
Abstract
N-methyl-D-aspartate receptors (NMDARs) play essential roles in vital aspects of brain functions. NMDARs mediate clinical features of neurological diseases and thus, represent a potential therapeutic target for their treatments. Many findings implicated the GluN2B subunit of NMDARs in various neurological disorders including epilepsy, ischemic brain damage, and neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease, Huntington’s chorea, and amyotrophic lateral sclerosis. Although a large amount of information is growing consistently on the importance of GluN2B subunit, however, limited recent data is available on how subunit-selective ligands impact NMDAR functions, which blunts the ability to render the diagnosis or craft novel treatments tailored to patients. To bridge this gap, we have focused on and summarized recently reported GluN2B selective ligands as emerging subunit-selective antagonists and modulators of NMDAR. Herein, we have also presented an overview of the structure–function relationship for potential GluN2B/NMDAR ligands with their binding sites and connection to CNS functionalities. Understanding of design rules and roles of GluN2B selective compounds will provide the link to medicinal chemists and neuroscientists to explore novel neurotherapeutic strategies against dysfunctions of glutamatergic neurotransmission.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amino acid glutamate (Glu) mediates most excitatory neurotransmission in the brain and spinal cord [1]. Glu belongs to a characteristic excitatory neurotransmitter for the mammalian central nervous system (CNS). Upon axon firing, Glu is released into the synaptic cleft and binds to presynaptic and postsynaptic Glu-activated receptors to initiate neuronal response [2, 3].
Glu-activated receptors belong to two main classes (a) ionotropic Glu receptors (iGluRs), responsible for the fast signal transmission; and (b) metabotropic Glu receptors (mGluRs), producing slower signal transduction through secondary messenger systems [1, 2].
Glu acts through iGluRs by forming cation-permeable ion channels and divided into three important subgroups based on ligand selectivity studies: (1) α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (GluA1 through GluA4), (2) kainic acid (KA) receptors (GluK1 through GluK5), and (3) NMDARs [3, 4]. Thus, overstimulation of the glutamatergic neurotransmission and unchecked activation of Glu receptors have significant relevance in the clinical manifestations of CNS diseases [5, 6].
NMDAR subunit composition and their operation in CNS
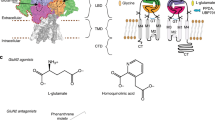
NMDARs are Glu-gated ion channels with a critical role in neurological processes [6]. Heterotetrameric NMDAR consists of four subunits. Different NMDAR subunits have been cloned into three types: (1) two subunits of glycine-binding GluN1, (2) two subunits of Glu-binding GluN2, and/or (3) GluN3 subunits. GluN1 subunit has eight splice variants (GluN1a-h), the GluN2 subunit is encoded by four different genes (GluN2A through 2D), and the GluN3 subunit is encoded by two different genes (GluN3A and GluN3B). Each subunit consists of an amino-terminal domain (ATD), ligand-binding domain (LBD), transmembrane domain (TMD), and intracellular carboxy-terminal domain (CTD). Binding sites for endogenous agonists Glu and glycine are in GluN1 and GluN2 (or GluN3) subunits [7,8,9]. Binding sites for phencyclidine [1-(1-phenyl cyclohexyl) piperidine, PCP] and Mg2+ are also located within the ion channel. ATD have binding sites for Zn2+, H+, polyamines, and ifenprodil [10].
NMDAR has characteristic features that leads to its importance in several neurological disorders (Fig. 1). These features includes (1) two endogenous agonists (Glu and glycine) for opening of the ligand-gated ion channel; (2) Mg2+ ions block the ion channel at resting membrane potential, which is removed by depolarization of the surrounding membrane; (3) extraordinary alliance between the agonism of Glu–glycine and voltage-dependent Mg2+ blockade results in the development of neuronal connections; (4) control over the influx of Ca2+ ions; and (5) several binding sites for ligands to modulate opening state of the ion channel [7,8,9,10,11,12].

Functional subunits and structure of diheteromeric NMDARs. Endogenous agonist glutamate binds to the glutamate binding site; glycine and d-serine bind to a glycine modulatory site of NMDARs
Under physiological conditions, NMDARs mediates slow Ca2+ permeable synaptic current by binding to endogenous ligand Glu and co-agonist glycine, when voltage-dependent block by extracellular Mg2+ is relieved. Ca2+ ions have beneficial effects on neuronal development in the CNS. However, overstimulation of NMDARs causes increased transmembrane flux of Ca2+, leading to neuronal death characterized by a progressive decline in cognition functions [13,14,15,16,17].
Elevated Ca2+ concentration through an opening of NMDARs also leads to excitotoxicity by increasing the concentration of neurotransmitter Glu. Excitotoxicity has a crucial role in the development and progression of neurodegenerative disorders such as epilepsy, Alzheimer’s disease (AD), Parkinson’s disease (PD), and other multiple CNS disorders [17,18,19,20,21,22,23].
An involvement of NMDARs in the process of excitotoxicity supports the hypothesis that a blockade or modulation of receptor results in neuroprotective effects and develop subunit selective probes to treat multiple CNS diseases. Thus, NMDARs represents a promising target for development of novel neurotherapeutic tools for the treatment of neurological diseases.
Role of NMDAR in neurodegenerative disorders
Alzheimer's disease
AD is a complex multifactorial neurodegenerative disorder characterized by inevitable loss of memory, cognitive abilities, and eventually an independent living ability [24]. By 2020, because of COVID-19, number of fatalities from AD increased by 17% [25]. The etiology of AD is not clear yet and several pathological events occur during disease progression such as amyloid-β (Aβ) deposition, tau accumulation, cellular oxidative stress, acetylcholine depletion, dysfunctional autophagy, dysfunction of NMDAR, and a neuroinflammation [4, 26,27,28,29]. These multifactorial neuropathological conditions lead to the synaptic dysfunction and subsequent neuronal death characterized by decline in neurocognitive abilities (Fig. 2). Possible neurotherapeutic targets involved in the AD are illustrated as Fig. 3.

Neurological events occur during progression of the AD

Therapeutic targets involved in the AD
Role of NMDAR in AD
NMDAR-mediated glutamatergic neurotransmission is essential for the synaptic plasticity and neuronal survival. Overactivation of NMDARs causes synaptic dysfunction by perturbing synaptic Ca2+ handling NMDAR subunits (Fig. 4). Thus, alteration in function of NMDAR is one of the mechanisms influencing the AD [30, 31].

Overactivation of NMDAR leading to AD [32]
NMDAR selective ligands as neurotherapeutics for AD
Memantine is the only clinically approved NMDAR antagonists and acts by preventing NMDAR-induced excitotoxicity. It preferentially functions as an antagonist of non-synaptic NMDARs [33, 34]. First GluN2B-selective candidate (Ifenprodil), blocks GluN2B receptor-mediated currents and have shown 400-fold lower affinity for GluN2A subunit. It proved as a use-dependent, voltage-independent, and non-competitive antagonist of GluN2B-NMDAR [28]. Neramexane (an alkyl-cyclohexane analogue) is a non-competitive NMDAR antagonist indicating long-term memory enhancement in adult rats analogous to the memantine (Fig. 5) [35]. Neramexane is structurally related to a memantine (developed by Merz Pharma), acts as a NMDAR antagonist and exhibits neuroprotective activities [36]. It also acts through nicotinic acetylcholine receptor by antagonizing α9α10 subtype in chronic tinnitus and sensorineural hearing loss [36, 37].

Epilepsy
Epilepsy is a complex neurological disorder resulting in an enduring predisposition to engender excessive neuronal activity that manifest in seizures. It is the second most chronic neurological condition affecting 1% people around the globe; however, existing antiepileptic drugs (AEDs) target only symptoms rather than the underlying cause [17, 18, 38].
Epilepsy is connected to alterations in central inhibitory γ-aminobutyric acid (GABA) and excitatory glutamatergic neurotransmission. Overstimulation of glutamatergic transmission and unchecked activation of Glu receptors have significant relevance in the clinical manifestations of epilepsy [39]. AEDs functions mainly by blocking Glu-induced excitotoxicity, activating voltage-operated Na+ and/or Ca2+ channels, activation of K+ channels, and through enhancement of GABA-mediated inhibition (Fig. 6).

Specific targets for epilepsy [40]
Role of NMDARs in epilepsy
NMDARs are activated by binding of the endogenous ligand Glu and co-agonist glycine, at their LBDs and removal of Mg2+ ion block by slight depolarization of the postsynaptic membrane (Fig. 7). Under physiological conditions, receptor activation at the NMDAR causes opening of an ion channel and permits cationic influx along their concentration gradient, resulting in membrane depolarization. Because of Ca2+ permeability, overactivation of NMDARs causes excitotoxicity leading to a neurodegeneration, which largely explains the role of NMDARs in epilepsy [41].

Function of NMDAR as an ion channel and mechanism of action for functional AEDs [41]
NMDAR selective ligands for the treatment of epilepsy
Felbamate (a dicarbamate derivative) is a clinically approved AED acting as non-competitive GluN2B antagonist. The drug is approved by the Food and Drug Administration for the treatment of focal seizures and Lennox-Gastaut syndrome. It inhibits voltage-sensitive Na+/Ca2+ channel with potentiation of GABA-induced chloride currents and eventual inhibition of NMDAR [42].
Magnesium sulphate is a non-competitive GluN2 antagonist. Mg2+ enters the brain and influences CNS by inhibiting NMDAR. Remacemide is a non-competitive GluN2B antagonist and acts as a potential AED with its active metabolite (desglycinyl-remacemide). It also acts as a voltage gated sodium channel blocker [43]. When used alone, remacemide has shown side effects such as vertigo, drowsiness, and gastrointestinal symptoms.
Amantadine acts by binding to NMDARs, it increases the rate of ion channel closure and useful as an adjunct in the treatment of refractory absence seizures (Fig. 8). It also acts as a nicotinic antagonist and dopamine agonist. It is also used as an antiviral agent for the prophylactic or symptomatic treatment of influenza A [44].

