
Abstract
Every year, more cases of sepsis appear in intensive care units. The most frequent complication of sepsis is septic encephalopathy (SE), which is also the essential determinant of mortality. Despite many years of research, it still is not known at which stage of sepsis the first signs of SE appear; however, it is considered the most frequent form of encephalopathy. Patients have dysfunction of cognitive abilities and consciousness, and sometimes even epileptic seizures. Despite intensive treatment, the effects of SE remain for many years and constitute an important social problem. Numerous studies indicate that changes in the brain involve free radicals, nitric oxide, increased synthesis of inflammatory factors, disturbances in cerebral circulation, microthromboses, and ischemia, which cause considerable neuronal destruction in different areas of the brain. To determine at what point during sepsis the first signs of SE appear, different experimental models are needed to detect the aforementioned changes and to select the proper therapy for this syndrome.
Similar content being viewed by others
Introduction
Sepsis is defined as the excessive inflammatory reaction of an organism to an infection [1, 2]. It is not a disease in itself, but rather is a systemic inflammatory response due to infection, burn, trauma, or other factors [3]. Sepsis and its complications are the most frequent cause of high mortality in the intensive care unit (ICU), estimated at about 750,000 cases annually in the USA [4, 5], and detected sepsis cases make up 75 % of all illnesses treated in the ICU [4, 6]. Additionally, despite considerable progress in diagnosis and treatment, a high degree of mortality is still noted, and the morbidity rate has been increasing annually from about 1.5 % to as much as 8 % [7]. The high mortality rate persists among patients treated for sepsis, even 1 month to 1 year after they leave the ICU [8]. This indicates that further examination of the effects of medical treatment for sepsis is necessary, especially considering that long-lasting disturbances in organ function may appear, both physically—as, for example, with dyspnea—and mentally, as fatigue or depression [9, 10].
Because the nervous system is susceptible to many different factors, it is not surprising that the intensive inflammatory response of sepsis affects brain function. Liver or renal dysfunction accompanying sepsis may result in encephalopathy [11, 12]; however, sepsis may result in encephalopathy even in the absence of systemic organ failure. Sepsis is typically regarded as being caused by infectious factors, such as bacteria, viruses, or fungi; however, encephalopathy may also occur with metabolic disorders [11], exposure to toxins [12] or radiation [13], injury [14, 15], disturbances in blood flow [16], and other factors. Among the many complications of sepsis, septic encephalopathy (SE) is considered the most frequent [4, 18], and it is estimated that 9–71 % of patients with diagnosed sepsis exhibit symptoms of encephalopathy [4, 19, 20]. Although SE has been described as a reversible syndrome, studies indicate long-lasting cognitive and depressive disturbances in patients after the sepsis resolves [21, 22]. Recovery from these cognitive and mental symptoms is often slow. The mortality of SE remains high and correlates with the intensity of the course of SE, as determined by the Glasgow Coma Scale [23–25], suggesting that nervous system dysfunction is the pivotal factor determining sepsis mortality.
Diagnosis of Septic Encephalopathy
A major problem that still exists is the inability to properly recognize the signs of SE, because septic patients are usually sedated, which masks neurologic disturbances. The diagnosis of SE requires the recognition of brain dysfunction, which depends on using clinical, electrophysiologic, or biochemical criteria [26–29]. Helpful diagnostic tools for determining mental state and predicting the markers of SE course and mortality are clinical scales such as the Glasgow Coma Scale, the Confusion Assessment Method for the ICU, and Adaption to the Intensive Care Environment [30–33]. In nonsedated patients, the diagnosis is simpler, and the Confusion Assessment Method for the ICU, which uses acute symptoms of mental changes, inattention, and disorganized thinking to indicate encephalopathy or delirium, may be applied [34, 35]. The Adaption to the Intensive Care Environment scale is based on a patient’s visual reaction to different stimulants, allowing the clinician to estimate the patient’s degree of consciousness or comprehension [20, 32]. In turn, the Glasgow Coma Scale or the Richmond Agitation–Sedation Scale may be applied in sedated patients [31, 36].
