
Abstract
The bloating behavior of metal droplets and the dephosphorization behavior of bloated droplets at 1853 K (1580 °C) were investigated using X-ray fluoroscopy coupled with constant volume pressure change measurements and chemical analysis of quenched samples. The effect of sulfur content on dephosphorization kinetics was studied during the decarburization period. The slag foamed during the reaction forming a foamy layer over a dense layer. After a short incubation period, the droplets became bloated due to internal decarburization. The bloated droplets floated from the dense slag into the foamy slag. The behavioral changes are directly related to the effect of sulfur on the incubation time for swelling. The dephosphorization reaction was very fast; droplets with low sulfur contents experienced phosphorus reversion shortly after entering the foamy slag, while those with higher sulfur content took a longer time to swell and went through reversion before they entered the foam. The dephosphorization rate and maximum phosphorus partition were higher at lower CO evolution rates because the dynamic interfacial oxygen potential increased with the decreasing oxygen consumption rate. The rate controlling step for dephosphorization was initially a combination of mass transport in both the metal and the slag. As the iron oxide in the slag was depleted, the rate control shifted to mass transport in slag.
Similar content being viewed by others

Introduction
During oxygen steelmaking, metal droplets created by the impact of the oxygen jet are ejected into the slag, where they are decarburized and dephosphorized by reaction with reducible oxides. Several researchers have studied aspects of droplet behavior relevant to steelmaking including, decarburization,[1,2,3,4,5,6] droplet generation,[7,8,9,10,11,12,13,14,15,16] size distribution,[17,18] and residence time.[19] Other workers have developed models[20,21,22,23] and conducted plant trials,[24,25,26,27,28] which considered the role of droplets in the overall process kinetics. Previous work in the authors’ laboratory[6] showed that droplet swelling, caused by CO formation inside the droplet, increased the droplet residence time in the slag, thereby favoring decarburization. This work formed the basis of the Bloated Droplet Model, which has since been employed in an overall BOF model by workers at Swinburne University in Australia.[20,21] Notwithstanding very recent work, which suggests bubbles formed on the outer surface of droplets can offer similar increases in residence time, convincing experimental evidence for bloated droplets has been reported by several researchers.[5,6,29] Given the importance of droplet swelling in controlling residence time and the fact that dephosphorization is only favored when slag and metal are in contact, data on dephosphorization of bloated droplets are required for the proper inclusion of dephosphorization in BOF process models.
Dephosphorization of hot metal has been studied extensively but for brevity only a subset of this work will be reviewed here. Research on phosphorus partitioning in the BOF has been reviewed by Basu et al.[30] More recently, phosphorus equilibrium between liquid iron and BOF type slag has been investigated by Assis et al.,[31] who further developed a corrected phosphorus partition correlation by combining other researchers’ work. Several studies have been conducted on the dephosphorization kinetics of iron–carbon melts using oxidizing slag.[32,33,34,35] Mori et al.[32] and Wei et al.[33] suggested that the rate of dephosphorization was under mixed control by mass transport in both slag and metal phase. Monaghan et al.[34] proposed that the rate determining step was mass transfer in the slag. Assuming mass transfer in both slag and metal was rate controlling, Manning and Fruehan[35] showed that the mass transfer parameter (kA) decreased as the reaction proceeded and appeared to be a function of interfacial tension, which changed with respect to time. Employing IMPHOS pilot plant data, Hewage et al.[36] investigated the dephosphorization kinetics in the BOF and found that dephosphorization cannot be explained by a simple first-order equation with either static equilibrium or dynamic equilibrium values. These workers also stressed the importance of transient parameters, such as surface area, volume, residence time, and mass transfer coefficient.
The present work seeks to develop a detailed quantitative understanding of the interplay between dephosphorization and decarburization of bloated droplets, and to investigate the effect of decarburization rate on a range of important factors in dephosphorization. Chen and Coley[6] showed that the rate of CO evolution during droplet swelling was primarily controlled by the nucleation of CO bubbles within the droplet. They also demonstrated that the rate of CO evolution, as a function of sulfur in the metal, went through a maximum at approximately 0.011 wt pct S. This suggests that although CO evolution was controlled primarily by nucleation, bubble growth also played a role; nucleation increased with the decreasing surface tension caused by sulfur, whereas bubble growth decreased due to the surface poisoning effect of sulfur. The optimum combination of increasing nucleation rate and decreasing growth rate occurred at approximately 0.011 w pct S. Given this observation, it should be possible by using droplets with different sulfur contents to determine the dephosphorization rate as a function of CO evolution rate.
Experimental Procedure
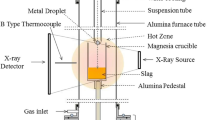
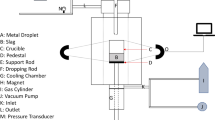
A resistance-heated vertical tube furnace (See Figure 1) with an 80-mm-diameter alumina working tube was used for all experiments. The tube ends were sealed with O-rings and water-cooled stainless steel caps. The furnace was equipped with X-ray imaging to observe the swelling of droplets in-situ and a pressure transducer to measure gas evolution using the constant volume pressure increase technique (CVPI).

