outcome needs be considered. The following section discusses the therapies that are found successful in preventing early brain injury in animals and the clinical trials that have used similar treatments against delayed developing complications (Fig. 5).

Therapeutic strategies against early brain injury after SAH: A battery of compounds working via different pathways has been examined against early brain injury after experimental SAH. Many of them have also been tested against delayed vasospasm and DIC. See text for explanation. ECE endothelin-converting enzyme, PKC protein kinase C, ERK1/2 extracellular signal-regulated kinase, ET-1 endothelin-1, NO nitric oxide, NOS nitric oxide synthase, GSNO S-nitrosoglutathione, SNP sodium nitroprusside, GTN nitroglycerin, eNOS endothelial nitric oxide synthase
Calcium Channel Blockers
Blockade of dihydropyridine-type calcium channel is found beneficial against SAH. Nimodipine is the most common agent used for this purpose. Animal studies show that nimodipine used 30 min to 6 h after SAH attenuates constriction and improves cerebral blood supply [96]. In SAH patients, nimodipine reduces the incidence of ischemic complications and the risk of poor outcome. Nimodipine is approved for use in SAH patients in the USA [1]. Current clinical practices call for oral administration within 4 days after SAH ictus for 21 days [97, 98].
The mechanisms underlying the beneficial effects of nimodipine in SAH patients are not clear. However, it is clear that reversal of delayed vasospasm is not one of them, as little reduction in angiographic vasospasm in patients on nimodipine is found [1]. Recovery of CBF and vasodilation, leading to cerebral protection, observed in animals, may explain nimodipine’s benefits, but remains to be established in SAH patients.
Endothelin-1 Antagonism
At least four approaches that block ET-1-mediated constriction of cerebral arteries are studied after SAH. These include: (1) blocking ET-1 biosynthesis [99, 100], (2) reducing extracellular ET-1 levels [101], (3) blocking ET-1 receptors [102, 103], and (4) inhibiting upregulation of endothelin receptors [104]. ET-1 receptor blockade has provided the most promising results. In animal studies, ET-1 receptor antagonists recover CBF when used 60 and 120 min after SAH [102]. In clinical trials, ET antagonist, Clazosentan, prevents vasospasm but does not improve the quality of life, supporting dissociation between the two measures [103]. As ET-1-mediated constriction contributes to brain injury beginning minutes after SAH, perhaps, a treatment strategy that prevents this contribution is warranted to maximize the benefits, improved quality of life, of ET-1 antagonism.
Magnesium Sulfate
A number of investigators have studied the effect of increasing cerebral magnesium against brain injury after SAH. Animal studies find that magnesium pretreatment decreases the duration of ischemic depolarization and reduces ischemic brain lesions upon acute SAH [105]. Clinical studies have so far examined the safety of magnesium treatment within the first 72 h after SAH. These small pilot studies report that continuous intravenous infusion of magnesium to obtain serum magnesium levels of 1.6–2.3 mmol/L or a rise of CSF magnesium level to 11% to 21% for 10 or 14 days is well tolerated [106]. Encouraged by the results of pilot studies, a large randomized, placebo-controlled, double-blind, multicenter phase III clinical trial (IMASH) was conducted [107]. The results could not confirm clinical benefits of intravenous magnesium infusion over placebo in SAH patients [108]. This failure may have resulted from the low CSF penetration of peripherally infused magnesium or a requirement of an even earlier administration to protect brain against injury.
Antioxidants
Antioxidants successfully prevent oxidative stress and decrease early brain injury in animals after SAH [109, 110]. However, clinical studies with the focus on delayed brain injury have not found these compounds effective [111, 112].
Methylprednisolone (a synthetic glucocorticoid) and tirilazad mesylate (a 21-aminosteroid) are the most studied antioxidants. In animals, methylprednisolone used early (immediately or 30 min) after SAH attenuates CBF reduction and a rise in cerebral resistance [113]. In addition, it prevents vasoactive prostanoid and ecosinoid release [109], reduces lipid peroxidation, and preserves an antioxidant enzyme system [114]. In a recent clinical study, methylprednisolone used within 24 to 48 h after SAH for 3 days improved 1-year functional outcome [111]. This study supports the idea that treating early brain injury after SAH improves outcome.
Similarly, tirilazad, when used in animals within 3 h after SAH prevents CBF and CPP changes [110], protects microvascular endothelium and blood–brain barrier [115]. In clinical trials, tirilazad therapy that began within 34–48 h after SAH and continued for 10 days showed improved outcome and decreased mortality in poor grade (grades IV and V) male patients only [116]. However, these results could not be reproduced and a meta-analysis that included five randomized placebo-controlled trials found no evidence that tirilazad reduces the risk of death or disability after SAH [112].
Free radical scavengers—ebselen and edaravone—are also examined against vasospasm in SAH patients. Little preclinical data exist on the efficacy of ebselen during the early phase of SAH. Experimental data on edaravone (MCI-186) show that its use within 24 h after SAH decreases lipid peroxidation (decreases malondialdehyde and increases SOD activity), decreases caspase-3 activation, increases 48 h survival, and improves neurological outcome [117]. In humans, ebselen and edaravone treatment beginning 4 days after SAH is associated with a trend towards lower incidence of vasospasm, cerebral infarction, and delayed ischemic injury [118].
