
Abstract
The distribution of hepatitis B virus (HBV) in the populations of island Southeast Asia is of medical and anthropological interest and is associated with an unusually high genetic diversity. This study examined the association of this HBV genetic diversity with the ethnogeography of the populations of the Indonesian archipelago. Whole genome analysis of 21 HBV isolates from East Nusa Tenggara and Papua revealed two recently reported HBV/B subgenotypes unique to the former, B7 (7 isolates) and B8 (5 isolates), and uncovered a further novel subgenotype designated B9 (4 isolates). Further isolates were collected from 419 individuals with defined ethnic backgrounds representing 40 populations. HBV/B was predominant in Austronesian-language-speaking populations, whereas HBV/C was the major genotype in Papua and Papua-influenced populations of Moluccas; HBV/B3 was the predominant subgenotype in the western half of the archipelago (speakers of the Western Malayo-Polynesian [WMP] branch of Austronesian languages), whereas B7, B8 and B9 were specific to Nusa Tenggara (Central Malayo-Polynesian (CMP)). The result provides the first direct evidence that the distribution of HBV genotypes/subgenotypes in the Indonesian archipelago is related to the ethnic origin of its populations and suggests that the HBV distribution is associated with the ancient migratory events in the peopling of the archipelago.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis B virus (HBV) is a major cause of liver diseases, particularly in Asia. Genetic variability of HBV plays an important role in the development to chronic hepatitis B and is associated with the clinical outcome and response to treatment [17, 38, 56]. Eight HBV genotypes, A to H, have been identified [2, 24, 25, 27, 30, 31, 36, 45], with genotypes B and C being predominant among Asian populations. Very recently, two new additional HBV genotypes, HBV/I and HBV/J, were proposed for isolates collected from Laos and Japan, respectively [14, 51].
Eight subgenotypes have been reported for the Asian HBV genotype B (HBV/B), each with different geographical predominance: B1 in Japan, B2 in China, B3 in Indonesia, B4 in Vietnam, B5 in the Philippines, B6 in the Arctic indigenous population, and B7 and B8 in eastern Nusa Tenggara islands of Indonesia [28, 29, 32, 34, 41, 42]. Similarly, HBV genotype C (HBV/C) has been classified into six geographically related subgenotypes: C1, C5 and C6 in Southeast Asia, C2 in East Asia [13, 24, 29, 41, 55], C3 mostly in the Pacific, and C4 in the Aborigines of Northeast Australia [32, 46].
The distribution of HBV genotypes and subgenotypes in the populations of island Southeast Asia is of particular interest. The Indonesian part of the archipelago alone consists of approximately 17,500 islands and is home to 230 million people of more than 500 ethnic populations, inhabiting around 6,000 islands [48]. The main origins of these populations are believed to be two major waves of ancient migration: the initial peopling of the archipelago by modern humans 60,000 years before present (yBP) and the arrival of Austronesian languages speakers around 5,000 yBP [3]. Information regarding the distribution of HBV genotypes/subgenotypes amongst the ethnic populations of the archipelago, therefore, might yield knowledge of anthropological significance. Such information is of medical importance, as this ethnically diverse region is now the major source of migrant populations in the more developed countries.
Our recent study suggests that the HBV genotype/subgenotype distribution in this archipelago is complex and indeed associated with the ethnic background of the populations rather than with geographical locations [34]. For example, HBV/B3 is found mainly in ethnic populations of the western half of the archipelago, while HBV/B7 is associated with ethnic populations of the Nusa Tenggara islands of the eastern half. A recent nationwide study of HBV molecular epidemiology in Indonesia showing the geographical specificity of distribution of HBV genotypes/subgenotypes also indicated a possible association with the ethnological origins of the populations [28]. This study was aimed to provide evidence that the distribution of HBV genotypes/subgenotypes is indeed related to the ethnogeographical structure of the Indonesian populations, in a study involving a large number of subjects with carefully defined ethnic backgrounds representing 40 ethnic populations. Our results demonstrate the association of HBV genotypes/subgenotypes with the ethnological origins of the populations of the Indonesian archipelago.
Materials and methods
Serum samples and ethnic populations
A total of 440 serum samples that were positive for HBsAg (310 men and 130 women; mean age, 40.2 ± 5.2 years) were obtained from asymptomatic carriers (263 samples), HBV-related liver disease patients (158 samples) who never received antiviral therapy, and blood donors (19 samples). The samples were collected from 20 geographical locations (Table 1). None of the participants was co-infected with either hepatitis C virus or human immunodeficiency virus. The ethnic background of the individuals from whom the samples were obtained was carefully documented and ascertained for at least three previous generations, both maternally and paternally, as described previously [26].
Ethnic populations were selected to represent the clustering of their genetic and linguistic affinities based on the mapping of human genetic diversity in Asia by the HUGO Pan-Asian SNP Consortium [12, 33, 52]: the Austronesian languages-speaking populations of western islands of Indonesia (Sumatra, Kalimantan and Java), the Austronesian-speaking populations of the islands of Sulawesi and the Nusa Tenggara archipelago, and the Papua and Papuan-speaking eastern island populations. The origins and characteristics of the individuals from whom the HBV isolates were obtained are shown in Table 1. Samples from Indonesians of Chinese ethnic origin were collected in three big cities (Jakarta, Surabaya and Medan). The study was approved by the Eijkman Institute Research Ethics Commission (EIREC No. 23/2007).
