
Abstract
MicroRNAs (miRNAs) play an important role in plant growth, development and responses to stresses. Rice blast is one of the most devastating diseases in rice production. However, little is known regarding the effects of miRNAs response to rice blast. Herein, by deep sequencing small RNA from the susceptible line ZhongerRuanzhan and its space-induced blast resistant mutant line H4 under normal conditions and upon Magnaporthe oryzae (M. orzyzae) infection, several known miRNAs were detected and their expression profiles were found to be negatively correlated with their targets. And, a total of 50 novel miRNAs induced by M. oryzae infection were also identified from four small RNA libraries. Moreover, 176 putative targets of 23 novel miRNAs, which are involved in the various functions, were validated by two degradome analysis. Six novel miRNAs were selected for further validation with qRT-PCR analysis and the results showed that their expression levels were associated with blast response. The knowledge obtained in this study will help us understanding the functions of miRNAs and their targets in regulating blast resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice blast is one of the most serious and devastating diseases that cause huge economic losses in rice production. The development of resistant lines is closely related to maintenance and improvement of rice grain yield and quality. To date, multiple regulatory factors that mediate rice resistance to diseases have been identified by a combination of genetic, biochemical and high-throughput sequencing approaches. However, the relevant regulatory mechanisms and the complex regulatory networks still need to be further elucidated.
MicroRNAs (miRNAs) are one of the most abundant small RNAs (sRNAs) in plants and their typical lengths are 18-25 nucleotides (Li et al. 2014). They are a group of endogenous, non-coding sRNAs involved in regulating gene expression mainly via repressing the translation or mediating the cleavage of target mRNA at the post-transcriptional level (Campo et al. 2013). In addition to their role in regulating gene expression associated with various plant developmental processes, such as organ morphogenesis, signal transduction pathway, responses to environmental stresses (Lu et al. 2007), miRNAs have also emerged as a potentially important means to be applied in the studies on the complex regulatory networks being operated during plant-fungus interactions (Jones-Rhoades and Bartel 2004; Nunes et al. 2011; Zhao et al. 2012a, b).
The discovery and functional analysis of miRNAs related to rice blast have been performed to a certain extent. Host miRNAs are involved in the interactions between rice and rice blast. It has been demonstrated that the expression profiles of miRNAs are modulated by blast stress and M. oryzae-derived elicitors could induce the expression of a number of miRNAs in rice (Campo et al. 2013; Wu et al. 2009; Zhu et al. 2008). Accumulating evidence has demonstrated that a number of miRNAs are involved in rice immunity against the blast fungus. For example, osa-miR7695 was reported to mediate the negative regulation of natural resistance-associated macrophage protein 6 (OsNramp6) to rice blast (Campo et al. 2013). And, overexpression of miR160a and miR398b enhanced plant resistance by restraining the fungal growth (Li et al. 2014). Moreover, it has been verified recently that osa-miR169 negatively regulates rice immunity by differentially repressing its target genes, the NF-YA family members (Li et al. 2017). These studies have indicated that miRNAs play an important role in rice immunity against M. oryzae.
To date, the next-generation sequencing technologies have provided high-throughput quantitative expression profiles with high accuracy that can greatly improve the identification of novel miRNAs. These technologies have been widely used in the identification of rice miRNAs (Addo-Quaye et al. 2008; German et al. 2008; Li et al. 2011; Li et al. 2016; Sunkar et al. 2008; Zhang et al. 2010). Moreover, degradome sequencing and/or parallel analysis of RNA ends (PARE) provide the efficient tools to identify sRNA targets by directly and globally identifying the residues of sRNA-directed target cleavage (Deng et al. 2006; Morin et al. 2008; Zhu et al. 2008). Herein, in order to identify novel miRNAs and miRNA-targets modulated by biotic stress, we sequenced four small RNAs and two degradome sequencing libraries. As the result, a total of 50 novel miRNAs and 176 miRNA-targets were identified in rice during M. oryzae infection. Among them, the expression profiles of six selected novel miRNAs were further validated by qRT-PCR and the target genes of four miRNAs were detected through degradome sequencing. Therefore, our data has provided the valuable information for investigating the miRNAs induced by rice blast and the interactions with their targets. Furthermore, further research on rice-blast fungus plant-pathogen system will enable us to reveal the molecular mechanisms and to gain new insights into the related regulatory factors, and to apply new findings in rice resistance breeding.
Materials and Methods
Plant material
One susceptible line ZhongerRuanzhan (ZE) and its space-induced blast resistant mutant line H4 were used in this study. H4 contains the resistance gene of Pik-H4, an allele at the Pik locus in the ZE background (Xiao et al. 2011). The M. oryzae race GD0193, one of the primary M. oryzae race in Guangdong Province, in incompatible with H4 but compatible with ZE (Additional files S1). Fourth-leaf-stage rice seedlings grown under natural light in a greenhouse at 28 °C were used for inoculation of the rice blast fungus. For fungal inoculation, freshly prepared M. oryzae spores (1 × 105 conidia/ ml, containing 0.02% v/v gelatin).