Antiplatelet Agents
Surprisingly, only little animal and more clinical data are available on the use of antiplatelet agents after SAH. Acetyl salicylic acid (aspirin) and ticlopidine are the most common antiplatelet agents studied after SAH. In vitro and in vivo experimental studies demonstrate that Aspirin prevents prolonged vasoconstriction produced by blood [119]. Meta-analysis of the published data shows a trend towards better outcome in patients treated with antiplatelet agents compared with patients who received no antiplatelet agent [120]. In a small study, ticlopidine, used after cisternal drainage, reduced platelet aggregation and improved functional outcome [121]. At least two studies have examined the outcome in patients who used aspirin prior to or soon after SAH. The results are contradictory. One study found increased hemorrhage size and poor outcome in women who consumed more than 15 adult aspirin tablets per month prior to SAH [122]; the other study found a significantly reduced risk of cerebral infarction in patients with significant urinary salicylate level 48 h after SAH [123]. Clearly, a larger study is needed to ascertain the effect of dose and timing of aspirin intake on the outcome after SAH.
Nitric Oxide
The effect of increasing NO bioavailability or prolonging the duration of NO-mediated mechanisms against early brain injury is examined. Methods used to increase NO bioavailability include intracarotid infusion of NO-saturated saline [124], administration of an NO donor [23, 124–127], and increase eNOS expression and/or activity [128]. S-nitrosoglutathione and nitroglycerin, NONOate, glyceroltrinitrate, and diazeniumdiolate are some of the NO donors studied after SAH [23, 125, 126]. Studies with NO donor find that its early use recovers CBF, dilates large and small cerebral vessels, and prevents excitotoxic glutamate release after SAH [23, 46]. Approaches used to increase eNOS expression and activity such as pre-SAH statin use [129] have also produced beneficial effects—reduction in the intensity of arterial spasm 2 days after SAH [129]. Similarly, patients who were taking statin before SAH exhibit decrease risk of symptomatic vasospasm and significantly lower incidence of cerebral infarctions [130, 131].
Another approach that prolongs NO-mediated mechanisms is inhibition of cyclic guanosine 3′,5′-monophosphate (cGMP) degradation by phosphodiesterase [132]. cGMP is a mediator of many of NO-induced effects. In animals, inhibition of phosphodiesterase activity improves NO-mediated vasodilatation [132]. In SAH patients, papaverine, a non-specific phosphodiesterase inhibitor, is routinely used to dilate constricted arteries during aneurysm surgery and for the treatment of cerebral vasospasm [1]. The short half-life and risks associated with this agent, however, outweigh its benefits [133].
Overall, it appears that increasing NO level at the vascular bed is beneficial after SAH, and the sooner this is done the better the outcome is. These experimental findings remain to be duplicated in a clinical setting.
Statins—Acute Treatment
Statins are hydroxymethylglutaryl coenzyme A reductase inhibitors and potent inhibitors of cholesterol synthesis. In addition, statins inhibit platelet aggregation, reduce excitotoxic effects of glutamate, prevent endothelial and neuronal apoptosis, reduce inflammation, enhance angiogenesis, and upregulate and activate endothelial nitric oxidase synthase and nitric oxide production.
The effect of acute (within minutes from ictus for animals and within 3 days for humans) statin treatment against early and delayed brain injury after SAH has been examined. In animals, simvastatin injected at 30 min and 24 and 48 h after SAH reduced perivascular granulocyte migration and basilar artery vasospasm [134]. Animal studies also show that the protective effects of statin are rapidly lost upon its withdrawal [135]. In contrast to animals, some clinical studies do [136] and others do not [137] find acute statin treatment beneficial in SAH patients. In most of these studies, therapy began within 72 h after SAH and continued for 14 days, and delayed outcomes such as vasospasm and delayed ischemic injury were evaluated. Recently, two separate groups conducted a meta-analysis on the effect of statin on SAH outcome [138, 139]. The results are inconsistent. One study found that post-SAH statin use decreases the overall incidences of delayed vasospasm, delayed ischemic injury, and mortality [139] , while the other found that this treatment does not improve neurological outcomes [138]. Consequently, at present, the effect of post-SAH statin use on outcome remains unresolved. Perhaps a large randomized trial that studies the effect on early brain injury in addition to the delayed complications as an outcome is warranted before a decision on acute statin use after SAH can be made.
Summary
Therapies against SAH are designed to treat vasospasm with the ultimate goal of preventing delayed ischemic injury and improving outcome. The success of these therapies in reducing incidence of delayed vasospasm without reduction in delayed ischemic injury and improved quality of life indicates that treating vasospasm alone may not achieve this goal. The results of animal and human studies indicate that mechanisms leading to brain injury activate minutes after SAH and may contribute to the pathogenesis of delayed ischemic injury. Therefore, a therapy that is directed towards inhibiting early brain injury may prove more beneficial in preventing delayed ischemic injury and improving quality of life in this setting.
