
Abstract
In the frame of translational breast cancer research, eligibility criteria for formalin-fixed paraffin-embedded tissue (FFPE) material processing for gene expression studies include tumor cell content (TCC) and sample site (primary vs metastatic tumors). Herein we asked whether the observed differences in gene expression between paired samples with respect to TCC and sample site also have different clinical significance. We assessed ESR1, ERBB2, MAPT, MMP7, and RACGAP1 mRNA expression with real time PCR in paired samples before (NMD) and after macrodissection (MD) from 98 primary tumors (PMD, PNMD) and 72 metastatic lymph nodes (LNMD, LNNMD), as well as from 93 matched P (mP) and LN (mLN). TCC range was 2.5–75 % in the NMD series and 28–98 % in the MD and in the mP/mLN series. The prognostic effect of these markers, individually or in clusters, remained stable between paired PMD/NMD. In comparison, cluster classification failed in the LNNMD group with lower TCC. In the mP/mLN cohort, RACGAP1 mRNA expression was of prognostic significance when tested in mLN samples (p < 0.001). Similarly, luminal B, HER2, and triple negative tumors were of dismal prognosis when classified in the LN component of the same series (mLN, overall survival: p = 0.013, p = 0.034, and p = 0.007, respectively). In conclusion, the clinical relevance of the RNA markers examined may be affected by TCC in metastatic LN samples but not in primary tumors, while it differs between primary tumors and matched metastases. These data will facilitate the design of translational studies involving FFPE sample series.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Translational studies are increasingly performed on RNA extracted from formalin-fixed paraffin-embedded (FFPE) tissue material with quantitative real-time PCR (qPCR) because it yields analytically accurate results even with degraded molecular templates, such as those from FFPE tissues [1]. Conceivably, sampling is of outmost importance for obtaining reliable and reproducible results that will be translated into clinical practice. The initial workflow involves pathologists who select tissue material from FFPE tissue banks and evaluate tissue eligibility for RNA extraction processing. Tumor cell content (TCC) and site of tumor sample, e.g., for breast cancer usually primary tumors vs lymph node metastases, are two major parameters that determine tissue sample eligibility for translational studies. In fact, apart from paraffin block availability, these two parameters are major limiting factors for obtaining the large sample series necessitated for the evaluation of the markers of interest.
With respect to TCC, a number of studies have shown that gene expression profiles in normal, cancer, and distinct elements within each tissue compartment from the same section may considerably vary [2–7]. However, the impact of molecular sample TCC on the evaluation of gene expression markers for their effect on patient outcome has mostly not been addressed in translational studies, perhaps with the exception of one [2]. Currently, limiting TCC rates for gene expression assessments broadly vary in the research setting. Minimal TCC ranges from 10 [2], 20 [8], 30–50 [9–15], to 70 % [16, 17]. TCC% cutoffs have been validated individually for diagnostic gene expression applications: 75 % for the classifier PAM50 (http://www.aruplab.com/guides/ug/tests/2004700.jsp), 50 % for Oncotype DX (http://www.oncotypedx.com/en-US/Breast.aspx), or 30 % for EndoPredict [18]. For large FFPE samples series but also in the diagnostic setting, the usually applied method for increasing TCC is macrodissection, i.e., procurement of tissue fragments from unstained sections with a scalpel [19]. In comparison to the more precise but costly and time-consuming laser microdissection [20], macrodissection is an almost no-cost approach. However, it is still an extra step in the whole procedure of extracting DNA/RNA from FFPE sections, meaning extra time and labor to spend in the course of a large-scale project.
In addition, although several studies have reported variable rates on the concordance of classic breast cancer parameters (hormone receptor and HER2 status) in primary tumors and metastatic lymph nodes with slide-based methods (IHC, mRNA ISH, FISH, CISH) [21–28], knowledge regarding mRNA expression in the same context is limited. In translational studies, however, tissue material from metastatic lymph nodes may occasionally be the only source for tumor geno/phenotyping.
With the above questions still open, the present study emerged as a necessity for understanding whether TCC and assessment in primary tumors vs metastatic lymph nodes would affect the prognostic significance of gene expression markers in the frame of translational research. Focused on these issues, we reevaluated the clinical impact of selected gene expression markers previously published [10, 13, 29–31] or currently under investigation by our group. Paired samples were prepared from whole sections (non-macrodissected, NMD) and from procured tissue fragments (macrodissected, MD) from routinely processed breast carcinoma tissues. The mRNA markers assessed were ESR1 (6q25.1, estrogen receptor-alpha [ER]); ERBB2 (17q21.1, v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, [HER2]); MAPT (17q21.1, microtubule-associated protein tau); MMP7 (11q21-q22, metalloproteinase-7); and RACGAP1 (12q13.12, Rac GTPase-activating protein 1). The role of ESR1 and ERBB2 in breast cancer has been extensively studied and the expression of both genes is used in molecular breast cancer subtyping [32] and in prognostic multigene signatures [33, 34]. In breast cancer, MAPT expression seems to be an independent favorable prognostic parameter [31] influenced by ER and may be predictive of response to taxanes [35]. MMP7, one out of many matrix metalloproteases that are involved in the breakdown of extracellular matrix in normal physiological processes and in wound healing, has been shown to promote breast cancer cell invasiveness in vitro [36]. RACGAP1, a GTPase-activating protein, is essential for the induction of cytokinesis [37] and may therefore promote cancer growth. The impact of the same mRNA markers on patient outcome was further examined in paired primary and metastatic lymph node samples.
