
Abstract
This paper reviews reported methods of the electrochemical deposition of nickel layers which are used as target materials for accelerator production of medical radioisotopes. The review focuses on the electrodeposition carried out from aqueous electrolytes. It describes the main challenges related to the preparation of suitable Ni target layers, such as work with limited amounts of expensive isotopically enriched nickel; electrodeposition of sufficiently thick, smooth and free of cracks layers; and recovery of unreacted Ni isotopes from the irradiated targets and from used electrolytic baths.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nuclear medicine is a fast-growing area of the medical sciences which has found numerous applications in modern laboratories and medical institutions. Its development strongly depends on availability of radioactive isotopes suitable for the medical applications. There are several methods of production of the latter, including nuclear reactions between target nuclei and high-energy charged particles acting as projectiles in medical accelerators [1,2,3,4,5]. Nickel isotopes (Table 1) are often applied as the target nuclei and, depending on the isotope and the projectile type and energy, their reactions produce radioisotopes of copper, cobalt and nickel which have medical importance. Some of these radionuclides are frequently applied in today’s nuclear medicine while the others are considered for the future applications (Table 2). Thus, 64Ni(p,n) reaction [2,3,4, 17,18,19, 25, 30,31,32,33,34,35,36,37,38,39,40,41,42,43,44, 62,63,64] is the main method of production of 64Cu, an isotope frequently used in positron emission tomography (PET) studies [5, 18, 20, 30, 64, 65] and also applied in internal radiotherapy [20, 44, 66, 67]. Another method of 64Cu production using Ni targets is based on 64Ni(d,2n) reaction [16, 20, 34, 40, 58]. Other PET radioisotopes which can be produced by means of nuclear reactions of nickel nuclei include 60Cu, 61Cu, 62Cu, 55Co and 57Ni [5, 9,10,11,12,13,14, 16,17,18,19,20,21,22, 24,25,26, 28,29,30, 34, 35, 41, 45, 61, 65, 66, 68,69,70]. 64Ni(α,p) is one of the reactions which can be used for production of therapeutic 67Cu radioisotope [15, 22, 23, 44, 60]. Nuclear reactions of nickel can also produce 57Co and 58Co which are radiolabelling agents used in radiotracer medical studies (e.g. Schilling test [71, 72]) [5, 10, 11, 16, 41, 69]. Finally, 64Ni(p,α) reaction produces 61Co that was suggested for use in medical molecular imaging [17]. The reactions of Ni nuclei with protons, deuterons or α particles are the primary or secondary methods of production of the medical radioisotopes mentioned above. Even if not considered as the primary method of the radioisotope production, the nuclear reactions of Ni may constitute an attractive alternative to the main production route, depending on the particle accelerator capabilities (projectile type and energy) and the isotope (target) availability. Except for the 57Ni case, the reactions listed in Table 2 allow production of carrier-free medical radioisotopes, i.e. free from other isotopes of the same element [23].
Preparation of Ni targets suitable for the use in the particle accelerators is one of the crucial steps in the process of production of the radioisotopes in question. Electrochemical deposition on a metallic substrate from an aqueous bath is the method most often applied for preparation of the nickel targets [4, 73]. This procedure offers several advantages over other methods of the accelerator targets production. It is relatively simple and does not require sophisticated and expensive equipment. The method is highly efficient [74] and the process can be conveniently controlled by applying required current density or cathodic potential. Electrodeposited layers do not require further post-plating processing [75]. Electrochemical plating allows working with minute amounts of the material and produces deposits with a wide range of thicknesses, although the maximum available thickness is often limited by peeling off of the deposit [73, 74]. Other than the latter disadvantages of the electrochemical deposition include possibility of contamination of the target with impurities from the electrolyte bath which are co-deposited or co-precipitated with the deposited metal [74]. Further on, the electrochemically plated deposits may have non-uniform thickness [31, 46, 74] and when the deposition is carried out from a solution with low metal content, the process can be time consuming (e.g. [2, 8, 10, 13, 17, 19, 45, 48, 59,60,61,62,63,64,65,66]).
Electrochemical deposition of nickel and its alloys is a well-established procedure which has been covered by numerous reviews and monographs (e.g. [76,77,78]). These papers deal with preparation of Ni deposits for, e.g. protective, electronic or decorative purposes or for applications in catalysis. This paper focuses on electrochemical deposition of nickel layers from aqueous baths intended exclusively for application in accelerator systems as targets for production of medical radioisotopes. A discussion of the electrodeposition of Ni targets from non-aqueous electrolytic baths (ionic liquids) can be found elsewhere [79].
Target requirements
The overall procedure of the production of the medical radioisotopes involves several steps, including the target preparation, irradiation of the target in an accelerator, separation of the reaction products and recovery of non-reacted target isotopes from the target material and purification of such obtained isotopes [12, 13, 17, 31, 33, 34, 46, 52,53,54, 80]. This paper focuses on the first stage of the procedure, i.e. the target preparation.
The accelerator targets used for production of the medical isotopes must meet certain medical and technical requirements [34]. Therefore, the overall approach to the electrochemical deposition of such materials may differ from the one applied for, e.g. protective layers plating. The most obvious property of the targets used for production of the medical radioisotopes is their very high chemical purity [1, 75, 81, 82]. The final product containing generated radioisotope must meet respective medical standards of purity [81, 82] and this aspect must be addressed in composition of the electrodes and the electrolyte bath used for the target deposition. Thus, the bath should not contain elements that can be co-deposited or co-precipitated with nickel and must be prepared using chemicals of appropriate purity [34]. It should be also free from toxic reagents [75]. If required, the reagents can be subjected to additional purification. As an example, solutions containing Ni ions can be purified using, e.g. ion exchange columns prior to completing the bath preparation procedure [49, 83]. The bath composition is simplified and usually includes only necessary components, such as supporting electrolyte (improves ionic conductivity of the electrolyte), pH regulators and hydrogen evolution inhibitors. Other components of typical plating baths, such as surface levellers, brighteners, accelerators and suppressors [76], are usually omitted. The components of the plating system, such as glassware and electrodes, should be subjected to a careful cleaning according to well-established procedures [17, 34, 49, 55, 83]. It is also recommended to work with a closed system where the electrolytic cell is emptied using a stream of a high-purity neutral gas [50]. Finally, the nickel deposits are also to be cleaned and dried [17, 31, 46, 50, 84].
Very important requirements in respect to the accelerator targets are imposed by parameters of the nuclear reaction of interest [3, 17, 36]. An example of the energy influence on the cross sections (σ) is shown in Fig. 1 for 64Ni(p,n) reaction [40, 85], which is commonly applied for production of 64Cu for PET studies and radiotherapy. Data are shown for the beam normal to the target, i.e. the target-beam angle equals 0°. The most desired projectile energy is the one which provides an acceptably high rate of the desired process (sufficiently high σ value, e.g. maximum seen in Fig. 1) with simultaneous reduction of probability of unwanted nuclear reactions [16, 17, 33, 35]. Figure 1 presents the case when all the incident protons hitting the target atoms have the same energy equal to the respective value given in the abscissa axis. However, the projectiles energy decreases continuously with the distance passed inside the target as a result of their interactions with the target’s atoms [86, 87]. Thus, sufficiently thick target is able to reduce high energy of the incident projectiles (descending section of the curve from Fig. 1) to the level corresponding to the highest σ values (maximum in Fig. 1). On the other hand, when the deposit is too thick, the energy of the projectile deep inside the layer drops to the value below the threshold level and the nuclear reaction is not observed. This leads to a poor utilisation of the expensive target material. Consequently, when the layer thickness is too small, the high-energy projectiles may reach the substrate beneath the Ni layer. As a result of the latter, undesired nuclear reactions of the substrate may take place making the latter highly radioactive. In this case, only a small fraction of the beam energy is used for production of the medical radioisotope. Finally, too thin target will not reduce high energy of the incident projectiles to the required level corresponding to the highest σ values.

Calculation of the optimal thickness of the target layer to be deposited requires determination of the stopping power of the projectiles inside the layer in question. This parameter represents energy loss of a charged projectile inside the matter [88] and depends both on the penetrated material and on the projectile (type and actual energy). Accurate calculations of the optimal layer thickness require application of a dedicated software (e.g. [89, 90]), as it was done in, e.g. [36, 91]. Figure 2 shows how the energy of a charged projectile decreases with the distance travelled inside a solid nickel layer. Data were calculated using SRIM application [92] for incident 11 MeV and 15 MeV protons (beam normal to the target). This figure also shows cross section values for 64Ni(p,n)64Cu reaction taken from Fig. 1 and plotted as a function of the average energy reached by the proton at a specified depth inside the nickel target. It follows that the effective Ni thickness, which reduces incident projectile energy by 90%, is equal to ca. 260 μm and ca. 450 μm for 11 and 15 MeV protons, respectively (Fig. 2). When the target-beam angle increases, the target penetration depth decreases accordingly [34, 36, 93, 94]. According to several authors, the range of the incident protons energies, optimal for the 64Ni(p,n)64Cu reaction, is from 8 or 9 to 12 MeV [33, 35, 40]. This corresponds to the highest cross section values seen in Fig. 1. It follows from Fig. 2 that the nickel thickness required to reduce energy of 11 MeV photons to 8 and to 9 MeV is equal to ca.77 and ca. 112 μm, respectively. A further increase in the layer thickness leads to a further decrease in the proton energy which eventually falls to the magnitude corresponding to considerably low cross section values (Fig. 1). Under such conditions the 64Cu yield still increases with the Ni thickness but with a slower rate [17]. Nevertheless, higher thicknesses of Ni layers, which allow beam energy reduction down to, e.g. 6 MeV, are also used in practice for production of 64Cu [55]. Some authors suggest that the optimal layer thickness should be equal to the maximum range of the projectiles in the selected material [36, 94]. This approach can be analysed using threshold energy for the specified nuclear reaction. The threshold proton energy for 64Ni(p,n)64Cu reaction is equal to ca. 2.5 MeV [36] and this value is reached at the depth of ca. 249 and ca. 439 μm for the incident 11 and 15 MeV projectiles, respectively (Fig. 2). Typical thickness of the Ni layers reported for the use in the medical accelerators vary from few to 200–300 μm, depending on the projectile type and energy and on the target-beam angle [17, 36, 41, 95].

Lines: proton energy vs. thickness of the nickel layer for the incident protons with the energy (E0) of 11 MeV (top panel) and 15 MeV (bottom panel) calculated using SRIM application [92]. Points: cross section of 64Ni(p,n)64Cu reaction taken from Fig. 1 and plotted as a function of the proton energy at a specified depth value; the lines are meant as a guide to the eye
Preferred are smooth and homogenous deposits with uniform thickness which are free of cracks, craters, etc. [34, 75]. Polycrystalline coatings, formed in a layer-by-layer manner or containing randomly oriented small size grains, meet these requirements [55, 62, 74, 95, 96]. Computer simulations reported in [97] show that application of targets, composed with crystals properly oriented in respect to the beam, may enhance the reaction yield through antichanneling phenomenon. Electrodeposition of Ni monocrystals, with the thickness even up to 100 μm or more, can be accomplished using well-oriented single crystal substrates, e.g. Cu [98,99,100,101]. This approach, however, seems to be hardly applicable for production of the accelerator targets in question. When exposed to air and moisture, the electrodeposited Ni undergoes surface oxidation which results in formation of surface nickel oxides and/or hydroxides [102]. This layer, however, does not create a significant barrier for energetic charged particles which are able to penetrate bulk of the sample.
Most of the discussed nuclear reactions use natural nickel isotopes of low abundance (Table 1). The probability of a nuclear reaction is directly proportional to the amount of the target nuclei in the irradiated material [88]. It increases when the latter is enriched with the isotope of interest. As an example, the target used in 64Ni(p,n)64Cu reaction, enriched with 64Ni up to the level of 100%, provides probability of the reaction almost 100 times greater than the target made with natural nickel (0.93% of 64Ni). Clearly, nickel used for preparation of the targets used for 64Cu production should be enriched as much as possible with the required isotope. Isotopically enriched Ni can be very expensive and usually only a small amount of such material is accessible for the target preparation. Here, the electrochemical deposition is one of the methods most suitable for work with minute amounts of the material to be converted into the target. If possible, unreacted nickel isotopes should be recovered from the irradiated target and from depleted electrolytic baths.
Thermal stability is another one target property imposed by conditions of the nuclear reactions carried out in an accelerator. Under vacuum conditions, the target experiences a thermal shock due to a fast rise in the temperature when subjected to a beam of high-energy projectiles [95]. The temperature may rise up to 200–500 °C and can be maintained for several hours [17, 43, 95]. After completing the irradiation, the target is quickly cooled down [95]. The nickel layers cannot crack or peel under such conditions. This requires high-quality nickel deposits which are free of cracks and which exhibit a very good adhesion to the substrate [34, 75, 95]. Separate tests of the target stability under conditions of fast heating and subsequent fast cooling are helpful here [34, 55, 95].
Bath preparation
The nickel can be purchased as a fresh reagent or can be recovered from the irradiated target or from a used electrolytic bath. Isotopically enriched nickel is expensive and usually accessible in limited amounts. As a result, its electrochemical deposition is typically carried out using a small volume (from few to 10 ml) of the electrolytic bath containing low or moderate concentration of the isotope (from few tens of mM to ca. 0.6 mM). The nickel concentration is selected in such a way that the deposition of all the nickel contained in the bath produces the metal layer with the required thickness.