Schematic diagram of the Furnace[37]: 1. Support Rod 2. Slag 3. X-ray Generator 4. Image Intensifier 5. Working tube 6. Iron Pellet
Fe-C-P-S droplets were made as follows: an appropriate amount of electrolytic iron, FeP and FeS were melted together to prepare master Fe-P-S alloys using an arc melter under an argon atmosphere. The master alloys were homogenized at 1173 K (900 °C) for 24 hours in a sealed tube furnace under flowing purified argon. After heat treatment, four samples were taken from different positions in the master alloy analyzed using inductively coupled plasma optical emission spectroscopy (ICP-OES). The master alloys were then rolled into sheet for convenient use. Finally, individual droplets were prepared by melting an appropriate quantity of master alloy along with an appropriate quantity of graphite in a vacuum arc melter. The oxygen content in the droplets fabricated in this manner was 50 ± 20 ppm.[38] Three sample droplets were taken from each batch of droplets to confirm the concentration of phosphorus using ICP-OES, and carbon and sulfur using LECO combustion analysis. Slags were prepared by mixing the reagent-grade Al2O3, CaO, and SiO2, and the mixture was held at 1073 K (800 °C) for 6 hours to remove moisture; FeO was added to the dry mixture to prepare slags of a desired composition. This mixture of slag was used directly for all experiments without premelting. Approximately 25 g of slag, mixed as described above, was placed in a 45-mm-diameter alumina crucible and then placed in a furnace which was sealed and flushed with deoxidized argon (copper turnings 873 K (600 °C)). The crucible was raised into the hot-zone of the furnace and, after reaching the desired temperature, was held for one hour to allow homogenization. The metal droplet was dropped into the molten slag via a dispensing tube as shown in Figure 1. A hole at the bottom of the dispensing tube was sized to ensure that the droplet must melt before passing through. When the droplet melted and entered the slag, the time was set to zero. Videos were recorded by X-ray Fluoroscopy (GE OEC Medical Systems, Inc.), and a differential pressure transducer, maximum measurable range of 2 psi, was used to measure the pressure change caused by the gas evolution during the reaction. An oxygen sensor was installed at the exhaust gas outlet to monitor the oxygen partial pressure in the furnace. The support rod shown in Figure 1 was held in place by an O-ring and a collar. By unscrewing the collar, the crucible could be dropped into the quench chamber (water-cooled stainless steel) within 1 second. Samples were quenched at different reaction times and taken for chemical analysis of phosphorus. Metal samples were dissolved in acid using a microwave digester and analyzed by ICP-OES. The slag composition for all experiments was 35 SiO2, 32 CaO, 17 Al2O3, and 16 wt pct FeO. Table I summarizes the composition of metal droplets used in experiments. All experiments were conducted at the temperature of 1853 K (1580 °C).
Experimental Results
Measurement of CO Evolution Rate
Figure 2(a) shows the total amount of CO gas generated during reactions between slag and droplets as a function of time and droplet sulfur content. It was found that the total amount of CO generated increased with an increase in sulfur content of the droplet. For example, the total CO gas produced for droplets with 0.007, 0.014, and 0.021 wt pct S were 9.23 × 10−4, 1.12 × 10−3, and 1.44 × 10−3 moles, respectively. It is also noteworthy that the initial stage of reaction, where the rate of CO generation was low, was extended with increasing droplet sulfur content. The peak CO evolution rate, taken from the maximum slope of each curve shown in Figure 2(a), was plotted in Figure 2(b) as a function of sulfur content. This figure shows that CO generation rate increases from 5.82 × 10−5 mole/s to a maximum of 1.04 × 10−4 mole/s for droplets with 0.014 wt pct S. The sulfur content at the peak, 0.014 wt pct, is higher than that (0.011 wt pct) previously reported by the authors.[39] This discrepancy is believed to be related to a change in the production technique of the droplets. The mechanism by which sulfur affects decarburization behavior will be discussed in Section IV.

(a) CO gas generation with time. (b) Peak decarburization rate as a function of metal sulfur content
X-ray Fluoroscopy Observations of Droplets
A series of X-ray images, depicting the typical bloating behavior of droplets containing 0.014 wt pct S, are presented in Figure 3(a) through (h). Once the droplet melted, ~30 seconds, and fell into the molten slag, the time was set to zero. Within the next 13 seconds, the initial decarburization step takes place. During this period, the droplet was covered by a gas halo and started to float out of the dense slag. In the meantime, a foamy slag layer was formed on top of the original dense slag. The proportion of foamy slag increased at the expense of the dense slag as the reaction proceeded. During this initial period, the total increase in droplet volume was less than three times the original value.