Materials and methods
Patients and tissues
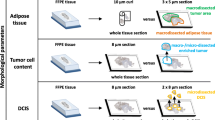
For the purposes of the present study, the clinical records and tissue material from 369 patients that had participated in the clinical trial HE10/97 conducted by the Hellenic Cooperative Oncology Group were retrieved. Patient and treatment characteristics have previously been published [38]; briefly, all patients had received dose-dense sequential epirubicin (E) and CMF with or without the addition of paclitaxel (T). From this clinical cohort, 349 patient cases, 442 paraffin blocks, and 527 RNA samples were included in the present study according to (a) availability of gene expression data for all five mRNA markers examined, (b) matched non-macrodissected (NMD) and macrodissected (MD) RNA samples, and (c) matched primary/lymph node RNA samples. Gene expression was analyzed in three series of matched RNA samples: (a) MD vs NMD from primary tumors (P); (b) MD vs NMD from metastatic lymph nodes [39]; and (c) matched P and LN samples (mP, mLN). The outline of these study groups is shown in Fig. 1; detailed patient demographics, clinical data, and standard tumor characteristics for all paired series are presented in ESM_1 (ESM_1_1). All breast carcinomas were centrally assessed with immunohistochemistry (IHC) for ER, PgR (scored according to [40]), and HER2, as well as with FISH for ERBB2 gene amplification (scored according to [41]). All patients had signed an informed consent form permitting the use of their biologic material for research purposes. The study was approved by the Bioethics Committee of the Aristotle University of Thessaloniki School of Medicine.

Outline of paired sample groups. P, primary tumor; LN, metastatic lymph node; MD, macrodissected; NMD, non-macrodissected. a The entire HE10/97 sample series with available RNA expression data are shown (in total, 442 FFPE tumor blocks from 349 patients). Matched P and LN samples were available in 93 cases. b Paired sample groups and overlapping are shown. The matched MD/NMD P series included 92 and the matched MD/NMD LN series included 72 sample pairs. Out of the MD/NMD P series, 41 (13 + 28a) MD samples were included in the P component of the matched P and LN series. Similarly, out of the matched MD/NMD LN series, 48 (20 + 28a) MD samples were included in the LN component of the matched P and LN series. Four samples (PMD, PNMD, LNMD, and LNNMD) were available in 28 cases (28a). A subset of P (N = 157) and LN samples (N = 22) shown in a was not eligible for paired sample analysis, as shown in b
Tissue sections were macrodissected where possible in cases with <75 % tumor cells in the whole section in order to increase tumor cell content (TCC) in the molecular sample. Samples were thus distinguished as MD (macrodissected) and NMD (non-macrodissected, whole sections) and are referred to as such throughout this manuscript. Histologic components were recorded as continuous variables (ESM_1_2). More details on manual macrodissection are described in ESM_1.
RNA extraction and mRNA expression investigations
RNA extraction from 527 tissue samples was performed using a fully automated silica-coated magnetic bead-based method in combination with a liquid handling robot (VERSANT Tissue Preparation System, Siemens Healthcare Diagnostics), as previously described [8, 42]. Details on RNA extraction and processing with reverse transcription quantitative real time PCR (RT-qPCR) are described in ESM_1. The assays used for ESR1, ERBB2, MAPT, MMP7, and RACGAP1 mRNA expression and their performance characteristics are shown in ESM_1_3. Relative quantification (RQ) values were assessed linearly as (40–dCT), whereby dCT = triplicate mean (CTtarget − CTRPL37A).
Statistics
This study involved paired sample analyses regarding TCC%, sample site, and RQ values. TCCNMD represents the percentage of neoplastic cells in the whole section and TCCMD, the percentage of neoplastic cells in the dissected tissue area. deltaTCC variables were calculated as TCCMD − TCCNMD for matched PMD/NMD and LNMD/NMD samples, and as TCCmLN − TCCmP for the corresponding matched samples.