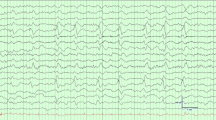
Another method used to obtain significant information about a patient’s mental status is electroencephalography; it is among the most sensitive diagnostic tools, and disturbances detected by electroencephalography correlate well with SE severity [37, 38, 39••, 40]. Young et al. [41] ascertained that the electroencephalogram is more sensitive than classic clinical criteria that define the state of consciousness; as disease severity increases, electroencephalogram recordings change from normal to excessive theta waves, followed by predominant delta waves, triphasic waves, and finally, suppression or burst suppression. In turn, Oddo et al. [42] observed periodic epileptiform discharges as well as seizure activity on electroencephalogram recordings in 22 % of SE patients, but in two thirds of the patients, the electroencephalographic abnormalities did not correlate with clinical observations. However, the aforementioned electroencephalographic abnormalities were more frequent in SE patients and positively correlated with the mortality or frequency of multiorgan dysfunction [43].
Neuroimaging changes are also helpful in detecting SE. Because of its ease and convenience, computed tomography of the brain is used most often [44], although more information is obtained by cranial magnetic resonance imaging (MRI). MRI is particularly helpful in excluding other diseases. MRI in SE may show cerebral infarction, leukoencephalopathy, and vascular edema [45]. Although these observations are hardly specific to SE [46], postmortem examinations show more frequent changes in magnetic resonance images of SE patients, most likely reflecting disturbances in the blood–brain barrier and white matter destruction, along with microvascular edema [45, 46].
Significant changes appear in the cerebral circulation with SE, and transcranial Doppler (TCD) sonography has been used to monitor cerebral vasomotor reactivity in the condition [39••, 47–49]. TCD sonography makes it possible to study cerebral arterial vessel reactivity to different parameters as increasing extracellular CO2 partial pressure or decreasing pH, using the brain’s inherent ability to maintain a constant blood flow despite changes in cerebral perfusion pressure [50, 51]. Often, disturbances in vascular autoregulation, especially in the early stages of SE [39••], suggest that hemodynamic phenomena play a crucial role in the course of SE [52]. However, TCD sonography cannot be entirely relied on as a diagnostic tool because in some instances of SE no differences in cerebral perfusion have been detected [48, 49]. Perhaps these differences are the result of the different rates of the course of inflammatory processes affecting the function of the cerebral vessel endothelium [49].
The ability to detect qualitative and quantitative differences of specific substances in tissue or blood may serve as a diagnostic tool, assist in prognosis, help in the selection of the appropriate therapy, and assist in the development of suitable research models to better recognize disease mechanisms. Unfortunately, examinations conducted over many years indicate there are no unambiguous or specific markers of SE. Most of the markers, such as increased serum levels of S-100β protein or nonspecific enolase, indicate only pathologic processes in the brain, not their nature [27, 28], and do not correlate with SE severity. Moreover, in some cases, there are no changes in the S-100β protein level in cerebrospinal fluid despite an increased level in the serum [46], disqualifying it as a marker of SE. Certainly, the discovery and use of other markers in the future remain challenges worthy of further research.
Clinical Symptoms of Septic Encephalopathy
The main features of encephalopathy are disturbances of consciousness, impaired cognitive function, personality changes, lack of concentration, and depressive symptoms [17, 21, 53]. Clinical symptoms observed during SE affect 8–70 % of patients with diagnosed sepsis [54] and include inattention, confusion, and considerable excitation, which may lead to stupor and coma. The first symptoms usually appear in the early stage of sepsis, often before other organ disturbances are diagnosed [55, 56], and represent the severest symptoms, such as weakness, anorexia, malaise, and concentration deficits. Sometimes convulsions, myoclonus, or asterixis may be observed, as well as focal or generalized seizures, although less frequently than in other encephalopathies [19, 57]. As a result, ill patients fall into delirium with acute impairment of consciousness, which appears in up to 82 % of mechanically ventilated patients [34]. Delirium is associated with several adverse outcomes, including increased morbidity and mortality, prolonged hospitalization, and poor surgical outcome, and the longer it lasts, the greater the probability of long-lasting behavioral disorders [58•].
