
Abstract
Professor Kurt Jellinger is well known for his seminal work on the neuropathology of age-associated neurodegenerative disorders, particularly Lewy body diseases. However, it is less well known that he also contributed important insights into the neuropathological features of several paediatric neurometabolic diseases, including Alpers–Huttenlocher syndrome, a syndrome of mitochondrial disease caused by POLG mutations, and infantile neuroaxonal dystrophy, a phenotype resulting from PLA2G6 mutations. Despite these rare diseases occurring in early life, they share many important pathological overlaps with age-associated Lewy body disease, particularly dysregulation of α-synuclein. In this review, we describe several neurometabolic diseases linked to Lewy body disease mechanisms, and discuss the wider context to pathological overlaps between neurometabolic and Lewy body diseases. In particular, we will focus on how understanding disease mechanisms in neurometabolic disorders with dysregulated α-synuclein may generate insights into predisposing factors for α-synuclein aggregation in idiopathic Lewy body diseases.
Similar content being viewed by others

Introduction
The Lewy body diseases (LBD) include Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB), all of which lie on a clinical spectrum of motor and cognitive symptoms (Jellinger and Korczyn 2018). PD and PDD present with motor symptoms such as rest tremor, bradykinesia, and unsteady gait that can progress into cognitive impairment, whilst DLB typically present with cognitive features that often develop into parkinsonian features similar to PD (McKeith et al. 2017). The characteristic pathological feature of LBD is intraneuronal inclusions of the protein α-synuclein termed Lewy bodies (McKeith et al. 2017; Spillantini et al. 1997). In addition to the presence of Lewy bodies, LBD cases are also characterised by nigral depigmentation and striatonigral dopaminergic denervation that is thought to underlie the extrapyramidal features that characterise clinical parkinsonism (Jellinger 2012).
The direct pathogenic relevance of Lewy bodies to the neuronal dysfunction and degeneration that characterises LBD is not known (Outeiro et al. 2019). Nevertheless, the observations that mutations in the α-synuclein gene SNCA cause familial PD (Polymeropoulos et al. 1997), the presence of α-synuclein in Lewy bodies (Spillantini et al. 1997), and downregulation of α-synuclein levels reducing risk of developing idiopathic PD (Mittal et al. 2017), suggest an important role for α-synuclein in pathological processes in LBD. Despite the putative role of α-synuclein in LBD, many questions remain, including why it aggregates, how (if at all) it induces neuronal dysfunction and degeneration, and thus its suitability as a target for candidate therapeutics.
Neurometabolic diseases typically arise as a result of mutations that induce perturbations in cellular metabolism that prominently affect neurons. Most are very rare and often affect individuals at earlier stages of life than age-associated neurodegenerative disorders; however, they often share overlaps with neuropathological changes observed in individuals with LBD. The present review will discuss metabolic diseases that share neuropathological overlaps with LBD in terms of α-synuclein aggregation, and will focus on PLA2G6-associated neurodegeneration (PLAN), POLG-associated neurodegeneration, Niemann–Pick Type C1, and Krabbe disease. In the present review, we will discuss what is known about shared mechanisms between the neurometabolic diseases of interest and idiopathic Lewy body disease, with a particular focus on whether the proposed pathways may be implicated in α-synuclein dysregulation in idiopathic Lewy body disease.
PLA2G6-associated neurodegeneration
PLAN is a group of disorders that fall under the umbrella of neurodegeneration with brain iron accumulation and are caused by mutations in the PLA2G6 gene, the protein product of which is thought to have a role in lipid membrane homeostasis and remodelling (Burke and Dennis 2009). PLAN is typically divided into four sub-types on the basis of clinical features and age of onset: infantile neuroaxonal dystrophy, atypical neuroaxonal dystrophy, adult-onset dystonia-parkinsonism, and autosomal recessive early onset parkinsonism (Guo et al. 2018). Neuroaxonal dystrophy was previously termed “Seitelberger’s disease”, and Prof Kurt Jellinger worked with Prof Franz Seitelberger to study this disease (Jellinger et al. 1968).
Infantile neuroaxonal dystrophy (INAD) presents between 6 months and 3 years of age, with rapidly progressing developmental delay or regression followed by muscle hypotonia and spasticity, leading to a complete loss of voluntary muscle control and death usually by the age of 5–10 years old (Babin et al. 2018). Atypical neuroaxonal dystrophy is characterised by ataxia, rigidity, and spasticity, with later onset from 3 years old to late teens, and slower progression than infantile neuroaxonal dystrophy (Guo et al. 2018). In contrast to infantile neuroaxonal dystrophy, both PLAN sub-types that onset in adulthood, adult-onset dystonia-parkinsonism and autosomal recessive early onset parkinsonism, are characterised by later onset between 20 and 40 years old, a slower rate of clinical deterioration, and responsiveness to dopaminergic agents (Guo et al. 2018). Adult-onset dystonia-parkinsonism is characterised by parkinsonian extrapyramidal features, though there is some heterogeneity in presentation, with neuropsychiatric features such as depression preceding motor symptoms in some cases (Karkheiran et al. 2015).