Typical behavior of a metal droplet with 0.0014 wt pct S observed by X-ray fluoroscopy at 1853 K (1580 °C)
After this incubation period, the droplet swelled rapidly and floated out of the dense slag. During this stage, the volume increased by over 14 times its original value; the corresponding increase in the droplet/slag interfacial area was over 5 times. During this period, the bloated droplet remained in the foamy slag. The fast decarburization ceased after 24 seconds, and the droplet sank back into the dense slag. The reactive droplet continued to form CO gas as it fell to the bottom of the crucible. The foamy slag began to collapse at the end of the fast decarburization period, lasting approximately 40 seconds before collapsing completely.
The change in the volume of droplets as a function of time and sulfur content is presented in Figure 4. This figure shows that as the sulfur content of droplets increased from 0.007 to 0.021 wt pct, the incubation time for swelling increased from 7 seconds to approximately 32 seconds. It also can be seen that droplets behaved similarly up to 0.018 pct S. The droplets with 0.021 wt pct S showed a much lower maximum volume but a much longer period of swelling. These droplets also had the longest incubation time and, despite the fact that they showed similar peak decarburization rates to the droplets with 0.007 wt pct S, the peak volume was considerably lower. This observation may be related to the fact that the rates of gas generation and escape were fairly close and this relatively small difference was responsible for the change in volume; a small fractional change in the rate of generation represents a fairly large change relative to the gas retained in the droplet.

Variation of the volume of bloated droplets with respect to sulfur content and time
Dephosphorization of Bloated Droplets
The change of phosphorus content of the droplet as a function of time and sulfur content is shown in Figure 5. It is worth noting at this point that these data are somewhat sparse, particularly when one considers any single condition. This is a direct consequence of the difficulty of quenching droplets at small time intervals, a weakness that is compounded when one considers that most of the dephosphorization is complete within a few seconds. The authors acknowledge this weakness and would not choose this technique in a study focused entirely on dephosphorization. However, the current work is focused on the interplay between dephosphorization and decarburization of bloated droplets. As such there was little choice but to actually study bloated droplets, despite any experimental difficulties that approach might present. As seen below in section IV, the data show a remarkably consistent pattern. The data points shown in Figure 5(a) prior to the reversion point are the mean of at least three different experiments. The error bars represent the standard error of the mean. According to Figures 5(a) through (e), droplets were dephosphorized first on entering the dense slag and subsequently exhibited one of two types of reversion behavior. Figure 5(a) shows a droplet that entered the foamy slag prior to reversion. Figure 5(e) shows the behavior of a droplet that remained in the dense slag for longer time and started to go through reversion in the dense slag. It is interesting to note that when the droplets, which have undergone phosphorus reversion in the foamy slag, sink back into the dense slag, dephosphorization starts again. It seems reasonable to assume that, depending on the extent to which dephosphorization has progressed prior to the droplet entering the foamy slag, a wide range of behaviors might be observed. This is seen in Figures 5(a) through (e) which show apparently random dephosphorization behavior as a function of droplet sulfur content. However, when one considers that the droplet sulfur content will control, the incubation time for swelling and thereby the time the droplet enters the foamy slag, and the rate of CO formation which will control the dynamic interfacial oxygen potential and the depletion of the slag FeO, it is possible to rationalize the apparently disparate behavior. Figures 5(a) and (b) show that droplets with low sulfur content dephosphorized primarily in the dense slag and experienced phosphorus reversion shortly after entering the foamy slag. Alternatively, those with higher sulfur content shown in Figures 5(c), (d), and (e) (0.014, 0.017, and 0.021 wt pct S) went through reversion before they entered the foam. Comparing the data presented in Figure 5 with that in Figure 2 suggests that for droplets with sulfur 0.017 wt pct and less, the minimum phosphorus content increased with the increasing CO evolution rate. The above observations are supported by the detailed X-ray images of droplets at each stage of reaction and agree with the established understanding of dephosphorization of iron–carbon alloys.[32,33,34]

Dephosphorization as a function of time and droplet sulfur content: (a) 0.007 wt pct S, (b) 0.011 wt pct S, (c) 0.014 wt pct S, (d) 0.017 wt pct S, and (e) 0.021 wt pct S
In order to present a clear view of the interplay between droplet decarburization and dephosphorization behavior, a schematic diagram is shown in Figure 6. This figure illustrates droplet behavior in terms of typical CO generation and dephosphorization during the course of an experiment, generalizing the behavior shown in Figures 2 and 5.

Schematic diagram of droplet behavior in terms of CO generation and dephosphorization as a function of time
Figure 6 shows that the behavior of droplets can be separated into three sequential stages. During Stage I, corresponding to the incubation period for droplet swelling, droplets were dephosphorized in the dense slag. During Stage II, droplets swelled because of the high CO evolution rate and floated into the foaming slag. In this study, droplets typically experienced phosphorus reversion during stage II because of high CO evolution rate and depletion of FeO in foaming slag, both contributing to a lower interfacial oxygen potential. Whether phosphorus reversion occurred in the dense slag or the foamy slag depended on the sulfur content of the droplet. Stage III was initiated when the rate of decarburization subsided, and the droplet sank back into the dense slag. The dense slag not having been depleted of FeO, and the decarburization of the droplet having decreased, would combine to offer a higher interfacial oxygen potential allowing renewed dephosphorization. This phenomenon is observed in the cases shown in Figures 5(a) and (e).