Increasing data indicate the significant risk of long-lasting neurocognitive changes, which may have a considerable influence on patients’ quality of life [22, 59–61]. These changes are varied, but mainly encompass psychomotor activity, visual and functional memory, verbal fluency, and visual construction [62]. It is estimated that about 45 % of patients who recover from sepsis still show cognitive dysfunction 1 year after hospitalization [63], and the dysfunction may persist even longer [61, 64, 65•]. Moreover, many patients who have had sepsis show depressive signs and anxiety disorders [66], with symptoms affecting up to 58 % of former ICU patients [64]. This creates a huge social problem because, depending on the degree of impairment, a tremendous burden may be placed on family members and caregivers [62].
Pathophysiology of Septic Encephalopathy
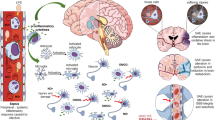
The cause of SE is still poorly understood, although many mechanisms for its formation and development have been proposed [67]. Although bacterial infection is one of the most frequent causes of sepsis, the vast majority of studies have not found bacteria present in the nervous system, which indicates that the cause of SE does not result in the direct infection of the brain with microorganisms, but has a different basis. Among the factors involved in SE are oxidative stress [68], increased cytokine and proinflammatory factor levels [69], disturbances in cerebral circulation [70], changes in blood–brain barrier permeability [71], injury to the brain’s vascular endothelium [72–74], altered levels of neurotransmitters [3], changes in amino acid levels [75], and bacterial endotoxins leaking through the blood–brain barrier [76]. However, the brain dysfunction observed during SE most likely is the result of a combination of these factors, with the onset of one factor leading to the activation of others.
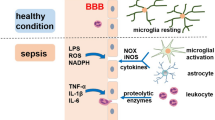
Bacterial endotoxins such as lipopolysaccharide (LPS) are primary factors that can initiate a considerable inflammatory reaction in an organism. In blood, LPS creates a complex with circulating LPS-binding protein (LBP), which, after binding to the membrane-bound CD14 receptor expressed constitutively by neutrophils and monocytes/macrophages, activates the immune system. In the brain, this type of receptor has been found in microglial cells [77]; therefore, these receptors may react to the appearance of bacterial toxins. Endothelial and smooth muscle cells do not possess the membrane-bound CD14 receptor but become activated by soluble CD14 receptor circulating in the blood. Through Toll-like receptors 2 and 4 [78, 79], the LPS–LBP–CD14 complexes stimulate the synthesis of proinflammatory cytokines such as interleukin-1β, interleukin-6, and tumor necrosis factor α [67], and initiate the synthesis and secretion of other inflammatory factors, reactive oxygen radicals [68], and nitric oxide [80]. The influx of monocytes and neutrophils to inflamed tissue increases, and the inflammatory reaction spreads through adjoining tissues [67]. The serum concentration of acute-phase proteins, such as protein C, increases, and the mobilization of the complement cascade system leads to the appearance of C3a and C5a components [81], which further increase production of proinflammatory cytokines [11]. The coagulation cascade overactivates, leading to disseminated intravascular coagulopathy, which disturbs the hemodynamic equilibrium and finally leads to microvessel thrombosis [82, 83]. Consequently, there is impairment of blood–brain barrier integrity, allowing the influx of other active compounds that stimulate the inflammatory response and spread it through the brain.
The blood–brain barrier strictly regulates the microenvironment of the nervous system, controls the blood flow through brain capillaries, and protects against the influx of harmful substances circulating in the blood. It is formed by tightly connected endothelial cells of brain vessels, which closely cooperate with astrocytes and pericytes [71]. It is known that during SE, blood–brain barrier integrity is compromised [18, 74, 84], which disrupts ionic homeostasis and allows transport of cytokines and inflammatory cells into the brain, directly or indirectly, resulting in neuronal loss [85, 86]. Blood–brain barrier permeability is also augmented by overexpression of inducible nitric oxide synthase in brain vessel endothelium [87], and increased pinocytosis [71] allows active substances to cross the blood–brain barrier despite preservation of the continuity of tight junctions between endothelial cells. Loss of blood–brain barrier impermeability leads to a disruption in water transport to the brain, which is tightly regulated by aquaporin 4 [88], resulting in perivascular edema, destruction of astrocyte endfeet [74], and secondary damage to nerve tissue [45]. This results in a decrease in diffusion through microvessel walls, a decrease in oxygen and nutrient use, and removal of harmful metabolites [74]. Because the cerebral blood autoregulation mechanism is disturbed during SE, the drop in blood pressure in patients with sepsis may directly affect the cerebral vascular bed, leading to hypoperfusion and, consequently, neuronal degeneration as a result of hypoxia–ischemia [89]. Because the brain consumes a large quantity of oxygen but has a weak antioxidant defense, it is susceptible to injury during sepsis. Because hypoxic–ischemic damage to the brain is a common feature of many brain diseases, its occurrence in SE patients does not make it a specific change; however, the disturbances in cerebral microcirculation observed during sepsis progression may play a crucial role in SE pathogenesis [47, 72]. Moreover, microcirculatory disorders are often connected to the upregulation of inflammatory genes such as tumor necrosis factor α, interleukin-1β, and inducible nitric oxide synthase transcripts, which also suggests a potential relationship between brain inflammation and blood flow disturbances.