The electrodeposition depends on numerous factors and by varying respective of them one may modify the process. These factors affect the process at its various stages and it is worth to summarise briefly proposed mechanisms of the electrochemical formation of coherent electrodeposits. Detailed discussion of this problem is out of scope of this paper and the readers are referred elsewhere, e.g. [103,104,105,106,107,108,109]. The electrodeposition process starts with reduction of the metal cations and formation of neutral adatoms at the substrate surface. The adatoms migrate across the surface until they reach energetically favourable sites where they form 2D or 3D nuclei (islands) together with other adatoms. The nucleation sites can be either located at the substrate terraces or formed by the substrate surface imperfections, such as kinks or edges [109, 110]. After reaching the critical size, the nuclei become thermodynamically stable and grow with the deposition time. Their growth can be accompanied by continuous formation of new nuclei (progressive nucleation with low nucleation rate) or the number of the nuclei may remain almost unchanged since the beginning of the process (instantaneous case with a high nucleation rate). The nuclei grow along the surface imperfection or at the terraces, depending on the substrate surface structure and the electrolyte composition. They may grow through a direct discharge of the metal cations at the nuclei surface and incorporation of such generated atoms into the nuclei lattice [111]. An alternative model assumes reduction of the metal cations at the surface of the bare substrate following surface diffusion of such formed adatoms towards the nuclei [111]. When the plating time passes, the nuclei coalesce to form continuous deposit: 2D nuclei produce a single monolayer of the metal atoms, while the 3D entities generate a few-atomic layer thick coating. Further formation of thicker, coherent deposits proceeds according to two mechanisms [112, 113]. Layer-by-layer grow includes incorporation of newly formed adatoms into kinks, steps and edges. The metal cations may be discharged directly at these surface imperfections or may diffuse from the discharge sites located at the terraces towards edges or steps [112]. New monoatomic layer grows in direction along and outside of the step. This process takes place when the substrate surface is stepwise, i.e. contains relatively smooth terraces with steps, and when the mismatch between crystal lattices of the substrate and the deposited metal is not too high [112]. Each new layer reproduces general arrangement of the original steps and edges [113]. The other mechanism (normal to the surface growth) includes formation and growth of subsequent 3D nuclei on top of the previously formed deposit layer [112]. It is followed by coalescence of the nuclei to form next continuous layer and subsequent formation of the new nuclei on the top of the latter [112]. This process takes place at surfaces of higher roughness where numerous nucleation sites can be formed in a stochastic manner [112]. It has been reported that various types of the nucleation and various mechanisms of the layer growth may occur simultaneously and the final structure of the nickel electrodeposit is a result of competition between various growth mechanisms [113].
The course of the abovementioned processes taking place during the nickel electrodeposition depends on numerous parameters. Thus, the electrode potential not only controls the charge transfer kinetics, including transition from the kinetic to the diffusional control of the process [103, 114, 115], but also influences number of the nucleation sites and the nucleation rate (e.g. [106, 116]). Further on, occurrence of parallel to the electrodeposition electrochemical reactions, such as hydrogen evolution, also depends on the electrode potential [103, 104, 117, 118]. As a consequence, the deposit properties (texture, microstructure) can be strongly affected by the deposition potential [119, 120].
The bath composition also has a strong influence on the deposition process. The bath may contain components additional to the deposited metal cations, supporting electrolyte and pH regulators. These additives affect the plating process through interaction with the substrate surface (adsorption–desorption), interaction with the nickel cations (complexation) and incorporation into the deposit [103, 114, 115, 121]. Thus, wetting agents diminish surface tension of the electrolyte, reduce undesired effects due to formation of H2 bubbles and improve homogeneity and microstructure of the Ni coating [75, 115, 122,123,124,125]. Other surfactants act as levellers and affect the deposit morphology and crystalline structure (size and preferred orientation), improve uniformity of the deposit thickness and reduce its roughness [107, 114, 115, 124, 126,127,128,129].
Organic complexing agents affect several aspects of the electrodeposition process [117, 118, 130, 131]. Formation of complexes with Ni cations improves microstructure and surface morphology of the deposit [120, 132,133,134], promotes specific orientation of the crystallites [132], changes nickel deposition rate [130, 132, 133] and increases microhardness of the coating [134], although it may decrease the overall faradaic efficiency [120, 132,133,134]. Properly selected complexes should exhibit respectively low stability; otherwise, electrodeposition may require higher cathodic polarisation as compared to the bath free from the complexing agent [75, 117]. In summary, the bath composition and pH affects the nucleation rate and number of the nuclei [106, 116, 135, 136], the deposit microstructure and texture [120, 127, 135, 137,138,139] and the layer growth rate [140].
The first step in the bath preparation procedure depends on the chemical form of the raw nickel material. When the nickel is in the form of salts, e.g. chlorides or sulphates [34, 39, 52, 55, 91, 95], the bath preparation usually starts with dissolution of the salt in water or in an electrolyte solution followed by addition of other soluble bath components, such as pH regulators or hydrogen evolution inhibitors. The salt can be subjected to a purification procedure which involves several steps of dissolution and precipitation of the nickel compounds [34]. However, isotopically enriched nickel is most often available in the form of a metallic powder [13, 31, 34, 38, 55, 68] and the bath preparation procedure requires selection of a proper method of the powder dissolution. Concentrated nitric acid dissolves metallic Ni powder easily and is usually considered as the solvent of the first choice [12, 17, 19, 31, 34, 38, 41, 43, 46,47,48,49,50,51, 56, 62, 68, 73, 96]. Such prepared electrolytic baths contain nitrates which may seriously affect the plating process. Although presence of a small amount of these anions can be beneficial for the deposit quality, too high concentration of the nitrates reduces faradaic efficiency of the deposition [141]. Nitrate ions may undergo cathodic reduction during the electrodeposition process leading to unwanted precipitation of Ni(OH)2 [142, 143]. Finally, presence of the nitric acid in the bath may result in dissolution of the nickel deposit when the latter is left under open circuit conditions or accidentally subjected to anodic polarisation [144, 145]. Therefore, various procedures aimed at removal or reduction of concentration of nitrates/HNO3 in the solutions obtained from the metal dissolution have been proposed (Table 3) [12, 17, 19, 31, 34, 38, 41, 46,47,48,49, 51, 56, 62, 68, 96, 149]. They include several steps, such as dissolution, precipitation, filtration and evaporation. Synthesis of a very high-purity Ni salt requires application of several such steps aimed to remove traces of the acids used during the procedure [62]. When the solid product is to be obtained, the final step may include precipitation of a high-purity Ni salt using solvent displacement crystallisation with an organic solvent, such as acetone [150, 151]. These procedures are, however, time consuming and require additional experimental set-ups dedicated to, e.g. safe acid evaporation. As an example, vacuum evaporation of the acid is sometimes carried out for as long as 24 h [62]. Solvents less often applied for dissolution of the metallic Ni include HCl and H2SO4 [10, 33, 34, 37, 55, 57, 59, 84]. The dissolution can be facilitated by using oxidising agents, such as H2O2 [10, 57, 59]. After completing the process, H2O2 can be catalytically decomposed using metallic Pt [57]. Table 3 also includes procedures of removal of excess chlorides/HCl in the course of the electrolytic bath preparation [10, 13, 55].
Application of the procedures listed in the Table 3 leads to obtaining a solid nickel salt (NiSO4 or NiCl2) or an aqueous solution containing Ni2+ (NiSO4 or Ni(NO3)2 in H2SO4(aq)). These crude products are subjected to a further treatment in the course of the bath preparation. It includes dissolution in water (in the case of solid salts), dilution with H2O, pH settings by adding respective buffering agents and, sometimes, addition of H3BO3 and other components of the supporting electrolyte. Two most often applied aqueous electrolytic baths used for deposition of the Ni targets are the alkaline one, usually with initial pH of 9–11 [12, 17, 19, 31,32,33, 43, 46, 47, 49,50,51,52, 56, 68, 83, 91, 96], and the slightly acidic one, typically with initial pH of 3-5 [10, 39, 41, 50, 51, 55, 62, 91, 95]. Application of the baths with other pH values, such as neutral [39, 43] or strongly acidic [38], is rarely reported. A long-time nickel electrodeposition from acidic baths (initial pH of 3.0–4.6) is accompanied by a pH decrease to the values of ca. 1.3–1.7 [55, 91]. These pH changes can be compensated by addition of pH regulators (e.g. NH4OH) during the deposition process [84].
Published compositions of the electrolytic baths used for electroplating of Ni targets used in medical accelerators are listed in Table 4 together with the deposition conditions and other parameters, such as efficiency and the deposit thickness. The alkaline baths are prepared by addition of NH4OH and NH4Cl or (NH4)2SO4 (or Na2SO4) [12, 17, 19, 31,32,33,34, 43, 46, 47, 49,50,51,52, 56, 68, 91, 96], which act as pH regulators and supporting electrolyte. In some cases, also H3BO3 is added [34, 52, 91]. In these solutions, the Ni2+ cation forms a series of complexes with NH3 with stoichiometry depending on the concentration of both components of the complex and on pH [91, 116, 152,153,154]. Formation of such complexes increases solubility of Ni(OH)2 which otherwise strongly decreases with the increasing pH [143, 155,156,157]. These complexes have a blue colour [143] which allows to track progress in the deposition process by means of colorimetric analysis of Ni content in the bath. This can be done optically or using a UV-vis spectrophotometer (main absorption maxima at ca. 612 and 373 nm [158]). Thus, when the whole Ni contained in the bath is to be deposited the plating process is continued until the electrolyte becomes colourless [17, 31,32,33,34, 38, 46, 49, 50]. It should be stressed that in the alkaline baths the NH3 can be electrochemically oxidised in reaction taking place on the anode surface. These processes lead to formation of N2 [159,160,161], nitrates [161, 162] or chlorinated organic compounds when the chlorides are present in the electrolyte [163].
The slightly acidic baths are prepared by dissolution of Ni salts in water or by respective treatment of the acidic solutions obtained from the metal dissolution, e.g. dilution with water (Table 4). The next steps may include addition of H3BO3 [10, 39, 55, 91] and inorganic salts containing NH4+ [51] which also act as a supporting electrolyte and which may improve quality of the deposit [76]. Although both chlorides and sulphates which may be present in these baths can form complexes with nickel(II) cations [156], it was reported that at low pH values and for sufficiently low concentration of the anions, the Ni2+ exists mainly as an aqueous complex in such electrolytes [152, 164,165,166]. These complexes are green (main absorption maxima at ca. 719 and 393 nm [152, 158, 167]), and, similarly to the alkaline bath case, the colour of the electrolyte can be used to track the progress in the deposition process. Ni2+ complexes with chlorides and sulphates prevail at high concentration of the anions, especially in excess to the nickel cations [116, 120, 165]. Although sulphates/bisulphates apparently do not affect significantly the form of the Ni2+ ions in the electrolyte [152], the anion identity influences the kinetics of the cation reduction [78, 168]. This effect is attributed to formation of complexes between partially discharged Ni cations (Ni+) and the anions. These complexes act as intermediates in the electrochemical reduction of Ni2+ [168]. The anion identity (chloride or sulphate/bisulphate) also influences the density of the nucleation sites and the nickel nucleation rate [169]. Presence of chlorides in the acidic bath increases electrical conductivity of the electrolyte [152], affects hydrogen evolution [138, 168], reduces stress inside the deposited nickel [170] and improves quality of the latter [91, 168]. It is also convenient to use the chloride bath when the nickel is recovered from the irradiated target by means of dissolution in HCl [38]. Such obtained solution or solid nickel salt already contains chlorides and can be easily converted to the electrolytic bath [33, 38, 55]. The chlorides, however, are anodically oxidised on both the Pt [171] and graphite [172, 173] anodes. Further on, the presence of the chlorides enhances anodic dissolution of platinum which may take place under strong anodic polarisation [174,175,176], i.e. under conditions often applied in the target deposition procedures. Such dissolved platinum can contaminate the nickel deposit.
Boric acid is the only other than the supporting electrolyte or the pH regulators addition often present in electrolytic baths used for the Ni target electrodeposition [10, 13, 34, 39, 52, 55, 91]. Dissociation of this weak acid strongly depends on the acidity. Thus, for pH below ca. 8–9 and for the acid concentrations in the range of 10−1 M, the undissociated H3BO3 is the prevailing form of the reagent present in the electrolyte [177]. This reagent has a strong influence on the nickel electrodeposition, although the literature is not fully consistent about its role [77, 78]. According to various reports, it prevents formation of Ni(OH)2 [178,179,180,181,182], improves deposit morphology and adhesion to the substrate [55, 76, 168, 183, 184], reduces number of cracks in the deposit [168], catalytically enhances Ni electrodeposition [179], regulates pH near the electrode surface [76, 180, 184] and suppresses hydrogen evolution [184]. The mechanism of the boric acid influence on the nickel electroplating seems to be not fully explained; it might be related to pH changes near the electrode surface (buffering agent) [178, 179, 183], surface adsorption [183] or formation of soluble complexes with the nickel cations [137, 177, 185]. Results of studies on H3BO3 influence on the electroplating of Ni targets for production of medical radioisotopes were reported in [91]. It was confirmed that under conditions of the target electrodeposition, the presence of the boric acid in the bath improves quality of the deposit (lower surface roughness and lack of pitting), although it has no evident and clear influence on the plating efficiency [55, 91]. Auger electron spectroscopy analysis of the surface and layers next to the surface of Ni deposited from a bath containing H3BO3 shows that boron is present only in the bottom layers of the deposit, i.e. in those being in contact with the substrate [91]. NiB alloys are not formed during nickel electrodeposition from acidic baths containing H3BO3 [55, 186].
