
Abstract
LiFePO4/C and LiYb0.02Fe0.98PO4/C composite cathode materials were synthesized by simple solution technique. The samples were characterized by X-ray diffraction, scanning electron microscope, and thermogravimetric–differential thermal analysis. Their electrochemical properties were investigated by cyclic voltammetry, four-point probe conductivity measurements, and galvanostatic charge and discharge tests. The carbon-coated and Yb3+-doped LiFePO4 sample exhibited an enhanced electronic conductivity of 1.9 × 10−3 Scm−1, and a specific discharge capacity of 146 mAhg−1 at 0.1 C. The results suggest that the improvement of the electrochemical performance can be attributed to the ytterbium doping, which facilitates the phase transformation between triphylite and heterosite during cycling, and the conductivity improvement by carbon coating.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mineral triphylite, having the formula of LiFePO4 and showing an ordered olivine structure is believed to be one of the most promising replacements of the cathode materials currently being used, such as LiCoO2 and LiMn2O4 [1]. Goodenough and co-workers first reported olivin-type LiFePO4 cathode material in 1997 [2]. This material has the advantages of being nontoxic, inexpensive, and environmentally friendly [3–5]. Moreover, it has an interesting theoretical specific capacity of about 170 mAh g−1, a good cycling stability, and a technically attractive flat voltage versus current profile of 3.45 V versus Li/Li, due to the two-phase extraction–insertion reaction. This reaction mechanism makes the theoretical capacity difficult to reach; the area of the interface between the LiFePO4–FePO4 phases, where the insertion or deinsertion of Li ions takes place, is limited. Furthermore, the FePO4 produced has a very low conductivity [6–8]. The low electronic conductivity and low lithium-ion diffusivity of LiFePO4 restrict its commercial applications.
Various synthesis and processing approaches have been employed to overcome this problem. Initially, additives were introduced to synthesize LiFePO4/conductive material composites, such as dispersed carbon [9–14], metal powders [15, 16], and intrinsically conducting polymers [17]. However, the resuls have shown improvements on bulk conductivity without changes on any LiFePO4 lattice parameters. Another method used was doping, where Li ions (on the M1 site) and/or Fe ions (on the M2 site) were substituted. It was reported that low-level doping of LiFePO4 by a range of supervalent ions (e.g., Mg2+, Al3+,Ti4+, Zr4+, Nb5+) increases the electronic conductivity to values greater than 10−2 Scm−1 at room temperature [18]. In addition, investigations of doping of other heteroatoms indicate that the electrochemical properties of LiFePO4 could be improved by this modification [19–23].
Among these methods, expensive Fe2+ salts were often employed as the precursors, which increases the cost of the LiFePO4. For large-scale industrialization, inexpensive Fe3+ salts as raw materials and noncomplex craft should be proposed to prepare LiFePO4 [24]. In this paper, we present a simple method for preparing LiFePO4/C using cheap Fe(NO3)3 instead of Fe2+ salts as the iron source, and polyethylene glycol (PEG), a high temperature reducer and a carbon source. In this study, the synthesis and characterization of ytterbium-doped LiYb0.02Fe0.98PO4/C cathode material and the improved electrochemical performances will be demonstrated.
Experimental
Virginal and 2 % doped LiYb0.02Fe0.98PO4/C samples were prepared via a simple solution method using analytical grade reagents as starting materials. The stoichiometric amounts of Fe(NO3)3 × 9H2O (Merck 98 %), NH4H2PO4 (Merck 98 %) and LiNO3 (Riedel-de Haen 95) were dissolved in distilled water. For the doped sample, the stoichiometric amounts of Yb2O3 solution in HNO3 were added to the bare solution.This mixture was stirred slowly until a clear solution formed, followed by heating at 80 °C for 4 h with constant agitation to form a homogeneous suspension. Then, the gel was heated at 300 °C in air for 3 h to remove the water and for decomposition of nitrate in the absence of reductive atmosphere.