As a generalized inflammatory reaction progresses, nitric oxide, cytokines, and prostaglandins modulate neurotransmission in the brain, especially regarding the β-adrenergic system, γ-aminobutyric acid (GABA)ergic synapses, central muscarinic cholinergic regulation, corticotropin-releasing factor, adrenocorticotropic hormone, vasopressin synthesis, medullary autonomic center output, and the monoaminergic, glutamatergic, and neurotrophic systems, leading to behavioral changes [69]. Increased levels of tyrosine, tryptophan, and phenylalanine have been observed in serum and cerebrospinal fluid, attenuating neurotransmitter synthesis. Moreover, prolonged exposure to LPS disrupts synaptic transmission and excitability of the hippocampal pyramidal neurons that are part of the emotional and behavioral systems [90]. LPS drives the considerable decrease in the density of cells in the hippocampal CA1/CA2 regions, the cell loss in the prefrontal cortex, and the reduction in cholinergic innervation in the cortex [90], which may contribute to memory deficits. Moreover, in patients with SE, the concentration of aromatic amino acids in the brain increases, and these substances may play a role as “false” neurotransmitters and/or disturb neurotransmitter synthesis [75]. This may lead to derangement of the quantitative relationship between aromatic amino acids and branched-chain amino acids, causing a decrease in the concentrations of norepinephrine, dopamine, and serotonin in the brain, whereas GABA levels remain unchanged, which may be the reason for mental abnormalities in the early stages of sepsis [17, 75].
During SE, brain damage has been observed most often in the cortex, but sometimes also in other structures, even the spinal cord [17]. The most frequent changes in the brain are ischemic lesions, especially in the autonomic system nuclei. Perivascular edema, swelling of astrocyte endfeet, and signs of apoptosis may be observed. Neurons have shrunken nuclei and damaged cell membranes [74], and astrocytes, microglia, and perivascular macrophages show a high degree of activity [91], which undoubtedly is evidence of an inflammatory process.
Models of Septic Encephalopathy
Through years of research, several animal models of sepsis and SE have been created; the main ones are endotoxemia induction, bacterial or viral inoculation, and cecal ligation and puncture (CLP). The simplest method, and the one used most often to develop an inflammatory response, is endotoxemia induction. Bacterial endotoxins in the form of LPS injected intravenously or intraperitoneally cause symptoms similar to those of sepsis, mainly in the vascular system, which influences the nervous system [80]. After endotoxemia induction, the concentration of proinflammatory cytokines increases, but with a temporal profile different from that in humans, which is the weakness of this method. However, this method allows precise control of the concentration of endotoxin in the serum and detection of disturbances in the organism’s function depending on the LPS dose [92]. Another important advantage of this method is that it may be used in human research [37].
Another method of developing a multiorgan inflammatory state is the administration of cultures of live bacteria, instead of their endotoxins [93]. This approach allows precise selection of the particular bacterial strain, as well as determination of the proper concentration of live bacteria, depending on the phenomenon studied. However, a disadvantage of this method is that the bacteria must be administered in large doses, because a large portion of them are removed by the immune system; an individual’s likelihood of having a reaction to a particular pathogen also plays a role. On the other hand, one advantage is the possibility of inducing an infection with a concrete pathogen and in a particular organ, such as the lungs (inhalation) or kidneys (injection) [93].