DISCUSSION
Effect of Sulfur on Droplet Swelling
Figure 4 shows that the incubation time for droplet swelling rises with an increase in sulfur content. In order to examine this effect further, the incubation time is plotted in Figure 7 as a function of the activity of sulfur in the droplet. Here, the sulfur activity, h s , was calculated using Wagner’s interaction parameter formalism and data from Hino and Ito.[40]

The effect of sulfur on incubation time for droplet swelling
The linear relationship shown in Figure 7 is consistent with sulfur-blocking reaction sites for oxygen transfer to the droplet. The delay of droplet swelling also has a strong influence on dephosphorization, shown in Figure 5. Comparing Figures 5 and 7 offers one possible explanation for the effect of sulfur on slowing the rate of dephosphorization. Longer incubation times for droplet swelling represent longer times with less stirring in the droplet which could lead to lower mass transfer rates for phosphorus. It is also possible that lower dephosphorization rates at higher sulfur levels are directly related to the surface poisoning effect of sulfur.
Effect of Sulfur on CO Evolution
As a surface active element, sulfur decreases the surface tension of metal, thereby reducing the energy barrier for CO nucleation and decreasing the minimum carbon content required to nucleate CO bubbles. The increase in the total CO production with increasing sulfur levels (Figure 2(a)) can be attributed to this effect.
As far as the rate of CO generation is concerned, sulfur has two opposing effects: on the one hand, it will lower the surface tension of the metal and increase the rate of gas bubble nucleation, and on the other hand, sulfur will have a negative effect on bubble growth by blocking reaction sites. Previous work in the authors’ laboratory[6] has shown that the peak rate of CO formation is proportional to the metal volume, which implies a strong influence of nucleation in controlling the rate. Although it is clear that the number of gas bubbles formed will depend on nucleation rate, the size of these bubbles at the point where they escape from the droplet will depend on their subsequent growth rate. Increasing the sulfur will decrease the growth rate. These two competing effects explain the maximum in gas generation rate seen in Figure 2(b): at lower sulfur levels, the effect of sulfur in enhancing bubble nucleation dominates and at higher sulfur contents its effect on bubble growth is dominant. The current work has been conducted to elucidate the effect of sulfur on dephosphorization of iron carbon droplets. Some might consider the range of sulfur used in this case to be limited; however, this range was chosen as the range over which the most dramatic effect on CO generation has been observed. Several researchers[37,41] have shown that sulfur at these relatively modest levels can cause a significant increase in the CO formation rate. Depending on temperature and carbon content, the CO evolution rate increases by between a factor of two and a factor of five when sulfur increases from 0.003 to 0.013 wt pct. The rate then subsides with increasing sulfur above 0.013 wt pct. This phenomenon is shown in Figure 2 and is remarkably consistent amongst several different researchers; the sulfur level corresponding to the peak CO generation rate varies by not more than 0.003 wt pct. The variation in sulfur content in the current work was chosen specifically to span this peak, aiming for lower CO generation rates at both high and low sulfur contents as well as high CO generation rates at the peak. It is well established in the literature that the interfacial oxygen potential, which creates the driving force for dephosphorization, is set by the balance of oxygen supply in the slag and consumption by reaction with carbon in the metal. Therefore, it is reasonable to expect a decrease in dephosphorization extent, and possibly rate, at higher CO generation rates. The current work was specifically aimed at probing this effect for the case of bloated droplets.
The observation that faster CO evolution rates decrease the phosphorus partition (Figures 2 and 5) is consistent with the authors’ previous work[39] and agrees with the theory that the oxygen potential at the slag–metal interface is set by the balance between oxygen supply by the slag and oxygen consumption by carbon in the metal.[33] In apparent contradiction to this, the metal phosphorus content at the reversion point, for droplets containing 0.021 wt pct S, is much higher than at 0.007 wt pct S although the peak CO generation rates are similar. It appears that a more detailed discussion about the effect of sulfur on phosphorus partition is required, and in addition to considering the rate of CO generation, one should also consider whether the droplet is resident in the dense slag or the foamy slag.