Leaves of H4 and ZE were collected at 0 h, 24 h after inoculation (HAI), respectively. Four small RNA libraries (Table 1) were constructed with the small RNA extracted from the collected leaves and subjected to high throughput sequencing. For two degradome library construction, equal amounts of RNA samples (water-treated and pathogen-treated) of ZE isolated at 0 h and 24 h after inoculation were mixed to generate one degradome library S, and equal amounts of RNA samples (water-treated and pathogen-treated) of H4 isolated at 0 h and 24 h after inoculation were mixed to generate another degradome library R. All the samples were immediately frozen in liquid nitrogen, and stored at −80 °C for further use.
Construction and sequencing of sRNA and degradome libraries
Small RNA library construction and Illumina sequencing were performed as described (Mi et al. 2008). Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA), and sRNAs were separated and enriched by size fractionation with 15% denaturing PAGE. Purified 18-30 nt sRNAs were reversely transcribed after ligating 5’and 3’adaptors, and subsequently sequenced using a Solexa high-throughput sequencer (Illumina, USA) at BGI, Shenzhen, China.
The quantity and purity of the isolated total RNA were examined by using Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number > 7.0. Approximately 20 μg of total RNA were used to prepare degradome library as reported previously (Li et al. 2015; Addo-Quaye et al. 2008; Hafner et al. 2008). The single-end sequencing (36 bp) was performed on an Illumina Hiseq2500 at the LC-BIO (Hangzhou, China) following the vendor’s recommended protocol.
Analysis of target gene expression through microarray
Analysis of gene expression profiles was performed on the GeneChip rice genome array (Affymetrix, Santa Clara, CA). The plant materials were sampled at 0 h and 24 h after inoculation. The expression profiles of corresponding target genes were clustered by Cluster 3.0 to explore the functions of novel miRNAs. The cluster analysis was performed by adapting the hierarchical, Median Center (gene), and average linkage program.
Analysis of gene expression profiles with qRT-PCR
Total RNA was extracted from 100 mg of rice seedlings with Trizol (Invitrogen, Carlsbad, CA, USA), and purified RNA was reversely transcribed into cDNA using PrimeScript™RT Reagent Kit (Takara, Dalian, China). The qRT-PCR assay was performed using the Applied Biosystems TaqMan® microRNA Assay (Foster City, CA, USA). A two-step assay was performed on a RoterGene-6000 (Corbett Research, Australia) according to commercial protocols. After being normalized by reference gene U6 RNA, relative quantification of expression levels of novel miRNA was calculated with 2-ΔΔCT method (Livak and Schmittgen 2001) using the manual threshold cycle setting. All samples and standards were run in triplicate.
Results
Deep sequencing of four sRNA libraries
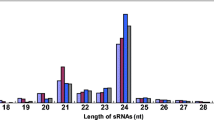
To identify the miRNAs elicited by blast fungus, four libraries were constructed (Table 1) and the results were shown in Table 2, with the lengths ranging from 10 to 30 nt. A majority of the redundant sRNAs were 21–24 nt in length (Fig. 1a), consistent with the distribution of the typical sizes by dicer-derived products (Eulgem and Somssich 2007).


Primary analysis of sequencing data from wild-type and space- induced mutant rice lines treated with Magnaporthe oryzae. a Size distribution of sRNAs signatures. b Common and specific unique sRNA between different libraries. c Annotation of sRNA signatures of the four libraries
These sRNA libraries showed variations among different rice lines and treatments (Fig. 1b). Remarkable differences existed between two lines (ZE and H4) under one kind of treatment (mock or blast infection), as it demonstrated that the number of 79216 (3.15%) and 97743 (10.73%) unique sRNAs were found upon mock- and blast-treated libraries in different lines, respectively. However, the most significant changes were induced by the blast fungus in the ZE library, which shared only 54782 (2.72%) unique sRNAs with library A, though library A was the most abundant one; In contrast, the smallest difference was elicited by the blast pathogen in the resistant mutant library B, which shared 123609 (11.94%) unique sRNAs with library D. Therefore, these results suggested that diverse expression profiles of sRNAs might be related to blast resistance of rice lines.
Using the Short Oligonucleotide Analysis Package (SOAP)(Beijing Genomics Institute)(BGI) to match with the sRNA reads, the known rice sRNAs accounted for 85.46% (A, 7476714), 79.35% (B, 9647324), 77.70% (C, 7721715) and 76.10% (D, 8476386) of the rice genome, respectively. After removing other RNA categories matched to NCBI Genbank, Rfam database, known rice miRNA precursor, repeat associated RNA and siRNA, the remaining reads: 1681359(A); 3829741(B); 3182403(C); and 3970132(D), were used for further analysis, as shown in Fig. 1c.