References
Bederson JB, Connolly ES Jr, Batjer HH, Dacey RG, Dion JE, Diringer MN, Duldner JE Jr, Harbaugh RE, Patel AB, Rosenwasser RH (2009) Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 40:994–1025
Alaraj A, Charbel FT, Amin-Hanjani S (2009) Peri-operative measures for treatment and prevention of cerebral vasospasm following subarachnoid hemorrhage. Neurol Res 31:651–659
Kassell NF, Sasaki T, Colohan AR, Nazar G (1985) Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke 16:562–572
Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller E, Zauner A, Dorsch N, Clark J, Ono S, Kiris T, Leroux P, Zhang JH (2009) Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res 31:151–158
Wilkins RH, Alexander JA, Odom GL (1968) Intracranial arterial spasm: a clinical analysis. J Neurosurg 29:121–134
Cahill J, Calvert JW, Zhang JH (2006) Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 26:1341–1353
Friedrich V, Flores R, Muller A, Sehba FA (2010) Escape of intraluminal platelets into brain parenchyma after subarachnoid hemorrhage. Neuroscience 165:968–975
Stein SC, Browne KD, Chen XH, Smith DH, Graham DI (2006) Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery 59:781–787, discussion 787–788
Nau R, Haase S, Bunkowski S, Bruck W (2002) Neuronal apoptosis in the denate gyrus in humans with subarachnoid hemorrage and cerebral hypoxia. Brain Pathol 12:329–336
Simeone FA, Ryan KG, Cotter JR (1968) Prolonged experimental cerebral vasospasm. J Neurosurg 29:357–366
Kapp J, Mahaley MS Jr, Odom GL (1968) Cerebral arterial spasm. 2. Experimental evaluation of mechanical and humoral factors in pathogenesis. J Neurosurg 29:339–349
Nornes H, Magnaes B (1972) Intracranial pressure in patients with ruptured saccular aneurysm. J Neurosurg 36:537–547
Arutiunov AI, Baron MA, Majorova NA (1970) Experimental and clinical study of the development of spasm of the cerebral arteries related to subarachnoid hemorrhage. J Neurosurg 32:617–625
Arutiunov AI, Baron MA, Majorova NA (1974) The role of mechanical factors in the pathogenesis of short-term and prolonged spasm of the cerebral arteries. J Neurosurg 40:459–472
Schwartz AY, Masago A, Sehba FA, Bederson JB (2000) Experimental models of subarachnoid hemorrhage in the rat: a refinement of the endovascular filament model. J Neurosci Methods 96:161–167
Fisher CM, Kistler JP, Davis JM (1980) Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 6:1–9
Bederson JB, Germano IM, Guarino L (1995) Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke 26:1086–1091
Asano T, Sano K (1977) Pathogenetic role of no-reflow phenomenon in experimental subarachnoid hemorrhage in dogs. J Neurosurg 46:454–466
Nornes H (1973) The role of intracranial pressure in the arrest of hemorrhage in patients with ruptured intracranial aneurysm. J Neurosurg 39:226–234
Hayashi T, Suzuki A, Hatazawa J, Kanno I, Shirane R, Yoshimoto T, Yasui N (2000) Cerebral circulation and metabolism in the acute stage of subarachnoid hemorrhage. J Neurosurg 93:1014–1018
Heuer GG, Smith MJ, Elliott JP, Winn HR, LeRoux PD (2004) Relationship between intracranial pressure and other clinical variables in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg 101:408–416
Jakubowski J, Bell BA, Symon L, Zawirski MB, Francis DM (1982) A primate model of subarachnoid hemorrhage: change in regional cerebral blood flow, autoregulation carbon dioxide reactivity, and central conduction time. Stroke 13:601–611
Sehba FA, Ding WH, Chereshnev I, Bederson JB (1999) Effects of S-nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke 30:1955–1961
Weir B, Grace M, Hansen J, Rothberg C (1978) Time course of vasospasm in man. J Neurosurg 48:173–178
Grosset DG, Straiton J, McDonald I, Bullock R (1993) Angiographic and Doppler diagnosis of cerebral artery vasospasm following subarachnoid haemorrhage. Br J Neurosurg 7:291–298
Grote E, Hassler W (1988) The critical first minutes after subarachnoid hemorrhage. Neurosurgery 22:654–661
Schmieder K, Moller F, Engelhardt M, Scholz M, Schregel W, Christmann A, Harders A (2006) Dynamic cerebral autoregulation in patients with ruptured and unruptured aneurysms after induction of general anesthesia. Zentralbl Neurochir 67:81–87
Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, Vajkoczy P, Lauritzen M, Dirnagl U, Bohner G, Strong AJ (2009) Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132:1866–1881
Gwag BJ, Canzoniero LM, Sensi SL, Demaro JA, Koh JY, Goldberg MP, Jacquin M, Choi DW (1999) Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neuroscience 90:1339–1348
Dreier JP, Windmuller O, Petzold G, Lindauer U, Einhaupl KM, Dirnagl U (2002) Ischemia triggered by red blood cell products in the subarachnoid space is inhibited by nimodipine administration or moderate volume expansion/hemodilution in rats. Neurosurgery 51:1457–1465, discussion 1465–1457