The limited number of the chemical reagents present in the electrolytic baths listed in the Table 4 helps to reduce the risk of contamination of the deposited targets. Reported results of chemical analysis reveal that such prepared Ni targets are free of significant quantities of metallic impurities [91, 96]. Before starting the plating procedure, the bath, especially the one intended to be mixed by stirring during the deposition process, can be subjected to filtration in order to remove undissolved solid matter [34].
Recovery of Ni isotopes
The irradiated target usually contains a significant amount of non-reacted Ni isotopes [4, 12, 16, 17, 19, 25, 31,32,33, 38, 41,42,43, 46, 50, 52, 54,55,56, 60, 80, 83, 93] that can be recovered and used for preparation of a new electrolytic bath. This can be accomplished providing that the purity of such obtained reagents is satisfying [54, 80]. In the first step of the recovery process, the irradiated target is dissolved using aggressive reagents, such as concentrated HCl, HNO3, often at elevated temperatures. Such obtained solution is subjected to ion exchange chromatography aimed at separation of the medical radioisotope from other reagents, including unreacted nickel [16, 17, 25, 31,32,33, 38, 41, 43, 46, 48,49,50, 52, 54, 56, 60, 80, 83]. As an alternative, one may separate Ni and Cu using selective liquid–liquid extraction [93] or using spontaneous electrodeposition with, e.g. Ni and Pt electrodes in a two compartment electrochemical cell [84]. Conditions applied in the recovery step are imposed mainly by the medical purity requirements and discussion of details of this procedure is out of scope of this paper. Instead, we focus on the eluted or extracted fraction containing the unreacted nickel. Several methodologies have been proposed here. In a simplest approach, one may directly convert the nickel containing eluate into the plating solution by diluting or preconcentrating, setting pH, adding respective chemicals, such as H3BO3 [33]. More complex approaches involve additional purification of the Ni containing fraction [47, 49]. Thus, it can be passed through an additional ion exchange column [31, 32, 38, 46, 49, 52, 80] and/or subjected to evaporation of the liquid and dissolution of such formed solid Ni salt. The latter evaporation–dissolution procedure can be applied once [17, 31, 38, 46, 48, 55] or repeated multiple times [12, 41, 47, 49, 50, 54, 56]. When the acid used for the dissolution of the nickel target and elution of the ion exchange columns contain the same type of anion as the one present in the intended bath, e.g. chlorides, one may easily convert the solution obtained in the recovery procedure into the plating solution by diluting or preconcentrating, setting pH, adding respective chemicals, such as H3BO3 [31, 33]. The final composition of the baths containing recovered Ni and the deposition parameters are the same as for the solutions prepared using the fresh reagents [12, 17, 31, 34, 41, 46, 47, 49, 50].
A nickel recovery from the electrolytic baths is another source of the valuable isotopes which can be used for preparation of new electrolytes. A method of the metal recovery from an acidic bath after a failed deposition attempt has been described in [187]. It is based on quantitative adsorption of Ni ions on an ion exchange resin. The resin is introduced into a chromatographic column followed by elution of nickel. The eluate can be further purified using another ion exchange column. When the resin and eluents are properly selected, one obtains a Ni2+ solution with a very low content of metallic impurities and additives originally present in the bath, such as H3BO3. Nickel can be also recovered from used baths which are depleted with the metal due to its consumption in the plating process [32]. Several such baths can be jointly reprocessed to obtain one with a sufficiently high Ni concentration [32].
Target electrodeposition
The nickel layers intended to be used as accelerator targets are deposited on a substrate which must possess good electrical and thermal conductivity [2, 32, 34, 74, 188]. The substrate must be relatively inexpensive, must be available at the required purity level and must provide a good adhesion of the deposited layer [32, 34, 188]. It must be also chemically inert when in contact with aggressive solvents, such as concentrated HCl, H2SO4, HNO3 or aqua regia. These reagents are used for dissolution of the irradiated target layer containing generated medical isotopes and unreacted nickel [10, 12, 13, 17, 27, 31,32,33, 37,38,39, 41, 43, 46,47,48,49,50, 52, 53, 55, 56, 60, 62, 80, 83, 84, 91, 93, 96, 188]. Typical substrates used here include solid (disc, foil etc.) polycrystalline Au [13, 17, 19, 25, 32,33,34, 38, 42, 47,48,49, 51, 53, 55, 56, 60, 83, 188], Pt [34, 46, 50], Ag [12, 37, 39] and Cu, the latter covered with a thin protective layer of Au or Ag [34, 41, 52, 93, 95]. The latter are intended to prevent the copper from dissolution when the target is in contact with the acids. Such protective layers are usually obtained by electrodeposition [34, 52, 95]. It should be bearing in mind, however, that practical application of Cu substrates covered with Au or Ag can be complicated by intermixing of the atoms at the interface between the copper and the protective layer. Cu atoms which enter the protective layer diffuse towards the Ni deposit and may contaminate the latter [34, 189,190,191,192,193]. Such interface mixing (interdiffusion) and subsequent diffusion inside the solid state are accelerated at elevated temperatures [189, 190] which can be reached by the target during the irradiation. Silver has the highest electrical and thermal conductivity amongst the most common substrate materials [32, 146] but is less chemically stable than Au or Pt and may dissolve when in contact with HCl [32]. Further on, adhesion of Ni deposits to Ag substrate may be relatively weak [194]. Platinum has better mechanical properties than gold but exhibits a lower electrical and thermal conductivity [32, 146]. It has been reported that thin gold foil is more suitable for use as the substrate for the accelerator targets than massive Au discs [32]. Less often reported substrates include Rh [84], graphite and Nb [34].
Apart from the chemical and physical properties of the substrate, one must also consider possible nuclear reactions taking place when the beam reaches the substrate material. The selection is based on a compromise between safety of the personnel and time required to deactivate the irradiated substrate. Preferred are substrates which do not produce significant amounts of radioisotopes and, if any, generate only short-lived radionuclides which do not emit very harmful radiation [32, 84]. Short half-life (τ1/2) increases the radionuclide-specific activity and dose delivered to the personnel and the chemical reagents, but, on the other hand, it allows reducing the time between the irradiation and the next use of the same substrate. As an alternative, one may allow generation of very long-lived radioisotopes which activity is very small due to a very long τ1/2. Here, we analyse 64Ni(p,n)64Cu reaction as an example. Typical energy of the protons used for the 64Cu production is not higher than 20 MeV. We assume that the nickel layer is thick enough, so the energy of the protons which hit the substrate is reduced to the value not exceeding 15 MeV [90]. Most important reactions of such protons with typical substrates are collected in Table 5. It follows that gold meets the abovementioned requirements better than Pt and Ag: it produces radioisotopes with shorter τ1/2 (faster deactivation of the target) in reactions with lower cross sections (lower amount of produced radionuclides) as compared to Pt and Ag [32]. Rh is a very expensive metal but its irradiation with protons with energy up to 15 MeV produces almost exclusively short-lived 103Pd which does not emit harmful radiation [84]. Niobium activation with protons produces short-lived 93mMo which decays to very long-lived 93Mo. The latter, however, emits only low-energy electrons and X-rays [7]. Apparently, application of niobium as the substrate has no other advantages [34].
The cathode (substrate) is flat (e.g. disc shaped) and usually located at the bottom of the electrolyte cell [10, 12, 46, 47, 50, 55, 62, 68, 96, 208], although it can be arranged also as a component of the wall of the cell [39]. The exposed area of the target (typically 0.5–1 cm2) can be limited using, e.g. dedicated o-rings, perforated plugs, masking plates or other components of the cell [10, 12, 13, 46, 49, 55, 83]. In automated systems, the substrate is arranged as a component of an element (a transport shuttle) which is automatically transferred between various sections of the system used for production of the radioisotopes [38, 46, 50]. The substrate is to be subjected to a proper cleaning/pre-treatment procedure in order to provide good adhesion of the deposit [37, 38, 49, 95]. These procedures include both chemical (dissolution of surface contaminants; etching) as well as mechanical (fine sanding) treatment [31, 32, 34, 37, 38, 49, 95, 209]. In contrast to the substrates made with noble metals, less noble materials, such as Cu, may require removal of native surface oxides/hydroxides. The separation/recovery of the elements from the irradiated target can be accomplished using electrochemical anodic dissolution of the target layer [10, 17, 33, 50] which also requires good electronic conductivity of the substrate. It is worth to stress that the electrochemical plating can be applied also for production of self-supporting nickel layers that can be used as targets in medical accelerators. A thick nickel layer is plated on an easily soluble substrate, e.g. Cu [29, 149, 209]. The Ni layer remaining after the dissolution of such substrate acts as the target, providing that its thickness and mechanical stability are sufficiently high [29, 149, 209]. This approach, however, has not been applied in production of medical isotopes.
Ni target electroplating is typically carried out in a two-electrode system with the substrate used for the deposition acting as the cathode and with a non-consumable anode (counter electrode) made with various materials. The anode material should be stable under strong anodic polarisation and should be available at an appropriate purity level [17]. Most often used anodes are made from platinum [10, 12, 13, 31, 33, 34, 38, 39, 41, 46, 48, 50, 51, 55, 56, 62, 83, 91, 95, 96] or graphite [17, 19, 32, 47, 49, 51, 68]. Both these materials are available at a high-purity level [34]. They are relatively electrochemically stable but differ in respect to mechanical properties and electrochemical activity. Graphite and platinum rods or wire can be arranged as a rotating element which is used for stirring the electrolyte and removal of generated H2 [10, 13, 32, 41, 47, 49, 62, 84]. Apart from the rods, the platinum is available also in the form of foils and wires. In contrast to the graphite, the Pt anode can be easily formed in various shapes designed to improve the electrodeposition process. This includes high real surface electrodes (e.g. gauze wire, spiral or platinised Pt, i.e. with the surface expanded by electrochemical deposition of a high surface area Pt layer [210]), which are required for reduction of the anodic polarisation, and structures which surround the cathode and provide more uniform distribution of the deposition currents at the substrate [76]. Platinum is more electrochemically active than the graphite and may behave as a stronger catalyst of other than oxygen evolution reactions, e.g. unwanted redox processes of the supporting electrolyte [211]. Both the platinum and graphite undergoes anodic corrosion, especially in acidic solutions under strong anodic polarisation [173, 212,213,214]. A significant undesired anodic dissolution of platinum takes place in acids in the presence of, e.g. halides [174,175,176, 214]. The platinum and graphite anodes are active in electrochemical oxidation of Cl− [171,172,173] and NH3 [161]. The reactions of the latter may result in formation of unwanted nitrates [161].
The electrodeposition is usually carried out either in a galvanostatic (controlled constant current) [32, 33, 37, 39, 41, 48, 49, 51, 55, 56, 62, 91, 95, 96] or potentiostatic (controlled constant voltage) modes [10, 13, 17, 34, 38, 46, 47, 50, 55, 68]. Thus, the electrode is polarised by applying selected current or voltage value. In the latter case, the potential difference (voltage) is typically measured between the cathode and the anode. When possible, a separate reference electrode, such as saturated calomel electrode or Hg|Hg2SO4, can be used in potential measurements [57, 91]. Most of the authors report electrodeposition using a single value of the current (galvanostatic) or voltage (potentiostatic). As compared to the galvanostatic mode, the potentiostatic deposition allows better control of the deposit morphology which can be determined by the electrode potential [119, 215]. Such obtained deposits are more uniform in morphology [119] and are composed with grains with narrower size distribution [216] as compared to the galvanostatic plating. Studies on the electrodeposition of Ni films for applications in microelectronics reveal that the potentiostatic plating produces deposits with quality better than for the galvanostatic mode, especially at low temperatures [217]. Long-time electroplating of the target which is carried out until all the nickel cations are deposited is accompanied by a continuous decrease in Ni2+ concentration in the bath. This is linked to the transition from kinetic to diffusional control of the process and indicates decreasing rate of the plating. Under such conditions, the rate of the potentiostatic plating drops to a very low value. When the potentiostating plating is carried out using an acidic bath, one may stabilise the current by continuous addition of an acid (e.g. HCl) in drop-wise amounts during the deposition [38]. As an alternative, a gradual increase in the applied voltage allows maintaining the required current value [68]. On the other hand, the galvanostatic mode is more convenient when one wants to control amount of the charge passed and to impose the reaction rate at initial stages of the process [75], while strict control of the deposit morphology is not necessary. Ni target electrodeposition using complex current or voltage programmes is rarely reported. Only few papers describe Ni target deposition using pulsed current: in [37], a saw tooth current ramp was applied, while in [83], “regular” pulsed electrodeposition with a square wave (on–off) current programme was used.