RQ values were used as continuous variables throughout this study. For comparisons of individual mRNA expression between paired samples, deltaRQ variables were calculated as follows: \( \mathrm{deltaR}{{\mathrm{Q}}_{\mathrm{P}}}=\left( {\mathrm{RQ}\;{{\mathrm{P}}_{\mathrm{MD}}}} \right)-\left( {\mathrm{RQ}\;{{\mathrm{P}}_{\mathrm{NMD}}}} \right) \); \( \mathrm{deltaR}{{\mathrm{Q}}_{\mathrm{LN}}}=\left( {\mathrm{RQ}\;\mathrm{L}{{\mathrm{N}}_{\mathrm{MD}}}} \right)-\left( {\mathrm{RQ}\;\mathrm{L}{{\mathrm{N}}_{\mathrm{N}\mathrm{MD}}}} \right) \); and \( \mathrm{deltaR}{{\mathrm{Q}}_{{\mathrm{P}/\mathrm{LN}}}}=\left( {\mathrm{RQ}\;\mathrm{mLN}} \right)-\left( {\mathrm{RQ}\;\mathrm{mP}} \right) \).
TCC percentage, deltaTCC, percentage of normal glandular breast structures, epithelial hyperplasia, and in situ carcinoma component were correlated with RQ and deltaRQ values using regression analysis. RQ values were compared against nominal values (ER/PgR IHC and HER2 status) using the Mann–Whitney test and for bivariate correlations with the Spearman’s test. RQ values were also compared for the same gene in paired samples with the Wilcoxon signed ranks test. deltaRQ variables were evaluated for changes in transcript levels between paired sample series with one-sample t test by taking into account the two-sided 95 % CI.
All above analyses concerned individual markers of gene expression. However, (a) molecules act in concert in biological systems, hence their ratios are important; and (b) changes in RQ values in paired samples might be due to changes in the expression of the reference gene rather than of the target gene. Therefore, RQ values of all markers were profiled for each sample group with hierarchical clustering by using the JMP v8.0.2 software (SAS). The number of clusters was selected based on the joint assessment of (a) the ability of the clusters to form meaningful biological patterns and (b) the cubic clustering criterion and the pseudo F-statistic. In order to describe clustered RQ values in matched paired groups, we used canonical discriminant analysis measuring the distance between clusters for each sample group and the contribution of each variable in the clustering process. Based on these results, clustering concordance was evaluated with simple Kappa statistics.
The main question addressed in this study was whether assessing mRNA expression markers in the above-described different sample series would yield a different prognostic impact for these markers. For this purpose, individual continuous ESR1, ERBB2, MAPT, MMP7, and RACGAP1 RQ variables from each sample group were initially submitted to univariate Cox analysis for correlations with patient disease-free (DFS) and overall survival (OS) that were calculated as previously described [30, 31, 38, 43].
Next, the discriminatory ability of the clusters of the above RQ values regarding DFS and OS was assessed by applying the C-index along with the 95 % CI [44, 45]. Clusters were compared against each other in each group with univariate Cox for assessing the predicted risk of events with 95 % CI’s.
Results
TCC% in paired sample series and impact on individual mRNA marker expression
The distribution of TCC% in all sample series examined is shown in Table 1 and in ESM_2 (ESM_2_1). The higher efficiency of MD in LN as compared to P samples was expected based on the more diffuse growth patterns of primary tumors as compared to metastatic foci in lymph nodes (examples are shown in ESM_2_2). Matched P and LN (mP and mLN) series included both MD and NMD samples. In comparison to the samples of the entire cohort (Table 1), TCC was >25 % in PMD and LNMD, as well as in mP and mLN samples.
Variations of relative quantification (RQ) values between paired samples are shown in Fig. 2 and in ESM_3 (ESM_3_1 and ESM_3_2). deltaRQ values appeared higher or lower up to more than 6 units corresponding to 6 cycles because RQ values were calculated linearly. Considering that 3 cycles correspond to a difference of tenfold in gene expression, the differences observed in individual matched pairs reached relative gene expression differences up to more than a hundredfold, in both directions.

Difference in the expression of individual ESR1, ERBB2, MAPT, MMP7, and RACGAP1 mRNA values in paired sample series. deltaRQ values are shown. P, primary tumor; LN, metastatic lymph node; MD, macrodissected; NMD, non-macrodissected. For P and LN MD/NMD pairs, deltaRQ = RQMD − RQNMD; for P/LN pairs, deltaRQ = RQmLN − RQmP. Although outliers were found in both the P and LN series, relative ESR1 and MMP7 mRNA expression was generally lower in PMD than in PNMD samples (a), while relative ESR1, ERBB2, and RACGAP1 expression appeared generally increased in LNMD as compared to LNNMD samples (b). In matched P/LN samples (c), MMP7 was expressed greater than tenfold lower in P as compared to matched LN metastases, while approximately 1/4 of ERBB2 and 1/3 of MAPT RQ values were more than twofold lower in matched P as well. One-sample t test 95 % CI and significant variability in deltaRQ values are shown
Macrodissection efficiency in increasing TCC% was related to higher RQ values in LNMD samples for ESR1, ERBB2, and MAPT, as well as in PMD samples for ERBB2 mRNA (ESM_3_3). In addition, in primary tumors, the extent of non-neoplastic breast tissue that was removed upon MD influenced ERBB2 and RACGAP1, while the extent of in situ carcinoma areas influenced MMP7 RQ values in PMD samples (ESM_3_4). The associations of individual marker mRNA expression with classic IHC parameters (ER, PgR, and HER2) were not altered in PMD as compared to PNMD samples (ESM_3_5) but varied between LNMD and LNNMD samples (ESM_3_6) and also between mP and mLN samples (ESM_3_7). When comparing all results from ESM_3_5, _3_6, and _3_7, it would be expected that marker associations be preserved in the PMD and mP, as well as in the LNMD and mLN sample groups. Such associations were indeed preserved for ESR1 and ERBB2 expression but not for MAPT, MMP7, and RACGAP1, indicating that the above cohorts were not comparable with each other.