The precursor powder was ground and an adequate amount of polyethylene glycol (PEG; mean molecular weight 40,000) was added into the obtained powder as a reducing agent and carbon source. Finally, the resulting slurry was heated in a tube furnace at 700 °C for 10 h under argon gas flow. During the final heating of the PEG-contained precursor, Fe3+ was reduced to Fe2+ with the help of hydrogen and carbon as the reducing agent. These were generated from the decomposition of PEG, which produces a strong reductive atmosphere. As a result of this process, the LiFePO4/C and LiYb0.02Fe0.98PO4/C samples were synthesized.
The crystallographic structure of the samples was characterized by an X-ray diffractometer (Bruker). The diffraction data were collected for 2 s at each 0.02° step width over a 2θ range from 10 to 60°. The Diffrac Plus and Win-Metric programs were used to obtain information about the crystal structures and lattice parameters of the prepared samples.
Thermogravimetric (TG) analysis of the samples were studied on a Perkin-Elmer Diamond model thermal analysis apparatus with a heating rate of 10 °C min−1 from ambient to 800 °C in an argon flow. The morphologies of the powders were observed using a scanning electron microscopy (LEO), operated at 20 kV. In order to measure the electronic conductivity, the powder was pressed into disc-shaped pellets at a pressure of 5 tons. Conductivity was measured by the four-point probe method, using an Iceblue Instrument (Kayseri, Turkey). The amount of carbon in the lithium iron phosphate was determined from the difference of weight loss t by heating at 700 °C for 2 h in air [25].
The electrochemical studies were carried out in two-electrode Teflon cells. The cells were fabricated by using the LiFePO4/C as a cathode and lithium foil as an anode. A glassfiber separator soaked in electrolyte separated the two electrodes. The electrolyte consisted of 1 M solution of LiClO4 dissolved in a ethylene carbonate/diethyl carbonate (EC/DEC; 1:1 ratio by volume). For the preparation of the cathode composite, a slurry mixed with 80 wt% of cathode active material, 15 wt% of carbon additive (including carbon black and carbon converted from PEG) and 5 wt% of polyvinylidene fluoride binder in 1-methyl-2-pyrolidinone (NMP) was pasted on the aluminium foil current collector with a diameter of 13 mm, followed by vacuum drying at 120 °C overnight in a vacuum oven and uniaxial pressing between two flat plates at 2 tons for 5 min. The electrode loading was about 5–7 mg of the cathode active material. The test cell performance was measured on a computer-controlled multichannel charge/discharge apparatus (MLab100, Wenking). All cells were assembled in an argon-filled dry glove box and tested at room temperature. The cell was cycled galvanostatically between 2.1 and 4.3 V at different current density (0.1–2 C). Cyclic voltammetry measurements were performed using an MLab100 Instrument at a scan rate of 0.01 mVs−1 between 2.2 and 4.4 V.
Results and discussions
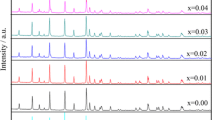
Figure 1 shows the XRD patterns of the LiFePO4/C and LiYb0.02Fe0.98PO4/C compounds. Both samples exhibited a single phase of LiFePO4 with an ordered olivine structure indexed to the orthorhombic Pnma space group. The obtained lattice parameters agreed well with those reported in the literature (JCPDS card no. 81–1,173). The detailed cell parameters of LiFePO4/C and LiYb0.02Fe0.98PO4/C composites were calculated and are listed in Table 1. No impurity phase has been detected, which indicates that the 2 % doping could not destroy the lattice structure. Also, there is no obvious diffraction response of the carbon because of its low content or amorphous state. The results obtained from combustion analysis (700 °C) showed that the amount of carbon in both LiFePO4/C and LiYb0.02Fe0.98PO4/C was 3 wt%. This residual carbon was uniformly dispersed in the compound, which improved the electroconductivity of the sample 2.7 × 10−2 Scm−1 presented in Table 1. According to the lattice parameters, the values of c are less affected than that of a and b. It has been reported that the lithium-ion intercalation–deintercalation proceeds through one-dimensional pathways along the c-axis [26].