The characteristic neuropathological features of individuals with PLA2G6 mutations are prominent neuroaxonal spheroids, iron deposition largely confined to the globus pallidus with variable affectation of the substantia nigra, and widespread neuronal loss (Kruer 2013). Lewy bodies are an invariant finding in all cases reported in the literature with confirmed genetic testing, and were notably present to a severe degree throughout the brain of 8 years old with clinical onset in infancy (Paisan-Ruiz et al. 2012). Lewy body diseases are normally associated with advancing age, and incidental Lewy bodies are typically only observed in individuals over the age of 60 (Frigerio et al. 2011; Outeiro et al. 2019); therefore, the presence and severity of Lewy body pathology in individuals as young as 8 years old suggests an association between Lewy body pathology and PLA2G6 mutations. As a result, and given that Lewy body formation in PLAN appear to follow Braak’s pathological staging scheme for Lewy body pathology in PD (Braak et al. 2003; Paisan-Ruiz et al. 2012), it has been suggested that INAD should be considered to lie on the α-synucleinopathy spectrum (Jellinger 2003). It is also notable that many cases of PLAN also manifested tau pathology, though this was a less invariant finding than Lewy body pathology as the 8 years old with severe Lewy body pathology did not manifest concomitant tau pathology (Paisan-Ruiz et al. 2012). Nevertheless, given the putative potentiating effect of α-synuclein on tau pathology (Bassil et al. 2021), and tau on α-synuclein pathology (Dasari et al. 2019), these interactions may contribute to pathological and clinical features of PLAN.
How PLA2G6 mutations induce the aggregation of α-synuclein is not clear, but as infantile neuroaxonal dystrophy is primarily characterised as an iron storage disorder, and iron dyshomeostasis has been implicated in α-synuclein aggregation (Xiao et al. 2018), one could speculate that elevated iron levels could be associated with α-synuclein aggregation in infantile neuroaxonal dystrophy. However, knockdown of PLA2G6 in drosophila led to changes in the composition of the phospholipid bilayer of neuronal cell membranes towards lipids with shorter acyl chains and increased membrane curvature, leading to the dissociation of α-synuclein from cell membranes and increased fibrillisation (Mori et al. 2019). Consistent with this proposition, PLA2G6 knockdown in mice led to α-synuclein aggregation in mitochondria (Sumi-Akamaru et al. 2016), perhaps due to altered membrane composition and subsequent dissociation of α-synuclein from mitochondrial membranes (Shen et al. 2014). Therefore, one could speculate that PLA2G6 mutations affect the membrane-binding propensity of α-synuclein, causing its dissociation and subsequent loss of alpha-helical structure, and aggregation in a concentration-dependent manner (Outeiro et al. 2019).
POLG-associated neurodegeneration
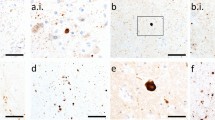
Mutations in POLG give rise to several clinical syndromes, including: Alpers–Huttenlocher syndrome (AHS), myocerebrohepatopathy spectrum (MCHS), myoclonic epilepsy myopathy sensory ataxia (MEMSA), ataxia neuropathy spectrum (ANS), and progressive external ophthalmoplegia (PEO) (Stumpf et al. 2013). Typically, AHS and MCHS onset in infancy or childhood, both with liver dysfunction and developmental delay, though AHS is also characterised by epilepsy, particularly in the occipital region, progressing to epilepsia partialis continua and status epilepticus (Wolf et al. 2009). MEMSA onsets in adolescence or adulthood and is characterised by epilepsy, myopathy and ataxia (Van Goethem et al. 2003). ANS onsets in early adolescence or adulthood and is characterised by ataxia and neuropathy, but without myopathy, and some may also have PEO (Van Goethem et al. 2004). Finally, PEO onsets in adulthood with progressive weakening of extraocular eye muscles leading to ptosis and reduced eye movement, though myopathy is often observed in these individuals (Van Goethem et al. 2001). As with many rare diseases, neuropathological data on individuals harbouring POLG mutations are limited by the relatively few reports in the literature. However, individuals with POLG mutations have varied pathological features, ranging from severe necrosis of the occipital cortex, particularly in AHS, to cortical laminar necrosis, with loss of cerebellar Purkinje cells an almost invariant finding across phenotypes (Fig. 1A.i., A.ii.).

In contrast to control cerebellum, which shows a clear layer of Purkinje cells (arrows) on H&E stain (A.i.), individuals with POLG mutations typically show a loss of Purkinje cells, with some dystrophic remnants of Purkinje cells (arrow), alongside associated Bergmann gliosis (arrowheads; A.ii.). The substantia nigra of a 59 year old male with a POLG mutation shows α-synuclein-immunoreactive Lewy bodies (arrowheads; B.i.), as does the temporal cortex of a 79 year old male with a POLG mutation (arrowheads; B.ii.). Krabbe disease patients show clusters of lipid-filled multi-nucleated globoid cells (arrowheads) and single mononucleated foamy macrophages (arrows) in the medial lemniscus of a 10 month old male (C.i.) and occipital white matter of a 9 month old male (C.ii.). α-Synuclein immunohistochemistry of Krabbe disease cases demonstrates spherical inclusions in frontal cortex of a 10 month old male (D.i.) and the putamen of a 12 month old female (D.ii.). Scale bars = 200 µm (A.i., B.i., B.ii.), 100 µm (A.ii., C.i., C.ii., D.i.), 50 µm (D.ii.)
POLG mutations have been linked to degeneration of the nigrostriatal system similar to PD with concomitant, though variable, parkinsonism. One study that conducted neuropathological examinations of four POLG mutation carriers identified severe deficiency of Complex I of the mitochondrial respiratory chain in substantia nigra neurons, but that this was not associated with clinical parkinsonism, whereas loss of nigral neurons was a good correlate of parkinsonian severity (Palin et al. 2013). In contrast, another study in six younger POLG cases confirmed severe Complex I deficiency and marked nigral neuronal loss but an absence of parkinsonian clinical features (Tzoulis et al. 2013). Although initial reports of parkinsonism in PEO cases indicated an absence of Lewy bodies in two cases, we have subsequently reported a higher prevalence of Lewy body pathology in a prospective series of older mitochondrial disease cases (Fig. 1B.i., B.ii.), particularly those with nuclear DNA mutations such as POLG, than in a comparable and older control population (Erskine et al. 2020). This study did not show consistently higher levels of Alzheimer-type pathology compared to that observed in normal ageing, and Lewy body pathology appeared to conform to Braak’s scheme for the distribution of Lewy bodies in PD (Braak et al. 2003). Consistent with the proposition that POLG mutations may increase vulnerability to Lewy body pathology, stem cells derived from a patient with a POLG mutation had increased levels of high molecular weight α-synuclein and higher levels of α-synuclein phosphorylated at serine 129 (Chumarina et al. 2019).