The strongest positive correlations of RQ values were observed between ESR1 and MAPT in all matched sample groups (all Spearman’s r > 0.5), while positive correlations between ERBB2 and RACGAP1 were encountered in LN samples only (all r > 0.35) (ESM_3_8). Negative correlations were observed between the RQ values of MMP7 and ESR1, as well as MAPT, with r values ranging between −0.23 and −0.35.
Clustering of ESR1, ERBB2, MAPT, MMP7, and RACGAP1 RQ values
Hierarchical clustering of the five mRNA markers was applied as described in the “Methods” section in the entire primary tumor and lymph node samples of the HE10/97 project and revealed four distinct categories of tumors in each series, with distinct characteristics (Fig. 3). Based on the above findings, for the biological characterization of clusters, except for the established roles of ESR1 and ERBB2 in breast cancer, we considered MAPT as a marker reflecting estrogen receptor activity [35] and RACGAP1 as a marker of proliferating cells [46]. Clusters were designated according to the established molecular subtypes of breast cancer as luminal A (LumA), luminal B (LumB), HER2-enriched (HER2), and triple negative (TN) (Fig. 3). These clusters were obtained for PMD and PNMD samples, for LNMD, and for mP and mLN samples. In the LNNMD series, the LumB cluster could not be formed, since it was represented by only one sample. Cluster discrimination in the paired sample series is shown in Fig. 4. Clusters LumA and LumB showed considerable overlapping in all groups, while HER2 and TN clusters were sharply distinguished. MMP7 RQ values had the lowest determinant role in cluster formation, while the role of RACGAP1 varied in the different sample groups. Detailed cluster statistics showing analogies between standardized values, driver genes, and discrimination ability are shown in ESM_4 (ESM_4_1 to ESM_4_4).

Biological relevance of the four-cluster model. Hierarchical clustering was set to define four clusters corresponding to LumA (ESR1 and MAPT high), LumB (ESR1 high and high RACGAP1/MAPT), HER2 (HER2 high/ESR1 and MAPT low), and TN (ESR1, HER2 and MAPT low) breast cancer subtypes, as shown in a and b. These clusters were initially identified in the entire series of primary tumor (P) and metastatic lymph node (LN) of the HE10/97 cohort. c The major analogies observed in b were preserved in paired sample series for LumA, LumB, HER2, and TN. LumB could not be identified in LNNMD samples. Standardized values correspond to mean of 0 and a standard deviation of 1

LumA, LumB, HER2, and TN clusters in paired primary and lymph node samples. Canonical discriminant structure is shown for macrodissected/non-macrodissected (MD/NMD) primary tumors (a), metastatic lymph nodes (LN) MD/NMD (b), and matched primary tumors and metastatic lymph nodes (mP/mLN) (c). Blue, LumA; red, LumB; green, HER2; brown, TN. Clusters in A and C followed the same pattern of discrimination and overlapping, which differed in the LN MD/NMD series
Cluster concordance in paired sample groups is shown in ESM_4_5. Importantly, although cluster concordance for paired groups ranged from only from 64 to 80 %, it did not statistically differ in samples with TCC <20 % vs those with original TCC ≥20 % within the same paired sample series (ESM_4_6). Finally, cluster associations with standard breast cancer markers, such as ER IHC and HER2 status determined in primary tumors only, were statistically significant for all comparisons (ESM_4_7).
Comparison of ESR1, ERBB2, MAPT, MMP7, and RACGAP1 mRNA expression, individually and clustered, with patient outcome
As shown in Table 2, no strongly significant differences were observed with respect to TCC% for ESR1, ERBB2, MAPT, MMP7, and RACGAP1 mRNA, when these markers were analyzed individually as continuous variables in paired PMD/NMD and LNMD/NMD samples. The only weak difference concerned the unfavorable prognostic effect of relatively high MMP7 in PMD, which was not observed in paired PNMD samples. This may be explained because MMP7 is a stromal marker, and PMD samples are expected to contain more tumor–stroma-specific mRNA than PNMD samples. However, hazard ratios and 95 % confidence intervals for MMP7 were similar in PMD and in PNMD. Thus, the observed difference in MMP7 statistical significance between PMD and PNMD samples was not considered as clinically significant. Similarly, the difference observed for relatively high MAPT as a favorable prognosticator for patient OS in mLN, but not in mP samples, was also not considered as clinically relevant. In this mP/mLN paired series, relatively high RACGAP1 was strongly associated with unfavorable DFS and OS only when examined in mLN samples (Table 2). However, these differences appeared to be paired sample cohort-specific, since, when examined in the entire HE10/97 population, RACGAP1 was an unfavorable prognostic parameter when examined in both primary tumor and in metastatic lymph node series.