The existence of a phosphorus reversion point, while the carbon oxidation reaction is still going on, offers an excellent opportunity to analyze the dynamic interfacial oxygen potential. It is not generally possible to measure the phosphorus partition at the slag–metal interface and therefore the partition cannot be used to calculate the local oxygen potential. However, at the reversion point, the rate of the forward reaction for dephosphorization equals that of the backward reaction; therefore, there will be no phosphorus concentration gradient in either the slag or the metal. One can assume the system is in equilibrium with regard to phosphorus and that bulk concentrations of phosphorus can be used to calculate L p at the interface as stated in Eq. [1].
where (pct P) represents phosphorus in the slag and [pct P] represents phosphorus in the metal. The subscripts e and i indicate equilibrium and interface, respectively. The measured values for L p at the reversion point are shown in Figure 8 as a function of peak decarburization rate. The solid line in this figure shows that the partition coefficient at reversion for droplets containing 0.017 wt pct S and less is inversely proportional to decarburization rate. Only the 0.021 wt pct S droplet deviates from this relationship. This latter observation may be related to the fact that this droplet went through reversion at a much longer reaction time when the foamy slag will have been much more depleted of FeO. The resulting lower rate of oxygen supply will have caused a much lower interfacial oxygen potential.

The effect of CO evolution rate on phosphorus partition coefficient
In general, it appears that CO evolution rate has a significant effect on L p; a faster decarburization rate leads to a faster oxygen consumption rate and a lower oxygen potential at the slag–metal interface. This decreases the driving force for dephosphorization which is consistent with the findings of other researchers.[32,33,34] By assuming that the relationship between decarburization rate and interfacial oxygen potential is maintained throughout the residence of the droplet in the slag, we can use the data obtained for the reversion point to calculate the oxygen potential at any time during the reaction. This assumption is reasonable for either foamy slag or dense slag but cannot account for the transition from one to the other because the slag mass transfer coefficient will be considerably lower in the foamy slag. This can also be used for individual droplets in the BOF as the relationship between individual droplets and slag is no different. It is worth noting two important differences for the BOF slag; due to the low basicity (approximately 0.8) and FeO depletion of the slag in this study, the phosphorus partition coefficient at the reversion point is very small, ~10−2. For droplets in oxygen steelmaking, the slag would not be depleted of FeO and the basicity would be more than three times higher. Assuming similar CO generation behavior and mass transfer coefficients in the slag, the oxygen potential for this study and typical steelmaking conditions can be calculated if we equate oxygen supply rate by FeO in the slag and oxygen consumption rate by the formation of CO in the metal:
By assuming that local equilibrium exists at the slag–metal interface, the following two reactions[40] can be coupled with Eq. [2] to calculate the oxygen potential:
The authors are currently developing an interfacial oxygen potential model including a detailed analysis of the oxygen supply side as well as consumption. This work will be presented in detail in a subsequent publication.[42] For this discussion, reasonable estimates of the slag mass transfer coefficient will be used to calculate the interfacial oxygen potential as a function of droplet decarburization rate. Molloseau and Fruehan[41] investigated decarburization behavior between iron–carbon droplets and slag containing FeO and found that the value of \( k_{\text{s}} \) is in the range of 1.3 × 10−3 and 1.3 × 10−2 cm/s. Taking the average value of \( k_{\text{FeO}} \)(5 .0 × 10−3 cm/s) for foamy slag and the industrial data for a typical slag composition,[24] the interfacial oxygen potential under typical steelmaking conditions was calculated by employing the CO generation behavior for droplets with 0.014 wt pct S. In Figure 9, these results are compared with similar calculations for the slag used in this study. It should be noted that for consistency with the industrial case, the calculations for both slags assumed no FeO depletion.

The effect of CO evolution rate on interfacial oxygen potential and phosphorus partition coefficient for this study and under steelmaking condition
Phosphorus partition coefficients were also included in Figure 9, calculated using Eq. [5]. The phosphate capacity \( C_{{{\text{PO}}_{4}^{3 - } }} \), was calculated using the optical basicity correlation developed by Mori:[43]
where \( K_{\text{P}}\,\)is the equilibrium constant for phosphorus gas in equilibrium with phosphorus in liquid iron, \( f_{\text{P}}\, \)is the Henrian activity coefficient for phosphorus in iron, and M is the molecular weight.
Figure 9 shows that without depletion of FeO in the slag, the interfacial oxygen potential in this study would maintain a relative high value and is close to typical steelmaking conditions. The phosphate capacity for the slag used in this study and that for a typical steelmaking slag were calculated to be 1.2 × 1017 and 1.9 × 1019, respectively. Due to the difference in phosphate capacity, the phosphorus partition coefficient of steelmaking slag is two orders of magnitude higher than that of the current slag as shown in Figure 9. This difference will have a major impact on the rate controlling step for the reaction which is discussed below.