The plating procedure usually requires deposition of all the nickel dissolved in the bath and this task is complicated by the fact that the metal concentration in the bath is usually low or moderate being limited by availability of the expensive nickel isotopes [51]. Under such conditions the deposition rate may quickly become limited by diffusion of nickel species towards the substrate (diffusion overpotential or concentration polarisation). Deposition of thick Ni layers from such baths is a time consuming process [2, 8, 10, 13, 17, 19 ,45, 59, 60–66]. It is, therefore, desired to develop the deposition procedure sufficiently fast as to ensure complete Ni deposition within a reasonable time. This requires an analysis of the factors which affect the plating efficiency. This includes the bath composition, the substrate surface pre-treatment, the applied current density or voltage, temperature and the electrolyte motion (stirring or recirculation). In some cases, the reports do not contain enough data required for complete scientific evaluation of the plating procedure. Nevertheless, data reported in Table 4 and supported by results of other works devoted to Ni electrodeposition for non-medical purposes allow drawing binding conclusions about selection of plating parameters suitable for preparation of the targets for the medical accelerators.
Influence of the nickel content in the bath
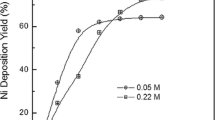
In general, the faradaic efficiency decreases with the decrease in the Ni concentration in the bath (Table 4). The plating and faradaic efficiency decrease with the deposition time due to the decreasing Ni content in the bath [51, 55, 91] and due to influence of the deposition time on pH of the bath which, in turn, affects the hydrogen evolution [91, 106]. Only at the beginning of the plating process the faradaic efficiency as high as 80% is observed [55]. Thus, even if as many as more than 85% of Ni is deposited during the first few hours of the process, the almost complete (quantitative) deposition of, say 97% of the nickel present in the bath requires up to 10–20 h [17]. As an example, deposition of ca. 100–130-μm-thick layers of Ni from baths containing 0.14–0.38 M of Ni requires from 6 to more than 20 h (estimated faradaic efficiency of 8–58%) (Table 4). For comparison, when the nickel concentration in an acidic bath is as high as ca. 1.7 M, the deposition of a nickel layer with the thickness of 108 μm takes ca. 2.5 h with the faradaic efficiency of ca. 74% [10]. Therefore, the nickel concentration in the bath should be respectively high, what can be achieved by reducing volume of the electrolyte used, if possible.
Influence of the bath pH
In general, the efficiency of the Ni electroplating increases with pH [48, 50, 51, 91, 95, 106, 134, 138, 158] and the target electrodeposition proceeds significantly faster and with a higher faradaic efficiency when an alkaline bath is used instead of an acidic one [50, 51, 91, 95]. Nevertheless, even in the case of the alkaline baths, the complete deposition of few tens to 100 mg of Ni takes many hours and the total faradaic efficiency lower than 50% is often observed (Table 4). Further on, the quality and mechanical stability of the nickel deposits deteriorates at high pH [39, 158] and poor reliability and reproducibility of the target deposition carried out in the alkaline baths is reported [48, 51]. Finally, the internal stress in Ni coatings strongly increases with the increasing pH of the bath [76]. The maximum current densities are comparable for the acidic and the alkaline baths (Table 4). The bath pH may affect the nickel electrodeposition in different ways. Thus, it has an influence on Ni2+ complexation: at high pH values, stable ammonia complexes predominate, while at low pH values, Ni2+ form stable complexes mainly with water or anions [116]. Identity of nickel complexes, which act as intermediates in the electroreduction process, is also pH dependent [116, 218]. The course of various eletrodeposition steps, such as adsorption at the electrode surface, decomposition of the complexes and the Ni2+ reduction (kinetics and thermodynamics), depend on composition of these Ni containing species, which, in turn, is pH governed [116, 136, 218, 219]. pH also affects adsorption of ions and hydrogen evolution [116, 136, 220]. All these factors lead to pH-dependent nucleation rate and nuclei density [116] and this, in turn, results in pH-dependent deposit microstructure and texture [215, 220].
Apparently, the alkaline baths are more often applied when the whole nickel is to be deposited and are more suitable when the deposition time and the faradaic efficiency play important roles as compared to the acidic electrolytes. Nevertheless, due to drawbacks of the deposition carried out from the alkaline baths, such as reported poor reproducibility and deposit quality, there is room for development of the acidic plating solutions.
Influence of the cathodic polarisation
Application of a strong cathodic polarisation of the substrate, i.e. high voltage or high current density, is probably the most obvious method of increasing the plating rate. An analysis of Table 4 shows that the faradaic efficiency increases with the voltage applied to the substrate. This approach, however, is limited by the fact that under too strong cathodic polarisation of the substrate, some undesired effects occur. Thus, under such conditions, the metal electroreduction takes place simultaneously to hydrogen evolution reaction, HER. As a result, a significant amount of the electricity is consumed by HER [33, 91] which lowers the faradaic efficiency of the target deposition, even down to a few percent (Table 4).
Further on, at too high current densities, the morphology of the deposit deteriorates [39, 55, 91, 95, 158, 209] and a high internal stress is developed [76, 184, 221]. Poor-quality deposits are obtained also when the current density is too low indicating that the applied current density should vary in a limited range [95]. The decrease in the deposit quality due to the increasing cathodic current can be attributed to the effects caused by the hydrogen evolution taking place parallel to the Ni deposition. It is assumed here that the HER contribution to the total current rises when the electrode polarisation becomes more cathodic. Presence of bubbles of gaseous hydrogen attached to the electrode surface leads to formation of the deposits with a high surface roughness and containing pores or cauliflower-like structures [117, 221]. Further on, the hydrogen bubbles can be entrapped inside the Ni deposit forming microvoids which may affect its mechanical properties [222]. The deposits formed under conditions of the excessive H2 bubbling are brittle [223] and this, in connection with non-uniform thickness of the rough deposits, limits application of high current densities for depositions of the accelerator targets.
Hydrogen absorption (uptake) by nickel, which may take place during electrochemical plating of this metal, is another process which may affect structure of the deposit and which importance rises with the increasing cathodic polarisation [78, 180, 223,224,225,226,227]. Atoms of the absorbed hydrogen occupy interstitial sites inside the nickel lattice and form a solid solution or a nickel hydride (bulk or subsurface), depending on the absorbed hydrogen concentration [78, 130, 228,229,230,231]. Desorption of hydrogen which was absorbed in the nickel coatings during the metal deposition as well as absorption–desorption of hydrogen in/from earlier prepared Ni layers lead to changes in the lattice constant of the metal [231, 232]. As a result, undesired stresses inside the metal lattice are generated [223, 226, 227, 233, 234]. This, in turn, may lead to formation of cracks in the deposit [234, 235] which make them unsuitable for the accelerator applications. The importance of the hydrogen absorption, which is so disadvantageous to the Ni deposit quality, increases with the cathodic polarisation [232] and becomes more important when certain absorption promoters are present in the electrolyte (e.g. thiourea, CN−, S2−, depending on pH) [138, 223, 227, 229, 232]. It follows then that such species should not be present in significant amounts in the baths used for plating the nickel targets.
Influence of other parameters
An analysis of Table 4 shows that a higher faradaic efficiency is achieved when the galvanostatic deposition is applied instead of the potentiostatic mode and when the Au substrate is used instead of Pt (complete Ni deposition from alkaline baths). The latter effect is attributed to the fact that the activity of Pt towards the hydrogen evolution reaction in alkaline electrolytes is higher than for Au [236, 237]. Studies carried out in the temperature range of 24–60 °C show that the target deposition efficiency is improved when temperature is raised [55, 95]. It should be stressed, however, that these tests were performed for relatively thin nickel target layers with the thickness of up to ca. 65 μm. The Ni targets obtained at 55 oC were found to have a better surface morphology [55, 95]. On the other hand, decorative and protective Ni coatings deposited from Watts bath reveal a decrease in the tensile strength and in the Vickers hardness when the bath temperature is increased up to ca. 50–60 oC [76]. The target deposition at elevated temperature carried out from a small volume bath may require continuous compensation of evaporated water using a dedicated pumping system [41].
Several studies on the electrochemical deposition of Ni targets reveal that the plating efficiency increases when the electrolyte is subjected to a motion [46, 50]. This can be done using a rotating anode [10, 13, 32, 41, 47, 49, 62], magnetic stirring rod [56], other rotating element [95] or using an electrolytic cell with recirculation of the electrolyte in a closed system [46, 50]. These procedures not only restore Ni2+ concentration near the electrode surface but also effectively remove H2 from the electrolyte [50]. The electrolyte stirring may affect morphology, texture and physical properties of the nickel deposits [41, 107, 134, 238, 239]. It has been reported that the deposit quality is improved when direction of the stirrer rotation is reversed during the plating process [41].
The quality of the deposit is determined also by the electrolytic cell geometry. The local plating rate and the deposit thickness distribution depend on the location of the anode in respect to the centre of the substrate (cathode) and resulting homogeneity of the electric field [46, 75]. Although the electrodeposition may lead to formation of cone-shaped Ni deposits with non-uniform thickness [31, 46], such targets also can be used in medical accelerators.
Suggested future works
It follows from the presented review that the electrochemical deposition of Ni targets for medical purposes is a well-established process which is routinely used in practice today. Nevertheless, several of its aspects need to be improved. It is suggested that the future works on this topic should address the following points:
- Improvement and simplification of the method of the nitrite removal from the solution obtained by the dissolution of the metallic Ni in HNO3. As an alternative, a novel method of nickel powder dissolution in a HNO3 free solvent may be also developed.
- Reduction of the electrodeposition time. Suggested solutions to the problem include application of a proper motion of the electrolyte during plating (stirring, circulation etc.) and selection of optimal current density or voltage.
- Increase in the faradaic efficiency of the process and, consequently, reduction of the plating time. Addition of proper hydrogen evolution inhibitors or Ni deposition accelerators can be helpful here [76, 78]. Special care must be taken in order to avoid introduction of substances which may contaminate the deposit.
- Addition of proper concentration of respective, non-toxic surfactants and complexing agents to the electrolyte bath in order to improve quality of the deposit (e.g. [57, 59, 75, 115, 124, 137, 240,241,242,243,244]. Such an approach may facilitate high current fast electrodeposition and may reduce the plating time. These additives or its reduction products, however, can be incorporated into the deposit leading to presence of unwanted elements in the target layer [75, 103, 107, 114, 115, 121, 123, 124, 245,243,244,245,249]. Therefore, separate studies must be performed in order to analyse the mechanism of the additives action (e.g. adsorption–desorption, reduction) and to evaluate its incorporation into the nickel deposit [103, 107, 115, 250, 251].
- Microstructure of the nickel deposits is significantly improved when complex current or voltage programmes, such as pulsed plating (e.g. [117, 127, 240, 248, 252,250,251,252,256] or different initial and final deposition parameters (current or voltage) [221], are applied. In the “regular” pulsed electrodeposition procedure, the cathodic polarisation is periodically switched on and off [257, 258]. During the “off” step, the concentration of the metal cations at the electrode surface undergoes relaxation, while hydrogen diffuses out of the electrode [252, 254, 257]. It is also suggested that Ni(OH)2 particles, which can be formed on the deposit surface, are desorbed during the “off” cycle [252, 254]. The reverse pulsed plating procedure comprises of alternatively applied cathodic and anodic polarisation steps [75, 117, 255, 257,255,256,260]. Apart from Ni2+ concentration relaxation and hydrogen removal from the electrode, the application of the anodic step also leads to a partial dissolution of the nickel deposit [75, 117, 255, 257, 258]. When the anodic polarisation conditions (time and current or potential) are properly selected, only most active surface features, such as low coordination-number adatoms, which form roughness increasing features, like pyramids or peaks, are dissolved. This produces smoother surfaces as compared to the direct current electrodeposition and “regular” pulsed approach [117, 245, 260]. The faradaic efficiency can be also improved when the reverse pulse plating is applied [245]. This may increase the plating rate through improving quality of the deposits obtained under conditions of strong cathodic polarisation of the substrate (applied high voltage or high current density). Such procedure, however, requires proper selection of several parameters, such as duration of respective steps, pulse frequency and applied cathodic and anodic polarisation. Further on, the reversed pulsed plating requires electrolytic bath with pH sufficiently low for nickel dissolution. Otherwise, the anodic polarisation may lead to surface passivation with nickel oxides or hydroxides instead of the dissolution. Finally, the equipment used in this type of the experiments is more sophisticated than that used for single current or voltage deposition.
- Addition of anodic and cathodic depolarisers reduces polarisation (overvoltage) of the anode and cathode (substrate), respectively [75, 118, 139, 248, 261]. Thanks to lowering overpotentials on the respective electrodes, they diminish undesired evolution of gases on both electrodes and dissolution of the anode material [139, 261]. Electrochemical reactions of such depolarisers, however, may result in unwanted effects. Thus, products of the electrochemical reaction of the depolariser may be incorporated into the plated layer or may cause additional chemical processes, such as those leading to precipitation of Ni(OH)2 when nitrates are used as the cathodic depolariser.