For cluster analysis with respect to patient outcome, cluster discrimination based on the C-index did not reveal any differences for both DFS and OS between paired groups, as shown in ESM_5 (ESM_5_1). Accordingly, no difference was observed in the prognostic relevance of the four clusters in PMD as compared to the PNMD group (Table 3 and Fig. 5a). The paired LNMD/NMD groups were practically not comparable for cluster performance, since LumB could not be formed in the LNNMD samples. Instead, LumA and especially TN tumors were overrepresented in this group, yielding statistically significant results (Table 3 and Fig. 5b). In comparison, the more accurately classified HER2 clusters were associated with worst prognosis in both LNMD and LNNMD series. Cluster comparisons for patient outcome in the mP/mLN series revealed the expected worse performance of LumB, HER2, and TN in comparison to LumA tumors only in the mLN series (Table 3 and Fig. 5c). Finally, in the entire HE10/97 P and LN cohorts, the same prognostic significance was revealed for the four clusters in P samples unrelated to TCC%, while comparable significance was observed in LN samples with higher TCC% only (ESM_5_2).

Comparison of overall patient outcome according to LumA, LumB, HER2 and TN classification of primary tumors and their metastases in lymph nodes. a Clusters are compared in paired groups from primary tumors (P), macrodissected (MD), and non-macrodissected (NMD). b Clusters are compared in paired metastatic lymph node (LN) MD and NMD samples. c Matched P and LN samples. Log-rank test significance is shown. Blue, LumA; red, LumB; green, HER2; brown, TN
Multivariate COX analysis was applied in each one of the paired groups and in the entire HE10/97 sample cohorts for observing the interference of the obtained clusters with standard clinicopathologic parameters (age, menopausal status, grade, tumor size, number of metastatic lymph nodes, chemotherapy regimen, hormonal therapy, ER and PgR IHC, and HER2 status) in paired sample series. The statistically significant results from this analysis are presented in ESM_5_3. The clinical significance of these findings should be assessed with caution, because small sample numbers for several categories and possible cohort specificity of the clusters yielded large confidence intervals, implying that the observed hazard ratios may not replicate in a future study. Besides cohort specificity of the findings, it should be noticed that calling a tumor as HER2-positive by IHC/FISH and as HER2-enriched by RQ-value clustering was not necessarily identical (ESM_4_7). Overall, though, hazard ratios for the clusters in the univariate (Table 3) and in the adjusted multivariate analysis (ESM_5_3) were either close to each other, or they were at least in the same direction (favorable or unfavorable).
Discussion
The purpose of this study was to investigate whether TCC in molecular samples affects the clinical relevance of broadly applied RNA markers in breast cancer research. Our data show that, independently of molecular sample TCC rate, RNA clusters with the markers examined yield the same prognostic information. This appears as a paradox but it is not, since clusters are, basically, ratios between marker measurements. Thus, although individual marker measurements do vary between matched samples with low and high TCC, as previously established [2–7], their analogies in such samples from the same tumor seem to be preserved. These results are in concordance with the only relevant published study so far [2], which employed fresh tissues from a limited number of patients, multiple sites from the same section, and microarray gene profiling. Our findings are also in line with a more recent study [47] showing that normal tissues in the presence of breast cancer may express the same ER-positive or ER-negative gene profiles as the hosted tumor, in a broad sense of field cancerization.
It is impossible and inapplicable to suggest a safe TCC cutoff for assessing RNA markers in primary tumor samples based on the results of the present study. For establishing an optimal cutoff, multiple RNA samples should have been prepared from every single histological sample with various but precise TCC rates (for example, 10, 30, 50, 70, and 100 %), involving the same tumor site (for example, tumor front). Further, in order to obtain adequate statistical power, the major breast cancer subtypes and the multiplicity of non-cancerous histological elements and tumor microenvironment, which would be contained in the 100–TCC% of the sample, should be considered in large numbers. To our knowledge, a study taking into account all of the above parameters has not yet been performed. Our data show that the 10 % TCC previously described [2] may not be irrelevant for assessing RNA markers in primary breast tumor samples. A safe conclusion from the present study may be that the commonly published 70 or 75 % TCC cutoff as an eligibility criterion for primary tumors seems overrated and results in the exclusion of large numbers of samples from translational study cohorts, thereby lowering the statistical power of such studies. Clearly, the low TCC allowance for RNA investigations should not apply for DNA studies [48].