Petzold GC, Einhaupl KM, Dirnagl U, Dreier JP (2003) Ischemia triggered by spreading neuronal activation is induced by endothelin-1 and hemoglobin in the subarachnoid space. Ann Neurol 54:591–598
van den Bergh WM, Algra A, van der Sprenkel JW, Tulleken CA, Rinkel GJ (2003) Hypomagnesemia after aneurysmal subarachnoid hemorrhage. Neurosurgery 52:276–281, discussion 281–272
Altura BT, Memon ZI, Zhang A, Cheng TP, Silverman R, Cracco RQ, Altura BM (1997) Low levels of serum ionized magnesium are found in patients early after stroke which result in rapid elevation in cytosolic free calcium and spasm in cerebral vascular muscle cells. Neurosci Lett 230:37–40
Berthon N, Laurant P, Fellmann D, Berthelot A (2003) Effect of magnesium on mRNA expression and production of endothelin-1 in DOCA-salt hypertensive rats. J Cardiovasc Pharmacol 42:24–31
Milhorat TH (1987) Acute hydrocephalus after aneurysmal subarachnoid hemorrhage. Neurosurgery 20:15–20
van Asch CJ, van der Schaaf IC, Rinkel GJ (2010) Acute hydrocephalus and cerebral perfusion after aneurysmal subarachnoid hemorrhage. AJNR Am J Neuroradiol 31:67–70
Kamiya K, Kuyama H, Symon L (1983) An experimental study of the acute stage of subarachnoid hemorrhage. J Neurosurg 59:917–924
Dorai Z, Hynan LS, Kopitnik TA, Samson D (2003) Factors related to hydrocephalus after aneurysmal subarachnoid hemorrhage. Neurosurgery 52:763–769, discussion 769–771
Enblad P, Valtysson J, Andersson J, Lilja A, Valind S, Antoni G, Langstrom B, Hillered L, Persson L (1996) Simultaneous intracerebral microdialysis and positron emission tomography in the detection of ischemia in patients with subarachnoid hemorrhage. J Cereb Blood Flow Metab 16:637–644
Berendes E, Walter M, Cullen P, Prien T, Van Aken H, Horsthemke J, Schulte M, von Wild K, Scherer R (1997) Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 349:245–249
Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J (1985) Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 18:211–216
Pennings FA, Bouma GJ, Ince C (2004) Direct observation of the human cerebral microcirculation during aneurysm surgery reveals increased arteriolar contractility. Stroke 35:1284–1288
Hatake K, Wakabayashi I, Kakishita E, Hishida S (1992) Impairment of endothelium-dependent relaxation in human basilar artery after subarachnoid hemorrhage. Stroke 23:1111–1116, discussion 1116–1117
Sehba FA, Flores R, Muller A, Friedrich V, Chen JF, Britz GW, Winn HR, Bederson JB (2010) Adenosine A(2A) receptors in early ischemic vascular injury after subarachnoid hemorrhage. J Neurosurg 113(4):826–834
Uhl E, Lehmberg J, Steiger HJ, Messmer K (2003) Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery 52:1307–1315, discussion 1315–1307
Sehba FA, Makonnen G, Friedrich V, Bederson JB (2007) Acute cerebral vascular injury occurs after subarachnoid hemorrhage and can be prevented by administration of a nitric oxide donor. J Neurosurg 106:321–329
Park KW, Metais C, Dai HB, Comunale ME, Sellke FW (2001) Microvascular endothelial dysfunction and its mechanism in a rat model of subarachnoid hemorrhage. Anesth Analg 92(4):990–996
Bevan JA, Bevan RD, Walters CL, Wellman T (1998) Functional changes in human pial arteries (300 to 900 micrometer ID) within 48 hours of aneurysmal subarachnoid hemorrhage. Stroke 29:2575–2579
Sehba FA, Mostafa G, Knopman J, Friedrich V Jr, Bederson JB (2004) Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J Neurosurg 101:633–640
Scholler K, Trinkl A, Klopotowski M, Thal SC, Plesnila N, Trabold R, Hamann GF, Schmid-Elsaesser R, Zausinger S (2007) Characterization of microvascular basal lamina damage and blood–brain barrier dysfunction following subarachnoid hemorrhage in rats. Brain Res 1142:237–246
Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH (2007) Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res 85:1436–1448
Germano A, d’Avella D, Imperatore C, Caruso G, Tomasello F (2000) Time-course of blood–brain barrier permeability changes after experimental subarachnoid haemorrhage. Acta Neurochir 142:575–580
Friedrich V, Flores R, Muller A, Sehba FA (2010) Luminal platelet aggregates in functional deficits in parenchymal vessels after subarachnoid hemorrhage. Brain Res 1354:179–187
Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH (2004) Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke 35:2412–2417
Dreier JP, Ebert N, Priller J, Megow D, Lindauer U, Klee R, Reuter U, Imai Y, Einhaupl KM, Victorov I, Dirnagl U (2000) Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg 93:658–666
Lee JY, He Y, Sagher O, Keep R, Hua Y, Xi G (2009) Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res 1287:126–135
Prunell GF, Svendgaard NA, Alkass K, Mathiesen T (2005) Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J Neurosurg 102:1046–1054