HBV genome sequencing
Viral DNA was extracted from 140 μL of HBsAg-positive serum using a QIAamp® DNA Mini Kit (QIAGEN Inc., Chatsworth, CA) according to the manufacturer’s instructions. HBV DNA was detected by nested PCR using Platinum® Taq DNA Polymerase (Invitrogen), targeting the conserved segment within the S gene, using primer sets described previously [36, 37, 58]: S2-1 (5′-CAAGGTATGTTGCCCGTTTG-3′, nt 455–474) and S1-2 (5′-CGAACCACTGAACAAATGGC-3′, nt 704–685) for the first round and S088 (5′-TGTTGCCCGTTTGTCCTCTA-3′, nt 462–471) and S2-2 (5′-GGCACTAGTAAACTGAGCCA-3′, nt 687–668) for the second round. Denaturizing, annealing and extension were carried out at 94°C for 30 s, 55°C for 30 s and 72°C for 1 min for both rounds of PCR (35 cycles for the first and 25 for the second round).
For whole-genome sequencing, five overlapping fragments were first amplified using primer sets described previously [35, 47, 49]: PS8-1 (5′-GTCACCATATTCTTGGGAAC-3′) and HS6-2 (5′-GCCAAGTGTTTGCTGACGCA-3′) for fragment A (nt 2,817–1,194), S2-1 and HB4R (5′-CGGGACGTAGACAAAGGACGT-3′) for fragment B (nt 487–1,434), HB5F (5′-GCATGGAGACCACCGTGAAC-3′) and S013 (5′-TCCACAGAAGCTCCAAATTCTTTT-3′) for fragment C (nt 1,256–1,941), PC1 (5′-CATAAGAGGACTCTTGGACT-3′) and HB9R (5′-GGATAGAACCTAGCAGGCAT-3′) for fragment D (nt 1,653–2,656), and HB10F (5′-CGCAGAAGATCTCAATCTCGG-3′) and T734 (5′-CTTCCTGACTGSCGATTGG-3′) for fragment E (nt 2,417–3,156). The amplification reaction was carried out for 35 cycles of denaturation at 94°C for 30 s, annealing at 55–59°C (depending on the primer pair used) for 30 s, and extension at 72°C for 60 s, and elongation at 72°C for 7 min. Amplification products were sequenced directly using Big Dye Terminator Reaction kits on an ABI 3130 Genetic Analyzer (ABI Perkin Elmer, Norwalk, CT, USA). The genome sequences were assembled and analyzed using BioEdit version 7.0.5 software.
For the sequencing of the preS2 region, semi-nested PCR was carried out employing primer sets PS1-1 (5′-CCTCCTGCCTCCACCAATCG-3′, nt 3125–3144) and t703 (5′-CAGAGTCTAGACTCGTGGTG-3′, nt 242–261) for the first round and PS1-1 and PS5-2 (5′-CTCGTGTTACAGGCGGGGTT-3′, nt 190–210) for the second round [49]. The PCR was carried out for 35 cycles of denaturation at 94°C for 30 s, annealing at 57–59°C (depending of the primer pair used) for 30 s, and extension at 72°C for 60 s, and elongation at 72°C for 7 min.
HBV genotype and subgenotype determination
Twenty-four complete genome sequences generated in this study, together with 141 obtained from GenBank (including Indonesian recently reported isolates) [28, 55] were aligned. A phylogenetic tree was constructed, and genetic distance was calculated by the 6-parameter method [40]. The genotypes and subgenotypes of the 24 new isolates were determined based on their phylogenetic co-clustering with the previously defined sequences. For the wider study of the HBV ethnogeographical distribution, genotype and subgenotype assignments were carried out for 654 isolates based on their preS2 sequences (440 sequences generated in this study and 214 sequences from GenBank) employing signatures of specific single nucleotide polymorphisms (SNPs) diagnostic for the various Asian HBV genotypes and subgenotypes, as reported previously [34] and developed further in this study (Table 2).
Results
HBV genotypes and discovery of another novel HBV/B subgenotype from the Nusa Tenggara islands
Phylogenetic analysis of the 24 complete HBV genome sequences obtained in this study (21 sequences from ethnic populations of the eastern region: Sumbanese [7], Flores [8], Alorese [1], and Papuan [Merauke 3, Jayapura 1 and Sentani 1]; and 3 sequences from ethnic populations of the western region: Javanesse [1] and Minang [2]), along with 141 sequences from GenBank, identified 3 HBV genotypes, 17 HBV/B, 6 HBV/C, and 1 HBV/D (Fig. 1). Of the 17 HBV/B isolates, 1 belonged to B3, 7 to B7, 5 to B8, and 4 to an unclassified cluster. The last of these was distinct from the existing HBV/B1-B8, with significant posterior probability (100). Phylogenetic trees constructed from ORFs P and S were consistent with that obtained from the complete genome although discordant with one constructed from ORF C, as observed previously, as the consequence of a recombination event involving this region [47]. Together with an intersubgenotype divergence of more than 4% for B1, B2, B4 and B6 (Table 3), we propose that the unidentified cluster represents a novel subgenotype, designated B9 (Fig. 1).