There are many potential mechanisms that could explain why individuals with POLG mutations would have increased risk of developing Lewy body pathology, including the long-established relationship between Complex I inhibition and α-synuclein aggregation (Cannon et al. 2009), excessive production of reactive oxygen species leading to the accumulation of degradation-resistant oxidised α-synuclein (Martinez-Vicente et al. 2008), and increasingly oxidised intracellular environments leading to increased unbound α-synuclein through reductions in its binding partners (Scarlata and Golebiewska 2014). The mitochondrial lipid cardiolipin has been demonstrated to stabilise monomeric α-synuclein on mitochondrial inner membranes (Ryan et al. 2018), but is highly vulnerable to peroxidation secondary to excessive production of reactive oxygen species (Paradies et al. 2014) that reportedly occurs in neuronal cells generated from POLG mutation carriers (Liang et al. 2020). Therefore, it is also plausible to suggest that altered mitochondrial membrane dynamics could lead to increased levels of unbound α-synuclein within neurons, somewhat similar to that proposed for PLA2G6 mutations.
Niemann–Pick type C1
Niemann–Pick disease is an umbrella term for a group of autosomal recessive lysosomal disorders characterised by the accumulation of undegraded lipids within affected cells (Santos-Lozano et al. 2015). Niemann–Pick type A and B differ from C and D on the basis of the affected mechanisms, with A and B resulting from sphingomyelinase deficiency and C and resulting from mutations in proteins involved in intracellular lipid and cholesterol trafficking (Sun, 2018). Niemann–Pick C1 results from mutations in NPC1, a lysosomal membrane protein, the loss of which induces defects in intracellular lipid trafficking and the accumulation of unesterified cholesterol and other sphingolipid species (Yu et al. 2014). Niemann–Pick C1 can present across childhood and adolescence, with early onset associated with more rapid progression, and typically involving hepatosplenomegaly, developmental delay, ataxia, hypotonia, and saccadic abnormalities (Sun 2018).
Neuropathologically, Niemann–Pick C1 is characterised by widespread accumulation of gangliosides and unesterified cholesterol in neurons, alongside meganeurites often larger than the soma from which they emanate (Zervas et al. 2001). There have been several reports of α-synuclein accumulation in Niemann–Pick C1 cases, from one case study reporting Lewy bodies (Chiba et al. 2014), to a case series of 12 individuals, of which nine had intraneuronal α-synuclein aggregation characterised as “pre-Lewy bodies”, whilst in two cases aged 23 and 32, it was typical Lewy body pathology (Saito et al. 2004). Therefore, although Niemann–Pick C1 appears associated with the accumulation of α-synuclein in most cases, it is not clear how this related to Lewy body formation, nor whether they conform to the distribution of Lewy body pathology in PD (Braak et al. 2003). However, given the young age of cases manifesting Lewy bodies, much younger than the age at which incidental Lewy body disease typically occurs, one could suggest an association between NPC1 mutations and α-synuclein aggregation. It is also notable that tau pathology was also observed in 10/12 Niemann–Pick cases with α-synuclein pathology noted previously, where its abundance appeared to closely mirror that of α-synuclein, and with which it was frequently co-localised (Saito et al. 2004).
There are several potential mechanisms that could underlie the increased levels of aggregated α-synuclein in Niemann–Pick Type C1. In addition to cholesterol, lipidomic analyses have demonstrated reductions of galactosylceramide alongside striking increases in cholesterol, glucosylceramide, lactosylceramide, gangliotriaosylceramide, and GM2 and GM3 gangliosides in Niemann–Pick type C1 (Vanier 1999). Glucosylceramide has been widely studied in the context of Lewy body diseases, where it has been demonstrated to precipitate the toxic conversion of α-synuclein (Zunke et al. 2018), and thus, its elevation in Niemann–Pick type C1 may contribute vulnerability to α-synuclein aggregation. GM2 gangliosides have also been associated with α-synuclein pathology in mouse models (Suzuki et al. 2003, 2007) and GM3 gangliosides have been demonstrated to induce α-synuclein fibrillisation in vitro (Gaspar et al. 2018). Therefore, there are several plausible links between lipids accumulated in Niemann–Pick type C1 and the aggregation of α-synuclein.
Krabbe disease
Krabbe disease is a rare autosomal recessive neurodegenerative disorder resulting from mutations in the GALC gene, which encodes the lipid-degrading lysosomal enzyme β-galactocerebrosidase (Graziano and Cardile 2015). 95% of cases have onset within the first 6 months of life, presenting with hyperirritability, hypersensitivity to external stimuli, and stiffness of limbs. Infantile cases progress quickly with marked psychomotor regression and hypotonicity, leading to a state of decerebrate posture and complete non-responsiveness, prior to death typically before the age of 2 years old (Hagberg et al. 1963). Later onset cases have been described, though their onset and progression is more variable, ranging from late infantile through to juvenile and even adult onset (Debs et al. 2013; Lyon et al. 1991). The classic neuropathological feature of Krabbe disease is spongiosis of the white matter and the presence of swollen lipid-laden macrophages termed globoid cells (Fig. 1C.i., C.ii.) (Itoh et al. 2002). The characteristic leukodystrophy of Krabbe disease is thought to result from the accumulation of the toxic lipid psychosine, a precursor of galactosylceramide, a substrate of the enzyme encoded by GALC (Spassieva and Bieberich 2016).