- Texture and microstructure of relatively thin Ni can be affected by selection of the substrate with a certain crystalline structure [108]. Also proper pre-treatment of the substrate surface, such as mechanical or electrochemical polishing, annealing and etching, improves properties of the electrodeposited nickel, e.g. roughness, microstructure, texture and tensile strength [262,260,264]. These approaches can be easily applied when the substrate has a form of, e.g. regular metal plate, but their applicability becomes limited when the substrate constitutes integral section of a complex transport shuttle used in automated systems.
- Deposition of the Ni target layers on different than the described above materials suitable for use in accelerator systems, e.g. Al [32, 68]. These substrates must be properly secured against dissolution during the isotopes recovery.
Conclusions
Reactions of nickel nuclei with high-energy charged particles (protons, deuterons, α particles) produce Cu, Co and Ni radioisotopes of medical importance. These reactions require a beam of high-energy charged particles, provided by an accelerator, and a respective nickel containing target to be bombarded by these projectiles. Typically applied are solid nickel targets prepared by electrochemical deposition on a conducting substrate. The targets must meet certain medical and technical conditions in order to be suitable for production of the medical isotopes. These requirements, in turn, must be addressed in the procedure of the electrochemical target plating. Preparation of the suitable Ni targets requires both electrolytic bath with proper composition and application of carefully selected deposition parameters. Several methods of the electrochemical deposition of the accelerator targets containing nickel layers are reviewed in this paper. They differ in respect to the electrolyte composition, including nickel concentration, and the deposition parameters. Different types of the raw nickel material, i.e. salts or metallic powder, are used for preparation of the bath. Further on, various methods of treatment of the isotopically enriched metallic Ni powder prior to its introduction to the bath are reported. It follows that the nickel layers with the thickness ranging from 100 to over 200 μm can be deposited from aqueous solutions containing low or moderate concentrations of the nickel ions. Properties of these layers, such as surface morphology, mechanical stability, cohesion and adhesion to the substrate, satisfy the requirements of the medical accelerators of the particles. Deposition of such targets, however, is time consuming and often requires at least several hours. It is suggested that certain aspects of the procedure of the electrochemical plating of the nickel targets can be improved in the future.
References
Hellborg R (2005) Electrostatic accelerators. Fundamentals and applications, Springer, Berlin
Synowiecki MA, Perk LR, Nijsen JFW (2018) Production of novel diagnostic radionuclides in small medical cyclotrons. EJNMMI Radiopharm Chem 3:3-1–3-25
Qaim SM (2017) Nuclear data for production and medical application of radionuclides: present status and future needs. Nucl Med Biol 44:31–49
Qaim SM, Spahn I (2018) Development of novel radionuclides for medical applications. J Label Compd Radiopharm 61:126–140
Boros E, Packard AB (2019) Radioactive transition metals for imaging and therapy. Chem Rev 119:870–901
De Laeter JR, Böhlke JK, De Bièvre P, Hidaka H, Peiser HS, Rosman KJR, Taylor PDP (2003) Atomic weights of the elements: review 2000. Pure Appl Chem 75:683–800
L’Annunziata MF (2012) Handbook of radioactivity analysis. Elsevier, Oxford
Nuclear structure & decay data, National Nuclear Data Center, Brookhaven National Laboratory (2020) https://www.nndc.bnl.gov/nudat2/
Amjed N, Hussain M, Aslam MN, Tárkányi F, Qaim SM (2016) Evaluation of nuclear reaction cross sections for optimization of production of the emerging diagnostic radionuclide 55Co. Appl Radiat Isot 108:38–48
Spellerberg S, Reimer P, Blessing G, Coenen HH, Qaim SM (1998) Production of 55Co and 57Co via proton induced reactions on highly enriched 58Ni. Appl Radiat Isot 49:1519–1522
Tárkány F, Szelecsenyi F, Kopecky P (1991) Excitation functions of proton induced nuclear reactions on natural nickel for monitoring beam energy and intensity. Appl Radiat Isot 42:513–517
Valdovinos HF, Hernandez R, Graves S, Ellison PA, Barnhart TE, Theuer CP, Engle JW, Cai W, Nickles RJ (2017) Cyclotron production and radiochemical separation of 55Co and 58mCo from 54Fe, 58Ni and 57Fe targets. Appl Radiat Isot 130:90–101
Radford LL, Fernandez S, Beacham R, El Sayed R, Farkas R, Benešová M, Müller C, Lapi SE (2019) New 55Co-labeled albumin-binding folate derivatives as potential PET agents for folate receptor imaging. Pharmaceuticals 12:166-1–166-12
Tárkányi FT, Ignatyuk AV, Hermanne A, Capote R, Carlson BV, Engle JW, Kellett MA, Kibédi T, Kim GN, Kondev FG, Hussain M, Lebeda O, Luca A, Nagai Y, Naik H, Nichols AL, Nortier FM, Suryanarayana SV, Takács S, Verpelli M (2019) Recommended nuclear data for medical radioisotope production: diagnostic positron emitters. J Radioanal Nucl Chem 319:533–666
Takács S, Aikawa M, Haba H, Komori Y, Ditrói F, Szűcs Z, Saito M, Murata T, Sakaguchi M, Ukon N (2020) Cross sections of alpha-particle induced reactions on natNi: production of 67Cu. Nucl Inst Methods Phys Res B 479:125–136
Zweit J, Smith AM, Downey S, Sharma HL (1991) Excitation functions for deuteron Induced reactions in natural nickel: production of No-carrier-added 64Cu from enriched 64Ni targets for positron tomography emission. Appl Radiat Isot 42:193–197
Avila-Rodriguez MA, Nye JA, Nickles RJ (2007) Simultaneous production of high specific activity 64Cu and 61Co with 11.4 MeV protons on enriched 64Ni nuclei. Appl Radiat Isot 65:1115–1120
Bailey DL, Townsend DW, Valk PE, Maisey MN (2005) Positron emission tomography. Basic sciences, Springer, London
McCarthy DW, Bass LA, Cutler PD, Shefer RE, Klinkowstein RE, Herrero P, Lewis JS, Cutler CS, Anderson CJ, Welch MJ (1999) High purity production and potential applications of copper-60 and copper-61. Nucl Med Biol 26:351–358
Ahmedova A, Todorov B, Burdzhiev N, Goze C (2018) Copper radiopharmaceuticals for theranostic applications. Eur J Med Chem 157:1406–1425
Sitarz M, Cussonneau JP, Matulewicz T, Haddad F (2020) Radionuclide candidates for β+γ coincidence PET: an overview. Appl Radiat Isot 155:108898-1–108898-12
Shuza Uddin M, Kim K, Nadeem M, Sudár S, Kim G (2018) Measurements of excitation functions of α-particle induced reactions on natNi: possibility of production of the medical isotopes 61Cu and 67Cu. Radiochim Acta 106:87–93
Knapp FF, Dash A (2016) Radiopharmaceuticals for therapy. Springer, New Delhi
Spellerberg S, Scholten B, Spahn I, Bolten W, Holzgreve M, Coenen HH, Qaim SM (2015) Target development for diversified irradiations at a medical cyclotron. Appl Radiat Isot 104:106–112
Nickles RJ, Avila-Rodriguez MA, Nye JA, Houser EN, Selwyn RG, Schueller MJ, Christian BT, Jensen M (2008) Sustainable production of orphan radionuclides at Wisconsin. Q J Nucl Med Mol Imaging 52:134–139
Ali SKI, Khandaker MU, Dababneh S, Kassim HA (2018) Evaluation of production cross-sections for 61Cu non-standard PET radionuclide via light-ion-induced nuclear reactions on Co, Ni, Zn targets. Nucl Inst Methods Phys Res B 436:221–235
Tolmachev V, Lundqvist H, Einarsson L (1998) Production of 61Cu from a Natural Nickel Target. Appl Radiat Isot 49:79–81
Strangis R, Lepera CG (2007) Production of 61Cu by deuteron irradiation of natural Ni. In: Proc 18th International Conference on Cyclotrons and Their Applications Cyclotrons Giardini Naxos 246-247
Muramatsu H, Shirai E, Nakahara H, Murakami Y (1978) Alpha particle bombardment of natural nickel target for the production of 61Cu. Int J Appl Radiat Isot 29:611–614
Smith SV, Jones M, Holmes V (2011) Production and selection of metal PET radioisotopes for molecular imaging. In: Singh N (ed) Radioisotopes - applications in bio-medical science. InTech, Rijeka
van der Meulen NP, Hasler R, Blanc A, Farkas R, Benešová M, Talip Z, Müller C, Schibli R (2019) Implementation of a new separation method to produce qualitatively improved 64Cu. J Label Compd Radiopharm 62:460–470
Thieme S, Walther M, Pietzsch HJ, Henniger J, Preusche S, Mäding P, Steinbach J (2012) Module-assisted preparation of 64Cu with high specific activity. Appl Radiat Isot 70:602–608
Ometáková J, Rajec P, Csiba V, Leporis M, Štefečka M, Vlk P, Galamboš M, Rosskopfova O (2012) Automated production of 64Cu prepared by 18 MeV cyclotron. J Radioanal Nucl Chem 293:217–222
Cyclotron produced radionuclides: emerging positron emitters for medical applications: 64Cu and 124I (2016). IAEA Radioisotopes and Radiopharmaceuticals Reports No. 1, IAEA, Vienna
Szelecsényi F, Blessing G, Qaim SM (1993) Excitation functions of proton induced nuclear reactions on enriched 61Ni and 64Ni: possibility of production of No-carrier-added 61Cu and 64Cu at a small cyclotron. Appl Radiat Isot 44:575–580
Kambali I (2014) Calculated radioactivity yields of Cu-64 from proton-bombarded Ni-64 targets using SRIM codes. Atom Indonesia 40:129–134
Poniger SS, Tochon-Danguy HJ, Panopoulos HP, O'Keefe GJ, Peake D, Rasool R, Scott AM (2012) Automated production of 124I and 64Cu using IBA Terimo and Pinctada metal electroplating and processing modules. AIP Conf Proc 1509:114–119
Burke P, Golovko O, Clark JC, Aigbirhio FI (2010) An automated method for regular productions of copper-64 for PET radiopharmaceuticals. Inorg Chim Acta 363:1316–1319
Soenarjo S, Rahman WY, Tryyanto S (2011) Simulations on nickel target preparation and separation of Ni(II)-Cu(II) matrix for production of radioisotope 64Cu. J Iptek Nukl Ganendra 14:1–9
Aslam MN, Sudár S, Hussain M, Malik AA, Shah HA, Qaim SM (2009) Charged particle induced reaction cross section data for production of the emerging medically important positron emitter 64Cu: a comprehensive evaluation. Radiochim Acta 97:669–686
Sadeghi M, Amiri M, Roshanfarzad P, Avila M, Tenreiro C (2008) Radiochemical studies relevant to the no-carrier-added production of 61,64Cu at a cyclotron. Radiochim Acta 96:399–402
Kume M, Carey PC, Gaehle G, Madrid E, Voller T, Margenau W, Welch MJ, Lapi SE (2012) A semi-automated system for the routine production of copper-64. Appl Radiat Isot 70:1803–1806
Thisgaard H, Jensen M, Elema DR (2011) Medium to large scale radioisotope production for targeted radiotherapy using a small PET cyclotron. Appl Radiat Isot 69:1–7
Qaim SM, Scholten B, Neumaier B (2018) New developments in the production of theranostic pairs of radionuclides. J Radioanal Nucl Chem 318:1493–1509
Tochon-Danguy HJ, Poniger SS, Sachinidis JI, Panopoulos HP, Scott AM (2012) Implementation of a solid target production facility. AIP Conf Proc 1509:176–180
Fiedler L, Kellner M, Oos R, Böning G, Ziegler S, Bartenstein P, Zeidler R, Gildehaus FJ, Lindner S (2018) Fully automated production and characterization of 64Cu and proof-of-principle small-animal PET imaging using 64Cu-labelled CA XII targeting 6A10 Fab. ChemMedChem 13:1230–1237
McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, Anderson CJ, Welch MJ (1997) Efficient production of high specific activity 64Cu using a biomedical cyclotron. Nucl Med Biol 24:35–43
Jeffery CM, Smith SV, Asad AH, Chan S, Price RI (2012) Routine production of copper-64 using 11.7 MeV protons. AIP Conf Proc 1509:84–90
Ohya T, Nagatsu K, Suzuki H, Fukada M, Minegishi K, Hanyu M, Fukumura T, Zhang MR (2016) Efficient preparation of high-quality 64Cu for routine use. Nucl Med Biol 43:685–691
Matarrese M, Bedeschi P, Scardaoni R, Sudati F, Savi A, Pepe A, Masiello V, Todde S, Gianolli L, Messa C, Fazio F (2010) Automated production of copper radioisotopes and preparation of high specific activity [64Cu]Cu-ATSM for PET studies. Appl Radiat Isot 68:5–13