Presently, the standard research model for sepsis, septic shock, and SE is CLP, which imitates peritonitis. In this method, the appendix is ligated and then punctured [84], allowing peritonitis to develop; the size of the perforation determines the sharpness and course of sepsis. CLP reproduces fairly well the hemodynamic and metabolic changes observed in sepsis and SE [94] and substantially imitates their course in humans; therefore, it now is the standard animal model of in vivo research on the inflammatory reaction.
Conclusion
SE is a highly complicated phenomenon in which various factors play a role. It is the most frequent type of encephalopathy seen in the ICU, and its appearance influences survival and mortality rates during treatment. Clinical sepsis arises mainly as a result of the activity of various bacterial products, which stimulates and establishes the inflammatory process; therefore, it is very difficult to simulate this syndrome in laboratory research. Unfortunately, little is known about what causes the changes that affect the brain and lead to reduced consciousness and behavioral symptoms as SE progresses. SE survivors may have long-lasting neurocognitive disturbances in the form of anxiety or depression, which are thought to result from inflammatory processes in the brain during sepsis that are caused by oxidative stress, inflammatory factors, blood–brain barrier injury, changes in cerebral circulation, and emboli of microvessels. These phenomena have a negative impact on the synthesis and secretion of neurotransmitters and stimulate the degeneration of neurons and their loss in different areas of the nervous system. Presently, an effective therapy developed specifically to treat SE does not exist; therefore, supportive therapy for the underlying disease is administered. Further studies are needed to determine the predispositions of sepsis and at what point during its progress inflammatory processes in the brain begin; it is hoped these studies will result in the detection of new markers to allow SE to be diagnosed very early so that the appropriate therapy can be initiated.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Vandijck D, Decruyenaere JM, Blot SI. The value of sepsis definitions in daily ICU-practice. Acta Clin Belg. 2006;61(5):220–6.
Vandijck DM, Reynvoet E, Blot SI, Vandecasteele E, Hoste EA. Severe infection, sepsis and acute kidney injury. Acta Clin Belg Suppl. 2007;2:332–6.
Green R, Scott LK, Minagar A, Conrad S. Sepsis associated encephalopathy (SAE): a review. Front Biosci. 2004;9:1637–41.
Frontera JA. Metabolic encephalopathies in the critical care unit. Continuum (Minneap Minn). 2012;18(3):611–39.
Sands KE, Bates DW, Lanken PN, et al. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA. 1997;278(3):234–40.
Esper A, Martin GS. Is severe sepsis increasing in incidence AND severity? Crit Care Med. 2007;35(5):1414–5.
Woltmann A, Hamann L, Ulmer AJ, Gerdes J, Bruch HP, Rietschel et. Molecular mechanisms of sepsis. Langenbecks Arch Surg. 1998;383(1):2–10.
Sasse KC, Nauenberg E, Long A, Anton B, Tucker HJ, Hu TW. Long-term survival after intensive care unit admission with sepsis. Crit Care Med. 1995;23(6):1040–7.
Brun-Buisson C. The epidemiology of the systemic inflammatory response. Intensive Care Med. 2000;26 Suppl 1:S64–74.
Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–50.
Kunze K. Metabolic encephalopathies. J Neurol. 2002;249(9):1150–9.
Dobbs MR. Toxic encephalopathy. Semin Neurol. 2011;31(2):184–93.
Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12(3):627–42.
Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76(5):886–99.
Baugh CM, Stamm JM, Riley DO, et al. Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav. 2012;6(2):244–54.
Badruddin A, Taqi MA, Abraham MG, Dani D, Zaidat OO. Neurocritical care of a reperfused brain. Curr Neurol Neurosci Rep. 2011;11(1):104–10.
Consales G, De Gaudio AR. Sepsis associated encephalopathy. Minerva Anestesiol. 2005;71(1–2):39–52.
Papadopoulos MC, Davies DC, Moss RF, Tighe D, Bennett ED. Pathophysiology of septic encephalopathy: a review. Crit Care Med. 2000;28(8):3019–24.
Davies NW, Sharief MK, Howard RS. Infection-associated encephalopathies: their investigation, diagnosis, and treatment. J Neurol. 2006;253(7):833–45.
Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med. 2007;33(6):941–50.
Streck EL, Comim CM, Barichello T, Quevedo J. The septic brain. Neurochem Res. 2008;33(11):2171–7.
Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest. 2006;130(3):869–78.
Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996;275(6):470–3.