NMDAR antagonists effective for the treatment of epilepsy [38]
Parkinson disease
PD is a neurodegenerative disease affecting elderly and worsen with the time [45]. Inhibition of dopamine synthesis and build-up of Lewy bodies (clumps of abnormal protein particles) are characteristic symptoms of PD, leading to the neuronal death from the substantia nigra [46]. These neuromolecular changes results into latent tremor, rigidity, and postural instability [47]. Non-motor symptoms including cognitive abnormalities, tiredness, and sleepiness also considered as comorbidities of PD [43]. Approaches for the management of PD are illustrated as Fig. 9.

Approaches for the management of PD [48]
Role of NMDAR in PD
Glutamatergic transmission plays a distinctive role in the pathophysiology of PD. The exacerbation of dyskinesia has been related to the hyperphosphorylation and subsequent overactivation of NMDARs [49]. Glu and glycine bind together to activate NMDAR; antagonists molecules interfere with binding of these endogenous ligands to limit NMDAR activity.
NMDAR ligands for the treatment of PD
Amantadine and memantine (NMDAR antagonists) are used as supplements with other PD treatments. These analogues blocks both acetylcholine and NMDARs, exhibiting their crucial role as PD therapeutics. Amantadine (NMDAR antagonist) is the only FDA-approved medication for the treatment of levodopa related dyskinesia in Parkinson's patients and to treat extrapyramidal side effects of medications. Currently no drugs are available that have significant effects on symptoms that develop in the later stages of PD. NMDAR ligands can also be used as a neuroprotective that slows the disease progression by blocking glutamatergic excitotoxicity and stimulating synaptogenesis with neurotrophic release [50].
Huntington’s disease
Huntington’s disease (HD) is a neurodegenerative disorder characterized by loss of striatal neurons. Huntingtin gene located on the short arm of chromosome 4p16.3 and is mainly responsible for the HD [51]. Disease predominantly develops in the middle of life and characterised by dementia with undesirable choreatic movements [52]. It is a neurological condition passed from one generation to the next generation. Figure 10 demonstrates the different targets involved in HD [53].

Therapeutic targets for HD [53]
Role of NMDARs in HD
Animal studies showed that injections of kainic or quinolinic acids produced lesions as seen in HD; supporting the hypothesis that NMDA receptor-mediated excitotoxicity may play a role in the etiology of disease [53].
For further research into HD, the role of PSD-95 was investigated. It is well-known scaffolding protein that binds to multiple cytoplasmic proteins, including normal huntingtin and GluN2-NMDARs. After binding, it causes receptor clustering in the post-synaptic membrane to result physiological inhibition of NMDAR activity. Additionally, the presence of mutant huntingtin protein causes a disruption in PSD-95 binding to NMDARs and resulting in excitotoxicity with ultimately increased neuronal cell death, which is consistent with HD [54].
NMDAR ligands in HD
Memantine reduces striatal cell death in PD. There have been no studies to evaluate memantine for cognition in HD [55]. Amantadine improves channel stability in tight spaces. To achieve symptomatic benefit for chorea, high doses of amantadine were required. Amantadine shown intolerable side effects such as hallucinations, confusion, insomnia, sleepiness, agitation, and anxiety at high dose [56, 57]. Riluzole inhibits NMDARs by acting as a GluN2B antagonist. It inhibits Glu release from corticostriatal terminals (Fig. 11). Riluzole is an approved ligand for the treatment of amyotrophic lateral sclerosis and thus, the target of interest for treating depression. It also binds to voltage-gated sodium channels, thereby prevents the propagation of action potential and the eventual axonal release of Glu [58]. The drug had no effect on chorea, behavioural and cognitive symptoms, or functional independence in a large randomized 3-year study [59].

Riluzole as a GluN2B selective NMDAR antagonist [59]
Ischemic stroke
The blockage of a brain blood vessel causes an ischemic stroke and make-up 87% of all strokes. Stroke is the third most common cause of disability and second most common cause of death. In cerebral ischaemia, the decreased blood flow to brain causes impairment of neuronal functions leading to the neuronal cell death [60]. Specific therapeutic strategies employed to cure ischemic stroke are given as Fig. 12.

Therapeutic strategies employed to cure ischemic stroke
Complex role of NMDARs in cerebral ischemia
Cerebral ischemia is specifically characterised by limited blood flow with reduced amount of oxygen and nutrients needed to maintain the ionic homeostasis. Ionic gradient disruption depolarizes cells and initiating the release of Glu (an excitatory neurotransmitter) into the synaptic cavity. Functional reuptake transporters are unable to remove the excess Glu due to reduced energy associated with cerebral ischemia. Glutamatergic receptors become overactive leading to excitotoxicity and ultimate ischemic brain injury [60, 61]. Thus, excitotoxicity is considered as one of the major pathogenic mechanisms underlying ischemic brain injury.
NMDARs plays a central role in excitotoxic neuronal death caused by ischemic stroke, but NMDAR blockers are not yet translated into clinical stroke treatments. However, NMDAR-associated signalling complexes has been identified as an important death-signalling pathways linked to NMDARs. Thus, development of ligands specifically regulating NMDAR pathways has received an immense attention by neuroscientists to treat cerebral ischaemia.
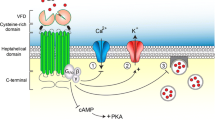
Distinct role of NMDARs in neuronal survival and death
NMDARs are excessively activated in ischemic stroke and initiate toxic cascades that kill neurons. Activation of NMDARs triggers both pro-survival or pro-death signalling pathways depending on subcellular locations or involvement of specific subunits of NMDARs. Stimulation of synaptic NMDAR results into the activation of pro-survival signalling pathways; however, activation of extra-synaptic NMDARs is related to pro-death pathways.
Synaptic NMDAR activation induces the expression of pro-survival genes and suppression of pro-death genes, eventually contributing to pro-survival effects. More specifically, synaptic NMDAR activation causes activation of phosphoinositide-3-kinase (PI3K)/Akt kinase pathway and cAMP-response element binding protein (CREB) dependent gene expression to promote expression of pro-survival genes. CREB induced expression of pro-survival genes triggers the protection of neurons against apoptotic insults. Activated extra-synaptic NMDARs decreases pro-survival signalling mediated by synaptic NMDARs, through dephosphorylation of NMDARs and inactivation of CREB to promote the expression of pro-death genes.
Both GluN2A and GluN2B subunits of NMDARs have shown characteristic role in the ischemic stroke. GluN2A-NMDARs are associated with pro-survival effects, whereas GluN2B-NMDARs are linked to pro-death signalling complexes. Thus, activation of either synaptic or extra-synaptic GluN2B-NMDARs results in excitotoxicity and neuronal apoptosis, whereas activation of GluN2A-NMDARs leads to neuronal survival and neuroprotection against ischemic insults [62,63,64].
NMDAR ligands in ischemic stroke
The non-competitive NMDAR antagonist (MK801) reduces ischemic cell death and prevents the growth of ischemia tolerance. Roles of NMDAR ligands in ischemia cell injury and ischemic tolerance are diverse (Fig. 13). Licostinel functions as a GluN2B antagonist. It is a potent competitive antagonist at the glycine site on the NMDAR. Phase II clinical trial have shown agitation, dizziness, conscious level depression with gastrointestinal upset for the licostinel. Renal toxicity in animals have prevented further clinical development of licostinel [63]. Selfotel is a Glu recognition site competitive antagonist of NMDAR. Stroke and traumatic brain injury trials have been conducted for the antagonist. Selfotel shown side-effects such as agitation, confusion, reduced consciousness level, hallucinations, and a hypertension. Clinical trials were halted due to the increased mortality. Aptiganel is a non-competitive NMDAR antagonist with high ion channel site affinity. Clinical trial was halted due to a higher mortality and inadequate therapeutic outcomes. Gavestinel acts as a competitive antagonist at the glycine site of NMDAR. In the trial, there was no evidence of benefit from gavestinel [64].