XRD patterns of LiFePO4/C and LiYb0.02Fe0.98PO4/C samples
The electrochemical performance of the electrode is greatly influenced by the particle size and the morphology of the particles [27]. The scanning electron microscopy (SEM) images of the LiFePO4/C and LiYb0.02Fe0.98PO4/C composites are shown in Fig. 2. It can be seen from Fig. 2 that the particle size of Yb-doped LiFePO4/C was smaller than bare LiFePO4/C. In general, the Yb-doped LiFePO4 powder shown less agglomeration and the scatter was more uniform than the undoped one. It seems that the Yb ions in the solution prevent particles from conglomerating and form secondary particles during calcinations, which may be conducive to shorten the lithium diffusion distance.

Scanning electron micrographs of LiFePO4/C (a) and LiYb0.02Fe0.98PO4/C (b) samples
The DTA/TG result of LiFePO4/C is shown in Fig. 3. The weight loss at temperatures between 50 and 150 °C corresponding to the endothermic peak is due to water vaporization and carbonization of PEG, respectively. The weight loss in the next step at temperatures between 150 and 240 °C resulted from the decomposition of nitrates in the precursor. The residual organic materials of PEG were also decomposed and carbonized below 380 °C. A corresponding exothermic peak associated with the crystallization of LiFePO4 was observed at around 460 °C. Therefore, the LiFePO4 precursor was calcined above 500 °C to obtain the crystallized phase [28]. In order to gain a further insight into the effect of doping on the electrochemical performance of LiFePO4/C, cyclic voltammetry was carried out. The CV curves of LiFePO4/C and LiYb0.02Fe0.98PO4/C samples were shown in Fig. 4. It can be seen from Fig. 4 that the two samples have similar redox peaks. The redox peaks of LiYb0.02Fe0.98PO4/C and LiFePO4/C were centered at 3.55/3.35 and 3.65/3.28 V, respectively. Appearently, the peak separation of LiYb0.02Fe0.98PO4/C was narrower and the peak shape was sharper than that of LiFePO4/C. These results may demonstrate that lithium insertion and extraction readily occurred in the LiYb0.02Fe0.98PO4/C material [29].

The DTA/TGA curve for LiFePO4/C over the temperature range from ambient to 800 °C at a heating rate of 10 °C min−1 in argon atmosphere at 200 ml min−1 flow rate

The first cyclic voltammetry profiles of LiFePO4/C and LiYb0.02Fe0.98PO4/C samples
The charge/discharge capacity of both LiFePO4/C was tested at 0.1 C in the voltage range between 2.2 and 4.2 V. Figure 5 shows the charge/discharge curves with a flat voltage plateau at around 3.4 V. This demonstrates that the two-phase redox reaction occurs via a first-order transition between FePO4 and LiFePO4 [30]. A reversible capacity of about 146 mAh/g was obtained for the LiYb0.02Fe0.98PO4/C carbon composite, which is higher than the capacity of undoped LiFePO4/C. The capacity of undoped LiFePO4/C, on the other hand, was observed as 136 mAh/g for the same condition. The higher discharge capacity for sample LiYb0.02Fe0.98PO4/C was explained in terms of particle size, [31, 32] and the lowest electrochemical polarization of Yb-doped LiFePO4/C which may suggest that the increased conductivity (1.9 × 10−2 Scm−1) is induced by the doping and carbon coating [33].

Initial charge and discharge curves of LiFePO4/C and LiYb0.02Fe0.98PO4/C samples
Rate capabilities of both bare and Yb-doped LiFePO4/C at different C rates were given in Fig. 6. The capacity of the Yb-doped LiFePO4/C was larger than that of the bare LiFePO4/C in all current densities. Also, the rate capability results were much better than those of the Yb-doped LiYb0.02Fe0.98PO4/C. The capacity drop with an increase in the current density from 0.1 to 1 C was 14 % for the ytterbium doping and 24 % for the undoped LiFePO4/C. Therefore, it seems that the ytterbium doping can be a powerful method to enhance the conductivity and preserve the capacity even at the high current densities.