Cahill J, Calvert JW, Marcantonio S, Zhang JH (2007) p53 may play an orchestrating role in apoptotic cell death after experimental subarachnoid hemorrhage. Neurosurgery 60:531–545, discussion 545
Sehba FA, Schwartz AY, Chereshnev I, Bederson JB (2000) Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. J Cereb Blood Flow Metab 20:604–611
Yatsushige H, Calvert JW, Cahill J, Zhang JH (2006) Limited role of inducible nitric oxide synthase in blood–brain barrier function after experimental subarachnoid hemorrhage. J Neurotrauma 23:1874–1882
Ng WH, Moochhala S, Yeo TT, Ong PL, Ng PY (2001) Nitric oxide and subarachnoid hemorrhage: elevated level in cerebrospinal fluid and their implications. Neurosurgery 49:622–626, discussion 626–627
Sehba FA, Mustafa G, Friedrich V, Bederson JB (2005) Acute microvascular platelet aggregation after subarachnoid hemorrhage. J Neurosurg 102:1094–1100
Flores R, Muller A, Friedrich V, and Sehba FA (2009) Neutrophils in early vascular injury after subarachnoid hemorrhage (SAH). Society for Neurosciences abstracts; Poster 51.5/I20
Moro MA, Almeida A, Bolanos JP, Lizasoain I (2005) Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med 39:1291–1304
Szabo C, Dawson VL (1998) Role of poly(ADP-ribose) synthetase in inflammation and ischaemia–reperfusion. Trends Pharmacol Sci 19:287–298
Pluta RM, Oldfield EH, Boock RJ (1997) Reversal and prevention of cerebral vasospasm by intracarotid infusions of nitric oxide donors in a primate model of subarachnoid hemorrhage. J Neurosurg 87:746–751
Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH (2005) Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. Jama 293:1477–1484
Pluta RM, Boock RJ, Afshar JK, Clouse K, Bacic M, Ehrenreich H, Oldfield EH (1997) Source and cause of endothelin-1 release into cerebrospinal fluid after subarachnoid hemorrhage. J Neurosurg 87:287–293
Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, Fritzinger M, Horn P, Vajkoczy P, Wendel-Wellner M, Ragoschke A, Kuehl S, Brunner J, Schurer L, Schmiedeck P, Hennerici M (2000) Endothelin-1 in subarachnoid hemorrhage: an acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke 31:2971–2975
Zimmermann M, Seifert V (1998) Endothelin and subarachnoid hemorrhage: an overview. Neurosurgery 43:863–875, discussion 875–866
Juvela S (2000) Plasma endothelin concentrations after aneurysmal subarachnoid hemorrhage. J Neurosurg 92:390–400
Ostrowski RP, Colohan AR, Zhang JH (2006) Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res 28:399–414
Kobayashi H, Ide H, Ishii H, Kabuto M, Handa Y, Kubota T (1995) Endothelin-1 levels in plasma and cerebrospinal fluid following subarachnoid haemorrhage. J Clin Neurosci 2:252–256
Josko J, Hendryk S, Jedrzejowska-Szypula H, Gwozdz B, Herman ZS, Gawlik R (1998) Influence endothelin ETA receptor antagonist—BQ-123—on changes of endothelin-1 level in plasma of rats with acute vasospasm following subarachnoid hemorrhage. J Physiol Pharmacol 49:367–375
Vikman P, Beg S, Khurana TS, Hansen-Schwartz J, Edvinsson L (2006) Gene expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J Neurosurg 105:438–444
Kasuya H, Weir BK, White DM, Stefansson K (1993) Mechanism of oxyhemoglobin-induced release of endothelin-1 from cultured vascular endothelial cells and smooth-muscle cells. J Neurosurg 79:892–898
Gaetani P, Marzatico F, Rodriguez y Baena R, Pacchiarini L, Vigano T, Grignani G, Crivellari MT, Benzi G (1990) Arachidonic acid metabolism and pathophysiologic aspects of subarachnoid hemorrhage in rats. Stroke 21:328–332
Marzatico F, Gaetani P, Cafe C, Spanu G, Rodriguez y Baena R (1993) Antioxidant enzymatic activities after experimental subarachnoid hemorrhage in rats. Acta Neurol Scand 87:62–66
Lin CL, Hsu YT, Lin TK, Morrow JD, Hsu JC, Hsu YH, Hsieh TC, Tsay PK, Yen HC (2006) Increased levels of F2-isoprostanes following aneurysmal subarachnoid hemorrhage in humans. Free Radic Biol Med 40:1466–1473
Endo H, Nito C, Kamada H, Yu F, Chan PH (2007) Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of Akt/glycogen synthase kinase-3beta survival signaling. J Cereb Blood Flow Metab 27:975–982
Kamii H, Kato I, Kinouchi H, Chan PH, Epstein CJ, Akabane A, Okamoto H, Yoshimoto T (1999) Amelioration of vasospasm after subarachnoid hemorrhage in transgenic mice overexpressing CuZn-superoxide dismutase. Stroke 30:867–871, discussion 872
Handa Y, Kubota T, Kaneko M, Tsuchida A, Kobayashi H, Kawano H (1995) Expression of intercellular adhesion molecule 1 (ICAM-1) on the cerebral artery following subarachnoid haemorrhage in rats. Acta Neurochir (Wien) 132:92–97
Lin CL, Dumont AS, Calisaneller T, Kwan AL, Hwong SL, Lee KS (2005) Monoclonal antibody against E selectin attenuates subarachnoid hemorrhage-induced cerebral vasospasm. Surg Neurol 64:201–205