Phylogenetic analysis of 24 new whole HBV genome sequences reveals a novel HBV/B subgenotype. A phylogenetic tree was constructed from 24 whole genome sequences generated in the present study (bold and italic), and 141 representative sequences retrieved from the GenBank database (8 HBV/A [A1 4, A2 4], 58 HBV/B [B1 4, B2 8, B3 12, B4 4, B5 6, B6 8, B7 11, and B8 5], 38 HBV/C [C1 5, C2 5, C3 2, C4 2, C5 7, C6 12, C7 5], 18 HBV/D [D1 4, D2 5, D3 5 and D4 4], 4 HBV/E, 7 HBV/F [F1 2, F2 5], 4 HBV/G and 4 HBV/H). Genetic distances were calculated by the six-parameter method [40], with the wooly monkey strain (WMHBV) AY2266578 as an outgroup. The length of the horizontal bar indicates the number of nucleotide substitutions per site, and the posterior probability values are indicated at the roots of the tree. The tree demonstrates a clear distinction between the East Asia and Southeast Asia HBV/B subgenotype groups (the non-recombinant type [42] is indicated by light shading, and the recombinant type by darker shading) and reveals that 17 out of the 24 new sequences belong to HBV/B (1 B3, 7 B7, 5 B8, and 4 belonging to a previously unidentified but distinct cluster), 5 HBV/C (2 C1 and 3 C6), and 1 HBV/D (D1). The unidentified cluster was separated from other HBV/B Southeast Asia isolates, with a good value for posterior probability, suggesting that it is of a novel HBV/B subgenotype, designated B9. Of the sequences retrieved from GenBank, 5 reported previously as B3 (AB493827, AB493828, AB493829, AB493830, and AB493831) [55] clustered with B7. Furthermore, one previously unidentified isolate (AB493834) [55] was found to cluster with B8
Subgenotype B9 was distinguished from other HBV/B subgenotypes by specific features seen in the region encoding HBsAg and HBcAg. In the part of the S gene (nt 155–832) encoding small surface protein (226 amino acid residues), HBsAg, two nucleotide substitutions were found in the B9 subgenotype isolate group that are not present in other isolate groups of B subgenotypes (Supplementary Figure 1). These nucleotide substitutions were 555A and 570T, both of which were silent mutations. Within their core regions, encompassing nt 1901-2452, six nucleotide substitutions that are not present in other isolate groups of HBV/B subgenotypes were identified: 49G, 207A, 214T, 228G, 229A, and 291A (Supplementary Figure 2). Three amino acid substitutions that are not present in other isolate groups of HBV/B subgenotypes were found: Val15, Leu72, and Lys77.
The phylogenetic relationship between Asian HBV genotypes/subgenotypes and their ethnic origin and geographical distribution was also demonstrated, with HBV/B1 and B2, which are found in Japan and China, respectively, clearly separated with the other HBV/B subgenotypes from island Southeast Asia (B3, B4, B5, B7, B8 and B9). Interestingly, HBV/C1, C2 and C5, which are predominant in Southeast and East Asia, formed a major cluster completely that was distinct from C6, which is specific to Papua. The other HBV/C subgenotypes, which is specific to the Oceanian (C3) and Aboriginal populations of northern Australia (C4), formed distant clusters.
Ethnogeographical distribution of HBV/B, HBV/C and HBV/D subgenotypes in the Indonesian archipelago
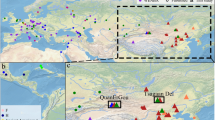
HBV genotypes/subgenotypes in this study were determined based on the diagnostic sites of SNPs in the preS2 sequence. The distribution of the genotypes of the 440 HBV isolates was 312 HBV/B (70.9%), 121 HBV/C (27.5%), and 7 (1.6%) HBV/D (Table 4). The distribution of the HBV genotypes and their subgenotypes showed distinct ethnic-related patterns in the prevalence of genotypes B, C and D (Fig. 2).

Ethnogeographical distribution of HBV genotypes/subgenotypes in the Indonesian archipelago. A total of 440 new HBV isolates were collected from 40 ethnic populations by a strict protocol to ensure the ethnic origins of their hosts to three previous generations (maternally and paternally) as described in Table 1. The isolates were genotypes/subgenotypes based on a set of diagnostic SNPs, as shown in Table 2. Three genotypes, B, C and D, and their subgenotypes were determined as shown in Table 4. Data on previously published Indonesian HBV isolates were added to the above, but only those from individuals of known ethnic origin (86) [References and number of isolates: 1 (8), 34 (54), 55 (13), 57 (11)]. Figure 2a shows the distribution of HBV genotypes in Indonesia in comparison with isolates from mainland Asia and Oceania derived from published data (3691) [References and number of isolates: 41 (100), 39 (720), 50 (332), 18 (146), 38 (367), 53 (382), 19 (209), 59 (776), 56 (211), 5 (220), 60 (67), 9 (62), 23 (51), 16 (48)]. Note that these data were from isolates with defined geographical but not ethnic origins. Figure 2b shows details of HBV/B and HBV/C subgenotypes, which are the main HBV genotypes in Indonesia. The genotypes/subgenotypes are as follows: A (yellow), B (shades of blue), C (shades of red) and D (green)
Of the 189 isolates from the islands of western Indonesia (Sumatra, Nias, Mentawai, Kalimantan, Java and Lombok islands), HBV/B accounted for almost 74.6% (141 isolates) followed by HBV/C (48 isolates; 25.4%). At the subgenotype level, B3 was by far the predominant one (70.9%), followed by B8 (9.9%), B9 (7.8%), and B5 (6.4%), while B2 and B7 represented only 2.8% and 2.1%, respectively, of the total HBV/B population. Of the HBV/C isolates, C1 was detected in 28 (58.3%) and C2 in 20 (41.7%) isolates. Thus, HBV/B and its subgenotype B3 were the predominant genotype and subgenotype in the islands of western Indonesia, with the exception of the Minang population of western Sumatra, in which HBV/C and its subgenotype C1 were the predominant genotype and subgenotype.