GluN2B subunit selective ligands for the treatment of cerebral ischemia
One of the major underlying causes for failure of NMDAR antagonists against cerebral ischemia may be the intricacy of NMDAR signalling. Given the roles of NMDARs in pro-survival and pro-death signalling in neurons, it would be better to develop NMDAR antagonists that selectively block only pro-death effects of NMDARs without disturbing pro-survival pathways. NMDAR antagonists failed in the treatment of cerebral ischemia due to poor tolerance and inadequate therapeutic time window of ligands [64].
In conclusion, GluN2B-NMDARs could be the vital target for treatment of cerebral ischemia. GluN2B-selective antagonists may have a distinct benefit of inhibiting the pro-death effects of NMDAR signalling without affecting the pro-survival functions associated with GluN2A subunit. However, it is challenging to segregate different NMDAR subunits, identify synaptic and extra-synaptic sites to selectively block extra-synaptic GluN2B subunit of NMDARs. Thus, development of GluN2B subunit selective antagonists with broad therapeutic time window would be urgently required for the plausible treatment of cerebral ischemia.
NMDAR inhibitory antibodies as an immunotherapeutic target
Antibody-based immunotherapies have been clinically successful in the treatment of cancer and inflammatory diseases [65, 66]. However, till-date, functional antibodies for the modulation of targets involved in CNS diseases has not been extensively explored. Immunotherapeutic studies are dominated by active or passive immunization approaches used to modify disease progression by targeting proteins implicated in the pathogenesis of neurodegenerative diseases [67]. Very few reports of antibody-based therapies in neurological diseases have been discussed compared to other diseases. An anti-amyloid antibody that targets beta-amyloid in the brain poses an intriguing possibility for delaying the age-of-onset of AD [68].
Tajima et al. identified functional antibodies targeting GluN1-GluN2B NMDAR that allosterically downregulates an ion channel activity [69]. These antibodies downregulate ion channel functions by binding to ATD of NMDAR and stabiles the receptor in non-active conformation. These subunit specific antibodies will act as neurotherapeutics by downregulating the function of NMDARs. However, further re-engineering strategies for tuning the efficacy and potency of inhibition will be required to develop site specific functional antibodies for the treatment of CNS diseases.
Young et al. also provided an overview of immunotherapeutic strategy for the treatment of stroke and epilepsy [70]. GluN1 antibodies have shown neuroprotective and anti-seizure response in rodent models of stroke and epilepsy, respectively. GluN1 immunotherapy boost the neuroprotective capability of endogenous antibodies through modulating NMDAR signalling at neurons and by maintaining the health of blood–brain barrier.
In conclusion, target specific antibodies could also act as scaffolds to deliver therapeutic antibodies into the brain by incorporating one arm with specificity against subunit of NMDAR and other produces the adequate neuropharmacological effect. Detailed understanding of antibody-based therapies coupled with site-specific targeting of NMDAR subunits could be required for the development of novel neurotherapeutics to treat neurological disorders.
Genetic evidence linking NMDAR to neurological diseases
Heterotetrameric NMDAR is assembled from seven different GRIN genes, namely two glycine-binding GluN1 subunits (encoded by GRIN1 gene) combined with two Glu (GRIN2A-D) or glycine binding (GRIN3A-B) subunits [71]. Till date, thousands of different GRIN variants have been discovered in paediatric patients, with majority of mutations concentrated within GRIN2A and GRIN2B genes [72, 73].
Pathogenic variants in GRIN genes causes severe brain disorders as identified in patients with AD, PD, epilepsy, attention deficit hyperactivity disorder, autism spectrum disorder, developmental delay, schizophrenia, and intellectual disability [74]. More specifically, GRIN1 mutations have a significant effect on a neuronal activity leading to focal dyscognitive, myoclonic, febrile, hypermotor, tonic-atonic, and generalized seizures [75]. For instance, Met641Leu de novo variant in GRIN1, and other de novo GRIN1 mutations were identified in severe early infantile spasms and encephalopathies [76]. GRIN2A constitutes a locus for mutations in patients with early onset of convulsions. GRIN2A gene present at human chromosome 16p13 leads to Rolandic epilepsy. Genetic studies also suggested that GRIN2A (GRIN2B was not involved) was a modifier gene for the PD [77, 78]. GRIN2B mutation is a rare cause of severe epileptic encephalopathy [79]. However, GRIN2B gene present at human chromosome 12p13 encoding GluN2B subunit was identified in patients with variable degree of ID, behavioural issues, and EEG abnormalities [80].
Till-date, no specific therapeutic options are available to combat GRIN associated diseases. Nevertheless, in gain-of-function mutations, FDA approved drug memantine (non-competitive NMDAR antagonist) has been extensively explored as a medication for associated neurological diseases [35, 81]. Loss-of-function mutations are harder to treat, as few subunit-selective drugs are helpful as a treatment [82].
Recent human genetic studies of heteromeric NMDAR mutations have shown that the GRIN associated mutations are a recurrent cause of severe and complex neurodevelopmental disorders. These highlights of genetic evidence can provide important clues in curating and functionally characterizing each mutation before treatment can be formulated [83]. Relation of a specific mutation to the alteration of NMDARs function may also provide an avenue to the targeted therapy for the treatment of brain diseases.
Role of GluN2B subunit of NMDAR in CNS diseases
Till date, it remains unclear which NMDAR subunit is critically involved in the synaptic transmission and influx of Ca2+ ions at many synapses. Given this lack of basic information about NMDAR subunits and their role in neurodegenerative diseases, there has been intense activity in developing subunit-selective probes that can be further used to obtain more important information about synaptic receptor identity and functions.
GluN2 subunit shows the differential temporal and spatial distribution in the CNS and thus provide an opportunity to develop region-specific modulators/antagonists based on compounds selective for one or another region of the GluN2 subunit. Diheteromeric NMDARs had multiple binding sites for extracellular small-molecule ligands as subunit-selective allosteric modulators. GluN1/GluN2 heterodimer with regulatory binding sites for small molecules are shown in Fig. 14. These specific sites for binding of allosteric modulators and antagonists will provide an idea to develop a subunit selective ligand that holds strong neurotherapeutic potential.

Despite of the potential to develop probes that can modulate NMDARs in specific brain structures afforded by GluN2 subunits, very few advances in developing subunit-selective antagonists or modulators have occurred between 2010 and 2021 [6, 7, 9].
GluN2B selective ligands
Discovery of an extensive class of GluN2B selective antagonists (prototypical analog ifenprodil, (1) has allowed a detailed exploration of a role of the GluN2B subunit in neurological indications [84, 85]. However, off-target effects of ligands in clinical testing have hindered approval for their human use (Fig. 15 [84,85,86,87,88,89]).

It has been accepted that the modulation/blockade of GluN2B/NMDARs has significant relevance for its clinical manifestations in several CNS diseases. Among NMDAR subunits, the GluN2B subunit has been more focused on basic and clinical neuroscience research over the past two decades, producing an overwhelming body of evidence that affects NMDAR and is effective in the prevention of adverse neurological symptoms in various models of neurological and neurodegenerative diseases.
Recent progress in the development of GluN2B selective ligands
In this review, we focused on GluN2B/NMDAR-targeted compounds and provided a brief overview of potent compounds synthesized, explored, and reported in recent years as GluN2B/NMDAR modulators or antagonists. The collected data could be helpful to yield therapeutically relevant insights into neurological diseases.
Irvine et al. (2019) designed and synthesized a series of novel naphthalene derivatives as NMDAR modulators [90]. Herein, we have outlined and discussed the effects of most active analogs from series of synthesized compounds (Fig. 16). 6-n-pentyl (10) and 6-isohexyl (11) 2-naphthoic acid derivatives have shown potentiating effects on specific subunits of NMDARs. Compounds 11 and 13 showed convincing positive allosteric modulatory activity at GluN2B subunit. Compound 10 enhanced NMDAR mediated cationic currents at diheteromeric NMDAR consisting of GluN1/GluN2A-D subunit combinations. Addition of 3-cyclopentylprop-1-yl at 6th position of naphthalene gave compound 12; led to a similar level of potentiating activity on GluN2A-D subunits when compared to compound 11, with the previous being more potent on GluN2A/NMDAR. In conclusion, these analogs will be helpful as a neurotherapeutic tool to study synaptic functions and could be used as a potential lead for the development of future drug candidate to treat schizophrenia and other CNS disorders (Fig. 16).

Naphthalene analogs as GluN2B selective NMDAR modulators [90]
Novel phenanthrenes were reported by Kudova et al. (2015) as NMDAR antagonists [91]. Here, the structure–activity relationship of pregnenolone sulfate analogs with structural modifications on steroidal D-ring has been described. For discussion, only potent NMDAR antagonists from reported analogs have been considered. When compared alkyl modifications on D-ring to acetyl moiety of pregnenolone sulfate; it was indicated that these modifications caused a decrease in IC50 at NMDARs. On the other hand, when 17-acetyl moiety of pregnanolone sulfate was replaced with hydrogen; more effective NMDAR inhibitors were produced. Comparison of IC50s of active compounds 14 (C17-ethyl, IC50 = 0.4 ± 0.1 μM), 15 (C17-isopropyl, IC50 = 0.16 ± 0.02 μM), 17 (C17-methyl, IC50 = 0.6 ± 0.1 μM), and 18 (C17-isobutyl, IC50 = 0.09 ± 0.01 μM) displayed admirable changes in their activities with respect to pregnanolone sulfate. In conclusion, the replacement of pregnanolone acetyl moiety with an alkyl substituent has a suboptimal effect on the ability of neurosteroids as inhibitors of NMDAR-induced cationic currents (Fig. 17).

Phenanthrene analogs as GluN1/GluN2B selective NMDAR antagonists [91]
Slavikova et al. (2016) developed and reported phenanthrenes as NMDAR antagonists [92]. Perhydrophenanthrene analogs with fully saturated phenanthrene skeleton analogous to the steroidal system were developed and then screened against NMDAR for possible activity. A preclinical assay was used to test the potential of these compounds to modify NMDAR cationic currents. These compounds were then screened selectively against recombinant GluN1/GluN2B subunits of NMDARs. In SAR studies, it was found that diverse polar substituents can entirely or partially degrade steroidal D-ring with the preservation of NMDAR antagonistic activity. Compound 27 was the most potent modulator of NMDAR from the library of perhydrophenanthrene analogs. An analogue (27) was also effective against GABA receptor. However, the exact role of synthesized molecules to minimize undesired psychomimetic side effects through GABA inhibition was not described. Thus, the steroidal D-ring is considered as a vital pharmacophore for both NMDAR/GABA selectivity and open-up the possibility of developing more receptor-specific neurotropic steroids as neurotherapeutics (Fig. 18).

Pregnanolone sulfate analogues as GluN1/GluN2B selective NMDAR antagonists [92]
Dey et al. (2018) reported 2-methyltetrahydro-3-benzazepin-1-ol as GluN2B selective NMDAR antagonists [93]. 2-methyltetrahydro-3-benzazepin-2-ols bind at the GluN1-1a/GluN2B interface like co-crystallized ligand (Ifenprodil). As seen in docking studies, phenyl moiety and 3-benzazepine were in the hydrophobic region of GluN1-1a/GluN2B NMDARs. Protonated amino and hydroxy groups of (R, R) cis-31 created two H-bonds with CONH2 moiety of Gln110 at GluN2B subunit. When compared to GluN2B affinity of (R, R) 29, the 4-phenylbutyl derivative (cis-31) has shown reduced affinity demonstrating the importance of a phenolic-OH moiety. The introduction of methyl moiety at the 2nd position also showed a reduction in GluN2B affinity (cis-31). However, reduced GluN2B affinity was compensated by homologation of 4-phenylbutyl (cis-31) to 5-phenylpentyl side chain (cis-32, Ki = 56 nM). In conclusion, an extension of the side chain was main contributing factor for high affinity as the 5-phenylpentyl group increased GluN2B affinity of cis-32 by 5-fold (Fig. 19).