Zhang LN, Wang XT, Ai YH, et al. Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008–2011. Chin Med J (Engl). 2012;125(5):828–31.
Huberlant V, Cosnard G, Hantson PE. Brain death in a septic patient: possible relationship with posterior reversible encephalopathy syndrome? Anaesth Intensive Care. 2009;37(6):1017–20.
Zauner C, Gendo A, Kramer L, et al. Impaired subcortical and cortical sensory evoked potential pathways in septic patients. Crit Care Med. 2002;30(5):1136–9.
Hsu AA, Fenton K, Weinstein S, Carpenter J, Dalton H, Bell MJ. Neurological injury markers in children with septic shock. Pediatr Crit Care Med. 2008;9(3):245–51.
Nguyen DN, Spapen H, Su F, et al. Elevated serum levels of S-100β protein and neuron-specific enolase are associated with brain injury in patients with severe sepsis and septic shock. Crit Care Med. 2006;34(7):1967–74.
Chelazzi C, Consales G, De Gaudio A. Sepsis associated encephalopathy. Curr Anaesth Crit Care. 2008;19:15–21.
Pytel P, Alexander JJ. Pathogenesis of septic encephalopathy. Curr Opin Neurol. 2009;22(3):283–7.
Schirmer-Mikalsen K, Vik A, Gisvold SE, Skandsen T, Hynne H, Klepstad P. Severe head injury: control of physiological variables, organ failure and complications in the intensive care unit. Acta Anaesthesiol Scand. 2007;51(9):1194–201.
De Jonghe B, Cook D, Griffith L, et al. Adaptation to the Intensive Care Environment (ATICE): development and validation of a new sedation assessment instrument. Crit Care Med. 2003;31(9):2344–54.
Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2(1):49.
Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA. 2001;286(21):2703–10.
Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370–9.
Ely EW, Truman B, Shintani A, et al. Monitoring sedation status over time in ICU patients: reliability and validity of the Richmond Agitation-Sedation Scale (RASS). JAMA. 2003;289(22):2983–91.
van den Boogaard M, Ramakers BP, van Alfen N, et al. Endotoxemia-induced inflammation and the effect on the human brain. Crit Care. 2010;14(3):R81.
Semmler A, Widmann CN, Okulla T, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84(1):62–9.
•• Schramm P, Klein KU, Falkenberg L, et al. Impaired cerebrovascular autoregulation in patients with severe sepsis and sepsis-associated delirium. Crit Care. 2012;16(5):R181. This article describes disturbances of cerebral blood flow during SE using a TCD sonography method. The authors measured blood velocity in the brain during the initial 4 days of SE and found impairment of brain vasculature autoregulation during the first 2 days, associated with signs of encephalopathy. They conclude that these disturbances are one of the trigger mechanisms for delirium in SE.
Kaplan PW, Rossetti AO. EEG patterns and imaging correlations in encephalopathy: encephalopathy part II. J Clin Neurophysiol. 2011;28(3):233–51.
Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9(1):145–52.
Oddo M, Carrera E, Claassen J, Mayer SA, Hirsch LJ. Continuous electroencephalography in the medical intensive care unit. Crit Care Med. 2009;37(6):2051–6.
Varelas PN, Hacein-Bey L, Hether T, Terranova B, Spanaki MV. Emergent electroencephalogram in the intensive care unit: indications and diagnostic yield. Clin EEG Neurosci. 2004;35(4):173–80.
Terborg C. Septische Enzephalopathie. Med Klin Intensivmed Notfmed. 2012;107(8):629–33.
Sharshar T, Carlier R, Bernard F, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 2007;33(5):798–806.
Piazza O, Cotena S, De Robertis E, Caranci F, Tufano R. Sepsis associated encephalopathy studied by MRI and cerebral spinal fluid S100B measurement. Neurochem Res. 2009;34(7):1289–92.
Taccone FS, Castanares-Zapatero D, Peres-Bota D, Vincent JL, Berre J, Melot C. Cerebral autoregulation is influenced by carbon dioxide levels in patients with septic shock. Neurocrit Care. 2010;12(1):35–42.
Pfister D, Siegemund M, Dell-Kuster S, et al. Cerebral perfusion in sepsis-associated delirium. Crit Care. 2008;12(3):R63.
