
Abstract
In order to enhance the structure determination process of macromolecular assemblies by NMR, we have implemented long-range pseudocontact shift (PCS) restraints into the data-driven protein docking package HADDOCK. We demonstrate the efficiency of the method on a synthetic, yet realistic case based on the lanthanide-labeled N-terminal ε domain of the E. coli DNA polymerase III (ε186) in complex with the HOT domain. Docking from the bound form of the two partners is swiftly executed (interface RMSDs < 1 Å) even with addition of very large amount of noise, while the conformational changes of the free form still present some challenges (interface RMSDs in a 3.1–3.9 Å range for the ten lowest energy complexes). Finally, using exclusively PCS as experimental information, we determine the structure of ε186 in complex with the HOT-homologue θ subunit of the E. coli DNA polymerase III.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudocontact shifts (PCS) are measured as the difference in chemical shifts between two NMR spectra, one of which is recorded with a paramagnetic center attached to the protein of interest. The presence of the paramagnetic center (usually a paramagnetic lanthanide; for a review on paramagnetic labeling techniques, see (Su and Otting 2010) changes the reference spectrum in several ways: Mainly, observed cross peaks are shifted, while active spins close to the paramagnetic probes (typically less than 5–10 Å) are no longer detected. The amount and direction of the shift in each dimension of the spectrum depends on multiple factors, including the vicinity of the spin to the lanthanide, and its position with respect to the anisotropic ∆χ-tensor. The ∆χ-tensor’s axial and rhombic components, as well as the relative orientation of the tensor frame to the protein, depend on the type of lanthanide used and on the surrounding electronic environment of the paramagnetic center (Bertini et al. 2002). This allows the measurement of several spectra by varying the lanthanide, which provides non-redundant information. Importantly, PCS can be measured up to distances of 40 Å from the paramagnetic center when a strong lanthanide such as Tb3+ or Dy3+ is being used, making this effect particularly suitable to obtain long-range inter-molecular information. Simple PCS-based rigid body docking concept was first demonstrated by Ubbink and coworkers (Ubbink et al. 1998). A more general method using lanthanide labeling techniques has been proposed (Pintacuda et al. 2006). The protocol has been recently reapplied in combination with chemical shift perturbation data (Saio et al. 2010). Atomic level details, necessary to precisely understand biomolecular interactions or to accurately design candidate drug compounds, can, however, only be disclosed using flexible docking approaches, such as the one offered by the data-driven docking package HADDOCK (Dominguez et al. 2003), which makes use of CNS as computational engine (Brunger et al. 1998). We present here the implementation of a PCS energy term into HADDOCK using the PARArestraints (Banci et al. 2004) module developed by Banci and coworkers, which we have ported into the structure calculation software CNS. We demonstrate that PCS alone are sufficient to accurately model the structure of a complex. We used as a test case the lanthanide-labeled N-terminal ε domain of the E. coli DNA polymerase III (ε186) in complex with the HOT domain. The active site of ε186 contains a pair of Mn2+/Mg2+ that can be substitute by a single lanthanide (Pintacuda et al. 2006). The unpaired electrons of the lanthanide induce in return intra-molecular PCS on nuclear spins in free ε186, as well as inter-molecular PCS when ε186 is bound to its protein partner. We investigate first the capability of PCS data to drive the docking. The protocol is then applied to model the structure of ε186 in complex with the HOT-homologue θ subunit of the E. coli DNA polymerase III.
Results and discussion
Docking with synthetic data: protocol
The performance of our PCS-driven flexible docking approach was first assessed on the ε186/HOT complex. Artificial PCS data were generated from the crystal structure (PDB id 2IDO, chains C and D) (Kirby et al. 2006) using the ∆χ-tensor parameters that best fit the available experimental PCS data for ε186 (Schmitz et al. 2006), assuming a single fixed location of the paramagnetic center. To keep the data set realistic, a generous flat random noise of ±0.15 ppm was added. The resulting PCSs range from −1.87 to 4.48 ppm. Furthermore, PCS that were not observed experimentally were removed. In total, five data sets were created: three for ε186 (Dy3+, Er3+ and Tb3+) and two for HOT (Dy3+ and Er3+). This synthetic data set matches the experimental data set available for the system ε186/θ in term of number of lanthanides used, number of PCSs observed, PCS value range and level of noise. We used the five data sets in the following docking runs. It is however to be noted that the Tb3+ data set is useful only to improve the location of the lanthanide, and does not help to drive the docking as it contains no intermolecular information.
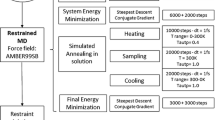
Two docking runs were performed: the first one from the bound forms of ε186 and HOT taken from 2IDO, the second one from the free forms consisting of the crystal structure of ε186 [PDB id 1J53 (Hamdan et al. 2002)] and the NMR ensemble of HOT [PDB id 1SE7 (DeRose et al. 2004)]. The axial and rhombic components were fitted against the noisy synthetic data of ε186 using the software Numbat (Schmitz et al. 2008), and entered into HADDOCK (the values are given in SI Table 1). Distance restraints were defined between the paramagnetic center and the coordinating residues of ε186 (Hamdan et al. 2002) to maintain the lanthanide ion at its known location. Flexible, disordered termini of the NMR structure of HOT were removed as they can obstruct the docking process. For each runs, 1,400 structures were calculated during the rigid body minimization stage; the 200 lowest score structures were subsequently subjected to a semi flexible simulated annealing in torsion angle space, followed by a final refinement with explicit solvent (water) according to the standard HADDOCK protocol (De Vries et al. 2007).