2-Methyltetrahydro-3-benzazepin-1-ol as GluN2B/NMDAR antagonists [93]
Tewes et al. (2015) synthesized four stereoisomeric 2-methyl-3-(4-phenylbutyl)tetrahydro-3-benzazepin-1-ols using multistep synthesis [94]. Among these derivatives, phenols exhibited higher GluN2B affinity than methyl ethers (Fig. 20). GluN2B affinity increased in the following order: (1R, 2S) < (1S, 2S) < (1S, 2R) < (1R, 2R). Stereoisomeric phenols (R, R)-43 and (S, R)-44 have shown the highest GluN2B affinity with excellent cytoprotective activity. Results proved the potential of these compounds as GluN2B selective NMDARantagonists. Molecular docking of 3-benzazepin-1-ols also supported the experimentally determined GluN2B affinities, as these compounds have shown prominent binding at N-terminal domain of GluN1-1b/GluN2B subunits. Benzyl ethers have shown high GluN2B binding affinity without cytoprotective effects. Reason for the high affinity of benzyl ethers at the GluN2B subunit was its binding at the lipophilic sub-pocket close to the ifenprodil binding site of NMDAR. These findings will be considered for the future development of GluN2B selective NMDAR antagonists.

Stereoisomeric 2-methyl-3-(4-phenylbutyl)tetrahydro-3-benzazepin-1-ols as GluN2B selective NMDAR antagonists [94]
Dey et al. (2016) designed and synthesized 3-benzazepines as GluN2B selective NMDAR antagonists [95]. The deconstruction approach analysed importance of phenolic-OH moiety for affinity and selectivity at GluN2B subunit. Phenolic-OH of 3-benzazepine-1,7-diol (46) was removed to obtain 3-benzazepin-1-ol (47) without other substituents at aromatic ring (Fig. 21). Insertion of N-triflyl protecting group was required to produce 3-benzazepin-1-ol (47). It was shown that the 3-benzazepin-1-ol (47) without phenolic-OH also interact with GluN2B subunit. However, the analogue has shown 5-fold reduced affinity compared to a phenol (46). Pharmacophore-based docking studies were performed to analyse the interactions of 3-benzazepin-1-ol (47) in the receptor binding pocket of GluN2B/NMDARs. Docking studies revealed the same binding pose with similar H-bond and hydrophobic interactions for docked 3-benzazepin-1-ol (47) compared to a crystallized keto-ifenprodil. In docking studies, the 3-benzazepin-1-ol (47) adopted binding mode analogous to a co-crystallized ligand. Computational data supported the affinity of outlined analogues, as phenyl ring of compound (47) was embedded in hydrophobic region of NMDAR through hydrophobic interactions with Tyr109 (GluN1) and Ile111 (GluN2B); protonated benzazepine also formed H-bond with >C=O group of Gln110 (GluN2B), like the protonated piperidine of a crystallized structure.

3-Benzazepin-1-ol as GluN2B selective NMDAR antagonists [95]
Benzazepine derivatives were structurally modified and synthesized by Dey et al. to target GluN2B subunits of NMDAR [96]. These compounds were screened for their abilities as selective NMDAR antagonists. Outlined compounds without substitutions at aromatic ring have exhibited very high GluN2B affinities. Characteristic GluN2B affinity of compounds 48 (Ki = 2.2 nM) and 49 (Ki = 6.0 nM) led to the conclusion that the interaction of phenolic-OH moiety was not essential for better GluN2B affinity. Phenylcyclohexyl derivative was found to be most effective GluN2B/NMDAR antagonist. 3-benzazepin-1-ol (48) with conformationally restricted phenylcyclohexyl substituent represented as effective GluN2B ligand with higher selectivity over non-NMDARs [PCP binding site (> 4500-fold), σ1 (37-fold), and σ2 receptors (25-fold)]. Eventually, the replacement of phenylcyclohexyl by benzylpiperidin-4-yl moiety (49) resulted into slightly decreased GluN2B affinity (Ki = 6 nM), but significantly reduced selectivity over σ1 and σ2 receptors (Fig. 22).

Benzazepines as GluN2B selective NMDAR antagonists [96]
Rath et al. (2017) synthesized 3-benzazepines as subunit selective GluN2B/NMDAR antagonists and determined their affinities using in vitro assays [97]. Potent methyl ethers (52 and 54) from series have shown high GluN2B affinity (52, Ki = 472 nM and 54, Ki = 606 nM). Replacement of methoxy group by hydroxyl substituent resulted into 3-fold increase in GluN2B affinity of phenol (53, Ki = 157 nM). Given structural modification was justified as potent compounds have shown higher GluN2B affinity than unmodified methyl ether (50). Moreover, compound containing hydroxymethyl side chain have shown analogous GluN2B affinity (54, Ki = 606 nM) compared to the lead analogue (50, Ki = 706 nM). Alteration in the phenolic-OH position and benzylic-OH replacement by vinyl group was well tolerated by GluN2B subunit with slight reduction of affinity (Fig. 23).

Structurally modified 3-benzazepines as GluN2B selective NMDAR antagonists [97]
Borgel et al. (2019) investigated pharmacokinetic parameters of enantiomerically pure GluN2B/NMDAR antagonists [98]. Log D values of these analogues were in the good range for penetration of blood–brain barrier to reach CNS [55 (Log D = 1.68), methyl ether (56, Log D = 2.46) and benzyl ether (57, Log D = 4.14)]. Benzyl ether (57) has shown the highest Log D value. Moreover, Log D value for well-known GluN2B antagonist (Ifenprodil) was described for the first time (Log D = 1.49). In conclusion, these compounds were proved as active GluN2B/NMDAR antagonists (Fig. 24).

Log D values for benzazepines effective as GluN2B selective NMDAR antagonists [98]
Ahmed et al. (2019) synthesized N-methyl-N-(3-phenylpropyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine (58) and N-(3-(4-fluorophenyl)propyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine (59) as plausible GluN2B active ligands. Later, these compounds were investigated for binding affinities towards GluN2B subunits and selectivity over non-NMDARs [99]. A robust radiosynthesis was established to afford [18F] (59) with moderate radiochemical yields, high molar activities, and excellent radiochemical purities. Although 6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amines showed high affinity and selectivity, these compounds lacked prominent polar features that would be required to label them as potential GluN2B/NMDAR imaging agents. These findings led to conclusion that the reported class of compounds would require further structural modifications to develop more efficacious GluN2B imaging agents (Fig. 25).

N-methyl-N-(3-phenylpropyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine (58) and N-(3-(4-fluorophenyl)propyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-7-amine (59) as GluN2B selective NMDAR antagonists [99]
3-Benzazepines with 4-phenylbutyl substituents at 3rd position bearing F-atom (in either β or γ-position) were synthesized by Thum et al. [100]. An impact of fluorine and substitution pattern on affinity and selectivity at GluN2B/NMDAR was evaluated using pharmacological assays. Phenolic-3-benzazepine (60) with fluorine atom at γ-position exhibited promising GluN2B affinity (Ki = 16 nM). Thus, the substitution of F-atom was an essential feature to increase affinity of these analogues as selective NMDAR antagonists (Fig. 26).

Phenolic-3-benzazepine (60) as GluN2B/NMDAR antagonists [100]
Wagner et al. (2019) designed and synthesized benzazepine derivatives as selective GluN2B/NMDAR antagonists [101]. Here, phenylbutyl substituent of potent GluN2B antagonist (61) was modified to uncover high GluN2B affinity fluorinated ligands. Modification was tried to develop fluorinated PET tracers for imaging of GluN2B subunits. Introduction of 2-fluoroethoxy moiety at ortho, meta, and/or para positions of terminal phenyl group (62) led to the considerably reduced affinity towards prominent ifenprodil binding site of GluN2B/NMDARs. From reported compounds, phenol with p-fluoroethoxy moiety at terminal phenyl ring was presented as effective GluN2B probe to treat CNS disorders (Fig. 27).

Structurally modified benzazepines as GluN2B selective NMDAR antagonists [101]
Lutnant et al. (2016) synthesized bioisosteric benzimidazolones as potent GluN2B/NMDAR antagonists [102]. Benzimidazolone was unsuitable bioisostere of phenol or methyl ether to become potential GluN2B antagonists. Tricyclic 7-annulenobenzimidazolone system was too rigid to induce a fit at ifenprodil binding site of NMDAR. However, the tricyclic amine (63) opened-up the possibility for several modifications at various ring positions of the skeleton structure. From series of tricyclic [7]-annulenobenzimidazolones, compound 63 (Ki = 697 nM) showed adequate binding affinity at GluN2B subunit of NMDARs. Additionally, the compound 63 (Ki = 549 nM) interact with σ2 receptors but with poor selectivity. These facts suggested that compound 63 will be utilized as lead for the development of selective GluN2B antagonists (Fig. 28).

Bioisosterically modified benzimidazolone (63) as GluN2B selective NMDAR antagonists [102]
Benzo[7]annulenes were structurally modified by Gawaskar et al. and screened to uncover their abilities as GluN2B selective NMDAR antagonists [103]. Benzo[7]annulen-7-amines were designed and synthesized through conformational restriction of potent GluN2B ligand [Ro 25-6981 (8)] and through removal of polar hydroxyl group. Structurally modified compounds have shown moderate GluN2B affinities (16–57 nM). Results revealed that polar substituents on Ro 25-6981 (8) were not essential for strong interaction with GluN2B/NMDARs. However, an insertion of nitro group at 2nd position of benzo[7]annulene scaffold increased GluN2B affinity up to 5–10-fold compared to unsubstituted derivatives (Fig. 29). Phenylpropylamine analogue [65 (Ki = 1.6 nM)] was proved as potent a GluN2B selective ligand. Docking studies also revealed that the phenylpropylamine (65) have shown additional H-bonding between protonated central amino group (65) and carbamoyl moiety of amino acid (Gln110) in ifenprodil binding pocket. Nitro analogues were able to bind at ifenprodil binding site with analogous orientation to that of co-crystallized ligand that may be the reason for high binding affinity of synthesized ligands.

Benzo[7]annulenes as GluN2B selective NMDAR antagonists [103]
To prove the hypothesis of beneficial effects of electron deficient aromatic systems on the GluN2B affinity, a set of pyridine annulenes were synthesized by Zscherp et al. [104]. Bioisosteric modification and replacement of nitrobenzene in compound 66 by pyridine (67) led to 200-fold reduced GluN2B affinity. These results proved that the high GluN2B affinity of compound 66 was only due to the electron withdrawing nitro group. Reduced GluN2B affinity of pyridine derivatives indicated that the pyridine ring was not an appropriate bioisosteric replacement for chlorobenzene and nitrobenzene. High affinity of compound 66 at GluN2B subunit was due to polar interactions of NO2 group with GluN2B/NMDAR. Active analogue (66) established H-bond network with the conserved water molecule in binding pocket of NMDAR; the interaction was responsible for high GluN2B affinity (Fig. 30).