Expression profiles of registered miRNAs and related sRNA signatures
Since some miRNAs were tissue-specific, time-specific or stress-induced, only 291, 210, 164 and 220 registered miRNAs were identified in libraries A, B, C and D respectively, as summarized in Table 3. Particularly, osa-miR162 and osa-miR168 were both significantly down-regulated in the wild-type, while showed an inconspicuous difference in the mutant (Fig. 2a). Osa-miR162 and osa-miR168, which target AGO protein and DCL1, respectively, are more liable to be altered by the pathogen in the wild-type plant than in the mutant. Similarly, registered miRNAs showed the greatest diversity between library A and C, 135 registered miRNAs were expressed specifically in library A. On the other hand, 186 registered miRNAs appeared synchronously in library B and D, as shown in Fig. 2b.



Common and specific registered miRNA in wild-type and space- induced resistant mutant elicited by Magnaporthe oryzae. a Expression analysis of osa-miR162 and osa-miR168 in different libraries. The expression of miRNAs in two samples (control and treatment) was normalized to produce the expression of transcript per million (TPM), and fold-change means log2 (latter TPM / former TPM). b Common and specific registered miRNAs between different libraries. c PCA (Principle component analysis) of expression of registered miRNAs between different libraries. A, mock-treated wild-type; B, mock-treated mutant; C, blast-treated wild-type; D, blast-treated mutant. d-e Expression profile of registered miRNA and targets in ZE and H4 libraries
The microarray analysis of rice genome expression was used for auxiliary identification of miRNA targets, which enable us to gain a better knowledge about miRNA functions (Sunkar et al. 2007). Therefore, we linked up with the expression profiles of miRNAs and their targets to further illustrate their relationships upon blast infection. As shown in Fig. 2c, the expression of miR164 was negatively correlated with its targets (LOC_Os12g05260, and LOC_Os04g40780) upon wide-type libraries. In addition, the expression profiles of miR528, miR535, miR156, miR166 and miR167 were negatively correlated with their targets to certain extents, respectively. Similar changes could also be seen in H4 as well (Fig. 2d), demonstrating certain degradative functions of miRNA- targets. Thus, they may be involved in the regulation of rice immunity against M. oryzae infection.
Identification and analysis of novel miRNAs elicited by M. oryzae infection
To identify the novel miRNAs elicited by the blast pathogen infection, the surrounding sequences of unnamed sRNAs were extracted and their secondary structures were predicted by using RNAfold and Mireap. After being validated by Mircheck and following the previously set criteria (Rajagopalan et al. 2006; Song et al. 2010), a total of 50 novel miRNAs were identified and classified into three categories, as shown in Table 4. Firstly, nine novel miRNAs, named T2-T10, occurred with miRNAs* synchronously in at least one library. Secondly, two sequences, T12 (osa-miR169c-a) and T13, were defined as the conserved miRNAs by alignment with osa-miRNA169 families and osa-miRNA818 families, separately, as shown in Fig. 3a, b. Thirdly, 39 unnamed novel miRNAs, from the 43 candidate miRNAs present in at least two libraries, were grouped into the third category and named T14-T52. Additionally, osa-miR2863b (T1) and osa-miR396i (T11) reported from a previous study were also found in our research (Zhao et al. 2012a, b).

Sequence analysis of novel miRNAs elicited by M. oryzae infection (a) Antisense sequences of miR169 family members (b) Conserved features of the novel rice miRNA T13 elicited by Magnaporthe oryzae. (c) Special origin of novel rice miRNAs elicited by Magnaporthe oryzae (d) Nucleotide bias at each position in novel rice miRNAs elicited by Magnaporthe oryzae
In plants, a majority of miRNAs are mainly derived from intergenic regions, and a minority of miRNAs is derived from introns or exons (Teune and Steger 2010). Based on The Institute for Genomic Research (TIGR) database, 29 novel miRNAs are positioned in intergenic regions of the rice genome, 16 and 5 novel miRNAs were produced in introns and exons, respectively. They may be related to most of the non-conservative miRNAs derived from the coding regions (Chen et al. 2011; Wei et al. 2011). Some miRNAs can be the transcripts derived from the cluster area of the same miRNA site. In this study, we identified four miRNAs in precursor of annotated miRNAs, as shown in Fig. 3c. T14 and T36 resided on the same arm of osa-miR169f and osa-miR167e. Moreover, T48 and T51 were derived from the complementary arms of osa-miR399d and osa-miR2864.
The lengths of novel miRNAs are mainly distributed in the range of 20 to 24 nt. It has been reported that 5 ‘U is a feature of plant miRNA, because AGO1 protein tends to be combined with U of the first base (Schreiber et al. 2011). In this study, 19 novel miRNAs started with U. In addition, the eighth base is mainly G associated with the identification of RISC (Mei et al. 1998), as shown in Fig. 3d. It is worth mentioning that there are nine miRNAs being 23 nt in length, and seven of which had a bias for G at the first position, suggesting that they may be a new type of rice miRNAs.