Only three infantile Krabbe disease cases have been evaluated for α-synuclein pathology in the literature, with all demonstrating Thioflavin-S and α-synuclein-positive spherical inclusions reminiscent of Lewy bodies in frontal cortical tissue (Fig. 1D.i., D.ii.) (Smith et al. 2014). These findings are notable both due to the very young age of the Krabbe disease patients in which α-synuclein pathology was observed, but also as variants in GALC are associated with increased risk of developing idiopathic Parkinson’s disease, this indicates a direct mechanistic link between Krabbe disease and age-associated Lewy body diseases (Kia et al. 2021; Li et al. 2018). Given that α-synuclein pathology has only been evaluated in one brain region in human Krabbe disease brains, it is not possible to determine the extent to which it overlaps with the distribution of Lewy body pathology in PD. Furthermore, no study to our knowledge has evaluated whether concomitant Alzheimer-type pathology is present in Krabbe disease brains.
There are several plausible mechanisms that may account for α-synuclein pathology in Krabbe disease; first, psychosine has been demonstrated to interact with, and induce the fibrillisation of, α-synuclein in vitro (Abdelkarim et al. 2018). However, psychosine also induces cholesterol mislocalisation and alterations to membrane curvature (D'Auria et al. 2017; Hawkins-Salsbury et al. 2013), which may promote α-synuclein membrane dissociation and subsequent accumulation.
Implications for idiopathic Lewy body disease
Rare neurometabolic diseases with α-synuclein pathology offer a potentially unique insight into the formation of Lewy bodies in idiopathic Lewy body disease, given that their aetiology is well known and thus mechanisms that may give rise to α-synuclein aggregation can be inferred. If processes affected in the described neurometabolic disease are also known to be perturbed in idiopathic Lewy body disease, then one could speculate that they may contribute to the formation of Lewy bodies and may be worthy of further study to better understand the pathogenesis of Lewy body disease.
There are conflicting reports about the role of variants in PLA2G6 and risk of PD, with some demonstrating an association (Liu et al. 2020a, b) and others not (Liu et al. 2020a, b). However, a study using cells obtained from idiopathic PD patients demonstrated deficient activity of iPLA2, the protein product of PLA2G6 (Zhou et al. 2016), and it has been reported to be located within Lewy bodies of idiopathic PD cases (Miki et al. 2017). Therefore, although limited, there is some evidence for alterations to PLA2G6-associated function in idiopathic LBD; however, given that Lewy bodies are an invariant finding in all PLAN cases in the literature, further understanding of how iPLA2 is altered in idiopathic Lewy body disease is a pressing issue.
Similar to PLA2G6, POLG variants are not thought to be associated with increased risk of developing idiopathic Lewy body disease (Bentley et al. 2014; Hudson et al. 2009; Tiangyou et al. 2006). However, POLG is a mitochondrial DNA polymerase, the dysfunction of which has a profound impact on the mitochondrial respiratory chain (Lax et al. 2016), and alterations to the mitochondrial respiratory chain have been consistently observed in PD and DLB (Hatton et al. 2020; Reeve et al. 2012; Schapira et al. 1990), which may point to some shared aetiological factors. In contrast to the mitochondrial respiratory chain, no study to our knowledge has investigated whether alterations to cardiolipin occur in Lewy body disease despite increasing interest in this area (Gilmozzi et al. 2020). Therefore, it remains unclear whether cellular changes resulting from POLG mutations that are putatively related to α-synuclein aggregation are also observed in Lewy body disease.
Natural variants in NPC1 are not associated with increased risk of PD (Ouled Amar Bencheikh et al. 2020; Zech et al. 2013); however, some of the substrates that accumulate in Niemann–Pick Type C1, such as cholesterol, have been identified as dysregulated in idiopathic PD (Huang et al. 2019; Park et al. 2021) and DLB (Bettcher et al. 2017; Bosco et al. 2006). As with Niemann–Pick type C1, there is evidence for alterations to GM3 gangliosides in Lewy body diseases, with studies demonstrating elevations in plasma (Chan et al. 2017) and cerebrospinal fluid (Huebecker et al. 2019) in PD. There is also evidence that upregulation of β-hexosaminidase, an enzyme responsible for degradation of GM2 gangliosides, protects dopaminergic neurons from α-synuclein toxicity, implying a role for GM2 gangliosides in α-synuclein-mediated neurodegeneration (Brekk et al. 2020). Furthermore, glucosylceramide, which is elevated in Niemann–Pick type C1, has been demonstrated to be elevated in PD (Mielke et al. 2013), where it has been demonstrated to induce α-synuclein aggregation (Zunke et al. 2018) and enhance neuronal susceptibility to α-synuclein toxicity (Henderson et al. 2020). Taken together, these findings suggest that whilst NPC1 is not a risk factor for idiopathic Lewy body diseases, there is evidence to suggest dysregulation of lipid species associated with Niemann–Pick Type C1.
Unlike PLA2G6, POLG, and NPC1, multiple large studies have consistently reported that GALC variants are associated with PD risk (Kia et al. 2021; Li et al. 2018). Furthermore, there is direct evidence that psychosine, the lipid that accumulates in Krabbe disease, directly interacts with α-synuclein and induces its fibrillisation in vitro (Abdelkarim et al. 2018; Smith et al. 2014), and is elevated in PD brain tissue lysates (Marshall et al. 2018). Taken together, these findings highlight plausible and direct mechanisms through which dysregulation of psychosine may contribute to α-synuclein pathology in Krabbe disease and idiopathic Lewy body disease.