Pyridine annulene (67) as GluN2B selective NMDAR antagonists [104]
Fluorinated benzo[7]annulen-7-amines were designed and synthesized as selective GluN2B/NMDAR antagonists by Thum et al. [105]. Diverse substituted benzo[7]annulen-7-amines were then utilized to develop structure–affinity relationships of GluN2B antagonists. Reported compounds have various fluorinated and nonfluorinated spacers with diverse substituents at amino group of benzo[7]annulen-7-amines. Results supported the addition of fluorine atom at the side chain to develop selective GluN2B/NMDAR antagonists. Methyl-ether with β-fluorinated phenylpropyl side chain (69, Ki = 9.2 µM) and bromo analogue with γ-fluorinated phenylbutyl chain (68, Ki = 14 µM) were found to be promising GluN2B ligands from series of synthesized compounds (Fig. 31).

Fluorinated benzo[7]annulen-7-amines as GluN2B selective NMDAR antagonists [105]
Gawaskar et al. (2017) designed and synthesized selective NMDAR antagonists utilizing structure-based drug design strategy [106]. When focused on a potent analogue, 2-chloroderivative (71) was recognised as one of the most promising ligands from reported compounds. Compound 71 has high affinity (Ki = 2.1 nM) and well supported by docking studies. A strong halogen bond interaction between compound 71 (Ki = 2.1 nM) and proximal amino acid Glu236; may be the reason behind affinity of outlined compound. Unexpectedly, the OBn derivative (72, Ki = 3.6 nm) also showed high GluN2B affinity in neuropharmacological assay and was able to occupy pocket of GluN2B/NMDAR through flexible benzyloxy substituent. Phenyl ring of benzyloxy group interact with positively charged guanidine of Arg115 at GluN2B subunit. Thus, the compound 72 was highly selective towards GluN2B subunit over σ1, σ2 receptors and PCP binding site (Fig. 32).

Potent benzo[7]annulen-7-amines as GluN2B selective NMDAR antagonists [106]
GluN2B/NMDAR is an effective target for chronic pain due to its involvement in the disease and distribution in CNS [107]. Anan and co-workers identified cyclohexanol-based compounds (73; IC50 = 7.0 nM and 74; IC50 = 7.1 nM) as selective GluN2B/NMDAR antagonists (Fig. 33). A combination of scaffold hopping approach, bioisosteric replacement, and optimization of pharmacokinetic profile resulted in identification of cyclohexanol-based compounds as effective GluN2B/NMDAR antagonists. These compounds were further optimized by controlling lipophilicity and screened to develop structure–activity relationship as GluN2B selective antagonists. Compounds 73 and 74 also demonstrated excellent analgesic activity in mouse formalin test at both early and late phases.

Cyclohexanols as GluN2B selective NMDAR antagonists [107]
Negative allosteric modulators (NAMs) of GluN2B/NMDARs have proved to be a potential pharmacological tool for treating chronic neurodegenerative diseases [108]. Buemi et al. designed and synthesized NAMs through computational studies and obtained effective GluN2B probes. Series of indoles utilizing lead compound (75, IC50 = 25 nM) were synthesized and further tested for efficacies through competition assay at GluN2B subunit of NMDAR. Compounds 76 (IC50 = 83 nM) and 77 (IC50 = 71 nM) were observed as moderately effective NAMs when compared to lead compound 75 (Fig. 34). Additionally, molecular docking studies have been executed for active compounds (75 and 76); results agreed with the biological data. Lead compounds 75 and 76 have shown contacts at GluN1/GluN2B ligand binding site of NMDARs. Presented results will be useful to design novel NAMs targeting at interface of GluN1/GluN2B subunit (Fig. 34).

Indoles as GluN2B selective NMDAR antagonists [108]
Temme et al. (2018) designed and developed benzo[7]annulene derivatives as an GluN2B/NMDAR antagonists [109]. Modification of reported GluN2B/NMDAR antagonists through replacing phenolic hydroxyl group (78) with hydroxymethyl moiety (79) resulted in the metabolically stable NAMs of GluN2B subunit. Phenolic-OH and hydroxymethyl moieties acts as H-bond donors making them more hydrophilic; but hydroxymethyl group was less prone to glucuronidation and sulfation during biotransformation in liver resulting metabolically more stable compounds.
Here, we have discussed most active analogue (79) from synthesized compounds by Temme et al. Previously reported phenol with phenylpropyl side chain (78; Ki = 21 nM) has exhibited lower GluN2B affinity and cytoprotective activity than phenylbutyl homolog (78a; Ki = 28 nM). Modified hydroxymethyl derivative have shown good affinity at GluN2B subunits (79, Ki = 101 nM) with high selectivity over other non-NMDAR binding sites. Compound 79 has 10-fold lower cytoprotective activity when compared to ifenprodil (Fig. 35). Thus, phenylpropyl derivative (79) with hydroxymethyl moiety represented as more selective GluN2B antagonist. Molecular docking also revealed that the H-bond between Glu236 and hydroxymethyl moiety (79) was responsible for stabilizing the closed NMDAR conformation.

Benzo[7]annulene as GluN2B selective NMDAR antagonists [109]
Temme et al. (2020) synthesized compounds bearing 3-benzazocines, tetralinamines and indanamines as selective NMDAR antagonists [110]. Eight-membered 3-benzazocine ring was helpful to increase GluN2B/NMDAR affinity as indicated by compound 80 (Ki = 32 nM). 3-Benzazocine analogue (80) was identified as most active GluN2B selective antagonist with moderate cytoprotective activity (IC50 = 0.89 µM). Compound (80) has also shown Ca2+ current inhibition (60% at 10 mM) in electrophysiological recordings. An indanamine derivative also shown good affinity at GluN2B subunit (81, Ki = 3.2 nM) with cytoprotective activity. Results indicated that phenyl substituents around ring-contracted tetralinamines (81a, Ki = 8.4 nM) and indanamines (81, Ki = 3.2 nM) were favourable for higher GluN2B affinity (Fig. 36).

3-Benzazocine (80), tetralinamine (81a) and indanamine (81) as GluN2B selective NMDAR antagonists [110]
Krausova et al. (2018) designed and developed novel GluN2B/NMDAR modulators [111]. Herein, library of compounds bearing C-3 hemiester moiety with Δ5- double bond was utilized to generate novel selective antagonists. Analogues formed by various modifications at position C-17 were evaluated for their abilities to modulate functional activity against NMDAR subunits. Results indicated that the structural modification of C-17 substituent of D-ring maintained positive modulatory effect against NMDARs. However, potentiating effect of these compounds exhibited dependency on length of C-3 substituents for each D-ring modification. These compounds were proved as positive modulators of recombinant GluN1/GluN2B subunits.
Compound 83 (EC50 = 7.4 uM) and 84 (EC50 = 1.8 μM) were considered as the most efficacious and potent modulator from reported compounds. In addition, compound 82 has a subunit-independent effect at recombinant NMDARs and has only minor inhibitory effect at non-NMDARs. These analogues will be modified at C-3 and/or at D-ring for further optimization of neuropharmacological properties to produce novel neuroactive compounds (Fig. 37).

Steroidal 3-hemiesters as GluN2B selective NMDAR antagonists [111]
Strong et al. (2017) designed and developed novel tetrahydroisoquinolines as positive allosteric NMDAR modulators derived from known class of GluN2C/D-selective tetrahydroisoquinoline analogues [112, 113]. Prototypical positive allosteric modulator (85, EC50 = 300 nm) was highly selective for GluN2C and GluN2D subunits than other NMDAR subunits.
Compounds reported were active at GluN2B, GluN2C, and GluN2D subunits of NMDAR. These compounds have shown distinct structural-activity relationship based on structural differences around isopropoxy functionality. An introduction of isopropoxy group and its modifications resulted into enhanced activity at GluN2B subunits (Fig. 38).

Modified analogues of tetrahydroisoquinolines [113]
Two important substituents as a methoxy group at meta position and an ethoxy group at para position (B-ring) were highlighted in Fig. 38. Changes to compound 86 (GluN2B; EC50 = 5.2 µM) have shown different effects on NMDAR potentiation than 85. These two functional groups were inactive when introduced at 85, however compound 87 (GluN2B subunit; EC50 = 5.3 µM) was only active at GluN2B subunit in presence of isopropoxy group on C-ring. These results suggested that the isopropoxy group (86) was able to revive potential at NMDAR that was absent in previous compound (85). Although, thioamide was inactive on compound 85 but it was essential for potent activity of tetrahydroisoquinoline with isopropoxy group [86; EC50 = 5.2 µM, Fig. 39]. S-(−)-enantiomer of compound 88 (GluN2B subunit; EC50 = 7.2 µM) was separated and shown activity only at GluN2B subunit. When S-(−)-enantiomer (88) was converted to thioamide (89); only S-(−)-89 was active at GluN2B subunit (EC50 = 0.55 µM), whereas R-(+)-89 was inactive at all NMDAR subunits.

Tetrahydroisoquinolines as GluN2B subunit selective positive allosteric modulators [113]
In conclusion, only compounds 88 and 89 have shown highest degree of selectivity and efficacy at GluN2B compared to other NMDAR subtypes. These results provided starting point for the development of selective positive allosteric modulators of GluN2B subunits.
Baumeister et al. (2019) synthesized and developed [7]annulene[b]thiophen-6-amine derivatives as NMDAR antagonists [114]. Bioisosteric replacement of benzene, methoxybenzene and aniline moieties of available potent GluN2B is selectively antagonized by the thiophene ring. The pharmacological data obtained for thiophene derivatives [(89) and (90)] indicated that the bioisosteric replacement of GluN2B ligands by using thiophene was well tolerated. [7]Annulenothiophene (89) without benzylic OH moiety revealed 8-fold higher GluN2B affinity (Ki = 26 nM) than with benzylic OH (90, Ki = 204 nM). Thiophene bioisostere showed preference to GluN2B subunit over σ receptors. However, overall data suggested that the benzylic-OH moiety does not seem to be essential for higher GluN2B subunit affinity (Fig. 40).