Promoter elements play an important role in regulatory network of plant miRNAs, and their binding sites of many transcription factors, such as MYC, ARF, and LFY etc., can often be found in miRNA promoter regions. In our study, diverse kinds of cis-elements of these novel miRNAs had been identified in their promoter regions, some of which are related to fungal trigger or specific hormone responses (Table 5). Among them, box-w1 and W-box, which belong to the fungal elicitor responsive elements, were found in the promoters of 19 novel miRNAs. Moreover, the motifs of CGTCA, TGA, TCA and TGACG responding to the stress induced by jasmonic acidmethyl (MeJA) and salicylic acid (SA) can be detected nearly in all the novel miRNAs except T33. Additionally, the upstream sequences of 27 novel miRNAs were embedded with TC-rich repeats, which are involved in defense and stress responsiveness. On the whole, a total of 50 novel miRNA genes were predicted to be associated with diverse transcription factors involved in defense against fungal infection and/or other stresses.
Target analysis of novel miRNAs
Degradome sequencing is an effective and primary way to find the targets of miRNAs. In our study, two libraries were constructed to further validate the novel miRNA targets. After having removed the 3’adaptor and the structural RNAs (rRNAs, tRNAs, snRNAs, and snoRNAs), 15060667 (99.53%) and 22988985(99.42%) clean reads from libraries S and R can be mapped to the genome, respectively. Among them, 5564896(99.49%) and 7268546(99.48%) unique mappable reads were produced, respectively (Table 6). Of which, 176 targets of 23 miRNAs were verified by degradome sequencing. Then, we gathered the miRNA-targets to conduct GO analysis and found that their functions were multifarious (Fig. 4a and Additional files S2), including protein binding, signal transduction, response to stress etc. Nevertheless, not all the novel miRNAs were assigned with targets by degradome sequencing. Thus, whether this was due to their low expression levels and/or due to their negative regulation via translational repression needs to be validated with further experiments.


Analysis of novel miRNAs-targets (a) GO Term of novel miRNAs targets (b) Relative expression levels of six novel rice miRNAs in Magnaporthe oryzae -treated mutant and wild-type lines. X-axis indicates the time after being treated with pathogen, and black lines and red lines represents wild type and the mutant, respectively
We further analyzed the expression profiles of six novel miRNAs of genes that target the receptor Ser/Leu protein kinase or transcription factors or those involved in diverse biotic stress response with qRT-PCR. Of which, four novel miRNAs-targets were detected by degradome sequencing while the other two novel miRNAs-targets were predicted by bioinformatics (Table 7). As shown in Fig. 4b, the novel miRNAs displayed different kinds of expression patterns after inoculation. The first 48 HAI should be a key time interval during which novel miRNAs were modulated by pathogen infection to regulate their targets in responding to biotic stress. Among the first 48 intervals, the expression levels of T19, T34 and T46 showed the unified changes between wild types and their mutations. Of the three intervals 6 h, 12 h and 48 h, expression profiles of T19 and T46 displayed fluctuation as the inoculation time went by, but the expression of T34 kept rising. Other three miRNAs, the expression levels of T4, T13 and T51 changed irregularly between wild types and their mutations, which may be subjected to the resistant/susceptible material between ZE and H4.
Discussion
Changes in Expression Profiles of sRNAs or miRNAs
It is an economic way to control the destructive plant diseases such as rice blast with resistant cultivars (Meyers et al. 2008). Therefore, a model of which major resistance gene prevents infection from a strain of M. oryzae carrying the corresponding avirulence gene (Zhao et al. 2012a, b) was proposed. However, the variations of the pathogen could overcome the resistance of the R gene and result in severe blast damage (Lu et al. 2008). To effectively address this challenge, the development of new sources of resistant germplasm and pyramiding different genes related to blast resistance are urgently needed. A mutant, H4, conferring stable and high-level resistance to blast at both the seedling and maturity stages was derived from a susceptible indica rice cv. ZE. H4 was found to be resistant to more isolates than twelve monogenic lines (Xiao et al. 2011), suggesting that H4 can be an excellent source for the development of blast resistant varieties.
In the current study, we attempted to explore the miRNAs induced by blast. It should be noted that under the mock-treated condition, H4 exhibited significantly diverse sRNA components different from those of the wild-type ZE, indicating that the variation in expression may be caused not only by stress but also by morphologic and material differences during normal growth. Since sRNA components of the wild-type are much more variable than those of the mutant after pathogen inoculation, the variation in sRNAs might also be another key factor in enhancing the resistance of mutant to the pathogen, and some sRNAs could be involved in the blast-stress response. For instance, Osa-miR162 and osa-miR168, which target AGO protein and DCL1, respectively, could interfere with the genesis of miRNAs (Morel et al. 2002; Zhou et al. 2007). The proteins required for sRNA biogenesis and function have been shown to be required for resistance to pathogens (Lindow et al. 2007). Herein, we found that blast-induced osa-miR162 and osa-miR168 were much more variable in the wild-type than in the mutant, and the variability was consistent with the greater changes in miRNA components or their expression levels in the wild-type (Fig. 2a). Consequently, the stable expression profiles of some miRNAs might help to enhance the blast resistance in the mutant, and is, therefore, feasible to identify the novel miRNAs that respond to the pathogen.