Dephosphorization Kinetics
It is well established that most slag–metal chemical reactions reach a local equilibrium at the interface. This is consistent with the assumptions of previous researchers who have all agreed that the kinetics of phosphorus oxidation are limited by mass transfer of phosphorus in the slag, metal, or both phases.[32,33,34,35] Given the relatively higher phosphorus partition coefficient in the early stages of the reaction, the reaction is more likely to be controlled by the mass transfer of phosphorus in the metal or by a combination of mass transfer in both the slag and metal. Rate equations for control by phosphorus transfer in the metal and slag are given, respectively, by Eqs. [6] and [7].
where \( W_{\text{s}} \) and \( W_{\text{m}} \)are the mass of slag and metal, respectively; \( k_{\text{s}} \) and \( k_{\text{m}} \) are the mass transfer coefficients for phosphorus in the slag and metal; \( \rho_{\text{m}} \) and \( \rho_{\text{s}} \)are the densities of metal and slag; \( [{\rm pct\,} P]_{i} \) and \( \left( {{\rm pct\,} P} \right)_{i} \)are phosphorus content in the metal and slag at the interface.
The rate of change of phosphorus in the metal is related to that in the slag by a simple mass balance:
.
By combining Eqs. [1] and [6] through [8], the rate equation for mass transfer controlled in both the slag and metal can be expressed as[35]
From Eq. [9], the overall mass transfer coefficient, \( k_{\text{o}} , \) with units of cm/s, can be written as
In order to estimate the overall mass transfer coefficient of phosphorus, \( k_{\text{o}} \), it is very common to normalize the driving force by integrating Eq. [9]. Since the surface area of the metal droplet \( A \). Eq. [9] changes with time, a time averaged area must be used in the integrated form of the rate equation. The time averaged area was calculated from the data shown in Figure 4 using the method developed by Rhamdhani et al.[38] By integrating Eq. [9], we can obtain Eq. [11]:
Setting C = \( \left( {1 + \frac{{W_{\text{m}} }}{{L_{\text{P}} W_{\text{s}} }}} \right)\frac{{\left[ {{\rm pct\,} P} \right]_{\text{b}} }}{{\left[ {{\rm pct\,} P} \right]_{\text{o}} }} - \frac{{W_{\text{m}} }}{{L_{\text{P}} W_{\text{s}} }} \) and B = \( \left( {\frac{{W_{\text{m}} }}{{\rho_{\text{m}} A}}\frac{1}{{1 + \frac{{W_{\text{m}} }}{{L_{\text{P}} W_{\text{s}} }}}}} \right) \), Eq. [11] becomes \( {\text{B}}\ln C = kt. \)
All data shown in Figure 5 are plotted in Figure 10 according to Eq. [11]. This figure shows that, with the exception of the droplets containing 0.021 wt pct S, the dephosphorization data for reaction times less than 6 seconds can be fitted to a single straight line, suggesting that the reaction has the same controlling step initially for droplets with 0.017 wt pct S and less. The droplets with 0.021 wt pct S show a much lower overall mass transfer coefficient, which suggests a possible different reaction mechanism.

Dephosphorization data from Fig. 5 are replotted as a function of time based on Eq. [11]
The slope of the solid straight line shown in Figure 10 represents the initial overall mass transfer coefficient k o, which is 0.0284 cm/s. Using this value and considering the changing surface area of bloated droplets (changed from 1.2 to 2 cm2 during the period represented by the solid straight line), the k o A at the initial stage were calculated to be between 0.0341 and 0.0568 cm3/s. Based on our calculations in this study, the mass transfer coefficient for the dense slag, k s, and L P can be assumed to be 0.01 cm/s and 10, respectively. Then k m during the initial stages of reaction was determined to be 0.085 cm/s. At a temperature of 1573 K (1300 °C), Wei et al.[44] estimated k m to be 0.05 cm/s where mechanical stirring was employed. Therefore, the value of 0.085 found in this study seems reasonable when one considers the gas stirring effect.
From Figure 10, it can be inferred that k o for droplets with 0.011 wt pct S (represented by the dotted line) show a decreasing value after around 5 seconds. This deviation is probably caused by the dramatic increase in decarburization rate which would be expected to decrease the interfacial oxygen potential. This in turn would lead to a lower L P. According to Eq. [10], a smaller L P would lead to a lower k o as evidenced by the change in slope of the dotted line shown in Figure 10. To quantify the change of L P before and after the point of deviation, \( L_{P} \) at t = 4 seconds and t = 7 seconds for droplets with 0.011 wt pct S were calculated using Eqs. [2] through [5]. The resulting values were used in Eq. [10] to determine k o at these two time points, assuming k s and k m to be 0.001 and 0.01 cm/s, respectively. The calculated results are given in Table II.
Table II shows that k o decreases by a factor of 4 because of the drop of L P. It also shows that the contribution to resistance from the slag side increases from 79 to 95 percent as L P drops from 6 to 1. This results in a shift in mechanism where the rate controlling step shifts from mixed mass transfer control in both slag and metal phase to almost pure slag mass transfer control. In Figure 10, droplets with 0.017 wt pct S (represented by the dashed line) show a more dramatic deviation compared to droplets with 0.011 wt pct S. This behavior can be understood if one considers the different decarburization rates of these two types of droplets where 0.017 wt pct S droplets have a higher decarburization rate than droplets with 0.011 wt pct S as shown in Figure 2(b). Higher decarburization rate would lead to a more dramatic decrease of interfacial oxygen potential, causing a bigger drop in L P. All droplets except those with the highest sulfur content 0.021 wt pct S appear to follow the same reaction mechanism in the initial stages. From analysis, this appears to be mixed control involving mass transport in the metal and the slag. The highest sulfur droplets follow a very different reaction path with a much slower reaction rate even in the earliest stage. The authors cannot offer a definitive explanation for this observation at present.