Relationship between capacity and discharge rate of the LiFePO4/C and LiYb0.02Fe0.98PO4/C samples
Conclusion
Phase-pure, olivine LiFePO4/C and ytterbium-doped LiYb0.02Fe0.98PO4/C materials with carbon contents of 3 wt% have been synthesized by a simple and cheap solution method using PEG as the carbon source. The cell performance was improved and the initial capacity of LiYb0.02Fe0.98PO4/C was 146 mAhg−1 obtained at 0.1 C. As a result, ytterbium doping is very promising for the improvement of the electrochemical performance of LiFePO4. Both discharge capacity and rate capability are well ameliorated.
References
L. Meng, X. Kai, L. Dezhan, P. Yi, Synthesis of LiFePO4 by one-step annealing under the vacuum condition. J. Mater. Sci. 40, 2639–2641 (2005)
A.K. Padhi, K.S. Nanjundaswamy, J.B. Goodenough, Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997)
Y. Cho, T. Fey, H.J. Kao, Physical and electrochemical properties of La-doped LiFePO4/C composites as cathode materials for lithium-ion batteries. Solid State Electrochem. 12, 815–823 (2008)
M.S. Whittingham, Insertion electrodes as SMART materials: the first 25 years and future promises. Solid State Ion. 134, 169–178 (2000)
A.R. Armstrong, P.G. Bruce, Synthesis of layered LiMnO2 as an electrode for rechargeable lithium batteries. Nature 381, 499–500 (1996)
S. Franger, F. Le Cras, C. Bourbon, H. Rouault, Comparison between different LiFePO4 synthesis routes and their influence on its physico-chemical properties. J. Power Sources 119–121, 252–257 (2003)
A. Yamada, S.C. Chung, K. Hinokuma, Optimized LiFePO4 for lithium battery cathodes. J. Electrochem. Soc. 148, A224–A229 (2001)
A.S. Andersson, J.O. Thomas, The source of first-cycle capacity loss in LiFePO4. J. Power Sources 97–98, 498–502 (2001)
N. Ravet, Y. Chouinard, J.F. Magnan, S. Besner, Electroactivity of natural and synthetic triphylite. J. Power Sources 97–98, 503–507 (2001)
S.T. Myung, S. Komaba, N. Hirosaki, H. Yashiro, Emulsion drying synthesis of olivine LiFePO4/C composite and its electrochemical properties as lithium intercalation material. Electrochim. Acta 49, 4213–4222 (2004)
K. Zaghib, J. Shim, A. Guerfi, P. Charest, Effect of carbon source as additives in LiFePO4 as positive electrode for lithium-ion batteries. Electrochem. Solid State Lett. 8, A207–A210 (2005)
A.D. Spong, G. Vitins, J.R. Owen, A solution–precursor synthesis of carbon-coated LiFePO4 for Li-ion cells. J. Electrochem. Soc. 152, A2376–A2382 (2005)
H. Liu, L.J. Fu, H.P. Zhang, J. Gao, C. Li, Effects of carbon coatings on nanocomposite electrodes for lithium-ion batteries. Electrochem. Solid State Lett. 9, A529–A533 (2006)
H. Goktepe, H. Şahan, F. Kılıç, Ş. Patat, Improved of cathode performance of LiFePO4/C composit using different carboxylic acids as carbon sources for lithium ion batteries. Ionics 16, 203–208 (2010)
F. Croce, A.D. Epifanio, J. Hassoun, B. Scrosati, A novel concept for the synthesis of an improved LiFePO4 lithium battery cathode. Electrochem. Solid State Lett. 5, A47–A50 (2002)