Mack WJ, Mocco J, Hoh DJ, Huang J, Choudhri TF, Kreiter KT, Lozier A, Opperman M, Poisik A, Yorgason J, Solomon RA, Mayer SA, Connolly ES (2002) Outcome prediction with serum intercellular adhesion molecule-1 levels after aneurysmal subarachnoid hemorrhage. J Neurosurg 96:71–75
Kubo Y, Ogasawara K, Kakino S, Kashimura H, Tomitsuka N, Sugawara A, Ogawa A (2008) Serum inflammatory adhesion molecules and high-sensitivity C-reactive protein correlates with delayed ischemic neurologic deficits after subarachnoid hemorrhage. Surg Neurol 69:592–596, discussion 596
Mocco J, Mack WJ, Kim GH, Lozier AP, Laufer I, Kreiter KT, Sciacca RR, Solomon RA, Mayer SA, Connolly ES Jr (2002) Rise in serum soluble intercellular adhesion molecule-1 levels with vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 97:537–541
Mathiesen T, Edner G, Ulfarsson E, Andersson B (1997) Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J Neurosurg 87:215–220
Gruber A, Rossler K, Graninger W, Donner A, Illievich MU, Czech T (2000) Ventricular cerebrospinal fluid and serum concentrations of sTNFR-I, IL-1ra, and IL-6 after aneurysmal subarachnoid hemorrhage. J Neurosurg Anesthesiol 12:297–306
Kasuya H, Shimizu T (1989) Activated complement components C3a and C4a in cerebrospinal fluid and plasma following subarachnoid hemorrhage. J Neurosurg 71:741–746
German JW, Gross CE, Giclas P, Watral W, Bednar MM (1996) Systemic complement depletion inhibits experimental cerebral vasospasm. Neurosurgery 39:141–145, discussion 145–146
Hirashima Y, Hamada H, Kurimoto M, Origasa H, Endo S (2005) Decrease in platelet count as an independent risk factor for symptomatic vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 102:882–887
Denton IC, Robertson JT, Dugdale M (1971) An assessment of early platelet activity in experimental subarachnoid hemorrhage and middle cerebral artery thrombosis in the cat. Stroke 2:268–272
Abumiya T, Fitridge R, Mazur C, Copeland BR, Koziol JA, Tschopp JF, Pierschbacher MD, del Zoppo GJ (2000) Integrin alpha(IIb)beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke 31:1402–1409, discussion 1409–1410
Okada Y, Copeland BR, Mori E, Tung MM, Thomas WS, del Zoppo GJ (1994) P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke 25:202–211
Clower BR, Yoshioka J, Honma Y, Smith RR (1988) Pathological changes in cerebral arteries following experimental subarachnoid hemorrhage: role of blood platelets. Anat Rec 220:161–170
Sun BL, Zhang SM, Xia ZL, Yang MF, Yuan H, Zhang J, Xiu RJ (2003) The effects of nimodipine on regional cerebral blood flow, brain water and electrolyte contents in rats with subarachnoid hemorrhage. Clin Hemorheol Microcirc 29:337–344
Dorhout Mees SM, Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, van Gijn J (2008) Calcium antagonists for aneurysmal subarachnoid hemorrhage. Stroke 39:514–515
Dorhout Mees SM, Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, and van Gijn J (2007) Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev CD000277
Onoda K, Ono S, Ogihara K, Shiota T, Asari S, Ohmoto T, Ninomiya Y (1996) Inhibition of vascular contraction by intracisternal administration of preproendothelin-1 mRNA antisense oligoDNA in a rat experimental vasospasm model. J Neurosurg 85:846–852
Caner HH, Kwan AL, Arthur A, Jeng AY, Lappe RW, Kassell NF, Lee KS (1996) Systemic administration of an inhibitor of endothelin-converting enzyme for attenuation of cerebral vasospasm following experimental subarachnoid hemorrhage. J Neurosurg 85:917–922
Yamaura I, Tani E, Maeda Y, Minami N, Shindo H (1992) Endothelin-1 of canine basilar artery in vasospasm. J Neurosurg 76:99–105
Clozel M, Watanabe H (1993) BQ-123, a peptidic endothelin ETA receptor antagonist, prevents the early cerebral vasospasm following subarachnoid hemorrhage after intracisternal but not intravenous injection. Life Sci 52:825–834
Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S, Pasqualin A (2008) Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 39:3015–3021
Beg SA, Hansen-Schwartz JA, Vikman PJ, Xu CB, Edvinsson LI (2006) ERK1/2 inhibition attenuates cerebral blood flow reduction and abolishes ET(B) and 5-HT(1B) receptor upregulation after subarachnoid hemorrhage in rat. J Cereb Blood Flow Metab 26:846–856
van den Bergh WM, Zuur JK, Kamerling NA, van Asseldonk JT, Rinkel GJ, Tulleken CA, Nicolay K (2002) Role of magnesium in the reduction of ischemic depolarization and lesion volume after experimental subarachnoid hemorrhage. J Neurosurg 97:416–422
McKee JA, Brewer RP, Macy GE, Borel CO, Reynolds JD, Warner DS (2005) Magnesium neuroprotection is limited in humans with acute brain injury. Neurocrit Care 2:342–351
Wong GK, Chan MT, Poon WS, Boet R, Gin T (2006) Magnesium therapy within 48 hours of an aneurysmal subarachnoid hemorrhage: neuro-panacea. Neurol Res 28(4):431–435