Discussion
The discussed neurometabolic diseases typically have a relatively young onset, in contrast to the age-associated presentation of idiopathic Lewy body diseases. Nevertheless, despite incidental Lewy bodies typically occurring over the age of 60 years (Gilmozzi et al. 2020), it is notable that many of the discussed metabolic diseases manifest α-synuclein aggregates at a much younger age. Given that Lewy body pathology occurs at a much younger age in individuals with particular neurometabolic diseases than occurs incidentally, one could quite reasonably suggest that α-synuclein aggregation is related to the underlying metabolic dysfunction that gives rise to the primary pathology. Therefore, understanding the processes that may contribute to α-synuclein aggregation in neurometabolic diseases, where causal disease mechanisms are already well characterised, may provide unique insights into pathological processes that contribute to Lewy body formation in idiopathic Lewy body disease.
The present discussion has highlighted four rare neurometabolic disorders with α-synuclein accumulation, all of which have alterations to particular lipid species that are also reportedly dysregulated in idiopathic Lewy body disease. Therefore, it is tempting to speculate that alterations to lipid metabolism may contribute to α-synuclein aggregation, either by direct lipid–protein interactions, as with psychosine or GM3 gangliosides, or indirectly by reducing membrane binding and thus increasing the abundance of free α-synuclein within neurons, as with PLA2G6 and cardiolipin. These are not trivial matters of enquiry as many candidate disease-modifying therapies in development for idiopathic Lewy body disease target the aggregation of α-synuclein. However, if α-synuclein aggregation in idiopathic Lewy body disease results from an underlying metabolic deficit, as it appears to do in some rare neurometabolic disorders, then such efforts are unlikely to be effective as they would not address the underlying metabolic dysfunction that led to α-synuclein aggregation, and which likely contributes to neural dysfunction and degeneration.
Although there is a renewed interest in the role of lipids in Lewy body diseases (Fanning et al. 2020), there is a pressing need to better understand changes to lipid species in post-mortem Lewy body disease brain tissue and how these may contribute to Lewy body formation. One potential barrier to this is that lipids are more difficult to study in brain tissue than proteins, and the methods involved often require tissue homogenates meaning that topographical information is lost compared to histological preparations. There is also a pressing need to better understand the extent to which α-synuclein aggregates in neurometabolic diseases recapitulate key pathogenic features of Lewy body diseases, such as increased levels of phosphorylation at serine 129 and the ability to seed aggregation of monomeric α-synuclein. Answering these questions may help gain further insights into α-synucleinopathies and determine the extent to which these diseases at the opposite ends of life share key pathogenic mechanisms.
References
Abdelkarim H, Marshall MS, Scesa G, Smith RA, Rue E, Marshall J, Bongarzone ER et al (2018) alpha-Synuclein interacts directly but reversibly with psychosine: implications for alpha-synucleinopathies. Sci Rep 8(1):12462. https://doi.org/10.1038/s41598-018-30808-9
Babin PL, Rao SNR, Chacko A, Alvina FB, Panwala A, Panwala L, Fumagalli DC (2018) Infantile neuroaxonal dystrophy: diagnosis and possible treatments. Front Genet 9:597. https://doi.org/10.3389/fgene.2018.00597
Bassil F, Meymand ES, Brown HJ, Xu H, Cox TO, Pattabhiraman S, Lee VM et al (2021) alpha-Synuclein modulates tau spreading in mouse brains. J Exp Med. https://doi.org/10.1084/jem.20192193
Bencheikh BOA, Senkevich K, Rudakou U, Yu E, Mufti K, Ruskey JA, Gan-Or Z et al (2020) Variants in the Niemann–Pick type C gene NPC1 are not associated with Parkinson’s disease. Neurobiol Aging 93:143 e141-143 e144. https://doi.org/10.1016/j.neurobiolaging.2020.03.021
Bentley SR, Shan J, Todorovic M, Wood SA, Mellick GD (2014) Rare POLG1 CAG variants do not influence Parkinson’s disease or polymerase gamma function. Mitochondrion 15:65–68. https://doi.org/10.1016/j.mito.2014.01.004
Bettcher BM, Ard MC, Reed BR, Benitez A, Simmons A, Larson EB, Mungas D et al (2017) Association between cholesterol exposure and neuropathological findings: the ACT study. J Alzheimers Dis 59(4):1307–1315. https://doi.org/10.3233/JAD-161224
Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P Jr, Kelly JW et al (2006) Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol 2(5):249–253. https://doi.org/10.1038/nchembio782
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Brekk OR, Korecka JA, Crapart CC, Huebecker M, MacBain ZK, Rosenthal SA, Hallett PJ et al (2020) Upregulating beta-hexosaminidase activity in rodents prevents alpha-synuclein lipid associations and protects dopaminergic neurons from alpha-synuclein-mediated neurotoxicity. Acta Neuropathol Commun 8(1):127. https://doi.org/10.1186/s40478-020-01004-6
Burke JE, Dennis EA (2009) Phospholipase A2 biochemistry. Cardiovasc Drugs Ther 23(1):49–59. https://doi.org/10.1007/s10557-008-6132-9
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT (2009) A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis 34(2):279–290. https://doi.org/10.1016/j.nbd.2009.01.016