A new type of 23 nt miRNA in rice
In plant, most of miRNAs are typically 21 nt or 22 nt in length. The 23 nt miRNAs are rare and it occurs only by bioinformatics prediction at an early stage (Willmann and Poethig 2007). However, in the past several years, some 23 nt rice miRNAs, such as osa-miR1863b.2 (Zhao et al. 2012a, b), the osa-miR2275c (Lu et al. 2008), the osa-miR5073 (Wei et al. 2006) and osa-miR5340 (Willmann and Poethig 2007), were registered in miRbase. In our study, nine novel miRNAs were identified from a large number of 23 nt sRNA signatures, which had been reported in Brassica napus. They might be a specific class of functional miRNAs lasting for certain period of time or under certain conditions (Vaucheret et al. 2004). Seven of the nine novel miRNAs in rice preferred to be initiated with a G bias, but they were inconsistent with the 23 nt signatures from B. napus, which preferred to end with an A bias (Vaucheret et al. 2004). This difference implicates that they may be related to specific functions in different organisms.
Moreover, the presence of 23 nt novel miRNAs was validated by the following lines of evidence: 1) two 23 nt miRNAs, T9 and T10, were supported by the presence of miRNA* and appropriate myeloid Elf-1 like factors (MEFIs), thus they are authentic miRNAs (Lampard et al. 2008; Ledent et al. 2002); and 2) T21 and T52 were matched to a registered EST; and 3) the MEFI values of T20 and T41 were 1.27 and 1.20, respectively. The precursors of T20 and T41 were derived from repetitive loci, but these sequences with predicted fold-back structures could still be annotated as authentic loci for miRNA in rice (Lampard et al. 2008). Thus, they could be annotated. However, the other three novel miRNAs without define evidence, T18, T24 and T39, might not be bona fide miRNAs.
MiRNA-targeted genes involved in resistance variation
Since more comprehensive functional genes are affected by the non-conserved miRNAs rather than by the conserved miRNAs (Sonnenfeld et al. 2005), genes with diverse functions were predicted to be the targets of the novel miRNAs. According to GO and KEGG analysis, the targeted genes may be involved in the blast-stress response pathway, and the novel miRNAs may play important roles in plant defense responses through regulation of their targeted genes.
Large families of basic helix–loop–helix (bHLH) transcription factors play essential roles in physiological and developmental processes (Fujiwara et al. 2006; Nishimura et al. 2009; Ono et al. 2001; Suharsono et al. 2002). In M. oryzae, Mstu 1, an APSES protein with highly conserved bHLH DNA binding domains, is important for pathogenicity and asexual development (Kim et al. 2012); OsRac1 is a key regulator involved in the basal resistance and defense responses (Rushton et al. 2010; Ryu et al. 2006; Wu et al. 2005). Rac Immunity 1(RAI1) encodes a putative bHLH transcription factor involved in the rice defense responses through regulating the elicitor-responsive genes encoding phenylalanine ammonia-lyase 1 (PAL1) and OsWRKY19 (Qiu and Yu 2009). In our study, LOC_Os10g42430.1 and LOC_Os04g41229.1 were indicated as the possible targets of T34 and T42, respectively, which encode bHLH transcription factors. In addition, the expression level of T34 was significantly increased in the 48 HAI, suggesting that T34 is induced by pathogen and responds to biotic stress by regulating its targets.
WRKY proteins, characterized by the WRKY domain that binds to a consensus cis-element termed W-box (TTGACT/C) (Shimono et al. 2007), are a family of zinc-finger transcription factors involved in plant response to pathogen infection and a variety of environmental stresses. To date, more than 100 WRKY genes have been identified in rice genome (Qiu et al. 2007), and many of them are involved in the immune response (Abbruscato et al. 2012). The expression of OsWRKY45 is markedly induced in response to ABA and positively regulates the resistance to blast and bacterial blight (Han et al. 2013). Overexpression of OsWRKY13 (Wei et al. 2013), OsWRKY22 (Chujo et al. 2007), OsWRKY30 (Wang et al. 2007), OsWRKY31 (Nishizawa et al. 2016), OsWRKY47 (Sunkar et al. 2006), OsWRKY53 (Sunkar et al. 2008) and OsWRKY89 (Dugas and Bartel 2008) clearly enhanced the resistance against blast fungus. In our study, Zinc-finger proteins LOC_Os06g47850.1 and WRKY DNA-binding protein LOC_Os02g08440, which were targeted by T31 and T34, repectively, were also identified, suggesting their possible roles in rice response to blast fungus.
Deposition of data
The sRNA sequences data and GeneChip rice genome array data are accessible through NCBI’s GEO Series accession number GSE36205 and GSE36013.
References
Abbruscato, P., Nepusz, T., Mizzi, L., Del, C. M., Morandini, P., Fumasoni, I., Michel, C., Paccanaro, A., Guiderdoni, E., Schaffrath, U., Morel, J. B., Piffanelli, P., & Faivre-Rampant, O. (2012). OsWRKY22, a monocot WRKY gene, plays a role in the resistance response to blast. Molecular Plant Pathology, 13(8), 828–841.
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., & Axtell, M. J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Current Biology, 18(10), 758–762.