Wong GK, Poon WS, Chan MT, Boet R, Gin T, Ng SC, Zee BC (2010) Intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage (IMASH): a randomized, double-blinded, placebo-controlled, multicenter phase III trial. Stroke 41:921–926
Gaetani P, Marzatico F, Renault B, Fulle I, Lombardi D, Ferlenga P, Rodriguez y Baena R (1990) High-dose methylprednisolone and ‘ex vivo’ release of eicosanoids after experimental subarachnoid haemorrhage. Neurol Res 12:111–116
Hall ED, Travis MA (1988) Effects of the nonglucocorticoid 21-aminosteroid U74006F on acute cerebral hypoperfusion following experimental subarachnoid hemorrhage. Exp Neurol 102:244–248
Gomis P, Graftieaux JP, Sercombe R, Hettler D, Scherpereel B, Rousseaux P (2010) Randomized, double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J Neurosurg 112:681–688
Zhang S, Wang L, Liu M, Wu B (2010) Tirilazad for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2:CD006778
Hall ED, Travis MA (1988) Attenuation of progressive brain hypoperfusion following experimental subarachnoid hemorrhage by large intravenous doses of methylprednisolone. Exp Neurol 99:594–606
Lombardi D, Gaetani P, Marzatico F, Cafe C, Rodriguez y Baena R (1992) Effect of high-dose methylprednisolone on anti-oxidant enzymes after experimental SAH. J Neurol Sci 111:13–19
Smith SL, Larson PG, Hall ED (1997) A comparison of the effects of tirilazad on subarachnoid hemorrhage-induced blood–brain barrier permeability in male and female rats. J Stroke Cerebrovasc Dis 6:389–393
Kassell NF, Haley EC Jr, Apperson-Hansen C, Alves WM (1996) Randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in Europe, Australia, and New Zealand [see comments]. J Neurosurg 84:221–228
Gao Y, Ding XS, Xu S, Wang W, Zuo QL, Kuai F (2009) Neuroprotective effects of edaravone on early brain injury in rats after subarachnoid hemorrhage. Chin Med J (Engl) 122:1935–1940
Munakata A, Ohkuma H, Nakano T, Shimamura N, Asano K, Naraoka M (2009) Effect of a free radical scavenger, edaravone, in the treatment of patients with aneurysmal subarachnoid hemorrhage. Neurosurgery 64:423–428, discussion 428–429
Linder M, Alksne JF (1978) Prevention of persistent cerebral smooth muscle contraction in response to whole blood. Stroke 9:472–477
Dorhout Mees SM, van den Bergh WM, Algra A, and Rinkel GJ (2007) Antiplatelet therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev CD006184
Toyoda O, Nakajima H, Nukui H (1990) Changes in coagulation and fibrinolysis and effects of ticlopidine and cisternal drainage in the acute phase following aneurysm rupture. Neurol Med Chir (Tokyo) 30:670–675
Iso H, Hennekens CH, Stampfer MJ, Rexrode KM, Colditz GA, Speizer FE, Willett WC, Manson JE (1999) Prospective study of aspirin use and risk of stroke in women. Stroke 30:1764–1771
Juvela S (1995) Aspirin and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurosurg 82:945–952
Afshar JK, Pluta RM, Boock RJ, Thompson BG, Oldfield EH (1995) Effect of intracarotid nitric oxide on primate cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg 83:118–122
Tierney TS, Clatterbuck RE, Lawson C, Thai QA, Rhines LD, Tamargo RJ (2001) Prevention and reversal of experimental posthemorrhagic vasospasm by the periadventitial administration of nitric oxide from a controlled-release polymer. Neurosurgery 49:945–951, discussion 951–943
Ito Y, Isotani E, Mizuno Y, Azuma H, Hirakawa K (2000) Effective improvement of the cerebral vasospasm after subarachnoid hemorrhage with low-dose nitroglycerin. J Cardiovasc Pharmacol 35:45–50
Raabe A, Zimmermann M, Setzer M, Vatter H, Berkefeld J, Seifert V (2002) Effect of intraventricular sodium nitroprusside on cerebral hemodynamics and oxygenation in poor-grade aneurysm patients with severe, medically refractory vasospasm. Neurosurgery 50:1006–1013, discussion 1013–1004
Luders JC, Weihl CC, Lin G, Ghadge G, Stoodley M, Roos RP, Macdonald RL (2000) Adenoviral gene transfer of nitric oxide synthase increases cerebral blood flow in rats. Neurosurgery 47:1206–1214, discussion 1214–1205
McGirt MJ, Lynch JR, Parra A, Sheng H, Pearlstein RD, Laskowitz DT, Pelligrino DA, Warner DS (2002) Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke 33:2950–2956
McGirt MJ, Blessing R, Alexander MJ, Nimjee SM, Woodworth GF, Friedman AH, Graffagnino C, Laskowitz DT, Lynch JR (2006) Risk of cerebral vasopasm after subarachnoid hemorrhage reduced by statin therapy: a multivariate analysis of an institutional experience. J Neurosurg 105:671–674
Parra A, Kreiter KT, Williams S, Sciacca R, Mack WJ, Naidech AM, Commichau CS, Fitzsimmons BF, Janjua N, Mayer SA, Connolly ES Jr (2005) Effect of prior statin use on functional outcome and delayed vasospasm after acute aneurysmal subarachnoid hemorrhage: a matched controlled cohort study. Neurosurgery 56:476–484, discussion 476–484
Sobey CG, Quan L (1999) Impaired cerebral vasodilator responses to NO and PDE V inhibition after subarachnoid hemorrhage. Am J Physiol 277:H1718–H1724