Chan RB, Perotte AJ, Zhou B, Liong C, Shorr EJ, Marder KS, Alcalay RN et al (2017) Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: a lipidomic analysis. PLoS ONE 12(2):e0172348. https://doi.org/10.1371/journal.pone.0172348
Chiba Y, Komori H, Takei S, Hasegawa-Ishii S, Kawamura N, Adachi K, Shimada A et al (2014) Niemann–Pick disease type C1 predominantly involving the frontotemporal region, with cortical and brainstem Lewy bodies: an autopsy case. Neuropathology 34(1):49–57. https://doi.org/10.1111/neup.12047
Chumarina M, Russ K, Azevedo C, Heuer A, Pihl M, Collin A, Roybon L et al (2019) Cellular alterations identified in pluripotent stem cell-derived midbrain spheroids generated from a female patient with progressive external ophthalmoplegia and parkinsonism who carries a novel variation (p.Q811R) in the POLG1 gene. Acta Neuropathol Commun 7(1):208. https://doi.org/10.1186/s40478-019-0863-7
Dasari AKR, Kayed R, Wi S, Lim KH (2019) Tau interacts with the C-terminal region of alpha-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry 58(25):2814–2821. https://doi.org/10.1021/acs.biochem.9b00215
D’Auria L, Reiter C, Ward E, Moyano AL, Marshall MS, Nguyen D, Bongarzone ER et al (2017) Psychosine enhances the shedding of membrane microvesicles: implications in demyelination in Krabbe’s disease. PLoS ONE 12(5):e0178103. https://doi.org/10.1371/journal.pone.0178103
Debs R, Froissart R, Aubourg P, Papeix C, Douillard C, Degos B, Sedel F et al (2013) Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis 36(5):859–868. https://doi.org/10.1007/s10545-012-9560-4
Erskine D, Reeve AK, Polvikoski T, Schaefer AM, Taylor RW, Lax NZ, Ng YS et al (2020) Lewy body pathology is more prevalent in older individuals with mitochondrial disease than controls. Acta Neuropathol 139(1):219–221. https://doi.org/10.1007/s00401-019-02105-w
Fanning S, Selkoe D, Dettmer U (2020) Parkinson’s disease: proteinopathy or lipidopathy? NPJ Parkinsons Dis 6:3. https://doi.org/10.1038/s41531-019-0103-7
Frigerio R, Fujishiro H, Ahn TB, Josephs KA, Maraganore DM, DelleDonne A, Ahlskog JE et al (2011) Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging 32(5):857–863. https://doi.org/10.1016/j.neurobiolaging.2009.05.019
Gaspar R, Pallbo J, Weininger U, Linse S, Sparr E (2018) Ganglioside lipids accelerate alpha-synuclein amyloid formation. Biochim Biophys Acta Proteins Proteom. https://doi.org/10.1016/j.bbapap.2018.07.004
Gilmozzi V, Gentile G, Castelo Rueda MP, Hicks AA, Pramstaller PP, Zanon A, Pichler I et al (2020) Interaction of alpha-synuclein with lipids: mitochondrial cardiolipin as a critical player in the pathogenesis of Parkinson’s disease. Front Neurosci 14:578993. https://doi.org/10.3389/fnins.2020.578993
Graziano AC, Cardile V (2015) History, genetic, and recent advances on Krabbe disease. Gene 555(1):2–13. https://doi.org/10.1016/j.gene.2014.09.046
Guo YP, Tang BS, Guo JF (2018) PLA2G6-associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol 9:1100. https://doi.org/10.3389/fneur.2018.01100
Hagberg B, Sourander P, Svennerholm L (1963) Diagnosis of Krabbe’s infantile leucodystrophy. J Neurol Neurosurg Psychiatry 26:195–198. https://doi.org/10.1136/jnnp.26.3.195
Hatton C, Reeve A, Lax NZ, Blain A, Ng YS, El-Agnaf O, Erskine D et al (2020) Complex I reductions in the nucleus basalis of Meynert in Lewy body dementia: the role of Lewy bodies. Acta Neuropathol Commun 8(1):103. https://doi.org/10.1186/s40478-020-00985-8
Hawkins-Salsbury JA, Parameswar AR, Jiang X, Schlesinger PH, Bongarzone E, Ory DS, Sands MS et al (2013) Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. J Lipid Res 54(12):3303–3311. https://doi.org/10.1194/jlr.M039610
Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, Lee VMY et al (2020) Glucocerebrosidase activity modulates neuronal susceptibility to pathological alpha-synuclein insult. Neuron 105(5):822 e827-836 e827. https://doi.org/10.1016/j.neuron.2019.12.004
Huang X, Sterling NW, Du G, Sun D, Stetter C, Kong L, Mailman RB et al (2019) Brain cholesterol metabolism and Parkinson’s disease. Mov Disord 34(3):386–395. https://doi.org/10.1002/mds.27609
Hudson G, Tiangyou W, Stutt A, Eccles M, Robinson L, Burn DJ, Chinnery PF (2009) No association between common POLG1 variants and sporadic idiopathic Parkinson’s disease. Mov Disord 24(7):1092–1094. https://doi.org/10.1002/mds.22310
Huebecker M, Moloney EB, van der Spoel AC, Priestman DA, Isacson O, Hallett PJ, Platt FM (2019) Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol Neurodegener 14(1):40. https://doi.org/10.1186/s13024-019-0339-z
Itoh M, Hayashi M, Fujioka Y, Nagashima K, Morimatsu Y, Matsuyama H (2002) Immunohistological study of globoid cell leukodystrophy. Brain Dev 24(5):284–290. https://doi.org/10.1016/s0387-7604(02)00057-8
Jellinger KA (2003) Neuropathological spectrum of synucleinopathies. Mov Disord 18(Suppl 6):S2-12. https://doi.org/10.1002/mds.10557
Jellinger KA (2012) Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 27(1):8–30. https://doi.org/10.1002/mds.23795
Jellinger KA, Korczyn AD (2018) Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 16(1):34. https://doi.org/10.1186/s12916-018-1016-8
Jellinger K, Seitelberger F, Rosenkranz W (1968) Infantile neuroaxonal dystrophy. Early form with preferential cerebellar involvement. Acta Neuropathol 10(2):123–131. https://doi.org/10.1007/BF00691306
Karkheiran S, Shahidi GA, Walker RH, Paisan-Ruiz C (2015) PLA2G6-associated dystonia-Parkinsonism: case report and literature review. Tremor Other Hyperkinet Mov (NY) 5:317. https://doi.org/10.7916/D84Q7T4W