Campo, S., Peris-Peris, C., Sire, C., Moreno, A. B., Donaire, L., Zytnicki, M., Notredame, C., Llave, C., & San, S. B. (2013). Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. The New Phytologist, 199(1), 212–227.
Chen, C. J., Liu, Q., Zhang, Y. C., Qu, L. H., Chen, Y. Q., & Gautheret, D. (2011). Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biology, 8(3), 538–547.
Chujo, T., Takai, R., Akimoto-Tomiyama, C., Ando, S., Minami, E., Nagamura, Y., Kaku, H., Shibuya, N., Yasuda, M., Nakashita, H., Umemura, K., Okada, A., Okada, K., Nojiri, H., & Yamane, H. (2007). Involvement of the elicitor-induced gene OsWRKY53 in the expression of defense-related genes in rice. Biochimica et Biophysica Acta, 1769(7-8), 497–505.
Deng, Y., Zhu, X., Shen, Y., & He, Z. (2006). Genetic characterization and fine mapping of the blast resistance locus Pigm(t) tightly linked to Pi2 and Pi9 in a broad-spectrum resistant Chinese variety. Theoretical and Applied Genetics, 113(4), 705–713.
Dugas, D. V., & Bartel, B. (2008). Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Molecular Biology, 67(4), 403–417.
Eulgem, T., & Somssich, I. E. (2007). Networks of WRKY transcription factors in defense signaling. Current Opinion in Plant Biology, 10(4), 366–371.
Fujiwara, M., Umemura, K., Kawasaki, T., & Shimamoto, K. (2006). Proteomics of Rac GTPase signaling reveals its predominant role in elicitor-induced defense response of cultured rice cells. Plant Physiology, 140(2), 734–745.
German, M. A., Pillay, M., Jeong, D. H., Hetawal, A., Luo, S., Janardhanan, P., Kannan, V., Rymarquis, L. A., Nobuta, K., German, R., De Paoli, E., Lu, C., Schroth, G., Meyers, B. C., & Green, P. J. (2008). Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nature Biotechnology, 26(8), 941–946.
Hafner, M., Landgraf, P., Ludwig, J., Rice, A., Ojo, T., Lin, C., Holoch, D., Lim, C., & Tuschl, T. (2008). Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods, 44(1), 3–12.
Han, M., Ryu, H., Kim, C., Park, D., Ahn, Y., & Jeon, J. (2013). OsWRKY30 is a transcription activator that enhances rice resistance to the Xanthomonas oryzae pathovar oryzae. Journal of Plant Biology, 56(4), 258–265.
Jones-Rhoades, M. W., & Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Molecular Cell, 14(6), 787–799.
Kim, S. H., Oikawa, T., Kyozuka, J., Wong, H. L., Umemura, K., Kishi-Kaboshi, M., Takahashi, A., Kawano, Y., Kawasaki, T., & Shimamoto, K. (2012). The bHLH Rac Immunity1 (RAI1) Is Activated by OsRac1 via OsMAPK3 and OsMAPK6 in Rice Immunity. Plant & Cell Physiology, 53(4), 740–754.
Lampard, G. R., Macalister, C. A., & Bergmann, D. C. (2008). Arabidopsis stomatal initiation is controlled by MAPK-mediated regulation of the bHLH SPEECHLESS. Science, 322(5904), 1113–1116.
Ledent, V., Paquet, O., & Vervoort, M. (2002). Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biology, 3(6), H30.
Li, T., Li, H., Zhang, Y. X., & Liu, J. Y. (2011). Identification and analysis of seven H(2)O(2)-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica). Nucleic Acids Research, 39(7), 2821–2833.
Li, Y., Lu, Y. G., Shi, Y., Wu, L., Xu, Y. J., Huang, F., Guo, X. Y., Zhang, Y., Fan, J., Zhao, J. Q., Zhang, H. Y., Xu, P. Z., Zhou, J. M., Wu, X. J., Wang, P. R., & Wang, W. M. (2014). Multiple rice microRNAs are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiology, 164(2), 1077–1092.
Li, T., Ma, L., Geng, Y., Hao, C., Chen, X., & Zhang, X. (2015). Small RNA and Degradome Sequencing Reveal Complex Roles of miRNAs and Their Targets in Developing Wheat Grains. PLoS One, 10(10), e139658.
Li, Z. Y., Xia, J., Chen, Z., Yu, Y., Li, Q. F., Zhang, Y. C., Zhang, J. P., Wang, C. Y., Zhu, X. Y., Zhang, W., & Chen, Y. Q. (2016). Large-scale rewiring of innate immunity circuitry and microRNA regulation during initial rice blast infection. Scientific Reports, 6, 25493.
Li, Y., Zhao, S., Li, J., Hu, X., Wang, H., Cao, X., Xu, Y., Zhao, Z., Xiao, Z., Yang, N., Fan, J., Huang, F., & Wang, W. (2017). Osa-miR169 Negatively Regulates Rice Immunity against the Blast Fungus Magnaporthe oryzae. Frontiers in Plant Science, 8, 2.
Lindow, M., Jacobsen, A., Nygaard, S., Mang, Y., & Krogh, A. (2007). Intragenomic matching reveals a huge potential for miRNA-mediated regulation in plants. PLoS Computational Biology, 3(11), e238.