In contrast, in the Moluccas and Papua, in the far east of the Indonesian archipelago, HBV/C was the predominant genotype (80%), followed by HBV/D (16.7%), and notably, only one HBV/B isolate was detected out of 30 examined. C1 was found in 37.5%, C2 in 20.8%, C5 in 12.5%, and C6 in 29.2% of the 24 HBV/C isolates. In the coastal populations of Papua, HBV/C6, a recently reported HBV/C subgenotype [24, 28, 55], was by far the major subgenotype (43.8%), with C2 being the second (31.3%) and HBV/D constituting 25%.
In between, in Sulawesi and the East Nusa Tenggara (Sumba, Flores and Alor) islands, all three of the HBV genotypes, B, C and D, were detected, in 147 (78.2%), 39 (20.7%), and 2 (1.1%), respectively, of the 188 isolates examined. More variation in HBV/B subgenotypes were detected: B3 in 12 (8.2%), B5 in 15 (10.2%), B7 in 64 (43.5%), B8 in 11 (7.5%) and B9 in 45 (30.6%). Of the HBV/C isolates, C1, C2 and C5 accounted for 21 (53.9%), 13 (33.3%) and 5 (12.8%) isolates, respectively.
The distribution of HBV genotypes/subgenotypes among Indonesians of Chinese ethnic origin was dominated by HBV/B2 (42.4% of the total 33 HBV isolates), followed by HBV/B3 as the second dominant HBV/B subgenotype (25.5%).
The 24 HBV complete genomes together with 416 preS2 sequences obtained in this study have been deposited in the GenBank database with accession numbers GQ358136 to GQ358159 and GU071282 to GU071721, respectively.
Discussion
Several new HBV/B subgenotypes have been discovered in recent years from studies in ethnic populations of Asia, in addition to the initial subgenotypes identified in Japanese (Bj/B1), Chinese (Ba/B2) and ‘Indonesian’ (B3) populations [32, 47]. Recent studies [28, 34] suggested that the eastern islands of the Indonesian archipelago have an unusually high genetic diversity of HBV/B. For example, two subgenotypes have been reported (B7 and B8) from this region, in addition to B3, which is dominant in the western half of the archipelago. This observation is in contrast with that of the Japanese and Chinese populations in East Asia, which exhibit only one HBV/B subgenotype for each population, B1 and B2, respectively [32, 42].
One more HBV/B subgenotype, B9, was discovered in the present study, in the East Nusa Tenggara islands of Indonesia. In addition to the posterior probability value (100), pairwise comparison of the HBV/B genome sequences (Table 3) revealed that the intersubgenotypic divergences of HBV/B9 against B1, B6, B2 and B4 were significantly higher than the suggested 4% as the distinguishing divergence for subgenotypes [20] (6.07 ± 0.49, 5.87 ± 0.29, 4.86 ± 0.37, and 4.82 ± 0.45, respectively). Although the divergences were less against B5, B8, B7 and B3 (3.43 ± 0.25, 3.22 ± 0.24, 3.21 ± 0.31, and 3.07 ± 0.24, respectively), we argue that consideration of the distinct geographical and host ethnicity association [21], in addition to the phylogenetic and genetic distance data, define the unidentified cluster as a distinct subgenotype. Consistent with the above arguments, B5, initially discovered in the Philippines [41] also shows only 3.2% nucleotide divergence from the better established B3 of western Indonesia (Table 3).
Subgenotype B9 branched at a position more distant than the ancestral point of B3 and B7 (Figure 1), suggesting that it is evolutionarily older than B3 and B7.
Amino acid substitution patterns in core proteins of B9 isolates (Val15, Leu72, Lys77) also distinguished them from other HBV/B subgenotypes. Two of the three substitutions occurred at known immune recognition sites: the immunodominant CD4 T-cell epitopes (amino acids 1–20) and the B-cell determinant (amino acid 74–89) [15, 21]. Further bioinformatics and experimental studies of B9 together with other HBV/B subgenotypes would be needed to understand the dynamic interactions between the virus and host immune system as well as natural selection in different host populations.