In comparison to primary tumors, TCC seemed to affect the clinical relevance of clusters in metastatic lymph nodes, although not of single markers. This condition may be RACGAP1-related, since this marker is expressed in lymphocytes as well [46] and will not be tumor-specific in a lymph node environment. However, the clinical relevance of these clusters in the LNMD and in the mLN groups was not the same, suggesting sample cohort bias, which is expected in fragmented sample series. Hence, TCC alone did not seem to determine the clinical relevance of the markers examined in the present study in metastatic lymph node samples.
Our data also suggest that it is ineligible to substitute for primary tumor samples with lymph node metastases and vice versa for translational study purposes, since the same RNA markers may have different clinical relevance when examined in each setting. Regional lymph node metastases are usually diagnosed simultaneously with the primary tumor and are, hence, not considered as a metachronous disease development. These regional metastases may not share the phenotypic characteristics of the primary tumor [22, 24–27, 49], one of the reasons being the evolution of different metastatic clones from a heterogeneous genetic background in the primary tumor [50]. Although, again, cohort bias may underlie the presented results from the mP and mLN series, our data indicate that when histologic material from both primary tumor and metastatic lymph nodes is available, it may be more informative, for example, to evaluate the HER2-enriched subtype in lymph nodes than in primary tumors. Whether such an approach should be integrated into practice for clinical decision-making is a question to be answered in prospective studies.
Concerning individual markers, RACGAP1 has recently been revealed as a proliferation marker associated with prognosis in breast cancer [51, 52]. Herein we show that its expression may undergo changes similar to those described for Ki-67 in metastatic lymph nodes vs primary tumors [24], which, at least in the present series, seemed ERBB2-related. In addition, MMP7 expression, a marker of epithelial–mesenchymal transition in colorectal cancer [53] and of invasiveness of breast cancer cells in vitro [36], may be associated with adverse outcome in a subset of primary breast carcinomas that needs to be defined.
Overall, in line with the previously described intrinsic characteristics of breast cancer [32, 54–56], the major genes determining the molecular subtypes in the four clusters were ESR1 and ERBB2, followed by the ER-dependent MAPT and by the proliferation marker RACGAP1. The stromal factor MMP7 did not significantly contribute in this rough subtype classification. The present study shows that, when examining RNA markers which are involved in pathways that are drivers in cancer cells but are of low activity in the coexisting non-cancer cells, such as in primary tumors surrounded by non-neoplastic breast tissue elements, TCC% may be of low importance for obtaining clinically relevant results. By contrast, when the same markers are examined in an environment where some of them may be expressed in non-cancerous cells, such as the proliferation pathway in lymph nodes bearing breast cancer metastases, TCC% may influence the prognostic significance of these markers. With the reservation that the results concerning “quantitative” or semiquantitative RNA markers, individually or in profiles, are overall cohort-specific in retrospective studies, our data may contribute to a more efficient and rational design of translational studies on FFPE tissues.
References
Paik S, Kim CY, Song YK, Kim WS (2005) Technology insight: application of molecular techniques to formalin-fixed paraffin-embedded tissues from breast cancer. Nat Clin Pract Oncol 2:246–254
Barry WT, Kernagis DN, Dressman HK et al (2010) Intratumour heterogeneity and precision of microarray-based predictors of breast cancer biology and clinical outcome. J Clin Oncol 28:2198–2206
Becette V, Vignaud S, Regnier C et al (2004) Gene transcript assay by real-time RT-PCR in epithelial breast cancer cells selected by laser microdissection. Int J Biol Markers 19:100–108
Finak G, Bertos N, Pepin F et al (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14:518–527
Ma XJ, Dahiya S, Richardson E et al (2009) Gene expression profiling of the tumour microenvironment during breast cancer progression. Breast Cancer Res 11:R7
Schobesberger M, Baltzer A, Oberli A et al (2008) Gene expression variation between distinct areas of breast cancer measured from paraffin-embedded tissue cores. BMC Cancer 8:343
Sugiyama Y, Sugiyama K, Hirai Y, Akiyama F, Hasumi K (2002) Microdissection is essential for gene expression profiling of clinically resected cancer tissues. Am J Clin Pathol 117:109–116
Hennig G, Gehrmann M, Stropp U et al (2010) Automated extraction of DNA and RNA from a single formalin-fixed paraffin-embedded tissue section for analysis of both single-nucleotide polymorphisms and mRNA expression. Clin Chem 56:1845–1853