[7]Annulene[b]thiophen-6-amines GluN2B selective NMDAR antagonists [114]
Baumeister et al. (2020) modified and synthesized [7]annuleno[b]thiophenes as GluN2B/NMDAR antagonists [115]. Higher GluN2B affinity was achieved with [7]annuleno[b]thiophenes with 3-phenylpropylamino (92, Ki = 6 nM) or 4-phenylbutylamino moieties (93, Ki = 9 nM; Fig. 41). Unfortunately, these structural modifications have shown high GluN2B affinity and slight σ receptor affinity. Preferred interaction with the ifenprodil binding site of GluN2B/NMDARs could only be observed in case of methylamines. Among the series of homologous secondary amines, the 4-phenylbutylamine has shown highest GluN2B affinity which is analogous to ifenprodil (Ki = 10 nM) and eliprodil (Ki = 13 nM).

[7]Annuleno[b]thiophenes GluN2B selective antagonists [115]
Conclusion
NMDARs are diverse in subunit composition, biophysical and pharmacological properties, interacting partners and subcellular localization. Subunit composition varies across CNS regions during neuronal development and neurological disease states. At fully mature synapses, NMDAR subunit contents depend on the neuronal activity.
Here, we have collected and described the recent data of potential selective ligands of GluN2B subunit of NMDARs. Arranged comprehension of distinct roles of GluN2B selective ligands is beneficial for both medicinal chemists and neuroscientists to define new strategies to counteract the deleterious effects of deregulated NMDAR functions.
Information presented in this review will be obliging to understand the structure–function relationship of potential GluN2B subunit selective ligands that will advance understanding of many aspects of brain function at molecular, cellular, and systems level and provide new treatment modalities. Moreover, taking stock of these subunit selective compounds, as can and has been done previously [17, 18], reminds us that the future is bright and that the persistent, thoughtful, creative, and collaborative efforts stand an excellent chance of solving the most stubborn and intractable neurological problems.
References
Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol 50:295–322. https://doi.org/10.1146/annurev.pharmtox.011008.145533
Bleakman D, Alt A, Nisenbaum ES (2006) Glutamate receptors and pain. Semin Cell Dev Biol 17:592–604. https://doi.org/10.1016/j.semcdb.2006.10.008
Ribeiro FM, Vieira LB, Pires RG, Olmo P, Ferguson SS (2017) Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res 115:179–191. https://doi.org/10.1016/j.phrs.2016.11.013
Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity, and disease. Nat Rev Neurosci 14:383–400. https://doi.org/10.1038/nrn3504
Horak M, Holubova K, Nepovimova E, Krusek J, Kaniakova M, Korabecny J, Vyklicky L, Kuca K, Stuchlik AJ, Soukup O (2017) The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog Neuropsychopharmacol Biol Psychiatry 75:54–62. https://doi.org/10.1016/j.pnpbp.2017.01.003
Hansen KB, Wollmuth LP, Bowie D, Furukawa H, Menniti FS, Sobolevsky AI, Swanson GT, Swanger SA, Greger IH, Nakagawa T, McBain CJ (2021) Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol Rev 73:298–487. https://doi.org/10.1124/pharmrev.120.000131
Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. https://doi.org/10.1124/pr.109.002451
Skrenkova K, Hemelikova K, Kortus S, Kaniakova M, Krausova B, Horak M (2019) Structural features in the glycine-binding sites of the GluN1 and GluN3A subunits regulate the surface delivery of NMDA receptors. Sci Rep 9:1–6. https://doi.org/10.1038/s41598-019-48845-3
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–62
Li L, Zhou Q (2015) GluN2B-NMDA receptors in Alzheimer’s disease: what do they got to do with AD? J Neurol Disord 3:1–3. https://doi.org/10.4172/2329-6895.1000e118
Ahmed H, Haider A, Ametamey SM (2020) N-methyl-d-aspartate receptor modulators: a patent review (2015–present). Expert Opin Ther Pat 30:743–767. https://doi.org/10.1080/13543776.2020.1811234
Jewett BE, Thapa B (2021) Physiology, NMDA receptors. StatPearls Publishing, Treasure Island
Ruppa KB, King DR, Olson E (2012) NMDA antagonists of GluN2B subtype and modulators of GluN2A, GluN2C, and GluN2D subtypes—recent results and developments. In: Annual reports in medicinal chemistry. Elsevier, Inc., pp 89–103. https://doi.org/10.1016/B978-0-12-396492-2.00007-2
Strong L, Jing Y, Prosser AR, Traynelis SF, Liotta DC (2014) NMDA receptor modulators: an updated patent review (2013–2014). Expert Opin Ther Pat 24:1349–1366. https://doi.org/10.1517/13543776.2014.972938
Vyklicky V, Korinek M, Smejkalova T, Balik A, Krausova B, Kaniakova M, Lichnerova K, Cerny J, Krusek J, Dittert I, Horak M, Vyklicky L (2014) Structure, function, and pharmacology of NMDA receptor channels. Physiol Res 63:191–203. https://doi.org/10.33549/physiolres.932678
Liu W, Jiang X, Zu Y, Yang Y, Liu Y, Sun X, Xu Z, Ding H, Zhao Q (2020) A comprehensive description of GluN2B-selective N-methyl-d-aspartate (NMDA) receptor antagonists. Eur J Med Chem 200:112447. https://doi.org/10.1016/j.ejmech.2020.112447
Ugale VG, Wani R, Khadse SC, Bari SB (2020) NMDA receptor antagonists: emerging drugs to treat neurodegenerative diseases. In: Torrens F, Mahapatra DK, Haghi AK (eds) Biochemistry, biophysics, and molecular chemistry applied research and interactions, Taylor & Francis, pp 136–174. https://doi.org/10.1201/9780429284175
Ugale VG, Dhote A, Narwade R, Khadse SC, Reddy PN, Shirkhedkar A (2021) GluN2B/N-methyl-d-aspartate receptor antagonists: advances in design, synthesis, and pharmacological evaluation studies. CNS Neurol Disord Drug Targets 20:822–862. https://doi.org/10.2174/1871527320666210309141627
Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC (2012) Novel NMDA receptor modulators: an update. Expert Opin Ther Pat 22:1337–1352. https://doi.org/10.1517/13543776.2012.728587
Hoyte LP, Barber A, Buchan AM, Hill MD (2004) The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med 4:131–136. https://doi.org/10.2174/1566524043479248
Ghasemi M, Schachter SC (2011) The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav 22:617–640. https://doi.org/10.1016/j.yebeh.2011.07.024
Ugale VG, Bari SB, Khadse SC, Reddy PN, Bonde CG, Chaudhari PJ (2020) Exploring quinazolinones as anticonvulsants by molecular fragmentation approach: structural optimization, synthesis and pharmacological evaluation studies. ChemistrySelect 5:2902–2912. https://doi.org/10.1002/slct.201904776
Ugale VG, Bari SB (2016) Structural exploration of quinazolin-4(3H)-ones as anticonvulsants: rational design, synthesis, pharmacological evaluation, and molecular docking studies. Arch Pharm 349:864–880. https://doi.org/10.1002/ardp.201600218
Nicoll RA, Roche KW (2013) Long-term potentiation: peeling the onion. Neuropharmacology 74:18–22. https://doi.org/10.1016/j.neuropharm.2013.02.010
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–366. https://doi.org/10.1126/science.1072994
Omar SH, Scott CJ, Hamlin AS, Obied HK (2017) The protective role of plant biophenols in mechanisms of Alzheimer’s disease. J Nutr Biochem 47:1–20. https://doi.org/10.1016/j.jnutbio.2017.02.016
Sanz-Clemente A, Nicoll RA, Roche KW (2013) Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19(1):62–75. https://doi.org/10.1177/1073858411435129
Regan MC, Romero-Hernandez A, Furukawa H (2015) A structural biology perspective on NMDA receptor pharmacology and function. Curr Opin Struct Biol 33:68–75. https://doi.org/10.1016/j.sbi.2015.07.012
Yu A, Lau AY (2018) Glutamate and glycine binding to the NMDA receptor. Structure 26:1035–1043. https://doi.org/10.1016/j.str.2018.05.004
Vieira M, Yong XLH, Roche KW, Anggono V (2020) Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J Neurochem 154(2):121–143. https://doi.org/10.1111/jnc.14970
Kumar A (2015) NMDA receptor function during senescence: implication on cognitive performance. Front Neurosci 9:473. https://doi.org/10.3389/fnins.2015.00473
Palop JJ, Mucke L (2010) Amyloid-Β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci 13(7):812–818. https://doi.org/10.1038/nn.2583
Deture MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14(1):32. https://doi.org/10.1186/s13024-019-0333-5
Mota SI, Ferreira IL, Rego AC (2014) Dysfunctional synapse in Alzheimer’s disease—a focus on NMDA receptors. Neuropharmacology 37:1039–1047. https://doi.org/10.1016/j.neuropharm.2013.08.013
Zoladz PR, Campbell AM, Park CR, Schaefer D, Danysz W, Diamond DM (2006) Enhancement of long-term spatial memory in adult rats by the noncompetitive NMDA receptor antagonists, memantine and neramexane. Pharmacol Biochem Behav 85(2):298–306. https://doi.org/10.1016/j.pbb.2006.08.011
Hanschmann A, Althaus M, Gebauer A, Parsons C, Danysz W (2010) Merz Pharma GmbH and Co KGaA, assignee. Novel combination of neramexane for treatment of neurodegenerative disorders. United States Patent Application US 12/452771, 29 July
Sahoo AK, Dandapat J, Dash UC, Kanhar S (2018) Features and outcomes of drugs for combination therapy as multi-targets strategy to combat Alzheimer’s disease. J Ethnopharmacol 215:42–73. https://doi.org/10.1016/j.jep.2017.12.015