A closer examination of the phylogenetic relationships of the 165 complete genome sequences (24 sequences from this study and 141 from the GenBank database) between the various Asian HBV genotypes/subgenotypes clearly revealed their ethnogeographical association (Fig. 1). HBV/B subgenotypes specific to East Asia (B1 in Japan and B2 in China) clearly separated from those of island Southeast Asia (B3, B4, B5, B7, B8 and B9). This observation revealed that HBV/B1 and B2 were the HBV/B subgenotypes specific to East Asia, while B3, B4, B5, B7, B8, and B9 specific to Southeast Asia.
To genotype and subgenotype a large number of HBV isolates in this study (440 isolates), we have utilized the sequence diversity of the preS2 sequence. We have shown previously that the sequence of the preS2 region—which is more variable than the S region, perhaps because it is subject to fewer functional constraints —can be used for reliable HBV/B and HBV/C subgenotyping on the basis of a set of diagnostic SNPs [34]. These diagnostic SNPs were determined from preS2 sequences of HBV isolates subgenotyped by phylogenetic analysis of whole genome sequences [34]. Additional diagnostic sites were identified from the 24 new whole genome sequences (Table 2). Thus, this study confirmed the usefulness of diagnostic SNPs of the preS2 sequence, particularly for analysis of large samples.
The results of our study provide the first direct evidence that the distribution of HBV genotypes/subgenotypes in the Indonesian archipelago is related to the ethnic origins of its populations. The genetic clustering of the ethnic populations of Indonesia has been defined as part of a recent large study on the genetic diversity of Asia by The HUGO Pan-Asian SNP Consortium [52]. The clustering is consistent with the ethnolinguistic structure of the Asian populations investigated [54]. Except for Papua, most of the ethnic populations of Indonesia are speakers of languages belonging to the Austronesian linguistic family. Our finding that HBV/B is the predominant genotype in the Indonesian archipelago, except in Papua and Papuan-influenced neighboring populations of Moluccas, where HBV/C was predominant, suggests that HBV/B is specifically associated with the Austronesian speakers, whereas HBV/C is the major genotype in Papua.
Of particular significance with respect to the origin of the ethnic populations is the association between the observed HBV/B subgenotypes and the linguistic subgroups of the Austronesian speakers. There are three Austronesian language subgroups in the Southeast Asian archipelago [4, 54]: Western Malayo-Polynesian (WMP; Sumatra, Java, Kalimantan, Sulawesi and the western islands of Nusa Tenggara), Central Malayo-Polynesian (CMP; the eastern islands of Nusa Tenggara and southern Moluccas) and South Halmahera West New Guinea (SHWNG). HBV/B3 is the major subgenotype in the Austronesian WMP speakers of the western half of Indonesia, whereas unique HBV/B subgenotypes—B7, B8, and B9—were observed in the populations of East Nusa Tenggara islands belonging to the Austronesian CMP linguistic subgroup.
The observation of HBV/B subgenotypes that are unique to the Indonesian ethnic populations and their distribution following the ethnolinguistic structure of the populations suggests that it is unlikely that these subgenotypes have been introduced in recent times. Rather, the result indicates that the origin of HBV distribution is associated with the ancient migratory events involved in the peopling of the archipelago. Archaeological and anthropological findings indicate that there were two major migratory events associated with the peopling of the Indonesian archipelago: the first occurred some 60,000 years before present (yBP) with the earliest arrival of modern humans in their continuing migration from Africa to Papua and Australia, while the second occurred around 3000–5,000 yBP as part of the diaspora of Austronesian-language-speaking populations [3, 11]. The Austronesian speakers replaced and perhaps assimilated most of the original Austromelanoid populations, but in the island of Papua New Guinea, the populations originating from the initial peopling event some 50,000 years earlier remain isolated, separated by extreme geographical features. The long isolation is reflected by the fact that there are more than 1,000 distinct languages spoken on the island, belonging to three language families [6, 7], in addition to the Austronesian languages spoken by the coastal populations.
It has been suggested that the history of HBV evolution in primates is a relatively recent event, with the divergence in humans and apes occurring only in the last 6,000 to 7,000 years [8, 61]. However, this suggestion is incompatible with the finding of HBV in isolated aboriginal populations in Papua New Guinea and Australia [10, 43, 46]. The ubiquitous distribution of HBV/C in East and Southeast Asia, Papua New Guinea and Australia argues for an early introduction HBV along with the initial peopling of the islands.
Following the above scenario, HBV/C, which was shown to be dominant in populations of mainland Asia and in indigenous populations of Papua and Australia (Fig. 2) but relatively low in the Austronesian-speaking populations, would have probably been introduced by the initial peopling of the archipelago. C1, which is the predominant HBV/C subgenotype in Indonesia, is also most prevalent in southern China [56]. The arrival of the Austronesian-speaking populations in the archipelago 3,000 to 5,000 yBP presumably displaced most HBV/C with the introduction of the Austronesian-associated HBV/B. The observation of the different spectrum of HBV subgenotypes associated with the WMP, CMP and SHWNG branches of the Austronesian-speaking population further supports the suggestion of co-migration of HBV/B and its human hosts and that the transmission of HBV in the distant past was mainly vertical, from mother to child, mimicking the transmission of the maternally inherited human mitochondrial DNA [34].