Cronin M, Sangli C, Liu ML et al (2007) Analytical validation of the Oncotype DX genomic diagnostic test for recurrence prognosis and therapeutic response prediction in node-negative, estrogen receptor-positive breast cancer. Clin Chem 53:1084–1091
Fountzilas G, Valavanis C, Kotoula V et al (2012) HER2 and TOP2A in high-risk early breast cancer patients treated with adjuvant epirubicin-based dose-dense sequential chemotherapy. J Transl Med 10:10
Mittempergher L, de Ronde JJ, Nieuwland M et al (2011) Gene expression profiles from formalin fixed paraffin embedded breast cancer tissue are largely comparable to fresh frozen matched tissue. PLoS One 6:e17163
Psyrri A, Kalogeras KT, Kronenwett R et al (2012) Prognostic significance of UBE2C mRNA expression in high-risk early breast cancer. A Hellenic Cooperative Oncology Group (HeCOG) Study. Ann Oncol 23:1422–1427
Skarlos P, Christodoulou C, Kalogeras KT et al (2012) Triple-negative phenotype is of adverse prognostic value in patients treated with dose-dense sequential adjuvant chemotherapy: a translational research analysis in the context of a Hellenic Cooperative Oncology Group (HeCOG) randomized phase III trial. Cancer Chemother Pharmacol 69:533–546
Sparano JA, Goldstein LJ, Childs BH et al (2009) Relationship between topoisomerase 2A RNA expression and recurrence after adjuvant chemotherapy for breast cancer. Clin Cancer Res 15:7693–7700
Urban P, Vuaroqueaux V, Labuhn M et al (2006) Increased expression of urokinase-type plasminogen activator mRNA determines adverse prognosis in ErbB2-positive primary breast cancer. J Clin Oncol 24:4245–4253
Brase JC, Schmidt M, Fischbach T et al (2010) ERBB2 and TOP2A in breast cancer: a comprehensive analysis of gene amplification, RNA levels, and protein expression and their influence on prognosis and prediction. Clin Cancer Res 16:2391–2401
Hui AB, Shi W, Boutros PC et al (2009) Robust global micro-RNA profiling with formalin-fixed paraffin-embedded breast cancer tissues. Lab Invest 89:597–606
Denkert C, Kronenwett R, Schlake W et al (2012) Decentral gene expression analysis for ER+/Her2- breast cancer: results of a proficiency testing program for the EndoPredict assay. Virchows Arch 460:251–259
Kristiansen G (2010) Manual microdissection. Methods Mol Biol 576:31–38
Rabien A (2010) Laser microdissection. Methods Mol Biol 576:39–47
Aoyama K, Kamio T, Nishikawa T, Kameoka S (2010) A comparison of HER2/neu gene amplification and its protein overexpression between primary breast cancer and metastatic lymph nodes. Jpn J Clin Oncol 40:613–619
Cardoso F, Di Leo A, Larsimont D et al (2001) Evaluation of HER2, p53, bcl-2, topoisomerase II-alpha, heat shock proteins 27 and 70 in primary breast cancer and metastatic ipsilateral axillary lymph nodes. Ann Oncol 12:615–620
D'Andrea MR, Limiti MR, Bari M et al (2007) Correlation between genetic and biological aspects in primary non-metastatic breast cancers and corresponding synchronous axillary lymph node metastasis. Breast Cancer Res Treat 101:279–284
Falck AK, Ferno M, Bendahl PO, Ryden L (2010) Does analysis of biomarkers in tumour cells in lymph node metastases give additional prognostic information in primary breast cancer? World J Surg 34:1434–1441
Santinelli A, Pisa E, Stramazzotti D, Fabris G (2008) HER-2 status discrepancy between primary breast cancer and metastatic sites. Impact on target therapy. Int J Cancer 122:999–1004
Simon R, Nocito A, Hubscher T et al (2001) Patterns of her-2/neu amplification and overexpression in primary and metastatic breast cancer. J Natl Cancer Inst 93:1141–1146
Strien L, Leidenius M, von Smitten K, Heikkila P (2010) Concordance between HER-2 and steroid hormone receptor expression between primary breast cancer, sentinel node metastases, and isolated tumour cells. Pathol Res Pract 206:253–258
Zheng WQ, Lu J, Zheng JM, Hu FX, Ni CR (2001) Variation of ER status between primary and metastatic breast cancer and relationship to p53 expression. Steroids 66:905–910
Ihnen M, Wirtz RM, Kalogeras KT et al (2010) Combination of osteopontin and activated leukocyte cell adhesion molecule as potent prognostic discriminators in HER2- and ER-negative breast cancer. Br J Cancer 103:1048–1056
Pentheroudakis G, Batistatou A, Kalogeras KT et al (2011) Prognostic utility of beta-tubulin isotype III and correlations with other molecular and clinicopathological variables in patients with early breast cancer: a translational Hellenic Cooperative Oncology Group (HeCOG) study. Breast Cancer Res Treat 127:179–193
Pentheroudakis G, Kalogeras KT, Wirtz RM et al (2009) Gene expression of estrogen receptor, progesterone receptor and microtubule-associated protein Tau in high-risk early breast cancer: a quest for molecular predictors of treatment benefit in the context of a Hellenic Cooperative Oncology Group trial. Breast Cancer Res Treat 116:131–143
Sorlie T, Perou CM, Tibshirani R et al (2001) Gene expression patterns of breast carcinomas distinguish tumour subclasses with clinical implications. Proc Natl Acad Sci U S A 98:10869–10874