K.S. Park, J.T. Son, H.T. Chung, S.J. Kim, C.H. Lee, Surface modification by silver coating for improving electrochemical properties of LiFePO4. Solid State Commun. 129, 311–314 (2004)
G.X. Wang, L. Yang, Y. Chen, J.Z. Wang, S. Bewlay, An investigation of polypyrrole-LiFePO4 composite cathode materials for lithium-ion batteries. Electrochim. Acta 50, 4649–4654 (2005)
S.Y. Chung, J.T. Bloking, Y.M. Chiang, Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 1, 123–128 (2002)
H. Liu, C. Li, Q. Cao, Y.P. Wu, R. Holze, Effects of heteroatoms on doped LiFePO4/C composites. J. Solid State Electrochem. 12, 1017–1020 (2008)
D.X. Gouveia, V. Lemos, J. Paiva, A.G. Filho, Spectroscopic studies of LixFePO4 and LixM0.03Fe0.97PO4 (M = Cr, Cu, Al, Ti). Phys. Rev. B 72, 24105–24110 (2005)
M. Abbate, S.M. Lala, L.A. Montoro, Ti-, Al-, and Cu-doping induced gap states in LiFePO4. Electrochem. Solid State Lett. 8, A288–A290 (2005)
G.X. Wang, S. Bewlay, S.A. Needham, H.K. Liu, R.S. Liu, Synthesis and characterization of LiFePO4 and LiTi0.01Fe0.99PO4 cathode materials. J. Electrochem. Soc. 153, A25–A31 (2006)
H. Goktepe, H. Şahan, A. Ulgen, Ş. Patat, Synthesis and electrochemical properties of carbon-mixed LiEr0:02Fe0:98PO4 cathode material for lithium-ion batteries. J. Mater. Sci. Technol. 27, 861–864 (2011)
Y. Linghui, L. Qiaosheng, W. Haihui, Synthesis of LiFePO4-C cathode materials using a green and low-cost method. Ionics 15, 689–692 (2009)
S. Yang, Y. Song, Y. Peter, M. Stanley, Reactivity stability and electrochemical behavior of lithium iron phosphates. Electrochem. Commun. 4, 239–244 (2002)
C.Y. Ouyang, S.Q. Shi, Z.X. Wang, X.J. Huang, L.O. Chen, First-principles study of Li ion diffusion in LiFePO4. Phys. Rev. B 69, 104303–104307 (2004)
S.L. Bewlay, K. Konstantinov, G.X. Wang, H.K. Liu, Conductivity improvements to spray-produced LiFePO4 by addition of a carbon source. Mater. Lett. 58, 1788–1791 (2004)
M.R. Yang, W.H. Ke, S.H. Wu, Preparation of LiFePO4 powders by co-precipitation. J. Power Sources 146, 539–543 (2005)
M. Pan, X. Lin, Z. Zhou, Electrochemical performance of LiFePO4/C doped with F synthesized by carbothermal reduction method using NH4F as dopant. J. Solid State Electrochem. 16, 1615–1621 (2012)
D. Shanmukaraj, G.X. Wang, Electrochemical studies on iFe1−x Co x PO4/carbon composite cathode materials synthesized by citrate gel technique for lithium-ion batteries. Mater. Sci. Eng. B 149, 93–98 (2008)
M. Higuchi, K. Katayama, Y. Azuma, Synthesis of LiFePO4 cathode material by microwave processing. J. Power Sources 119–121, 258–261 (2003)
H.C. Shin, W.I. Cho, H. Jang, Electrochemical properties of carbon-coated LiFePO4 cathode using graphite, carbon black, and acetylene black. Electrochim. Acta 52, 1472–1476 (2006)
C. Benoit, S. Franger, Chemistry and electrochemistry of lithium iron phosphate. Solid State Electrochem. 12, 983–987 (2008)
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Göktepe, H. Electrochemical performance of Yb-doped LiFePO4/C composites as cathode materials for lithium-ion batteries. Res Chem Intermed 39, 2979–2987 (2013). https://doi.org/10.1007/s11164-012-0811-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-012-0811-7