Kia DA, Zhang D, Guelfi S, Manzoni C, Hubbard L, Reynolds RH et al (2021) Identification of candidate Parkinson disease genes by integrating genome-wide association study, expression, and epigenetic data sets. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.5257
Kruer MC (2013) The neuropathology of neurodegeneration with brain iron accumulation. Int Rev Neurobiol 110:165–194. https://doi.org/10.1016/B978-0-12-410502-7.00009-0
Lax NZ, Grady J, Laude A, Chan F, Hepplewhite PD, Gorman G, Turnbull DM et al (2016) Extensive respiratory chain defects in inhibitory interneurones in patients with mitochondrial disease. Neuropathol Appl Neurobiol 42(2):180–193. https://doi.org/10.1111/nan.12238
Li G, Cui S, Du J, Liu J, Zhang P, Fu Y, Chen S et al (2018) Association of GALC, ZNF184, IL1R2 and ELOVL7 with Parkinson’s disease in Southern Chinese. Front Aging Neurosci 10:402. https://doi.org/10.3389/fnagi.2018.00402
Liang KX, Kristiansen CK, Mostafavi S, Vatne GH, Zantingh GA, Kianian A, Bindoff LA et al (2020) Disease-specific phenotypes in iPSC-derived neural stem cells with POLG mutations. EMBO Mol Med 12(10):e12146. https://doi.org/10.15252/emmm.202012146
Liu H, Wang Y, Pan H, Xu K, Jiang L, Zhao Y, Guo J et al (2020a) Association of rare heterozygous PLA2G6 variants with the risk of Parkinson’s disease. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2020.11.003
Liu H, Yao Y, Liu H, Peng Y, Ren J, Wu X, Fang Y et al (2020b) Lack of association between PLA2G6 genetic variation and Parkinson’s disease: a systematic review. Neuropsychiatr Dis Treat 16:1755–1763. https://doi.org/10.2147/NDT.S254065
Lyon G, Hagberg B, Evrard P, Allaire C, Pavone L, Vanier M (1991) Symptomatology of late onset Krabbe’s leukodystrophy: the European experience. Dev Neurosci 13(4–5):240–244. https://doi.org/10.1159/000112167
Marshall MS, Jakubauskas B, Bogue W, Stoskute M, Hauck Z, Rue E, Bongarzone ER et al (2018) Analysis of age-related changes in psychosine metabolism in the human brain. PLoS ONE 13(2):e0193438. https://doi.org/10.1371/journal.pone.0193438
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Cuervo AM et al (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118(2):777–788. https://doi.org/10.1172/JCI32806
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Kosaka K et al (2017) Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 89(1):88–100. https://doi.org/10.1212/WNL.0000000000004058
Mielke MM, Maetzler W, Haughey NJ, Bandaru VV, Savica R, Deuschle C, Liepelt-Scarfone I et al (2013) Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS ONE 8(9):e73094. https://doi.org/10.1371/journal.pone.0073094
Miki Y, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K (2017) PLA2G6 accumulates in Lewy bodies in PARK14 and idiopathic Parkinson’s disease. Neurosci Lett 645:40–45. https://doi.org/10.1016/j.neulet.2017.02.027
Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ, Scherzer CR et al (2017) beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357(6354):891–898. https://doi.org/10.1126/science.aaf3934
Mori A, Hatano T, Inoshita T, Shiba-Fukushima K, Koinuma T, Meng H, Hattori N et al (2019) Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and alpha-synuclein stability through membrane remodeling. Proc Natl Acad Sci USA 116(41):20689–20699. https://doi.org/10.1073/pnas.1902958116
Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, McKeith I et al (2019) Dementia with Lewy bodies: an update and outlook. Mol Neurodegener 14(1):5. https://doi.org/10.1186/s13024-019-0306-8
Paisan-Ruiz C, Li A, Schneider SA, Holton JL, Johnson R, Kidd D, Houlden H et al (2012) Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging 33(4):814–823. https://doi.org/10.1016/j.neurobiolaging.2010.05.009
Palin EJ, Paetau A, Suomalainen A (2013) Mesencephalic complex I deficiency does not correlate with parkinsonism in mitochondrial DNA maintenance disorders. Brain 136(Pt 8):2379–2392. https://doi.org/10.1093/brain/awt160
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2014) Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol 20(39):14205–14218. https://doi.org/10.3748/wjg.v20.i39.14205
Park JH, Lee CW, Nam MJ, Kim H, Kwon DY, Yoo JW, Kim DH et al (2021) Association of high-density lipoprotein cholesterol variability and the risk of developing Parkinson disease. Neurology 96(10):e1391–e1401. https://doi.org/10.1212/WNL.0000000000011553
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Nussbaum RL et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047. https://doi.org/10.1126/science.276.5321.2045
Reeve AK, Park TK, Jaros E, Campbell GR, Lax NZ, Hepplewhite PD, Turnbull DM et al (2012) Relationship between mitochondria and alpha-synuclein: a study of single substantia nigra neurons. Arch Neurol 69(3):385–393. https://doi.org/10.1001/archneurol.2011.2675
Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, Ryan SD et al (2018) Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat Commun 9(1):817. https://doi.org/10.1038/s41467-018-03241-9
Saito Y, Suzuki K, Hulette CM, Murayama S (2004) Aberrant phosphorylation of alpha-synuclein in human Niemann–Pick type C1 disease. J Neuropathol Exp Neurol 63(4):323–328. https://doi.org/10.1093/jnen/63.4.323
Santos-Lozano A, Villamandos Garcia D, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, Lucia A et al (2015) Niemann–Pick disease treatment: a systematic review of clinical trials. Ann Transl Med 3(22):360. https://doi.org/10.3978/j.issn.2305-5839.2015.12.04