Platz J, Barath K, Keller E, Valavanis A (2008) Disruption of the blood–brain barrier by intra-arterial administration of papaverine: a technical note. Neuroradiology 50:1035–1039
McGirt MJ, Pradilla G, Legnani FG, Thai QA, Recinos PF, Tamargo RJ, Clatterbuck RE (2006) Systemic administration of simvastatin after the onset of experimental subarachnoid hemorrhage attenuates cerebral vasospasm. Neurosurgery 58:945–951, discussion 945–951
Gertz K, Laufs U, Lindauer U, Nickenig G, Bohm M, Dirnagl U, Endres M (2003) Withdrawal of statin treatment abrogates stroke protection in mice. Stroke 34:551–557
Lynch JR, Wang H, McGirt MJ, Floyd J, Friedman AH, Coon AL, Blessing R, Alexander MJ, Graffagnino C, Warner DS, Laskowitz DT (2005) Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage: results of a pilot randomized clinical trial. Stroke 36(9):2024–2026
Vergouwen MD, Meijers JC, Geskus RB, Coert BA, Horn J, Stroes ES, van der Poll T, Vermeulen M, Roos YB (2009) Biologic effects of simvastatin in patients with aneurysmal subarachnoid hemorrhage: a double-blind, placebo-controlled randomized trial. J Cereb Blood Flow Metab 29:1444–1453
Kramer AH, Fletcher JJ (2010) Statins in the management of patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Neurocrit Care 12:285–296
Tapia-Perez JH, Sanchez-Aguilar M, Schneider T (2010) The role of statins in neurosurgery. Neurosurg Rev 33:259–270, discussion 270
Jackowski A, Crockard A, Burnstack G, Russel R, Kristek F (1990) The time course of intracranial pathophysiological changes following experimental subarachnoid hemorrhage in the rat. J Cereb Blood Flow Metab 10:835–839
Rasmussen G, Hauerberg J, Waldemar G, Gjerris F, Juhler M (1992) Cerebral blood flow autoregulation in experimental subarachnoid haemorrhage in rat. Acta Neurochir 119:128–133
Bederson JB, Levy AL, Ding WH, Kahn R, DiPerna CA, ALr J, Vallabhajosyula P (1998) Acute vasoconstriction after subarachnoid hemorrhage. Neurosurgery 42:352–360
Sarrafzadeh AS, Sakowitz OW, Kiening KL, Benndorf G, Lanksch WR, Unterberg AW (2002) Bedside microdialysis: a tool to monitor cerebral metabolism in subarachnoid hemorrhage patients? Crit Care Med 30:1062–1070
Sehba FA, Flores R, Ono K, Suzuki H, Sawada MS, and Kanzawa T (2008) Expression of cytokines and neurotrophins after acute subarachnoid hemorrhage. Annual Stroke Conference 2008, Session: 126, P327
Kamezaki T, Yanaka K, Nagase S, Fujita K, Kato N, Nose T (2002) Increased levels of lipid peroxides as predictive of symptomatic vasospasm and poor outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 97:1302–1305
Hirashima Y, Hayashi N, Endo S, Takaku A (1993) Sequential changes in the platelet count in patients with symptomatic vasospasm after subarachnoid hemorrhage. Neurol Med Chir (Tokyo) 33:220–224
Trojanowski T (1984) Early effects of experimental arterial subarachnoid haemorrhage on the cerebral circulation. Part I: Experimental subarachnoid haemorrhage in cat and its pathophysiological effects. Methods of regional cerebral blood flow measurement and evaluation of microcirculation. Acta Neurochir 72:79–94
Hubschmann OR (1987) The role of calcium in parenchymal cell injury in subarachnoid haemorrhage. Neurol Res 9:265–269
Sambrook MA, Hutchinson EC, Aber GM (1973) Metabolic studies in subarachnoid haemorrhage and strokes. II. Serial changes in cerebrospinal fluid and plasma urea electrolytes and osmolality. Brain 96:191–202
von Holst H, Mathiesen T (1990) Electrolyte concentrations in serum and CSF following subarachnoid haemorrhage. Br J Neurosurg 4:123–126
Yamaura I, Tani E, Saido TC, Suzuki K, Minami N, Maeda Y (1993) Calpain–calpastatin system of canine basilar artery in vasospasm. J Neurosurg 79:537–543
Yan J, Chen C, Hu Q, Yang X, Lei J, Yang L, Wang K, Qin L, Huang H, Zhou C (2008) The role of p53 in brain edema after 24 h of experimental subarachnoid hemorrhage in a rat model. Exp Neurol 214:37–46
Zubkov AY, Tibbs RE, Clower B, Ogihara K, Aoki K, Zhang JH (2002) Morphological changes of cerebral arteries in a canine double hemorrhage model. Neurosci Lett 326:137–141
Acknowledgments
This work was supported by the American Heart Association grant number GRNT4570012 (FAS), and the National Institutes of Health, grant numbers RO1 NS050576 (FAS) and NS053407 (JHZ), and by the Intramural Research Program (RMP) of the National Institutes of Stroke and Neurological Disorders, NIH.
Disclosure/Conflict of Interest
None
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sehba, F.A., Pluta, R.M. & Zhang, J.H. Metamorphosis of Subarachnoid Hemorrhage Research: from Delayed Vasospasm to Early Brain Injury. Mol Neurobiol 43, 27–40 (2011). https://doi.org/10.1007/s12035-010-8155-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-010-8155-z