Pfister D, Schmidt B, Smielewski P, et al. Intracranial pressure in patients with sepsis. Acta Neurochir Suppl. 2008;102:71–5.
Settakis G, Molnár C, Kerényi L, et al. Acetazolamide as a vasodilatory stimulus in cerebrovascular diseases and in conditions affecting the cerebral vasculature. Eur J Neurol. 2003;10(6):609–20.
Settakis G, Páll D, Molnár C, Bereczki D, Csiba L, Fülesdi B. Cerebrovascular reactivity in hypertensive and healthy adolescents: TCD with vasodilatory challenge. J Neuroimaging. 2003;13(2):106–12.
Szatmari S, Vegh T, Csomos A, et al. Impaired cerebrovascular reactivity in sepsis-associated encephalopathy studied by acetazolamide test. Crit Care. 2010;14(2):R50.
Lamar CD, Hurley RA, Taber KH. Sepsis-associated encephalopathy: review of the neuropsychiatric manifestations and cognitive outcome. J Neuropsychiatry Clin Neurosci. 2011;23(3):237–41.
Kafa IM, Bakirci S, Uysal M, Kurt MA. Alterations in the brain electrical activity in a rat model of sepsis-associated encephalopathy. Brain Res. 2010;1354:217–26.
Bolton CF, Young GB. Managing the nervous system effects of sepsis. Chest. 2007;131(5):1273–4.
Bolton CF, Young GB, Zochodne DW. The neurological complications of sepsis. Ann Neurol. 1993;33(1):94–100.
Weathers AL, Lewis SL. Rare and unusual … or are they? Less commonly diagnosed encephalopathies associated with systemic disease. Semin Neurol. 2009;29(2):136–53.
• Girard TD, Jackson JC, Pandharipande PP, et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–20. The authors tested the hypothesis that the duration of delirium in the ICU is a predictor of long-term mental impairment after critical illness. They found severe cognitive impairment in 62% of patients 3 months after critical illness and in 36% of patients 12 months after critical illness, which supports their thesis.
Bilbo SD, Newsum NJ, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Differential effects of neonatal handling on early life infection-induced alterations in cognition in adulthood. Brain Behav Immun. 2007;21(3):332–42.
Taylor AK, Cousins R, Butt WW. The long-term outcome of children managed with extracorporeal life support: an institutional experience. Crit Care Resusc. 2007;9(2):172–7.
Lazosky A, Young GB, Zirul S, Phillips R. Quality of life after septic illness. J Crit Care. 2010;25(3):406–12.
Rothenhäusler HB, Ehrentraut S, Stoll C, Schelling G, Kapfhammer HP. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: results of an exploratory study. Gen Hosp Psychiatry. 2001;23(2):90–6.
Hopkins RO, Weaver LK, Chan KJ, Orme JF. Quality of life, emotional, and cognitive function following acute respiratory distress syndrome. J Int Neuropsychol Soc. 2004;10(7):1005–17.
Hopkins RO, Weaver LK, Collingridge D, Parkinson RB, Chan KJ, Orme JF. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2005;171(4):340–7.
• Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304(16):1787–94. This is a prospective study on cognitive impairment and functional disability of SE survivors at least 1 year following SE insult. The authors conclude that severe sepsis in older people is associated with substantial new cognitive impairments and quality of life of survivors.
Scragg P, Jones A, Fauvel N. Psychological problems following ICU treatment. Anaesthesia. 2001;56(1):9–14.
Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112(4):460–7.
Berg RMG, Møller K, Bailey DM. Neuro-oxidative-nitrosative stress in sepsis. J Cereb Blood Flow Metab. 2011;31(7):1532–44.
Jacob A, Brorson JR, Alexander JJ. Septic encephalopathy: inflammation in man and mouse. Neurochem Int. 2011;58(4):472–6.
De Backer D. Hemodynamic management of septic shock. Curr Infect Dis Rep. 2006;8(5):366–72.
Nico B, Ribatti D. Morphofunctional aspects of the blood–brain barrier. Curr Drug Metab. 2012;13(1):50–60.
Taccone FS, Scolletta S, Franchi F, Donadello K, Oddo M. Brain perfusion in sepsis. Curr Vasc Pharmacol. 2013;11(2):170–86.
Sharshar T, Polito A, Checinski A, Stevens RD. Septic-associated encephalopathy - everything starts at a microlevel. Crit Care. 2010;14(5):199.