Livak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods, 25(4), 402–408.
Lu, S., Sun, Y. H., Amerson, H., & Chiang, V. L. (2007). MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. The Plant Journal, 51(6), 1077–1098.
Lu, W. H., Wang, X. Z., Zheng, Q., Guan, S. H., Xin, P., & Sun, Y. Q. (2008). Diversity and stability study on rice mutants induced in space environment. Genomics, Proteomics & Bioinformatics, 6(1), 51–60.
Mei, M., Qiu, Y., Sun, Y., Huang, R., Yao, J., Zhang, Q., Hong, M., & Ye, J. (1998). Morphological and molecular changes of maize plants after seeds been flown on recoverable satellite. Advances in Space Research, 22(12), 1691–1697.
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., Cao, X., Carrington, J. C., Chen, X., Green, P. J., Griffiths-Jones, S., Jacobsen, S. E., Mallory, A. C., Martienssen, R. A., Poethig, R. S., Qi, Y., Vaucheret, H., Voinnet, O., Watanabe, Y., Weigel, D., & Zhu, J. K. (2008). Criteria for annotation of plant MicroRNAs. Plant Cell, 20(12), 3186–3190.
Mi, S., Cai, T., Hu, Y., Chen, Y., Hodges, E., Ni, F., Wu, L., Li, S., Zhou, H., Long, C., Chen, S., Hannon, G. J., & Qi, Y. (2008). Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell, 133(1), 116–127.
Morel, J. B., Godon, C., Mourrain, P., Beclin, C., Boutet, S., Feuerbach, F., Proux, F., & Vaucheret, H. (2002). Fertile hypomorphic ARGONAUTE (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell, 14(3), 629–639.
Morin, R. D., Aksay, G., Dolgosheina, E., Ebhardt, H. A., Magrini, V., Mardis, E. R., Sahinalp, S. C., & Unrau, P. J. (2008). Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Research, 18(4), 571–584.
Nishimura, M., Fukada, J., Moriwaki, A., & Fujikawa, T. (2009). Mstul, an APSES transeription factor ,is required for appressorium-mediated infection in Magnaporthe grisea. Bioscience, Biotechnology, and Biochemistry, 8(73), 1779–1786.
Nishizawa, Y., Mochizuki, S., Yokotani, N., Nishimura, T., & Minami, E. (2016). Molecular and cellular analysis of the biotrophic interaction between rice and Magnaporthe oryzae-Exploring situations in which the blast fungus controls the infection. Physiological and Molecular Plant Pathology, 95, 70–76.
Nunes, C. C., Gowda, M., Sailsbery, J., Xue, M., Chen, F., Brown, D. E., Oh, Y., Mitchell, T. K., & Dean, R. A. (2011). Diverse and tissue-enriched small RNAs in the plant pathogenic fungus, Magnaporthe oryzae. BMC Genomics, 12, 288.
Ono, E., Wong, H. L., Kawasaki, T., Hasegawa, M., Kodama, O., & Shimamoto, K. (2001). Essential role of the small GTPase Rac in disease resistance of rice. Proceedings of the National Academy of Sciences of the United States of America, 98(2), 759–764.
Qiu, Y., & Yu, D. (2009). Over-expression of the stress-induced OsWRKY45 enhances disease resistance and drought tolerance in Arabidopsis. Environmental and Experimental Botany, 65(1), 35–47.
Qiu, D., Xiao, J., Ding, X., Xiong, M., Cai, M., Cao, Y., Li, X., Xu, C., & Wang, S. (2007). OsWRKY13 mediates rice disease resistance by regulating defense-related genes in salicylate- and jasmonate-dependent signaling. Molecular Plant-Microbe Interactions, 20(5), 492–499.
Rajagopalan, R., Vaucheret, H., Trejo, J., & Bartel, D. P. (2006). A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes & Development, 20(24), 3407–3425.
Rushton, P. J., Somssich, I. E., Ringler, P., & Shen, Q. J. (2010). WRKY transcription factors. Trends in Plant Science, 15(5), 247–258.
Ryu, H. S., Han, M., Lee, S. K., Cho, J. I., Ryoo, N., Heu, S., Lee, Y. H., Bhoo, S. H., Wang, G. L., Hahn, T. R., & Jeon, J. S. (2006). A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Reports, 25(8), 836–847.
Schreiber, A. W., Shi, B. J., Huang, C. Y., Langridge, P., & Baumann, U. (2011). Discovery of barley miRNAs through deep sequencing of short reads. BMC Genomics, 12, 129.
Shimono, M., Sugano, S., Nakayama, A., Jiang, C. J., Ono, K., Toki, S., & Takatsuji, H. (2007). Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell, 19(6), 2064–2076.
Song, C., Wang, C., Zhang, C., Korir, N. K., Yu, H., Ma, Z., & Fang, J. (2010). Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics, 11, 431.
Sonnenfeld, M. J., Delvecchio, C., & Sun, X. (2005). Analysis of the transcriptional activation domain of the Drosophila tango bHLH-PAS transcription factor. Development Genes and Evolution, 215(5), 221–229.