The other interesting finding in this study was the observation that HBV/B2, which is characteristic to the Chinese populations of mainland Asia and Taiwan [22, 32], was also dominant in Indonesians of Chinese ethnic origin, consistent with our previous observation [34]. Significantly, HBV/B3 was found to be the second major HBV/B subgenotype in the Indonesian Chinese. HBV/B3 has never been reported in the populations of China and Taiwan. Thus, this observation presumably reflects the social interactions between the indigenous and Chinese populations of Indonesia.
Several interesting deviations from the general pattern were observed, such as in the Austronesian WMP-speaking populations of Minang of West Sumatra, the Mandar, Kajang and Toraja of South Sulawesi, and the mixed Austronesian-Papuan populations of Alor in East Nusa Tenggara. Some of these deviations could be traced to more recent population interactions and movements within the archipelago.
Independent of the speculations about its origin, the finding of a specific association of HBV subgenotypes with the ethnic populations of the Indonesian archipelago is of epidemiological and medical relevance. A study of mutations that underlie beta thalassemia in Indonesia, for example, has also indicated a similar association with ethnic populations in the distribution of some 30 beta-globin mutations [44]. The distribution of many other diseases in the Indonesian archipelago is probably also determined to various degrees by the genetic clustering of its ethnic populations.
References
Achwan WA, Muttaqin Z, Zakaria E, Depamede SA, Mulyanto, Sumohardjo S, Tsuda F, Takahashi K, Abe N, Mishiro S (2007). Epidemiology of hepatitis B, C, and E viruses and human immunodeficiency virus infections in Tahuna, Sangihe-Talaud Archipelago, Indonesia. Intervirol 50:408–411
Arauz-Ruiz P, Norder H, Robertson BH, Magnius LO (2002) Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J Gen Virol 83:2059–2073
Bellwood P (1995) Austronesian prehistory in Southeast Asia: homeland, expansion and transformation. In: Bellwood P, Fox JJ, Tryon D (eds) The Austronesians. The Australian National University, Canberra, pp 96–111
Bellwood P (1997). Recent Indo-Malaysian prehistory: according to the languages. In: Prehistory of the Indo-Malaysian archipelago, 2nd edn. University of Hawai’i Press, Honolulu, pp 96–127
Ding X, Mizokami M, Yao G, Xu B, Orito E, Ueda R, Nakanishi M (2001) Hepatitis B virus genotype distribution among chronic hepatitis B virus carriers in Shanghai, China. Intervirology 44:43–47
Ethnologue. Irian Jaya (2000). In: Grimes BF, Grimes JE (eds) Ethnologue, languages of the world, vol 1, 14th edn. SIL International, Dallas, pp 469–486
Ethnologue. Papua New Guinea (2000). In: Grimes BF, Grimes JE (eds) Ethnologue, languages of the world, vol 1, 14th edn. SIL International, Dallas, pp 741–799
Fares MA, Holmes EC (2002) A revised evolutionary history of hepatitis B virus (HBV). J Mol Evol 54:807–814
Gandhe SS, Chadha MS, Arankalle VA (2003) Hepatitis B virus genotypes and serotypes in Western India: lack of clinical significance. J Med Virol 69:324–330
Hawkers RA, Boughton CR, Ferguson V, Vale TG (1981) The seroepidemiology of hepatitis in Papua New Guinea. II. A long-term study of hepatitis B. Am J Epidemiol 114(4):563–573
Higham CFW (2000) Prehistory, language and human biology: is there a consensus in East and Southeast Asia? In: Jin L, Seielstad M, Xiao C (eds) Genetic linguistic and archaeological perspectives on human diversity in Southeast Asia. World Scientific Publishing, Singapore, pp 3–15
Huang AS, Tan CYH (2010) Achieving scientific eminence within Asia. Science 329:1471–1472
Huy TT, Ushijima H, Quang VX, Luengrojanakul P, Kikuchi K, Sata T, Abe K (2004) Genotype C of hepatitis B virus can be classified into at least two subgroups. J.Gen Virol 85:283–292
Huy TTT, Ngoc TT, Abe K (2008) New complex recombination genotype of hepatitis B virus identified in Vietnam. J Virol 82:5657–5663
Ie SI, Thedja MD, Roni M, Muljono DH (2010) Prediction of conformational changes by single mutation in the hepatitis B virus surface antigen (HBsAg) identified in HBsAg-negative blood donors. Virol J 7:326
Jazayeri MS, Basuni AA, Cooksley G, Locarnini S, Carman WF (2004) Hepatitis B virus genotypes, core gene variability and ethnicity in the Pacific region. J Hepatol 41:139–146
Kao JH, Chen PJ, Lai MY, Chen DH (2000) Hepatitis B genotypes correlate with clinical outcome in patients with chronic hepatitis B. Gastroenterol 118:554–559
Kao JH, Chen PJ, Lai MY, Chen DS (2004) Hepatitis B virus genotypes and spontaneous hepatitis B e antigen seroconversion in Taiwanese hepatitis B carriers. J Med Virol 72:363–369