Docking with synthetic data: bound–bound scenario
The results are summarized in Fig. 1. The rigid body stage of the bound–bound run resulted in more than one third of structures below 1 Å interface-RMSD (Fig. 1, plain red squares), corresponding to “high quality”—three stars prediction in CAPRI nomenclature (Janin 2005). The i-RMSD [interface-RMSD, (Mendez et al. 2003)] is calculated over the interface atoms of the complex located within 10 Å from the partner molecule, between a given model and a reference model, in this case 2IDO. After flexible refinement, the structures slightly moved away from the reference crystal structure (reflected in the i-RMSD values) (Fig. 1, plain green triangles), a result of the force field used and the molecular dynamics simulations. Note however that the overall score (including electrostatic and van der Waal energies) does improve. The resulting 200 structures form a single cluster of which the lowest structures are of high quality (Fig. 1, blue disks). Quite remarkably, similar results are obtained with a level of noise of ±0.45 ppm (SI Fig. 1), indicating that the method is extremely noise-tolerant, well beyond the precision of the measurements (PCS are usually measured with 0.05 ppm accuracy).

ε/HOT interface RMSD (i-RMSD) for the various stages of the bound–bound and unbound–unbound HADDOCK runs. The stars correspond to the i-RMSD CAPRI criteria for acceptable, medium and high quality prediction
Docking with synthetic data: unbound–unbound scenario
The unbound–unbound docking run is challenging in several ways. While ε186 free and bound structures are similar (1.4 Å backbone RMSD), they exhibit a large conformational change at loop 157 K–162 G located at the edge of the interface (SI Fig. 2), and HOT experiences a more global conformational change between 3.2 and 3.6 Å for the NMR ensemble (SI Fig. 3). This range of conformational changes is already in what is considered challenging in the docking field (Andrusier et al. 2008; Bastard et al. 2011; Bonvin 2006; Zacharias 2008). Under those conditions, obtaining high quality predictions has proven difficult, even for docking software that handles flexible segments (Andrusier et al. 2008; Bastard et al. 2011; Bonvin 2006; Zacharias 2008). About one third of the structures produced by the rigid-body stage are below 4 Å i-RMSD, satisfying the acceptable—1 star criteria of CAPRI classification (Fig. 1 red squares) (Janin 2005). The next two refinement stages of the HADDOCK run improved the average i-RMSD of the ten lowest energy complexes by as much as 0.53 Å (with a maximum improvement of 0.96 Å), indicating that the PCS energy term is pulling in the right direction (Fig. 1 green unfilled triangles and blue unfilled circles). Higher quality prediction would probably require a better sampling of the conformational changes at the interface, which is notoriously difficult (Bonvin 2006). For a complex such as ε186/HOT, a HADDOCK run based on PCS restraints is thus expected to generate acceptable to high quality solutions.
Docking with experimentally observed PCS
We applied the same protocol to generate a model of the homologous ε186/θ complex, which has, up to now, only been studied in a plain rigid-body approach (Pintacuda et al. 2006; Schmitz et al. 2008). The datasets used are now the experimental one as published in (Schmitz et al. 2006, 2008). The starting structures of θ were taken from the NMR ensemble 2AXD (Keniry et al. 2006). The flexible termini (residues 1–9 and residues 70–76) were removed based on visual inspection of the ensemble. This choice was corroborated by the fact that PCS were observed only for residues in the 9–66 range on θ. Both the free form and bound (in complex with HOT) forms of ε186 were used in the docking. 1,400, 200 and 200 structures were calculated respectively in the three stages of HADDOCK. However, after the rigid body first stage, HADDOCK selected only complexes originating from the bound form of ε186. This indicates that binding mode of ε186 to θ is similar to that of the ε186/HOT complex. This was supported by the analysis of two additional docking runs using either the bound or the free form of ε186 as starting structure: comparison of the top ten structures of the two runs revealed that (i) the electrostatic energy is on average better by 14% when the HOT-bound starting structure of ε186 is used, and (ii), under the same conditions, the buried surface area increased by 19%. The correlations between the calculated and experimental PCS, together with a representation of the best ten structures, are shown in Fig. 2. The ensemble of ten structures has been deposited in the protein data bank (Berman et al. 2000) under the accession code 2XY8.