Jeffery C, Chan S, Cryer D, Asad A, RAPID Group, Price RI (2010) Optimisation of an electroplating process to prepare a solid target for (p,n) based production of copper-64. In: Proc. The 13th International Workshop on Targetry and Target Chemistry, Risø, July 26-28 Abs. No. 020
Al Rayyes AH, Ailouti Y (2013) Production and quality control of 64Cu from high current Ni target. World J Nucl Sci Technol 3:72–77
Toyota T, Hanafusa T, Oda T, Koumura I, Sasaki T, Matsuura E, Kumon H, Yano T, Ono T (2013) A purification system for 64Cu produced by a biomedical cyclotron for antibody PET imaging. J Radioanal Nucl Chem 298:295–300
Obata A, Kasamatsu S, McCarthy DW, Welch MJ, Saji H, Yonekura Y, Fujibayashi Y (2003) Production of therapeutic quantities of 64Cu using a 12 MeV cyclotron. Nucl Med Biol 30:535–539
Manrique-Arias JC, Avila-Rodriguez MA (2014) A simple and efficient method of nickel electrodeposition for the cyclotron production of 64Cu. Appl Radiat Isot 89:37–41
Alliot C, Michel N, Bonraisin AC, Bossé V, Laizé J, Bourdeau C, Mokili BM, Haddad F (2011) One step purification process for no-carrier-added 64Cu produced using enriched nickel target. Radiochim Acta 99:627–630
Adam Rebeles R, Van den Winkel P, Hermanne A, Tárkányi F (2009) New measurement and evaluation of the excitation function of 64Ni(p,n) reaction for the production of 64Cu. Nucl Instr Met Phys Res B 267:457–461
Hermanne A, Tárkányi F, Takács S, Kovalev SF, Ignatyuk A (2007) Activation cross sections of the 64Ni(d,2n) reaction for the production of the medical radionuclide 64Cu. Nucl Instr Met Phys Res B 258:308–312
Daraban L, Adam Rebeles R, Hermanne A (2009) Study of the excitation function for the deuteron induced reaction on 64Ni(d,2n) for the production of the medical radioisotope 64Cu. Appl Radiat Isot 67:506–510
Ohya T, Nagatsu K, Suzuki H, Fukada M, Minegishi K, Hanyu M, Zhang MR (2018) Small-scale production of 67Cu for a preclinical study via the 64Ni(α, p)67Cu channel. Nucl Med Biol 59:56–60
Alharbi AA, Alzahrani J, Azzam A (2011) Activation cross-section measurements of some proton induced reactions on Ni, Co and Mo for proton activation analysis (PAA) purposes. Radiochim Acta 99:763–770
Kim JY, Park H, Lee JC, Kim KM, Lee KC, Ha HJ, Choi TH, An GI, Cheon GJ (2009) A simple Cu-64 production and its application of Cu-64 ATSM. Appl Radiat Isot 67:1190–1194
Jalilian AR, Osso J Jr (2017) The current status and future of theranostic Copper-64 Radiopharmaceuticals. Iran J Nucl Med 25:1–10
Ikotun OF, Lapi SE (2011) The rise of metal radionuclides in medical imaging: copper-64, zirconium-89 and yttrium-86. Future Med Chem 3:599–621
Rowshanfarzad P, Sabet M, Jalilian AR, Kamalidehghan M (2006) An overview of copper radionuclides and production of 61Cu by proton irradiation of natZn at a medical cyclotron. Appl Radiat Isot 64:1563–1573
Szymański P, Frączek T, Markowicz M, Mikiciuk-Olasik E (2012) Development of copper based drugs, radiopharmaceuticals and medical materials. Biometals 25:1089–1112
Boschi A, Martini P, Janevik-Ivanovska E, Duatti A (2018) The emerging role of copper-64 radiopharmaceuticals as cancer theranostics. Drug Discov Today 23:1489–1501
Piel H, Qaim SM, Stöcklin G (1992) Excitation functions of (p,xn)-reactions on natNi and highly enriched 62Ni: possibility of production of medically important radioisotope 62Cu at a small cyclotron. Radiochim Acta 57:1–5
Suryanto H, Kambali I (2018) A novel method for 57Ni and 57Co production using cyclotron-generated secondary neutrons. Atom Indonesia 44:81–87
Qaim SM (2019) Theranostic radionuclides: recent advances in production methodologies. J Radioanal Nucl Chem 322:1257–1266
Bell TK, Bridges JM, Nelson MG (1965) Simultaneous free and bound radioactive vitamin B12 urinary excretion test. J Clin Pathol 18:611–613
Zuckier LS, Rao Chervu L (1984) Schilling evaluation of pernicious anemia: current status. J Nucl Med 25:1032–1039
Muggleton AHF (1979) Preparation of thin nuclear targets. J Phys E Sci Instrum 12:780–807
Stolarz A (2014) Target preparation for research with charged projectiles. J Radioanal Nucl Chem 299:913–931
Standardized high current solid targets for cyclotron production of diagnostic and therapeutic radionuclides (2004) Technical Reports Series No. 432 IAEA Vienna
Di Bari GA (2001) Electrodeposition of nickel. In: Schlesinger M, Paunovic M (eds) Modern Electroplating. Wiley, Hoboken
Dennis JK, Such TE (1993) Nickel and chromium plating. Woodhead, Cambridge
Oriňáková R, Turoňová A, Kladeková D, Gálová M, Smith RM (2006) Recent developments in the electrodeposition of nickel and some nickel-based alloys. J Appl Electrochem 36:957–972
Chotkowski M, Połomski D, Czerwinski K (2020) Potential application of ionic liquids for electrodeposition of the material targets for production of diagnostic radioisotopes. Materials 13:5069-1–5069-26
Hou X, Jacobsen U, Jørgensen JC (2002) Separation of no-carrier-added 64Cu from a proton irradiated 64Ni enriched nickel target. Appl Radiat Isot 57:773–777
Shivarudrappa V, Vimalnath KV (2005) High purity materials as targets for radioisotope production: needs and challenges. Bull Mater Sci 28:325–330
Manual for reactor produced radioisotopes (2003) IAEA-TECDOC-1340 Report. IAEA, Vienna
Szajek LP, Meyer W, Plascjak P, Eckelman WC (2005) Semi-remote production of [64Cu]CuCl2 and preparation of high specific activity [64Cu]Cu-ATSM for PET studies. Radiochim Acta 93:239–244
Zeisler SK, Pavan RA, Orzechowski J, Langlois R, Rodrigue S, van Lier JE (2003) Production of 64Cu on the Sherbrooke TR-PET cyclotron. J Radioanal Nucl Chem 257:175–177
TALYS-based evaluated nuclear data library, TENDL (2019) https://tendl.web.psi.ch/tendl_2019/tendl2019.html
Quashie EE, Correa AA (2018) Electronic stopping power of protons and alpha particles in nickel. Phys Rev B 98:235122-1–235122-6
Williamson CF, Boujot JP, Picard J (1966) Tables of range and stopping power of chemical elements for charged particles of energy 0.05 to 500 MeV, Report CEA-R 3042, Centre d'Études Nucléaires de Saclay, Commissariat à l’Energie Atomique, Paris
Vértes A, Nagy S, Klencsár Z, Lovas RG, Rösch F (2011) Handbook of nuclear chemistry. Springer, New York
Ziegler JF, Ziegler MD, Biersack JP (2010) SRIM – the stopping and range of ions in matter. Nucl Instr Met Phys Res B 268:1818–1823
Poignant F, Penfold S, Asp J, Takhar P, Jackson P (2016) GEANT4 simulation of cyclotron radioisotope production in a solid target. Phys Med 32:728–734
Rajec P, Csiba V, Leporis M, Štefečka M, Losonczi Pataky E, Reich M, Ometáková J (2010) Preparation and characterization of nickel targets for cyclotron production of 64Cu. J Radioanal Nucl Chem 286:665–670
SRIM - The stopping and range of ions in matter http://www.srim.org
Adam Rebeles R, Van den Winkel P, Hermanne A, De Vis L, Waegeneer R (2010) PC-controlled radiochemistry system for preparation of no-carrier-added 64Cu. J Radioanal Nucl Chem 286:655–659
Kambali I, Suryanto H, Setiawan H (2014) The stopping power and range of energetic proton beams in nickel target relevant for copper-64 production. J Radioisot Radiopharm 17:27–34
Sadeghi M, Amiri M, Gholamzadeh Z, Rowshanfarzad P (2009) Nickel electrodeposition on copper substrate for cyclotron production of 64Cu. Radiochemistry 51:628–632
Xie Q, Zhu H, Wang F, Meng X, Ren Q, Xia C, Yang Z (2017) Establishing reliable Cu-64 production process: from target plating to molecular specific tumor micro-PET imaging. Molecules 22:641-1–641-10
Bagli E, De Salvador D, Bacci L, Sgarbossa F, Bandiera L, Camattari R, Germogli G, Mazzolari A, Sytov A, Guidi G (2019) Enhancement of the inelastic nuclear interaction rate in crystals via antichanneling. Phys Rev Lett 123:044801-1–044801-6
Chodkowski MR, Weil R (1977) The effect of stress applied during electrodeposition of nickel monocrystals on their structure. J Electrochem Soc 124:1678–1681
Ogawa S, Mizuno J, Watanabe D, Fujita FE (1957) On the twin structure of nickel films electrodeposited on single crystals of copper. J Phys Soc Jap 12:999–1006
Lawless KR (1965) Growth and structure of electrodeposited thin metal films. J Vac Sci Technol 2:24–34
Souzis L, Weil R (1976) Residual stresses in electrodeposited nickel monocrystals. Surf Technol 4:41–58
Leygraf C, Wallinder IO, Tidblad J, Graedel T (2016) Atmospheric corrosion. Wiley, Hoboken
Paunovic M, Schlesinger M (2006) Fundamentals of electrochemical deposition. Wiley, Hoboken
Milchev A (2002) Electrocrystallization. Fundamentals of nucleation and growth. Kluwer, New York
Rashidi AM (2012) A galvanostatic modeling for preparation of electrodeposited nanocrystalline coatings by control of current density. J Mater Sci Technol 28:1071–1076
Lantelme F, Seghiouer A, Derja A (1998) Model of nickel electrodeposition from acidic medium. J Appl Electrochem 28:907–913
Amblard J, Epelboin I, Froment M, Maurin G (1979) Inhibition and nickel electrocrystallization. J Appl Electrochem 9:233–242
Gündiler I, Murr LE (1976) Fundamental studies of the structure and growth of electrodeposited nickel. Thin Solid Films 37:387–406
Staikov G, Lorenz WJ (1997) The role of crystal imperfections in electrochemical phase formation and growth. Can J Chem 75:1624–1634
Staikov G (2007) Electrocrystallization in nanotechnology. Wiley, Weinheim
Guo L, Searson PC (2010) On the influence of the nucleation overpotential on island growth in electrodeposition. Electrochim Acta 55:4086–4091
Gamburg YD, Zangari G (2011) Theory and practice of metal electrodeposition. Springer, London
Amblard J, Froment M, Maurin G, Spyrellis N, Trevisan-Souteyrand E (1993) Nickel electrocrystallization-from nucleation to textures. Electrochim Acta 28:909–915
Popov KI, Djokić SS (2002) Fundamental aspects of electrometallurgy. Kluwer, New York
Oniciu L, Mureşan L (1991) Some fundamental aspects of levelling and brightening in metal electrodeposition. J Appl Electrochem 21:565–574
Grujicic D, Pesic B (2006) Electrochemical and AFM study of nickel nucleation mechanisms on vitreous carbon from ammonium sulfate solutions. Electrochim Acta 51:2678–2690
Popov KI, Djokić SS, Nikolić ND, Jović VD (2016) Morphology of electrochemically and chemically deposited metals. Springer, Switzerland
Schlesinger M, Paunovic M (2001) Modern electroplating. Wiley, Hoboken
Tatiparti SSV, Ebrahimi F (2012) Potentiostatic versus galvanostatic electrodeposition of nanocrystalline Al–Mg alloy powders. J Solid State Electrochem 16:1255–1262
Rudnik E, Wojnicki M, Włoch G (2012) Effect of gluconate addition on the electrodeposition of nickel from acidic baths. Surf Coat Technol 207:375–388
Matsui I, Uesugi T, Takigawa Y, Higashi K (2013) Effect of interstitial carbon on the mechanical properties of electrodeposited bulk nanocrystalline Ni. Acta Mater 61:3360–3369
Chen YC, Kuo SL, Lee JL, Ke ST, Wong CH, Ger MD (2008) The influence of surfactant CTAB on the microstructure and material properties of nickel microelectroforming. Key Eng Mater 364-366:346–350
Schmitz EPS, Quinaia SP, Garcia JR, de Andrade CK, Lopes MC (2016) Influence of commercial organic additives on the nickel electroplating. Int J Electrochem Sci 11:983–997
Mohanty US, Tripathy BC, Singh P, Keshavarz A, Iglauer S (2019) Roles of organic and inorganic additives on the surface quality, morphology, and polarization behavior during nickel electrodeposition from various baths: a review. J Appl Electrochem 49:847–870
Abel S, Freimuth H, Lehr H, Mensinger H (1994) Defined crystal orientation of nickel by controlled microelectroplating. J Micromech Microeng 4:47–54
Belhamel K, Kheraz H, Ludwig R, Dzung Nguen TK, Allsop N, Al-Juaid SS (2010) Electrodeposition and morphology analysis of nickel nanoparticles from sulphate bath. e-J Surf Sci Nanotech 8:227–232