Kim H, Jee YM, Song BC, Shin JW, Yang SH, Mun HS, Kim HJ, Oh EJ, Yoon JH, Kim YJ, Lee HS, Hwang ES, Cha CY, Kook YH, Kim BJ (2007) Molecular epidemiology of hepatitis B virus (HBV) genotypes and serotypes in patients with chronic HBV infection in Korea. Intervirology 50:52–57
Kim HJ, Lee DH, Gwak GY, Choi MS, Lee JH, Koh KC, Paik SW, Yoo BC (2007) Analysis of the core gene of hepatitis B virus in Korean patients. Liver Int 27:633–638
Kramvis A, Kew M, Francois G (2005) Hepatitis B virus genotypes. Vaccine 23:2409–2423
Kramvis A, Arakawa K, Yu MC, Nogueira R, Stram DO, Kew MC (2008) Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J Med Virol 80:27–46
Lim CK, Tan JTM, Khoo JBS, Ravichandran A, Low HM, Chan YC, Ton SH (2006) Correlations of HBV genotypes, mutations affecting HBeAg expression and HBeAg/Anti-HBe status in HBV carriers. Int J Med Sci 3:14–20
Lusida MI, Nugrahaputra VE, Soetjipto R, Handajani M, Nagano-Fujii M, Sasayama T, Utsumi T, Hotta H (2008) Novel subgenotypes of hepatitis B virus genotypes C and D in Papua, Indonesia. J Clin Microbiol 46:2160–2166
Magnius LO, Norder (1995). Subtypes, genotypes and molecular epidemiology of the hepatitis B virus reflected by sequence variability of the S-gene. Intervirology 38:24-34
Marzuki S, Sudoyo H, Suryadi H, Setianingsih I, Pramoonjago P (2003) Human genome diversity and disease on the island Southeast Asia. In: Marzuki S, Verhoef J, Snippe H (eds) Tropical diseases: from molecule to bedside, Adv Exp Med Biol 531, 3–18. Kluwer Academic/Plenum Publishers, New York, pp 3–18
Miyakawa Y, Mizokami M (2003) Classifying hepatitis B virus genotypes. Intervirology 46:329–338
Mulyanto, Depamede SN, Surayah K, Tsuda F, Ichiyama K, Takahashi M, Okamoto H (2009) A nationwide molecular epidemiological study on hepatitis B virus in Indonesia: identification of two novel subgenotypes, B8 and C7. Arch Virol 154(7):1047–1059
Nagasaki F, Niitsuma H, Cervantes JG, Chiba M, Hong S, Ojima T, Ueno Y, Bondoe E, Kobayashi K, Ishii M, Shimosegawa M (2006) Analysis of the entire nucleotide sequence of hepatitis B virus genotype B in the Philippines reveals a new subgenotype of genotype B. J Gen Virol 87:1175–1180
Naumann H, Schaefer S, Yoshida CF, Gaspar AM, Repp R, Gerlich WH (1993) Identification of a new hepatitis B virus (HBV) genotype from Brazil that expresses HBV surface antigen subtype adw4. J Gen Virol 74:1627–1632
Norder H, Hammas B, Lufdahl S, Courouce AM, Magnius LO (1992) Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of corresponding hepatitis B virus strains. J Gen Virol 73:1201–1208
Norder H, Courouce AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO (2004) Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 47:289–309
Normile D (2009) SNP study supports southern migration route to Asia. Science 326:1470
Nurainy N, Muljono DH, Sudoyo H, Marzuki S (2008) Genetic study of hepatitis B virus in Indonesia reveals a new subgenotype of genotype B in east Nusa Tenggara. Arch Virol 153:1057–1065
Okamoto H, Imai M, Shimozaki M, Hoshi Y, Iizuka H, Gotanda T, Tsuda F, Miyakawa Y, Mayumi M (1986) Nucleotide sequence of a cloned hepatitis B virus genome, subtype ayr: comparison with genomes of the other three subtypes. J Gen Virol 67:2305–2314
Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai M, Miyakama Y, Mayumi M (1988) Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol 69:2575–2583
Okamoto H, Nishizawa T (1992). Non-B non-G hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus. http://www.freepatentsonline.com/EP1010759.html (with permission)
Olinger CM, Jutavijittum P, Hüschen JM, Yousukh A, Samountry B, Thammavong T, Toriyama K, Muller CP (2008) Possible new hepatitis B virus genotype, Southeast Asia. Emerg Infect Dis 14:1777–1780
Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, Okanoue T, Yotsuyagani H, Lino S (2001) A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Hepatology 33:218–223
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Sakamoto T, Tanaka Y, Orito E, Co J, Clavio, Sugauchi F, Ozasa A, Quino A, Ueda R, Sollano J, Mizokami M (2006). Novel subtypes (subgenotypes) of hepatitis B virus genotypes B and C among chronic liver disease patients in the Philippines. J Gen Virol 87:1873-1882
Sakamoto T, Tanaka Y, Simonetti J, Osiowy C, Berresen ML, Koch A, Kurbanov F, Sugiyama M, Minuk GY, McMahon BJ, Joh T, Mizokami M (2007) Classification of hepatitis B virus genotype into 2 major types based on characterization of a novel subgenotype in Arctic indigenous populations. J Infect Dis 196:1487–1492