Papadopoulos MC, Lamb FJ, Moss RF, Davies DC, Tighe D, Bennett ED. Faecal peritonitis causes oedema and neuronal injury in pig cerebral cortex. Clin Sci (Lond). 1999;96(5):461–6.
Basler T, Meier-Hellmann A, Bredle D, Reinhart K. Amino acid imbalance early in septic encephalopathy. Intensive Care Med. 2002;28(3):293–8.
Kondo S, Kohsaka S, Okabe S. Long-term changes of spine dynamics and microglia after transient peripheral immune response triggered by LPS in vivo. Mol Brain. 2011;4:27.
Nadeau S, Rivest S. Endotoxemia prevents the cerebral inflammatory wave induced by intraparenchymal lipopolysaccharide injection: role of glucocorticoids and CD14. J Immunol. 2002;169(6):3370–81.
Han F, Yu H, Tian C, et al. Role for Toll-like receptor 2 in the immune response to Streptococcus pneumoniae infection in mouse otitis media. Infect Immun. 2009;77(7):3100–8.
Ock J, Jeong J, Choi WS, et al. Regulation of Toll-like receptor 4 expression and its signaling by hypoxia in cultured microglia. J Neurosci Res. 2007;85(9):1989–95.
Ziaja M, Pyka J, Ciombor J, Płonka P. Kinetics of nitric oxide release in neonatal and mature rat brain during endotoxemia, as studied by diethyldithiocarbamate spin trapping. Curr Top Biophys. 2005;29(1–2):73–82.
Guo RF, Riedemann NC, Ward PA. Role of C5a-C5aR interaction in sepsis. Shock. 2004;21(1):1–7.
Kushimoto S, Gando S, Saitoh D, et al. Clinical course and outcome of disseminated intravascular coagulation diagnosed by Japanese Association for Acute Medicine criteria. Comparison between sepsis and trauma. Thromb Haemost. 2008;100(6):1099–105.
Weon YC, Marsot-Dupuch K, Ducreux D, Lasjaunias P. Septic thrombosis of the transverse and sigmoid sinuses: imaging findings. Neuroradiology. 2005;47(3):197–203.
Flierl MA, Stahel PF, Rittirsch D, et al. Inhibition of complement C5a prevents breakdown of the blood–brain barrier and pituitary dysfunction in experimental sepsis. Crit Care. 2009;13(1):R12.
Nishioku T, Dohgu S, Takata F, et al. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood–brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol. 2009;29(3):309–16.
Venturi L, Miranda M, Selmi V, et al. Systemic sepsis exacerbates mild post-traumatic brain injury in the rat. J Neurotrauma. 2009;26(9):1547–56.
Sharshar T, Gray F, Lorin de la Grandmaison G, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362(9398):1799–805.
Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52(3):447–56.
Guo MF, Yu JZ, Ma CG. Mechanisms related to neuron injury and death in cerebral hypoxic ischaemia. Folia Neuropathol. 2011;49(2):78–87.
Semmler A, Frisch C, Debeir T, et al. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol. 2007;204(2):733–40.
Sharshar T, Gray F, Poron F, Raphael JC, Gajdos P, Annane D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit Care Med. 2002;30(10):2371–5.
Yang F, Comtois AS, Fang L, Hartman NG, Blaise G. Nitric oxide-derived nitrate anion contributes to endotoxic shock and multiple organ injury/dysfunction. Crit Care Med. 2002;30(3):650–7.
Tanaka T, Sunden Y, Sakoda Y, Kida H, Ochiai K, Umemura T. Lipopolysaccharide treatment and inoculation of influenza A virus results in influenza virus-associated encephalopathy-like changes in neonatal mice. J Neurovirol. 2010;16(2):125–32.
Ziaja M. Sepsis and septic encephalopathy: characteristics and experimental models. Folia Neuropathol. 2012;50(3):231–9.
Compliance with Ethics Guidelines
Conflict of Interest
Marek Ziaja declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Infection
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ziaja, M. Septic Encephalopathy. Curr Neurol Neurosci Rep 13, 383 (2013). https://doi.org/10.1007/s11910-013-0383-y
Published:
DOI: https://doi.org/10.1007/s11910-013-0383-y