Role of Partition Ratio on Reaction Kinetics
It is well established that the balance between control by mass transfer in the slag and by mass transfer in the metal is heavily influenced by L P. From the discussion above, it is clear that under the conditions investigated in the current work mixed control operates, shifting to mass transport control in slag as phosphorus partition ratio decreases because of FeO depletion of the slag. This shifting balance is probably the reason why researchers are divided between mass transfer in the slag[34] and some form of mixed control.[32,33,34,35]
From the above discussion, the dynamic nature of the interfacial oxygen potential may offer a way to rationalize the range of different rate determining steps for dephosphorization reported in the literature. It is also worth noting that for a proper description of dephosphorization, a complete understanding of the factors affecting the dynamic interfacial oxygen potential is required. The current work has demonstrated the effect of sulfur on the dynamic interfacial oxygen potential for bloated droplets. The authors are developing a quantitative kinetic model for dynamic interfacial oxygen potential in bloated droplets, which will be presented in a subsequent publication.[42]
Conclusion
During reaction at 1853 K (1580 °C) between Fe-C-P-S droplets and CaO-SiO2-Al2O3-FeO slag, decarburization was shown to go through a period of relatively slow reaction, followed by a period of very rapid reaction during which the droplet became bloated. The incubation period for bloating increased in length with the increasing sulfur content in the droplet. The rate of decarburization during the bloating period was found to go through a maximum at 0.014 wt pct S. The interaction between the decarburization and dephosphorization was studied in detail using the effect of sulfur on controlling the decarburization rate. The dephosphorization went through reversion under all conditions. The phosphorus partition at the reversion point decreased with the increasing decarburization rate. From this study, the following conclusions may be drawn.
-
1.
The rate of CO formation as a function of sulfur content is strongly influenced by two competing effects; sulfur decreases the metal surface tension, thereby increasing the nucleation rate of gas bubbles, while also poisoning the bubble surface to decrease the bubble growth rate. These competing effects led to a maximum CO evolution rate observed at about 0.014 wt pct S.
-
2.
The incubation time for droplet swelling is extended by an increase in metal sulfur content most likely because the poisoning effect of sulfur hinders oxygen transfer into droplet, thereby delaying the onset of internal nucleation of CO.
-
3.
The phosphorus partition coefficient, measured at the reversion point, shows a strong inverse relationship with increasing CO evolution rate because the associated oxygen consumption decreases the interfacial oxygen potential.
-
4.
The complex dephosphorization–reversion–dephosphorization behavior of droplets depending on timing of their movement between the dense and foamy slag can be explained by considering the more rapid depletion of iron oxide from the foamy slag.
-
5.
Kinetic analysis of dephosphorization shows that in the current study the rate controlling step is first mixed control by mass transport in both metal and slag, which then shifts to mass transport in slag as phosphorus partition ratio decreases because of FeO depletion in the slag.
References
E.W. Mulholland, G.S.F. Hazeldean and M.W. Davies: Journal of The Iron and Steel Institute, 1973, vol. 211, pp. 632-39.
T. Gare and G.S.F. Hazeldean: Ironmaking and Steelmaking, 1981, no. 4, pp. 169-81.
H. Gaye and P.V. Riboud: Metall. Trans. B, 1977, no. 9, vol. 8B, pp. 409-15.
D.J. Min and R.J. Fruehan: Metall. Trans. B, 1992, vol. 23B, pp. 29-37.
C.L. Molloseau and R.J. Fruehan: Metall. Trans. B, vol. 33B, 2002, pp. 335-44.
E. Chen and K. S. Coley: Ironmaking and Steelmaking, 2010, vol. 37(7), pp. 541-45.
R.C. Urquhart and W.G. Davenport: Can. Metall. Quarterly, 1973, no. 4, vol. 12, pp. 507-16.
N. Standish and Q.L. He: ISIJ Int., 1989, no. 6, vol. 29, pp. 455-61
Q.L. He and N. Standish: ISIJ Int., 1990, no. 4, Vol. 30, pp. 305-09.
Q.L. He and N. Standish: ISIJ Int., 1990, no. 5, vol. 30, pp. 356–61.
G. Turner and S. Jahanshahi: Trans. ISIJ, 1987, vol. 27, pp. 734-39.
S.C. Koria and K.W. Lange: Ironmaking and Steelmaking, 1983, no. 4, vol. 10, pp. 160-68.
S.C. Koria and K.W. Lange: Metall. Trans., 1984, vol. 15B, pp. 109-16.