Sanders RC, Lewis D, Dyke T, Alpers MP (1992) Marker of hepatitis B infection in Tari district, Southern Highlands Province, Papua New Guinea. P N G Med J 35:197–201
Setianingsih I, Williamson R, Marzuki S, Harahap A, Tamam M, Forrest S (1998) Molecular basis of β-thalassemia in Indonesia: application to prenatal diagnosis. Mol Diagn 3:11–20
Stuvyer L, De Gendt S, Van Geyt C, Zoulim F, Fried M, Schinazi RF, Rossau R (2000) A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J Gen Virol 81:67–74
Sugauchi F, Mizokami M, Orito E, Ohno T, Kato H, Suzuki S, Kimura T, Ueda R, Butterworth LA, Cooksley WG (2001) A novel variant genotype C hepatitis B virus identified in isolates from Australian Aborigines: complete genome sequence and phylogenetic relatedness. J Gen Virol 82:883–892
Sugauchi F, Orito E, Ichida T, Kato H, Sakugawa H, Kakumu S, Ishida T, Chutaputti A, Lai CL, Ueda R, Miyakawa Y, Mizokami M (2002) Hepatitis B virus of genotype B with or without recombination with genotype C over the precore region plus the core gene. J Virol 76:5985–5992
Sugiono D (2008) The mapping of languages in Indonesia. Language Center Press, Jakarta, p 7
Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S (1998) Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full-length isolates. Arch Virol 143:2313–2326
Tangkijvanich P, Mahachai P, Komolmit P, Fongsarun J, Theamboonlers A, Poovorawan Y (2005) Hepatitis B virus genotypes and hepatocellular carcinoma in Thailand. World J Gastroenterol 11:2238–2243
Tatematsu K, Tanaka Y, Kurbanov F, Sugauchi F, Mano S, Maeshiro T, Nakayoshi T, Wakuta M, Miyakawa Y, Mizokami M (2009) A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J Virol 83(20):10538–10547
The HUGO Pan-Asian SNP Consortium (2009) Mapping human genetic diversity in Asia. Science 326:1541–1545
Toan NL, Song LH, Kremsner PG, Duy DN, Binh VQ, Koeberlein B, Kaiser S, Kandolf R, Toressi J, Bock CT (2006) Impact of the hepatitis B virus genotype and genotype mixtures on the course of liver disease in Vietnam. Hepatology 43:1375–1384
Tryon D (1995) Proto-Austronesian and the major Austronesian subgroups. In: Bellwood P, Fox JJ, Tryon D (eds) The Austronesians: historical and comparative perspectives. The Australian National University, Canberra, pp 17–35
Utsumi T, Lusida MI, Yano Y, Nugrahaputra VE, Amin M, Juniastuti, Soetjipto, Hayashi Y, Hotta H (2009). Complete genome sequence and phylogenetic relatedness of hepatitis B virus isolates in Papua, Indonesia. J Clin Microbiol 47:1842–1847
Wang Z, Hou J, Wen S, Tanaka Y, Cheng J, Kurbanov F, Wang L, Jiang J, Naoumov NV, Mizokami M, Qi Y (2007) Distribution and characteristics of hepatitis B virus genotype C subgenotypes in China. J Viral Hepat 14:426–434
Wibawa ID, Suryadarma IG, Mulyanto TsudaF, Matsumoto Y, Ninomiya M, Takahashi M, Okamoto H (2007) Identification of genotype 4 hepatitis E virus strains from a patient with acute hepatitis E and farm pigs in Bali, Indonesia. J Med Virol 79:1138–1146
Yuasa R, Takahashi K, Dien BV, Binh NH, Morishita T, Sato K, Yamamoto N, Isomura S, Yoshioka K, Ishikawa T, Mishiro S, Kakumu S (2000) Properties of hepatitis B virus genome recovered from Vietnamese patients with fulminant hepatitis in comparison with those of acute hepatitis. J Med Virol 61:23–28
Yuen MF, Sablon E, Tanaka Y, Kato T, Mizokami M, Doutreloigne J, Yuan HJ, Wong DKH, Sum SM, Lai CL (2004) Epidemiological study of hepatitis B virus genotypes, core promoter and precore mutations of chronic hepatitis B infection in Hong Kong. J Hepatol 41:119–125
Zeng G, Wang Z, Wen S, Jiang J, Wang L, Cheng J, Tan D, Xiao F, Ma S, Li W, Luo K, Naoumov NV, Jou J (2005) Geographic distribution, virologic and clinical characteristics of hepatitis B virus genotype in China. J Viral Hepat 12:609–617
Zhou Y, Holmes EC (2007) Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol 65:197–205
Acknowledgments
The authors are grateful to Dr. Julius and colleagues, Department of Internal Medicine, Faculty of Medicine, Andalas University, Padang, Indonesia, Dr. Soeyata and colleagues, Department of Internal Medicine, Faculty of Medicine, Sriwijaya University, Palembang, Indonesia, and Dr. Zein Patiiha, Department of Medicine, Ternate General Hospital, Ternate, Indonesia, for their invaluable contribution of blood samples to this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Thedja, M.D., Muljono, D.H., Nurainy, N. et al. Ethnogeographical structure of hepatitis B virus genotype distribution in Indonesia and discovery of a new subgenotype, B9. Arch Virol 156, 855–868 (2011). https://doi.org/10.1007/s00705-011-0926-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-011-0926-y