Hanaya R, Arita K (2016) The new antiepileptic drugs: their neuropharmacology and clinical indications. Neurol Med Chir 56(5):205–220. https://doi.org/10.2176/nmc.ra.2015-0344
Barker-Haliski M, White HS (2015) Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med 5(8):a022863. https://doi.org/10.1101/cshperspect.a022863
Davies JA (1995) Mechanisms of action of antiepileptic drugs. Seizure 4(4):267–271. https://doi.org/10.1016/S1059-1311(95)80003-4
Wang JX, Furukawa H (2019) Dissecting diverse functions of NMDA receptors by structural biology. Curr Opin Struct Biol 54:34–42. https://doi.org/10.1016/j.sbi.2018.12.009
French J, Smith M, Faught E, Brown L (1999) Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 52(8):1540–1545. https://doi.org/10.1212/wnl.52.8.1540
Schachter SC, Tarsy D (2000) Remacemide: current status and clinical applications. Expert Opin Investig Drugs 9(4):871–833. https://doi.org/10.1517/13543784.9.4.871
Golan DE, Armstrong EJ, Armstrong AW (2017) Principles of pharmacology: the pathophysiologic basis of drug therapy, 4th edn. Wolters Kluwer, Philadelphia, pp 698–700
Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray Chaudhuri K, Weintraub D (2021) Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers 7(1):47. https://doi.org/10.1038/s41572-021-00280-3
Willis AW (2013) Parkinson disease in the elderly adult. Mol Med 110:406–410
Jankovic J, Aguilar LG (2008) Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr Dis Treat 4(4):743–757. https://doi.org/10.2147/ndt.s2006
Lau CG, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Murphy J, Bennett MVI, Zukin RS (2009) Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem Soc Trans 37:1369–1374. https://doi.org/10.1042/BST0371369
Villmann C, Becker CM (2007) On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist 13(6):594–615. https://doi.org/10.1177/1073858406296259
Benarroch EE (2011) NMDA receptors: recent insights and clinical correlations. Neurology 76:1750–1757. https://doi.org/10.1212/WNL.0b013e31821b7cc9
Fan MMY, Raymond LA (2007) N-Methyl-d-aspartate receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol 81(5–6):272–293. https://doi.org/10.1016/j.pneurobio.2006.11.003
Parameshwaran K, Dhanasekaran M, Suppiramaniam V (2008) Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol 210(1):7–13. https://doi.org/10.1016/j.expneurol.2007.10.008
Banerjee A, Larsen RS, Philpot BD, Paulsen O (2016) Roles of presynaptic NMDA receptors in neurotransmission and plasticity. Trends Neurosci 39(1):26–39. https://doi.org/10.1016/j.tins.2015.11.001
Park H, Popescu A, Poo M (2014) Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron 84:1009–1022. https://doi.org/10.1016/j.neuron.2014.10.045
Gupta KR, Hiwase CP, Bhandekar NS, Umekar MJ (2020) Therapeutic approaches in Alzheimer’s disease: amyloid peptide inhibitors. Indian J Pharm Pharmacol 7:147–154. https://doi.org/10.18231/j.ijpp.2020.025
Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F (2012) New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs. Br J Clin Pharmacol 73:504–517. https://doi.org/10.1111/j.1365-2125.2011.04134.x
Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, Golas G, Simeonov DR, Holloman C, Tankovic A, Karamchandani MM, Schreiber JM, Mullikin JC, Tifft CJ, Toro C, Boerkoel CF, Traynelis SF, Gahl WA (2014) GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol 1:190–198. https://doi.org/10.1002/acn3.39
Lazar MA, McIntyre RS (2019) Novel therapeutic targets for major depressive disorder. In: Quevedo J, Carvalho A, Zarate A (eds) Neurobiology of depression. Academic, New York, pp 383–400
Song X, Jensen MO, Jogini V, Stein RA, Lee CH, McHaourab HS, Shaw DE, Gouaux E (2018) Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 556:515–519. https://doi.org/10.1038/s41586-018-0039-9
Tymianski M (2011) Emerging mechanisms of disrupted cellular signalling in brain ischemia. Nat Neurosci 14(11):1369–1373. https://doi.org/10.1038/nn.2951
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. https://doi.org/10.1016/j.pneurobio.2013.11.006
Cai SX (2006) Glycine/NMDA receptor antagonists as potential CNS therapeutic agents: ACEA-1021 and related compounds. Curr Top Med Chem 6(7):651–662. https://doi.org/10.2174/156802606776894465
Wu QJ, Tymianski M (2018) Targeting NMDA receptors in stroke: new hope in neuroprotection. Mol Brain 11:1–4. https://doi.org/10.1186/s13041-018-0357-8
Ikonomidou C, Turski L (2002) Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 1(6):383–396. https://doi.org/10.1016/s1474-4422(02)00164-3
Scott AM, Wolchok JD, Old LJ (2012) Antibody therapy of cancer. Nat Rev Cancer 12:278–287. https://doi.org/10.1038/nrc3236
Chan AC, Carter PJ (2010) Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol 10:301–316. https://doi.org/10.1038/nri2761
Lemere CA, Masliah E (2010) Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol 6:108–119. https://doi.org/10.1038/nrneurol.2009.219
DeMattos RB, Lu J, Tang Y, Racke MM, DeLong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD (2012) A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 76(5):908–920. https://doi.org/10.1016/j.neuron.2012.10.029
Tajima N, Simorowski N, Yovanno RA, Regan MC, Michalski K, Gómez R, Lau AY, Furukawa H (2022) Development and characterization of functional antibodies targeting NMDA receptors. Nat Commun 13(1):923. https://doi.org/10.1038/s41467-022-28559-3
Young D (2020) The NMDA receptor antibody paradox: a possible approach to developing immunotherapies targeting the NMDA receptor. Front Neurol 11:635. https://doi.org/10.3389/fneur.2020.00635
Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, Traynelis SF (2018) Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol 150:1081–1105. https://doi.org/10.1085/jgp.201812032
Xu XX, Luo JH (2018) Mutations of N-Methyl-d-Aspartate receptor subunits in epilepsy. Neurosci Bull 34:549–565. https://doi.org/10.1007/s12264-017-0191-5
Myers SJ, Yuan H, Kang J-Q, Tan FCK, Traynelis SF, Low C-M (2019) Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Research 8:1940. https://doi.org/10.12688/f1000research.18949.1
Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF (2015) Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol 88:203–217. https://doi.org/10.1124/mol.115.097998
Fry A, Fawcett K, Zelnik N, Yuan H, Thompson B, Shemer-Meiri L (2018) De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141:698–712. https://doi.org/10.1093/brain/awx358
Pironti E, Granata F, Cucinotta F, Gagliano A, Efthymiou S, Houlden H (2018) Electroclinical history of a five-year-old girl with GRIN1-related early-onset epileptic encephalopathy: a video-case study. Epileptic Disord 20:423–427
Hamza TH, Chen H, Hill-Burns EM, Rhodes SL, Montimurro J, Kay DM, Tenesa A, Kusel VI, Sheehan P, Eaaswarkhanth M (2011) Genome-wide gene–environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via Interaction with coffee. PLoS Genet 7(8):e1002237. https://doi.org/10.1371/journal.pgen.1002237
Yamada-Fowler N, Fredrikson M, Söderkvist P (2014) Caffeine interaction with glutamate receptor gene GRIN2A-Parkinson’s disease in Swedish population. PLoS ONE 9:e99294. https://doi.org/10.1371/journal.pone.0099294
Sharawat I, Yadav J, Saini L (2019) GRIN2B novel mutation: a rare cause of severe epileptic encephalopathy. Neurology 67:562–563. https://doi.org/10.4103/0028-3886.257986
Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortum F, Fritsch A, Pientka FK (2010) Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet 42:1021–1026. https://doi.org/10.1038/ng.677
Amador A, Bostick CD, Olson H, Peters J, Camp CR, Krizay D, Chen W, Han W, Tang W, Kanber A, Kim S, Teoh J, Sah M, Petri S, Paek H, Kim A, Lutz CM, Yang M, Myers SJ, Bhattacharya S (2020) Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain 143:2039–2057. https://doi.org/10.1093/brain/awaa147
Tang W, Liu D, Traynelis SF, Yuan H (2020) Positive allosteric modulators that target NMDA receptors rectify loss-of-function GRIN variants associated with neurological and neuropsychiatric disorders. Neuropharmacology 177:108247. https://doi.org/10.1016/j.neuropharm.2020.108247
Garcıa-Recio A, Santos-Go´ mez A, Soto D, Julia-Palacios N, García-Cazorla A, Altafaj X, Olivella M, (2021) GRIN database: a unified and manually curated repertoire of GRIN variants. Hum Mutat 42:8–18. https://doi.org/10.1002/humu.24141
Mony L, Kew JN, Gunthorpe MJ, Paoletti P (2009) Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol 157:1301–1317. https://doi.org/10.1111/j.1476-5381.2009.00304.x
Hansen KB, Furukawa H, Traynelis SF (2010) Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol 78:535–549. https://doi.org/10.1124/mol.110.067157
Stroebel D, Buhl DL, Knafels JD, Chanda PK, Green M, Sciabola S, Mony LP, Paoletti J, Pandit, (2016) A novel binding mode reveals two distinct classes of NMDA receptor GluN2B-selective antagonists. Mol Pharmacol 89:541–551. https://doi.org/10.1124/mol.115.103036
Jiménez-Sánchez L, Campa L, Auberson YP, Adell A (2014) The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modelling schizophrenia and treating refractory depression. Neuropsychopharmacology 39:2673–2680. https://doi.org/10.1038/npp.2014.123