Marchionni L, Wilson RF, Marinopoulos SS, et al (2007) Impact of gene expression profiling tests on breast cancer outcomes. Evid Rep Technol Assess (Full Rep):1–105
Paik S, Shak S, Tang G et al (2004) A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 351:2817–2826
Ikeda H, Taira N, Hara F et al (2010) The estrogen receptor influences microtubule-associated protein tau (MAPT) expression and the selective estrogen receptor inhibitor fulvestrant downregulates MAPT and increases the sensitivity to taxane in breast cancer cells. Breast Cancer Res 12:R43
Wang F, Reierstad S, Fishman DA (2006) Matrilysin over-expression in MCF-7 cells enhances cellular invasiveness and pro-gelatinase activation. Cancer Lett 236:292–301
Zhao WM, Fang G (2005) MgcRacGAP controls the assembly of the contractile ring and the initiation of cytokinesis. Proc Natl Acad Sci U S A 102:13158–13163
Fountzilas G, Skarlos D, Dafni U et al (2005) Postoperative dose-dense sequential chemotherapy with epirubicin, followed by CMF with or without paclitaxel, in patients with high-risk operable breast cancer: a randomized phase III study conducted by the Hellenic Cooperative Oncology Group. Ann Oncol 16:1762–1771
Zaczek A, Markiewicz A, Jaskiewicz J et al (2010) Clinical evaluation of developed PCR-based method with hydrolysis probes for TOP2A copy number evaluation in breast cancer samples. Clin Biochem 43:891–898
Hammond ME, Hayes DF, Dowsett M et al (2010) American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 28:2784–2795
Wolff AC, Hammond ME, Schwartz JN et al (2007) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 131:18–43
Bohmann K, Hennig G, Rogel U et al (2009) RNA extraction from archival formalin-fixed paraffin-embedded tissue: a comparison of manual, semiautomated, and fully automated purification methods. Clin Chem 55:1719–1727
Koutras AK, Kalogeras KT, Dimopoulos MA et al (2008) Evaluation of the prognostic and predictive value of HER family mRNA expression in high-risk early breast cancer: a Hellenic Cooperative Oncology Group (HeCOG) study. Br J Cancer 99:1775–1785
Harrell FE Jr, Lee KL, Mark DB (1996) Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med 15:361–387
Pencina MJ, D'Agostino RB (2004) Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat Med 23:2109–2123
Seguin L, Liot C, Mzali R et al (2009) CUX1 and E2F1 regulate coordinated expression of the mitotic complex genes Ect2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol 29:570–581
Graham K, Ge X, de Las Morenas A, Tripathi A, Rosenberg CL (2011) Gene expression profiles of estrogen receptor-positive and estrogen receptor-negative breast cancers are detectable in histologically normal breast epithelium. Clin Cancer Res 17:236–246
Kotoula V, Charalambous E, Biesmans B et al (2009) Targeted KRAS mutation assessment on patient tumour histologic material in real time diagnostics. PLoS One 4:e7746
Aitken SJ, Thomas JS, Langdon SP, Harrison DJ, Faratian D (2010) Quantitative analysis of changes in ER, PR and HER2 expression in primary breast cancer and paired nodal metastases. Ann Oncol 21:1254–1261
Torres L, Ribeiro FR, Pandis N, Andersen JA, Heim S, Teixeira MR (2007) Intratumour genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast Cancer Res Treat 102:143–155
Milde-Langosch K, Karn T, Muller V, Witzel I, Rody A, Schmidt M, Wirtz RM (2012) Validity of the proliferation markers Ki67, TOP2A, and RacGAP1 in molecular subgroups of breast cancer. Breast Cancer Res Treat Nov 8 PMID: 23135572
Pliarchopoulou K, Kalogeras KT, Kronenwett R, et al (2012) Prognostic significance of RACGAP1 mRNA expression in high-risk early breast cancer: a study in primary tumours of breast cancer patients participating in a randomized Hellenic Cooperative Oncology Group trial. Cancer Chemother Pharmacol Oct 25 PMID:23096218
Brabletz T, Jung A, Kirchner T (2002) Beta-catenin and the morphogenesis of colorectal cancer. Virchows Arch 441:1–11
Haibe-Kains B, Desmedt C, Loi S et al (2012) A three-gene model to robustly identify breast cancer molecular subtypes. J Natl Cancer Inst 104:311–325
Hu Z, Fan C, Oh DS, Marron JS et al (2006) The molecular portraits of breast tumours are conserved across microarray platforms. BMC Genomics 7:96
Paik S (2011) Is gene array testing to be considered routine now? Breast 20(Suppl 3):S87–S91
Acknowledgments
The authors wish to thank Evita Fragou and Dimitra Katsala for monitoring the clinical study, Maria Moschoni for coordinating the data management, and Thalia Spinari for tissue sample collection. Translational research was supported by a HeCOG research grant: HE TRANS_BR.
Conflicts of interest
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kotoula, V., Kalogeras, K.T., Kouvatseas, G. et al. Sample parameters affecting the clinical relevance of RNA biomarkers in translational breast cancer research. Virchows Arch 462, 141–154 (2013). https://doi.org/10.1007/s00428-012-1357-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-012-1357-1