Correlation between predicted and observed PCS for the top-ranking structure of the ε186/θ complex calculated with HADDOCK. The top four ε186/θ structures superimposed on ε (gold) are shown in ribbon representation (figure generated with PyMOL (DeLano 2002))
Conclusion
We have demonstrated that PCS alone are sufficient to generate accurate models of complexes in combination with flexible docking. This approach, implemented in HADDOCK, was applied to model the structure of the ε186/θ based on experimental PCS data. It is anticipated that recent progresses in paramagnetic labeling techniques (Su and Otting 2010) will increase the popularity of the PCS as a structural restraints source. The inherent flexibility of some paramagnetic tags can easily be modeled by allowing for variation in the distance restraints used to maintain the ∆χ-tensor in place. The flexible, PCS-driven docking protocol described here will be made available in a future release of HADDOCK and also implemented in the web server portal (De Vries et al. 2010).
References
Andrusier N, Mashiach E, Nussinov R, Wolfson HJ (2008) Principles of flexible protein–protein docking. Proteins 73(2):271–289
Banci L, Bertini I, Cavallaro G, Giachetti A, Luchinat C, Parigi G (2004) Paramagnetism-based restraints for Xplor-NIH. J Biomol NMR 28(3):249–261
Bastard K, Saladin A, Prevost C (2011) Accounting for large amplitude protein deformation during in silico macromolecular Docking. Int J Mol Sci 12(2):1316–1333
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Bertini I, Luchinat C, Parigi G (2002) Magnetic susceptibility in paramagnetic NMR. Prog NMR Spectrosc 40(3):249–273
Bonvin AM (2006) Flexible protein-protein docking. Curr Opin Struct Biol 16(2):194–200
Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Sect D 54:905–921
De Vries SJ, Van Dijk ADJ, Krzeminski M, Van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AMJJ (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69(4):726–733
De Vries SJ, van Dijk M, Bonvin AMJJ (2010) The HADDOCK web server for data-driven biomolecular docking. Nat Protoc 5(5):883–897
DeLano WL (2002) The PyMOL molecular graphics system. Palo Alto, CA
DeRose EF, Kirby TW, Mueller GA, Chikova AK, Schaaper RM, London RE (2004) Phage like it HOT: solution structure of the bacteriophage P1-encoded HOT protein, a homolog of the theta subunit of E-coli DNA polymerase III. Structure 12(12):2221–2231
Dominguez C, Boelens R, Bonvin AMJJ (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125(7):1731–1737
Hamdan S, Carr PD, Brown SE, Ollis DL, Dixon NE (2002) Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure 10(4):535–546
Janin J (2005) Assessing predictions of protein–protein interaction: The CAPRI experiment. Protein Sci 14(2):278–283
Keniry MA, Park AY, Owen EA, Hamdan SM, Pintacuda G, Otting G, Dixon NE (2006) Structure of the theta subunit of Escherichia coli DNA polymerase III in complex with the epsilon subunit. J Bacteriol 188(12):4464–4473
Kirby TW, Harvey S, DeRose EF, Chalov S, Chikova AK, Perrino FW, Schaaper RM, London RE, Pedersen LC (2006) Structure of the Escherichia coli DNA polymerase III epsilon-HOT proofreading complex. J Biol Chem 281(50):38466–38471
Mendez R, Leplae R, De Maria L, Wodak SJ (2003) Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins 52(1):51–67
Pintacuda G, Park AY, Keniry MA, Dixon NE, Otting G (2006) Lanthanide labeling offers fast NMR approach to 3D structure determinations of protein–protein complexes. J Am Chem Soc 128(11):3696–3702
Saio T, Yokochi M, Kumeta H, Inagaki F (2010) PCS-based structure determination of protein–protein complexes. J Biomol NMR 46(4):271–280
Schmitz C, John M, Park AY, Dixon NE, Otting G, Pintacuda G, Huber T (2006) Efficient chi-tensor determination and NH assignment of paramagnetic proteins. J Biomol NMR 35(2):79–87
Schmitz C, Stanton-Cook MJ, Su XC, Otting G, Huber T (2008) Numbat: an interactive software tool for fitting delta chi-tensors to molecular coordinates using pseudocontact shifts. J Biomol NMR 41(3):179–189
Su XC, Otting G (2010) Paramagnetic labelling of proteins and oligonucleotides for NMR. J Biomol NMR 46(1):101–112
Ubbink M, Ejdeback M, Karlsson BG, Bendall DS (1998) The structure of the complex of plastocyanin and cytochrome f, determined by paramagnetic NMR and restrained rigid-body molecular dynamics. Structure 6(3):323–335
Zacharias M (2008) Combining elastic network analysis and molecular dynamics simulations by hamiltonian replica exchange. J Chem Theory Comput 4(3):477–487
Acknowledgments
This work was supported by the Dutch Foundation for Scientific Research (NWO) through a VICI grant (no. 700.56.442) to A.M.J.J.B and by the European Community, FP7 Access to Research Infrastructures Bio-NMR project (grant number 261863), and e-Infrastructure “e-NMR” and “WeNMR” projects (grant numbers 213010 and 261572).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schmitz, C., Bonvin, A.M.J.J. Protein–protein HADDocking using exclusively pseudocontact shifts. J Biomol NMR 50, 263–266 (2011). https://doi.org/10.1007/s10858-011-9514-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9514-4