Vizi ES, Kisfal M, Lőrincz T (2013) Role of nonsynaptic GluN2B-containing NMDA receptors in excitotoxicity: evidence that fluoxetine selectively inhibits these receptors and may have neuroprotective effects. Brain Res Bull 93:32–38. https://doi.org/10.1016/j.brainresbull.2012.10.005
Schreiber JA, Schepmann D, Frehland B, Thum S, Datunashvili M, Budde T, Hollmann M, Strutz-Seebohm N, Wünsch B, Seebohm G (2019) A common mechanism allows selective targeting of GluN2B subunit-containing N-methyl-d-aspartate receptors. Commun Biol 2:1–4. https://doi.org/10.1038/s42003-019-0645-6
Irvine MW, Fang G, Sapkota K, Burnell ES, Volianskis A, Costa BM, Culley G, Collingridge GL, Monaghan DT, Jane DE (2019) Investigation of the structural requirements for N-methyl-d-aspartate receptor positive and negative allosteric modulators based on 2-naphthoic acid. Eur J Med Chem 15:471–498. https://doi.org/10.1016/j.ejmech.2018.12.054
Kudova E, Chodounska H, Slavikova B, Budesinsky M, Nekardova M, Vyklicky V, Krausova B, Svehla P, Vyklicky L (2015) A new class of potent N-methyl-d-aspartate receptor inhibitors: sulfated neuroactive steroids with lipophilic D-ring modifications. J Med Chem 58:5950–5966. https://doi.org/10.3390/ph15010049
Slavikova B, Chodounska H, Nekardova M, Vyklicky V, Ladislav M, Hubalkova P, Krausova B, Vyklicky L, Kudova E (2016) Neurosteroid-like inhibitors of N-methyl-d-aspartate receptor: substituted 2-sulfates and 2-hemisuccinates of perhydrophenanthrene. J Med Chem 59:4724–4739. https://doi.org/10.2174/1570159X15666170329091935
Dey S, Schepmann D, Wünsch B (2018) 2-Methyltetrahydro-3-benzazepin-1-ols—the missing link in SAR of GluN2B selective NMDA receptor antagonists. Bioorg Med Chem 26:501–508. https://doi.org/10.1016/j.bmc.2017.12.010
Tewes B, Frehland B, Schepmann D, Robaa D, Uengwetwanit T, Gaube F, Winckler T, Sippl W, Wünsch B (2015) Enantiomerically pure 2-methyltetrahydro-3-benzazepin-1-ols selectively blocking GluN2B subunit containing N-methyl-d-aspartate receptors. J Med Chem 58:6293–6305. https://doi.org/10.1124/mol.115.103036
Dey S, Schepmann D, Wünsch B (2016) Role of the phenolic-OH moiety of GluN2B-selective NMDA antagonists with 3-benzazepine scaffold. Bioorg Med Chem Lett 26:889–893. https://doi.org/10.1016/j.bmcl.2015.12.067
Dey S, Temme L, Schreiber JA, Schepmann D, Frehland B, Lehmkuhl K, Strutz-Seebohm N, Seebohm G, Wuensch B (2017) Deconstruction–reconstruction approach to analyse the essential structural elements of tetrahydro-3-benzazepine-based antagonists of GluN2B subunit containing NMDA receptors. Eur J Med Chem 29:552–564. https://doi.org/10.1016/j.ejmech.2017.06.068
Rath S, Schepmann D, Wünsch B (2017) Replacement of benzylic hydroxy group by vinyl or hydroxymethyl moiety at the 3-benzazepine scaffold retaining GluN2B affinity. Bioorg Med Chem 25:5365–5372. https://doi.org/10.1016/j.bmc.2017.07.059
Börgel F, Galla F, Lehmkuhl K, Schepmann D, Ametamey SM, Wünsch B (2019) Pharmacokinetic properties of enantiomerically pure GluN2B selective NMDA receptor antagonists with 3-benzazepine scaffold. J Pharm Biomed Anal 172:214–222. https://doi.org/10.1016/j.jpba.2019.04.032
Ahmed H, Haider A, Varisco J, Stanković M, Wallimann R, Gruber S, Iten I, Häne S, MüllerHerde A, Keller C, Schibli R (2019) Structure–Affinity Relationships of 2,3,4,5-tetrahydro-1H-3-benzazepine and 6,7,8,9-tetrahydro-5H-benzo-[7]-annulen-7-amine analogues and the discovery of a radiofluorinated 2,3,4,5-tetrahydro-1H-3-benzazepine congener for imaging GluN2B subunit-containing N-Methyl-d-aspartate receptors. J Med Chem 62:9450–9470. https://doi.org/10.1021/acs.jmedchem.9b00812
Thum S, Schepmann D, Ayet E, Pujol M, Nieto FR, Ametamey SM, Wünsch B (2019) Tetrahydro-3-benzazepines with fluorinated side chains as NMDA and σ1 receptor antagonists: synthesis, receptor affinity, selectivity and antiallodynic activity. Eur J Med Chem 177:47–62. https://doi.org/10.1016/j.ejmech.2019.05.034
Wagner M, Schepmann D, Ametamey SM, Wünsch B (2019) Modification of the 4-phenylbutyl side chain of potent 3-benzazepine-based GluN2B receptor antagonists. Bioorg Med Chem 27:3559–3567. https://doi.org/10.1016/j.bmc.2019.06.035
Lütnant I, Schepmann D, Wünsch B (2016) Benzimidazolone bioisosteres of potent GluN2B selective NMDA receptor antagonists. Eur J Med Chem 116:136–146. https://doi.org/10.1016/j.ejmech.2016.03.065
Gawaskar S, Schepmann D, Bonifazi A, Robaa D, Sippl W, Wünsch B (2015) Benzo-[7]annulene-based GluN2B selective NMDA receptor antagonists: surprising effect of a nitro group in 2-position. Bioorg Med Chem Lett 25:5748–5751. https://doi.org/10.1016/j.bmcl.2015.10.076
Zscherp R, Baumeister S, Schepmann D, Wünsch B (2018) Pyridine bioisosteres of potent GluN2B subunit containing NMDA receptor antagonists with benzo-[7]-annulene scaffold. Eur J Med Chem 157:397–404. https://doi.org/10.1016/j.ejmech.2018.08.003
Thum S, Schepmann D, Reinoso RF, Alvarez I, Ametamey SM, Wünsch B (2019) Synthesis and pharmacological evaluation of fluorinated benzo [7]annulen-7-amines as GluN2B-selective NMDA receptor antagonists. J Label Compd Radiopharm 62:354–379. https://doi.org/10.1002/jlcr.3718
Gawaskar S, Temme L, Schreiber JA, Schepmann D, Bonifazi A, Robaa D, Sippl W, Strutz-Seebohm N, Seebohm G, Wünsch B (2017) Design, synthesis, pharmacological evaluation and docking studies of GluN2B-selective NMDA receptor antagonists with a benzo [7]annulen-7-amine scaffold. ChemMedChem 12:1212–1222. https://doi.org/10.1002/cmdc.201700311
Anan K, Masui M, Hara S, Ohara M, Kume M, Yamamoto S, Shinohara S, Tsuji H, Shimada S, Yagi S (2017) Discovery of orally bioavailable cyclohexanol-based NR2B-selective NMDA receptor antagonists with analgesic activity utilizing a scaffold hopping approach. Bioorg Med Chem Lett 27:4194–4198. https://doi.org/10.1016/j.bmcl.2017.06.076
Buemi MR, De Luca L, Ferro S, Russo E, Sarro G, Gitto R (2016) Structure-guided design of new indoles as negative allosteric modulators (NAMs) of N-methyl-d-aspartate receptor (NMDAR) containing GluN2B subunit. Bioorg Med Chem 24:1513–1519. https://doi.org/10.1016/j.bmc.2016.02.021
Temme L, Frehland B, Schepmann D, Robaa D, Sippl W, Wünsch B (2018) Hydroxymethyl bioisosteres of phenolic GluN2B-selective NMDA receptor antagonists: design, synthesis, and pharmacological evaluation. Eur J Med Chem 144:672–681. https://doi.org/10.1016/j.ejmech.2017.12.054
Temme L, Bechthold E, Schreiber JA, Gawaskar S, Schepmann D, Robaa D, Sippl W, Seebohm G, Wuensch B (2020) Negative allosteric modulators of the GluN2B NMDA receptor with phenylethylamine structure embedded in ring-expanded and ring-contracted scaffolds. Eur J Med Chem 190:112138. https://doi.org/10.1016/j.ejmech.2020.112138
Krausova B, Slavikova B, Nekardova M, Hubalkova P, Vyklicky V, Chodounska H, Vyklicky L, Kudova E (2018) Positive modulators of the N-methyl-d-aspartate receptor: structure–activity relationship study of steroidal 3-hemiesters. J Med Chem 61:4505–4516. https://doi.org/10.1021/acs.jmedchem.8b00255
Santangelo RM, Ogden KK, Strong KL, Khatri A, Chepiga KM, Jensen HS, Traynelis SF, Liotta DC (2013) Synthesis, and structure–activity relationship of tetrahydroisoquinoline-based potentiators of GluN2C and GluN2D containing N-methyl-d-aspartate receptors. J Med Chem 56:5351–5381. https://doi.org/10.1021/jm400177t
Strong KL, Epplin MP, Bacsa J, Butch CJ, Burger PB, Menaldino DS, Traynelis SF, Liotta DC (2017) The structure–activity relationship of a tetrahydroisoquinoline class of N-methyl-d-aspartate receptor modulators that potentiates GluN2B-containing N-methyl-d-aspartate receptors. J Med Chem 60:5556–5585. https://doi.org/10.1021/acs.jmedchem.7b00239
Baumeister S, Schepmann D, Wünsch B (2019) Synthesis, and receptor binding of thiophene bioisosteres of potent GluN2B ligands with a benzo [7]annulene-scaffold. Med Chem Commun 10:315–325. https://doi.org/10.1039/C8MD00545A
Baumeister S, Schepmann D, Wünsch B (2020) Thiophene bioisosteres of GluN2B selective NMDA receptor antagonists: synthesis and pharmacological evaluation of [7]annuleno[b] thiophen-6-amines. Bioorg Med Chem 28:115245. https://doi.org/10.1016/j.bmc.2019.115245
Acknowledgements
Dr. Vinod G. Ugale acknowledges the financial support from Department of Science and Technology (DST)- Science and Engineering Research Board (SERB), New Delhi, India (Ref. No. TAR/2021/000140). VGU is thankful to the Principal Dr. S. J. Surana, R. C. Patel Institute of Pharmaceutical Education and Research, Shirpur, Maharashtra (India) for providing necessary facilities.
Author information
Authors and Affiliations
Contributions
Manuscript has been written with contributions from all authors.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ugale, V., Deshmukh, R., Lokwani, D. et al. GluN2B subunit selective N-methyl-D-aspartate receptor ligands: Democratizing recent progress to assist the development of novel neurotherapeutics. Mol Divers (2023). https://doi.org/10.1007/s11030-023-10656-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11030-023-10656-0