S.C. Koria and K.W. Lange: Ironmaking and Steelmaking, 1986, no. 5, vol. 13, pp. 236-40.
Subagyo, G.A. Brooks, K.S. Coley, and G.A. Irons: ISIJ Int., 2003, no. 7, vol. 43, pp. 983-89.
Subagyo, G.A. Brooks, and K.S. Coley: Steelmaking Conf. Proc., ISS, Warrendale, PA, 2002, vol. 85, pp. 749-62.
Subagyo, G.A. Brooks, and K. Coley: Can. Metall. Quarterly, 2005, vol. 44 [1], pp119-29.
J. Schoop, W. Resch, and G. Mahn: Ironmaking and Steelmaking, 1978, vol. 2, pp. 72-79.
G. A. Brooks, Y. Pan, Subagyo and K. S. Coley: Metall. Trans. B, 2005, vol. 36B, pp 525-35.
N. Dogan, G. A. Brooks and M. A. Rhamdhani: ISIJ Int., 2011, no. 7, vol. 51, pp. 1086-92.
N. Dogan, G. A. Brooks and M. A. Rhamdhani: ISIJ Int., 2011, no. 7, vol. 51, pp. 1093–01
C. Kattenbelt and B. Roffel: Metall. Trans. B, 2008, vol. 39B, pp764-69.
R. Sarkar, P Gupta, Somnath Basu, and Bharath Ballal: Metall. Trans. B, 2015, vol. 46, pp. 961-76.
C. Cicutti, M. Valdez, T. Perez, R. Donayo and J. Petroni: Latin Am. Appl. Res., 2002, vol. 32, pp. 237.
D.J. Price, L.D.Steelmaking: The Institution of Mining and Metallurgy, London, UK, 1974, pp. 8-15.
B. Trentini: Trans. Met. Soc. AIME, 1968, vol. 242, pp. 2377-88.
P. Kozakevitch: JOM, 1969, no. 7, vol. 22, pp. 57
A. Chatterjee, N.O. Lindfors, and J.A. Wester: Ironmaking and Steelmaking, 1976, no. 1, vol. 3, pp. 21-32
M. Pomeroy, E. Chen, K.S. Coley, and G. Brown: Proceedings of the 6th European Oxygen Steelmaking Conference, Stockholm, 2011, vol. 38, pp. 141–51.
S. Basu, A.K. Lahiri and S. Seetharaman: Revue de Métallurgie, 2009, Vol. 106, pp. 21-26.
A. Assis, M. Tayeb, S. Sridhar and R.J. Fruehan: Metall. Trans. B, 2015, vol. 46B, pp. 2255-63.
K. Mori, Y. Fukami and Y. Kawai: Trans. ISIJ, 1988, vol.28, 315-18.
P. Wei, M. Sano, M. Hirasawa, and K. Mori: ISIJ.Int., 1993, vol. 33, 479-87.
B. J. Monaghan, R.J. Pomfret, and K.S. Coley: Metall. Trans. B, 1998, vol. 29B, pp. 111-18.
C. P. Manning and R. J. Fruehan: Metall. Trans. B, 2013, vol. 44B, pp. 37-44.
A. K. Hewage, B. K. Rout, G. Brooks and J. Naser: Ironmaking and Steelmaking, 2016, no. 5, vol.43, pp. 358–70.
E. Chen: PhD Thesis, McMaster University, Hamilton, CA, 2011, pp. 54.
M.A.Rhamdhani, G.A.Brooks and K.S. Coley: Metall. Trans. B, 2005, vol. 36B, pp. 219-27.
K. Gu, B. Jamieson, N. Dogan, and K.S. Coley: AISTech Conference Proceedings, 2015, pp. 229–37.
M. Hino and K. Ito: Thermodynamic Data for Steelmaking, Tohoku University Press, Sendai. 2009, pp. 259-64.
C. L. Molloseau and R. J. Fruehan: Metall. Trans. B, 2002, vol. 33B, pp. 335-44.
K. Gu, N. Dogan, and K. Coley: Metall. Trans. B, DOI:10.1007/s11663-017-1000-2.
T. Mori: Transactions of the Japan Institute of Metals, 1984, vol. 25, No.11, pp. 761-71.
P. Wei, M. Sano, M. Hirasawa and K. Mori: Transactions ISIJ, 1988, Vol.28, pp. 637-44.
Acknowledgments
The authors thank member companies in McMaster Steel Research Centre and the Natural Science and Engineering Research Council of Canada (NSERC) for funding this project.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Manuscript submitted November 2, 2016.
Rights and permissions
About this article

Cite this article
Gu, K., Dogan, N. & Coley, K.S. The Influence of Sulfur on Dephosphorization Kinetics Between Bloated Metal Droplets and Slag Containing FeO. Metall Mater Trans B 48, 2343–2353 (2017). https://doi.org/10.1007/s11663-017-1002-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-017-1002-0