Scarlata S, Golebiewska U (2014) Linking alpha-synuclein properties with oxidation: a hypothesis on a mechanism underling cellular aggregation. J Bioenerg Biomembr 46(2):93–98. https://doi.org/10.1007/s10863-014-9540-5
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54(3):823–827. https://doi.org/10.1111/j.1471-4159.1990.tb02325.x
Shen J, Du T, Wang X, Duan C, Gao G, Zhang J, Yang H et al (2014) alpha-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res 1591:14–26. https://doi.org/10.1016/j.brainres.2014.09.046
Smith BR, Santos MB, Marshall MS, Cantuti-Castelvetri L, Lopez-Rosas A, Li G, Bongarzone ER et al (2014) Neuronal inclusions of alpha-synuclein contribute to the pathogenesis of Krabbe disease. J Pathol 232(5):509–521. https://doi.org/10.1002/path.4328
Spassieva S, Bieberich E (2016) Lysosphingolipids and sphingolipidoses: psychosine in Krabbe’s disease. J Neurosci Res 94(11):974–981. https://doi.org/10.1002/jnr.23888
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Stumpf JD, Saneto RP, Copeland WC (2013) Clinical and molecular features of POLG-related mitochondrial disease. Cold Spring Harb Perspect Biol 5(4):a011395. https://doi.org/10.1101/cshperspect.a011395
Sumi-Akamaru H, Beck G, Shinzawa K, Kato S, Riku Y, Yoshida M, Mochizuki H et al (2016) High expression of alpha-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol Commun 4:27. https://doi.org/10.1186/s40478-016-0298-3
Sun A (2018) Lysosomal storage disease overview. Ann Transl Med 6(24):476. https://doi.org/10.21037/atm.2018.11.39
Suzuki K, Iseki E, Katsuse O, Yamaguchi A, Katsuyama K, Aoki I, Kosaka K et al (2003) Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. NeuroReport 14(4):551–554. https://doi.org/10.1097/00001756-200303240-00004
Suzuki K, Iseki E, Togo T, Yamaguchi A, Katsuse O, Katsuyama K, Hirayasu Y et al (2007) Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol 114(5):481–489. https://doi.org/10.1007/s00401-007-0264-z
Tiangyou W, Hudson G, Ghezzi D, Ferrari G, Zeviani M, Burn DJ, Chinnery PF (2006) POLG1 in idiopathic Parkinson disease. Neurology 67(9):1698–1700. https://doi.org/10.1212/01.wnl.0000238963.07425.d5
Tzoulis C, Tran GT, Schwarzlmuller T, Specht K, Haugarvoll K, Balafkan N, Bindoff LA et al (2013) Severe nigrostriatal degeneration without clinical parkinsonism in patients with polymerase gamma mutations. Brain 136(Pt 8):2393–2404. https://doi.org/10.1093/brain/awt103
Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C (2001) Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 28(3):211–212. https://doi.org/10.1038/90034
Van Goethem G, Mercelis R, Lofgren A, Seneca S, Ceuterick C, Martin JJ, Van Broeckhoven C (2003) Patient homozygous for a recessive POLG mutation presents with features of MERRF. Neurology 61(12):1811–1813. https://doi.org/10.1212/01.wnl.0000098997.23471.65
Van Goethem G, Luoma P, Rantamaki M, Al Memar A, Kaakkola S, Hackman P, Van Broeckhoven C et al (2004) POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 63(7):1251–1257. https://doi.org/10.1212/01.wnl.0000140494.58732.83
Vanier MT (1999) Lipid changes in Niemann–Pick disease type C brain: personal experience and review of the literature. Neurochem Res 24(4):481–489. https://doi.org/10.1023/a:1022575511354
Wolf NI, Rahman S, Schmitt B, Taanman JW, Duncan AJ, Harting I, Bast T et al (2009) Status epilepticus in children with Alpers’ disease caused by POLG1 mutations: EEG and MRI features. Epilepsia 50(6):1596–1607. https://doi.org/10.1111/j.1528-1167.2008.01877.x
Xiao Y, Chen X, Huang S, Li G, Mo M, Zhang L, Xu P et al (2018) Iron promotes alpha-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J Neurochem 145(1):34–50. https://doi.org/10.1111/jnc.14312
Yu XH, Jiang N, Yao PB, Zheng XL, Cayabyab FS, Tang CK (2014) NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta 429:69–75. https://doi.org/10.1016/j.cca.2013.11.026
Zech M, Nubling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B, Winkelmann J et al (2013) Niemann–Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS ONE 8(12):e82879. https://doi.org/10.1371/journal.pone.0082879
Zervas M, Dobrenis K, Walkley SU (2001) Neurons in Niemann–Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol 60(1):49–64. https://doi.org/10.1093/jnen/60.1.49
Zhou Q, Yen A, Rymarczyk G, Asai H, Trengrove C, Aziz N, Bolotina VM et al (2016) Impairment of PARK14-dependent Ca(2+) signalling is a novel determinant of Parkinson’s disease. Nat Commun 7:10332. https://doi.org/10.1038/ncomms10332
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Mazzulli JR et al (2018) Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97(1):92 e110-107 e110. https://doi.org/10.1016/j.neuron.2017.12.012
Acknowledgements
DE is funded by an Alzheimer’s Research UK Fellowship (ARUK-RF2018C-005). Brain tissue provided for the images in this manuscript was provided by Newcastle Brain Tissue Resource, which is funded in part by a Grant from the UK Medical Research Council (G0400074), by NIHR Newcastle Biomedical Research Centre awarded to the Newcastle upon Tyne NHS Foundation Trust and Newcastle University, and by a Grant from the Alzheimer’s Society and Alzheimer’s Research UK as part of the Brains for Dementia Research Project. The authors would like to thank the tissue donors and their families for donating brain tissue for research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Erskine, D., Attems, J. Insights into Lewy body disease from rare neurometabolic disorders. J Neural Transm 128, 1567–1575 (2021). https://doi.org/10.1007/s00702-021-02355-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-021-02355-7