Suharsono, U., Fujisawa, Y., Kawasaki, T., Iwasaki, Y., Satoh, H., & Shimamoto, K. (2002). The heterotrimeric G protein alpha subunit acts upstream of the small GTPase Rac in disease resistance of rice. Proceedings of the National Academy of Sciences of the United States of America, 99(20), 13307–13312.
Sunkar, R., Kapoor, A., & Zhu, J. K. (2006). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell, 18(8), 2051–2065.
Sunkar, R., Chinnusamy, V., Zhu, J., & Zhu, J. K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends in Plant Science, 12(7), 301–309.
Sunkar, R., Zhou, X., Zheng, Y., Zhang, W., & Zhu, J. K. (2008). Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biology, 8, 25.
Teune, J. H., & Steger, G. (2010). NOVOMIR: De Novo Prediction of MicroRNA-Coding Regions in a Single Plant-Genome. Journal of Nucleic Acids, 2010(2010), 10. https://doi.org/10.4061/2010/495904.
Vaucheret, H., Vazquez, F., Crete, P., & Bartel, D. P. (2004). The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes & Development, 18(10), 1187–1197.
Wang, H., Hao, J., Chen, X., Hao, Z., Wang, X., Lou, Y., Peng, Y., & Guo, Z. (2007). Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Molecular Biology, 65(6), 799–815.
Wei, L. J., Yang, Q., Xia, H. M., Furusawa, Y., Guan, S. H., Xin, P., & Sun, Y. Q. (2006). Analysis of cytogenetic damage in rice seeds induced by energetic heavy ions on-ground and after spaceflight. Journal of Radiation Research, 47(3-4), 273–278.
Wei, L. Q., Yan, L. F., & Wang, T. (2011). Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biology, 12(6), R53.
Wei, T., Ou, B., Li, J., Zhao, Y., Guo, D., Zhu, Y., Chen, Z., Gu, H., Li, C., Qin, G., & Qu, L. J. (2013). Transcriptional profiling of rice early response to Magnaporthe oryzae identified OsWRKYs as important regulators in rice blast resistance. PLoS One, 8(3), e59720.
Willmann, M. R., & Poethig, R. S. (2007). Conservation and evolution of miRNA regulatory programs in plant development. Current Opinion in Plant Biology, 10(5), 503–511.
Wu, K. L., Guo, Z. J., Wang, H. H., & Li, J. (2005). The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Research, 12(1), 9–26.
Wu, L., Zhang, Q., Zhou, H., Ni, F., Wu, X., & Qi, Y. (2009). Rice MicroRNA effector complexes and targets. Plant Cell, 21(11), 3421–3435.
Xiao, W. M., Yang, Q. Y., Wang, H., Guo, T., Liu, Y., Zhu, X. Y., & Chen, Z. Q. (2011). Identification and fine mapping of a resistance gene to Magnaporthe oryzae in a space-induced rice mutant. Molecular Breeding, 3(28), 303–312.
Zhang, W., Gao, S., Zhou, X., Xia, J., Chellappan, P., Zhou, X., Zhang, X., & Jin, H. (2010). Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biology, 11(8), R81.
Zhao, J. P., Jiang, X. L., Zhang, B. Y., & Su, X. H. (2012a). Involvement of microRNA-mediated gene expression regulation in the pathological development of stem canker disease in Populus trichocarpa. PLoS One, 7(9), e44968.
Zhao, Y. T., Wang, M., Fu, S. X., Yang, W. C., Qi, C. K., & Wang, X. J. (2012b). Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production- and development-correlated expression and new small RNA classes. Plant Physiology, 158(2), 813–823.
Zhou, X., Wang, G., & Zhang, W. (2007). UV-B responsive microRNA genes in Arabidopsis thaliana. Molecular Systems Biology, 3, 103.
Zhu, Q. H., Spriggs, A., Matthew, L., Fan, L., Kennedy, G., Gubler, F., & Helliwell, C. (2008). A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Research, 18(9), 1456–1465.
Funding
This work was supported by the National Natural Science Foundation of China (31401722) and the Natural Science Foundation of Guangdong Province of China (2014A030313463).
Author information
Authors and Affiliations
Contributions
SYD, JXZ, QYY, HW, ZQC and JFW conceived and designed the experiments. SYD, JXZ and HL performed the experiments. SYD, JXZ and DYS analyzed the data. QQY, HW and ZQC contributed reagents and materials. SYD, JXZ, QQY, HW, ZQC and JFW wrote and modified the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This research does not include any animal and/or human trials.
Ethical approval
The authors bear all the ethical responsibilities of this manuscript.
Additional information
Shuangyu Dong, Jingxin Zhang are co-first author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dong, S., Zhang, J., Sun, D. et al. Identification of Magnaporthe oryzae-elicited rice novel miRNAs and their targets by miRNA and degradome sequencing. Eur J Plant Pathol 151, 629–647 (2018). https://doi.org/10.1007/s10658-017-1399-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-017-1399-y