Mishra AC, Thakur AK, Srinivas V (2009) Effect of deposition parameters on microstructure of electrodeposited nickel thin films. J Mater Sci 44:3520–3527
Rashkov S, Atanasov N (1975) Texture and morphology of nickel coatings deposited in the presence of surfactants. Electrodep Surf Treatm 3:105–120
Darrort V, Troyon M, Ebotht J, Bissieux C, Nicollin C (1995) Quantitative study by atomic force microscopy and spectrophotometry of the roughness and brightness of electrodeposited nickel in the presence of additives. Thin Solid Films 265:52–57
Mech K (2017) Influence of organic ligands on electrodeposition and surface properties of nickel films. Surf Coat Technol 315:232–239
Survila A (2015) Electrochemistry of metal complexes. Wiley, Weinheim
Ibrahim MAM, Al Radadi RM (2015) Role of glycine as a complexing agent in nickel electrodeposition from acidic sulphate bath. Int J Electrochem Sci 10:4946–4971
El Boraei NF, Ibrahim MAM (2018) Catalytic effect of L-proline on the reduction of Ni(II) ions during nickel electrodeposition from a Watts-type nickel bath. Surf Coat Technol 347:113–122
Bigos A, Wolowicz M, Janusz-Skuza M, Starowicz Z, Szczerba MJ, Bogucki R, Beltowska-Lehman E (2021) Citrate-based baths for electrodeposition of nanocrystalline nickel coatings with enhanced hardness. J Alloys Compd 850:156857-1–156857-12
Gómez E, Pollina R, Vallés E (1995) Morphology and structure of nickel nuclei as a function of the conditions of electrodeposition. J Electroanal Chem 397:111–118
Proud WG, Gomez E, Sarret E, Valles E, Müller C (1995) Influence of pH on nickel electrodeposition at low nickel(II) concentrations. J Appl Electrochem 25:770–775
Ciszewski A, Posluszny S, Milczarek G, Baraniak M (2004) Effects of saccharin and quaternary ammonium chlorides on the electrodeposition of nickel from a Watts-type electrolyte. Surf Coat Technol 183:127–133
Mirkova L, Maurin G, Monev M, Chr T (2003) Hydrogen coevolution and permeation in nickel electroplating. J Appl Electrochem 33:93–100
Subbaiah T, Singh P, Hefter G, Muir D, Das RP (2000) Electrowinning of copper in the presence of anodic depolarisers - a review. Mineral Proc Extract Metal Rev: Int J 21:479–496
Kang JX, Zhao WZ, Zhang GF (2009) Influence of electrodeposition parameters on the deposition rate and microhardness of nanocrystalline Ni coatings. Surf Coat Technol 203:1815–1818
Ibrahim MAM (2006) Black nickel electrodeposition from a modified Watts bath. J Appl Electrochem 36:295–301
Streinz CC, Hartman AP, Motupally S, Weidner JW (1995) The effect of current and nickel nitrate concentration on the deposition of nickel hydroxide films. J Electrochem Soc 142:1084–1089
Hall DS, Lockwood DJ, Bock C, MacDougall BR (2015) Nickel hydroxides and related materials: a review of their structures, synthesis and properties. Proc R Soc A 471:20140792-1–20140792-65
Khaled KF (2010) Electrochemical behavior of nickel in nitric acid and its corrosion inhibition using some thiosemicarbazone derivatives. Electrochim Acta 55:5375–5383
Stupnišek-Lisac E, Karšulin M (1984) Electrochemical behaviour of nickel in nitric acid. Electrochim Acta 29:1339–1343
Lide DR (ed) (2005) CRC Handbook of chemistry and physics. CRC Press, Boca Raton
Tóth G, Madarász A (2006) Structure of BRIJ-35 nonionic surfactant in water: a reverse Monte Carlo study. Langmuir 22:590–597
Ghuge PD, Mali NA, Joshi SS (2020) Extractive distillation configuration for nitric acid dehydration using sulfuric acid as a solvent. Ind Eng Chem Res 59:6183–6193
Arnison GTJ (1967) The production of thin self-supporting nickel foils using an electroplating method. Nucl Inst Methods 53:357–358
Moldoveanu GA, Demopoulos GP (2015) Organic solvent-assisted crystallization of inorganic salts fromacidic media. J Chem Technol Biotechnol 90:686–692
Moldoveanu GA, Demopoulos GP (2002) Producing high-grade nickel sulfate with solvent displacement crystallization. JOM 54:49–53
Wang J, Gabe DR, Hart AC, Crouch PC (2013) The chemistry of nickel electroplating solutions. Trans IMF 91:4–10
Yuan L, Ding ZY, Liu SJ, He YN (2017) Cathodic process of nickel electrodeposition from ammonia–ammonium chloride solutions. Rare Metals 2017:1–6
Varadwaj PR, Cukrowski I, Marques HM (2008) DFT-UX3LYP studies on the coordination chemistry of Ni2+. Part 1: six coordinate [Ni(NH3)n(H2O)6-n]2+ complexes. J Phys Chem A 112:10657–10666
Salhi R (2010) Recovery of nickel and copper from metal finishing hydroxide sludges by ammoniacal leaching. Mineral Proc Extract Metal 119:147–152
Plyasunova NV, Zhang Y, Muhammed M (1998) Critical evaluation of thermodynamics of complex formation of metal ions in aqueous solutions. IV. Hydrolysis and hydroxo-complexes of Ni2+ at 298.15 K. Hydrometal 48:43–63
Mattigod SV, Rai D, Felmy AR, Rao L (1997) Solubility and solubility product of crystalline Ni(OH)2. J Solut Chem 26:391–403
Skitał PM, Sanecki PT, Saletnik D, Kalembkiewicz J (2019) Electrodeposition of nickel from alkaline NH4OH/NH4Cl buffer solutions. Trans Nonferrous Metals Soc China 29:222–232
Mohd Adli N, Zhang H, Mukherjee S, Wu G (2018) Review—ammonia oxidation electrocatalysis for hydrogen generation and fuel cells. J Electrochem Soc 165:J3130–J3147
Le Vot S, Reyter D, Roué L, Bélanger D (2012) Electrochemical oxidation of NH3 on platinum electrodeposited onto graphite electrode. J Electrochem Soc 159:F91–F96
Zöllig H, Fritzsche C, Morgenroth E, Udert KM (2015) Direct electrochemical oxidation of ammonia on graphite as a treatment option for stored sourceseparated urine. Water Res 69:284–294
Cabeza A, Urtiaga A, Rivero MJ, Ortiz I (2007) Ammonium removal from landfill leachate by anodic oxidation. J Hazard Mater 144:715–719
Pérez G, Saiz J, Ibañez R, Urtiaga AM, Ortiz I (2012) Assessment of the formation of inorganic oxidation by-products during the electrocatalytic treatment of ammonium from landfill leachates. Water Res 46:2579–2590
Martí-Calatayud MC, García-Gabaldón M, Pérez-Herranz V, Ortega E (2011) Determination of transport properties of Ni(II) through a Nafion cation-exchange membrane in chromic acid solutions. J Membr Sci 379:449–458
Ji J, Cooper WC (1996) Nickel speciation in aqueous chloride solutions. Electochim Acta 41:1549–1560
Hankin A, Kelsall GH (2012) Electrochemical recovery of nickel from nickel sulfamate plating effluents. J Appl Electrochem 42:629–643
Petrova EV, Vorontsova MA, Manomenova VL, Rashkovich LN (2012) Some properties of aqueous solutions of α-NiSO4·6H2O. Crystallogr Rep 57:579–584
Oriňáková R, Strečková M, Trnková L, Rozik R, Gálova M (2006) Comparison of chloride and sulphate electrolytes in nickel electrodeposition on a paraffin impregnated graphite electrode. J Electroanal Chem 594:152–159
Sahari A, Azizi A, Schmerber G, Dinia A (2008) Nucleation, growth, and morphological properties of electrodeposited nickel films from different baths. Surf Rev Lett 15:717–725
Tsuru Y, Nomura M, Foulkes FR (2000) Effects of chloride, bromide and iodide ions on internal stress in films deposited during high speed nickel electroplating from a nickel sulfamate bath. J Appl Electrochem 30:231–238
Patil RS, Juvekar VA, Naik VM (2011) Oxidation of chloride ion on platinum electrode: dynamics of electrode passivation and its effect on oxidation kinetics. Ind Eng Chem Res 50:12946–12959
Quintero Pulido DF, ten Kortenaar MV, Hurink JL, Smit GJM (2019) Characteristics of halide oxidation at graphite electrode for use in halide batteries. Sust Energy Technol Asses 33:14–23
Randin JP (1981) Nonmetallic electrode materials. In: JO’M B (ed) Electrochemical materials science. Springer, New York
Wang Z, Tada E, Nishikata A (2014) In situ analysis of chloride effect on platinum dissolution by a channel-flow multi-electrode system. J Electrochem Soc 161:F845–F849
Geiger S, Cherevko S, Mayrhofer KJJ (2015) Dissolution of platinum in presence of chloride traces. Electrochim Acta 179:24–31
Shrestha BR, Tada E, Nishikataba A (2014) Effect of chloride on platinum dissolution. Electrochim Acta 143:161–167
Graff A, Barrez E, Baranek P, Bachet M, Bénézeth P (2017) Complexation of nickel ions by boric acid or (poly)borates. J Solut Chem 46:25–43
Dávalos CE, López JR, Ruiz H, Méndez A, Antaño-López R, Trejo G (2013) Study of the role of boric acid during the electrochemical deposition of Ni in a sulfamate bath. Int J Electrochem Sci 8:9785–9800
Ji J, Cooper WC, Dreisinger DB, Peters E (1995) Surface pH measurements during nickel electrodeposition. J Appl Electrochem 25:642–650
Song KD, Kim KB, Han SH, Lee HK (2003) A study on effect of hydrogen reduction reaction on the initial stage of Ni electrodeposition using EQCM. Electrochem Commun 5:460–466
Yin KM, Lin BT (1996) Effects of boric acid on the electrodeposition of iron, nickel and iron-nickel. Surf Coat Technol 78:205–210
Ritzert NL, Moffat TP (2016) Ultramicroelectrode studies of self-terminated nickel electrodeposition and nickel hydroxide formation upon water reduction. J Phys Chem C 120:27478–27489
Šupicová M, Rozik R, Trnková L, Oriňáková R, Gálová M (2006) Influence of boric acid on the electrochemical deposition of Ni. J Solid State Electrochem 10:61–68
Tsuru Y, Nomura M, Foulkes FR (2002) Effects of boric acid on hydrogen evolution and internal stress in films deposited from a nickel sulfamate bath. J Appl Electrochem 32:629–634
Tilak BV, Gendron AS, Mosoiu MA (1977) Borate buffer equilibria in nickel refining electrolytes. J Appl Electrochem 7:495–500
Bekish YN, Poznyak SK, Tsybulskaya LS, Gaevskaya TV (2010) Electrodeposited Ni–B alloy coatings: structure, corrosion resistance and mechanical properties. Electrochim Acta 55:2223–2231
Avila-Rodriguez MA, Aguilar-Ortiz EA, Jalilian AR, Manrique-Arias JC, Zarate-Morales A, Flores-Moreno A (2019) A simple and efficient method to recover isotopically enriched Ni-64 from electrolytic solutions. Appl Radiat Isot 145:55–58
Elomaa VV, Jurttila J, Rajander J, Solin O (2014) Automation of 64Cu production at Turku PET Centre. Appl Radiat Isot 89:74–78
Pinnel MR (1979) Diffusion-related behaviour of gold in thin film systems. Gold Bull 12:62–71
Ravi R, Paul A (2012) Diffusion mechanism in the gold-copper system. J Mater Sci Mater Electron 23:2152–2156
Pucic SP (1993) Diffusion of copper into gold plating. IEEE Instrumentation and Measurement Technology Conference:114–117
Li WH, Joelle Ong SW (2012) Cu diffusion in Ag-plated Cu leadframe packages. Microelectron Reliab 52:1523–1527
Lin TY, Pecht MG, Das D, Pan J, Wenhui Z (2005) The evaluation of copper migration during the die attach curing and second wire bonding process. IEEE Trans Compon Packag Technol 28:337–344
Okamoto N, Wang F, Watanabe T (2004) Adhesion of electrodeposited copper, nickel and silver films on copper, nickel and silver substrates. Mater Trans 45:3330–3333
Tárkányi F, Takács S, Andó L, Vera-Ruiz H, Shubin YN, Hermanne A (2002) Status of the database for production of medical radioisotopes of 103Pd 123,124I, 201TI by using Rh, Te and Tl targets. J Nucl Sci Technol 39:1318–1321
Sadeghi M, Bakhtiari M, Bakht MK, Anjomrouz M, Mokhtari L (2012) Overview of mercury radionuclides and nuclear model calculations of 195Hgm,g and 197Hgm,g to evaluate experimental cross section data. Phys Rev C 85:034605-1–034605-8
Ditrói F, Tárkányi F, Takács S, Hermanne A (2016) Activation cross sections of proton induced nuclear reactions on gold up to 65 MeV. Appl Radiat Isot 113:96–109
Showaimy H, Solieman AHM, Abdel Hamid AS, Khalaf AM, Saleh ZA (2019) Measurements of activation cross sections for proton induced reactions on natural platinum targets leading to the formation of gold radioisotopes. Radiat Phys Chem 157:97–101
Tárkányi F, Ditró F, Taká S, Csikai J, Hermanne A, Uddin MS, Hagiwara M, Baba M, Shubin YN, Dityuk AI (2004) Activation cross-sections of light ion induced nuclear reactions on platinum: proton induced reactions. Nucl Instr Met Phys Res B 226:473–489
Mirzaii M, Sadeghi M, Gholamzadeh Z (2009) Targetry for cyclotron production of no-carrier-added cadmium-109 from natAg(p,n)109Cd reaction. Iran J Radiat Res 6:201–206
Tárkányi F, Ditrói F, Takács S, Hermanne A, Baba M, Yuki H, Ignatyuk AV (2017) Investigation of activation cross sections of proton induced reactions on rhodium up to 70 MeV for practical applications. Nucl Instr Met Phys Res B 412:190–197
Sudár S, Cserpák F, Qaim SM (2002) Measurements and nuclear model calculations on proton induced reactions on 103Rh up to 40 MeV: evaluation of the excitation function of the 103Rh(p,n)103Pd reaction relevant to the production of the therapeutic radionuclide 103Pd. Appl Radiat Isot 56:821–831
Lawriniang B, Ghosh R, Badwar S, Vansola V, Santhi Sheela Y, Suryanarayana SV, Naik H, Naik YP, Jyrwa B (2018) Measurement of cross-sections for the 93Nb(p,n)93mMo and 93Nb(p,pn)92mNb reactions up to ~20 MeV energy. Nucl Phys A 973:79–88
Endo A, Tamura T, Yamaguchi Y (1999) Compilation of nuclear decay data used for dose calculations: data for radionuclides not listed in ICRP Publication 38, JAERI-Data/Code 99-035. JAERI, Ibaraki
Zeisler SK, Becker DW, Pavan RA, Moschel R, Rühle H (2000) A water-cooled spherical niobium target for the production of [18F]fluoride. Appl Radiat Isot 53:449–453
Shahid M, Kim K, Naik H, Zaman M, Yang SC, Kim G (2015) Measurement of excitation functions in proton induced reactions on natural copper from their threshold to 43 MeV. Nucl Instr Met Phys Res B 342:305–313
Michel R, Bodemann R, Busemann H, Daunke R, Gloris M, Lange HJ, Klug B, Krins A, Leya I, Lüpke M, Neumann S, Reinhardt H, Schnatz-Biittgen M, Herpers U, Schiekel T, Sudbrock F, Holmqvist B, Condé H, Malmborg P, Suter M, Dittrich-Hannen B, Kubik PW, Synal HA, Filges D (1997) Cross sections for the production of residual nuclides by low- and medium-energy protons from the target elements C, N, 0, Mg, Al, Si, Ca, Ti, V, Mn, Fe, Co, Ni, Cu, Sr, Y, Zr, Nb, Ba and Au. Nucl Instr Met Phys Res B 129:153–193
Mushtaq A, Qaim SM (1990) Excitation functions of α- and 3He-particle induced nuclear reactions on natural germanium: evaluation of production routes for 73Se. Radiochim Acta 50:27–31
Wang X, Han Y, Zhang J, Li Z, Li T, Zhao X, Liu W (2019) Influence of electropolished copper substrate on morphology of electroplating self-supporting Ni films. Nucl Inst Met Phys Res A 927:343–348
Feltham AM, Spiro M (1971) Platinized platinum electrodes. Chem Rev 71:179–193
Chen G (2004) Electrochemical technologies in wastewater treatment. Sep Purif Technol 38:11–41
Rueffer M, Bejan D, Bunce NJ (2011) Graphite: an active or an inactive anode? Electrochim Acta 56:2246–2253
Topalov AA, Cherevko S, Zeradjanin AR, Meier JC, Katsounaros I, Mayrhofer KJJ (2014) Towards a comprehensive understanding of platinum dissolution in acidic media. Chem Sci 5:631–638
Wei R, Fang M, Dong G, Ho JC (2017) Is platinum a suitable counter electrode material for electrochemical hydrogen evolution reaction? Sci Bull 62:971–973
Motoyama M, Fukunaka Y, Sakka T, Ogata YH (2006) Effect of surface pH on electrodeposited Ni films. J Electrochem Soc 153:C502–C508
Popov KI, Pavlović MG, Maksimović MD, Krstajić SS (1978) The comparison of galvanostatic and potentiostatic copper powder deposition on platinum and aluminium electrodes. J Appl Electrochem 8:503–514
Chung CK, Chang WT (2015) Electrochemical deposition and mechanical property enhancement of the nickel and nickel-cobalt films. In: Nee AYC (ed) Handbook of manufacturing engineering and technology. Springer, London
Chassaing E, Joussellin M, Wiart R (1983) The kinetics of nickel electrodeposition inhibition by adsorbed hydrogen and anions. J Electroanal Chem 157:75–88
Santana AIC, Díaz SL, Barcia OE, Mattos OR (2009) A kinetic study on nickel electrodeposition from sulfate acid solutions. II. Reaction modeling. J Electrochem Soc 156:D331–D335
Boubatra M, Azizi A, Schmerber G, Dinia A (2012) The influence of pH electrolyte on the electrochemical deposition and properties of nickel thin films. Ionics 18:425–432
Kirihara S, Umeda Y, Tashiro K, Honma H, Takai O (2014) Stress control in high speed nickel plating by use of step control current. Trans IMF 92:290–294
Ebrahimi F, Ahmed Z (2003) The effect of current density on properties of electrodeposited nanocrystalline nickel. J Appl Electrochem 33:733–739
Gabe DR (1997) The role of hydrogen in metal electrodeposition processes. J Appl Electrochem 27:908–915
Gómez E, Müller C, Pollina R, Sarret M, Vallés E (1992) Studies of electrodeposition of nickel: different nickel( II) and sulphonated additive concentrations. J Electroanal Chem 33:41–64
Fleischmann M, Saraby-Reintjes A (1984) The simultaneous deposition of nickel and hydrogen on vitreous carbon. Electrochim Acta 29:69–75
Armyanov S, Sotirova-Chakarova G (1992) Hydrogen desorption and internal stress in nickel coatings obtained by periodic electrodeposition. J Electrochem Soc 139:3454–3457
Raub CJ (1993) Hydrogen in electrodeposits: of decisive importance, but much neglected. Plat Surf Finish 80:30–38
Baranowski B (1959) The dependence of the cathodic hydrogen content in thin nickel layers on the current density and saturation temperature. Bull Acad Pol Sci Chim 7:897–905
Baranowski B (1999) Electrochemical formation of nickel hydride in alkaline solutions. J Electroanal Chem 472:182–184
Kleperis J, Wójcik G, Czerwiński A, Skowroński J, Kopczyk M, Bełtowska-Brzezińska M (2001) Electrochemical behavior of metal hydrides. J Solid State Electrochem 5:229–249
Fukai Y (2005) The metal–hydrogen system. Basic bulk properties, Springer, Berlin
Juškėnas R, Selskis A, Kadziauskienė V (1998) In situ X-ray diffraction investigation of nickel hydride formation during cathodic charging of Ni. Electrochim Acta 43:1903–1911
Grdeń M, Klimek K (2005) EQCM studies on oxidation of metallic nickel electrode in basic solutions. J Electroanal Chem 581:122–131
Boniszewski T, Smith GC (1963) The influence of hydrogen on the plastic deformation ductility, and fracture of nickel in tension. Acta Metall 11:165–178
Monev M (2016) Hydrogenation and cracking of nickel coatings electrodeposited in the presence of brighteners. Bulg Chem Commun 48:73–77
Sheng W, Myint MNZ, Chen JG, Yan Y (2013) Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ Sci 6:1509–1512
Quaino P, Juarez F, Santos E, Schmickler W (2014) Volcano plots in hydrogen electrocatalysis - uses and abuses. Beilstein J Nanotechnol 5:846–854
Mamaghani KR, Naghib SM (2017) The effect of stirring rate on electrodeposition of nanocrystalline nickel coatings and their corrosion behaviors and mechanical characteristics. Int J Electrochem Sci 12:5023–5035
Bozzini B (2000) Mass-transport effects on texture formation of nickel electrodeposits. Mater Chem Phys 66:278–285
Xuetao Y, Yu W, Dongbai S, Hongying Y (2008) Influence of pulse parameters on the microstructure and microhardness of nickel electrodeposits. Surf Coat Technol 202:1895–1903
Rashidi AM, Amadeh A (2010) Effect of electroplating parameters on microstructure of nanocrystalline nickel coatings. J Mater Sci Technol 26:82–86
Yuan L, Hu J, Ding Z, Liu S (2017) Electrochemical deposition of bright nickel on titanium matrix from ammoniacal solution in the presence of thiourea. Int J Electrochem Sci 12:7312–7325
Wu X, Gao Q, Li Z (2015) Effects of additives on morphology and hydrogen evolution activities of nickel films prepared by electrodepositing. Int J Electrochem Sci 10:8823–8833
Marikkannu KR, Surya Kala K, Paruthimal Kalaignan G, Vasudevan T (2008) Electroplating of nickel from acetate based bath – Hull cell studies. Trans IMF 86:172–176
Wasekar NP, Haridoss P, Seshadri SK, Sundararajan G (2016) Influence of mode of electrodeposition, current density and saccharin on the microstructure and hardness of electrodeposited nanocrystalline nickel coatings. Surf Coat Technol 291:130–140
Mockute D, Bernotiene G (2000) The interaction of additives with the cathode in a mixture of saccharin, 2-butyne-1,4-diol and phthalimide during nickel electrodeposition in a Watts-type electrolyte. Surf Coat Technol 135:42–47
Han Q, Liu K, Chen J, Wei X (2003) A study on the electrodeposited Ni–S alloys as hydrogen evolution reaction cathodes. Int J Hydrog Energy 28:1207–1212
Imaz N, García-Lecína E, Díez JA (2010) Corrosion properties of double layer nickel coatings obtained by pulse plating techniques. Trans IMF 88:256–261
Wojciechowski J, Baraniak M, Pernak J, Lota G (2017) Nickel coatings electrodeposited from Watts type baths containing quaternary ammonium sulphate salts. Int J Electrochem Sci 12:3350–3360
Franklin TC (1987) Some mechanisms of action of additives in electrodeposition processes. Surf Coat Technol 30:415–428
Hassani S, Raeissi K, Golozar MA (2008) Effects of saccharin on the electrodeposition of Ni–Co nanocrystalline coatings. J Appl Electrochem 38:689–694
El-Sherik AM, Erb U, Page J (1996) Microstructural evolution in pulse plated nickel electrodeposits. Surf Coat Technol 88:70–78
Tóth-Kádár E, Bakonyi I, Pogány L, Cziráki Á (1996) Microstructure and electrical transport properties of pulse-plated nanocrystalline nickel electrodeposits. Surf Coat Technol 88:57–65
Kollia C, Spyrellis N, Amblard J, Froment M, Maurin G (1990) Nickel plating by pulse electrolysis: textural and microstructural modifications due to adsorption/desorption phenomena. J Appl Electrochem 20:1025–1032
Kollia C, Spyrellis N (1993) Textural modifications in nickel electrodeposition under pulse reversed current. Surf Coat Technol 57:71–75
Saitou M, Oshikawa W, Mori M, Makabe A (2001) Surface roughening in the growth of direct current or pulse current electrodeposited nickel thin films. J Electrochem Soc 148:C780–C783
Chandrasekar MS, Pushpavanam M (2008) Pulse and pulse reverse plating-conceptual, advantages and applications. Electrochim Acta 53:3313–3322
Larson C, Farr JPG (2012) Current research and potential applications for pulsed current electrodeposition – a review. Trans IMF 90:20–29
Chan KC, Qu NS, Zhu D (1998) Crystallographic textures and magnetic properties of electroformed nickel. J Appl Electrochem 28:1095–1099
Qu NS, Chan KC, Zhu D (1997) Surface roughening in pulse current and pulse reverse current electroforming of nickel. Surf Coat Technol 91:220–224
Dennis JK, Such TE (1986) Nickel and chromium plating. Butterworths, London
Ebrahimi F, Ahmed Z (2003) The effect of substrate on the microstructure and tensile properties of electrodeposited nanocrystalline nickel. Mater Charact 49:373–379
Nezhad AHN, Rahimi E, Arefinia R, Davoodi A, Hosseinpour S (2020) Effect of substrate grain size on structural and corrosion properties of electrodeposited nickel layer protected with self-assembled film of stearic acid. Materials 13:2052-1–2052-14
Fan C, Celis JP, Roos JR (1992) Effect of substrate pretreatment on the porosity in thin nickel electrodeposits. Surf Coat Technol 50:289–294
Acknowledgements
The contribution of Martyna Mieszkowska, M.Sc., was realised within Project No POWR.03.02.00-00-I009/17-00 (Operational Project Knowledge Education Development 2014-2020 co-financed by European Social Fund).
Funding
This work was supported by Grants No. 501-D112-01-1120000 (501100299, 5011000357) and 501-D112-64-0185200-80 (Faculty of Chemistry, University of Warsaw).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mieszkowska, M., Grdeń, M. Electrochemical deposition of nickel targets from aqueous electrolytes for medical radioisotope production in accelerators: a review. J Solid State Electrochem 25, 1699–1725 (2021). https://doi.org/10.1007/s10008-021-04950-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-021-04950-w