
Abstract
Pediatric neoplasms in the central nervous system (CNS) are the leading cause of cancer-related deaths in children. Recent developments in molecular analyses have greatly contributed to a more accurate diagnosis and risk stratification of CNS tumors. Additionally, sequencing studies have identified various, often entity specific, tumor-driving events. In contrast to adult tumors, which often harbor multiple mutated oncogenic drivers, the number of mutated genes in pediatric cancers is much lower and many tumors can have a single oncogenic driver. Moreover, in children, much more than in adults, fusion proteins play an important role in driving tumorigenesis, and many different fusions have been identified as potential driver events in pediatric CNS neoplasms. However, a comprehensive overview of all the different reported oncogenic fusion proteins in pediatric CNS neoplasms is still lacking. A better understanding of the fusion proteins detected in these tumors and of the molecular mechanisms how these proteins drive tumorigenesis, could improve diagnosis and further benefit translational research into targeted therapies necessary to treat these distinct entities. In this review, we discuss the different oncogenic fusions reported in pediatric CNS neoplasms and their structure to create an overview of the variety of oncogenic fusion proteins to date, the tumor entities they occur in and their proposed mode of action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neoplasms in the central nervous system (CNS) account for the second most common cancer and are the leading cause of cancer related deaths in children [165]. However, overall survival and therapy response vary widely between and within different pediatric CNS tumor entities. Treatment options include resection followed by radio- and/or chemotherapy depending on the patient’s age. This intensive therapy often has detrimental and long-term side effects, while the overall survival remains low for many entities or subentities. This is partly due to the lack of molecularly stratified trials and entity specific treatments [162].
Pediatric CNS tumors comprise a vast and expanding spectrum of molecularly defined entities [137]. Histologically, these entities present with various morphological patterns that are not unique for a single entity, thus complicating an accurate diagnosis based on standard histopathology alone. The need for a proper diagnosis together with the need for adequate targeted treatment options calls for identification of specific driver events and genetic and molecular signatures that better define tumor types. Molecular characterization by means of next generation sequencing (NGS) has added to the better understanding of these tumor signatures and has emphasized the oncogenic differences between pediatric and adult cancers. While adult neoplasms have high numbers of somatic mutations, these often lack in pediatric neoplasms [236]. In contrast, pediatric tumors show a higher frequency of germline alterations, copy number alterations, and structural alterations such as enhancer hijacking events and gene fusions as possible oncogenic drivers [4]. While already being well-described in pediatric hematological neoplasms and sarcomas, fusion proteins are now also emerging as important oncogenic driver events in pediatric CNS neoplasms [7, 98, 109, 144, 147, 150, 176].
Fusion genes in neoplasms
The first oncogenic fusion gene was already detected in the 1980s in chronic myeloid leukemia, but it was not until the use of NGS technologies for detection of rearrangements in the cancer transcriptome that the majority of fusion genes has been detected [73, 114, 191, 203]. The chromosomal rearrangements causing these fusions can be either balanced or unbalanced. An unbalanced rearrangement is often the result of the deletion of part of a chromosome, which then combines two genes that were formerly separated by an interstitial chromosomal segment. Balanced rearrangements can occur due to translocations, insertions, or inversions of chromosomal parts. Both balanced and unbalanced aberrations can occur between genes on the same chromosome (intra-chromosomal) as well as between genes on separate chromosomes (inter-chromosomal). These different types of rearrangements have been extensively reviewed [148, 149].
The outcome of these rearrangements is either an altered expression of one of the gene products, or a new fusion product due to the combination of the transcripts of both genes. The first occurs when the coding sequence of one gene is placed next to the promoter sequence of another gene. Although this is not a fusion protein, it does lead to altered expression levels and is seen in, for example, medulloblastoma (e.g., DDX31–GFI1B), neuroblastoma (e.g., HAND2–MYC), multiple myeloma (e.g., PRDM1–MYC), Burkitt lymphoma (e.g., IG–MYC), embryonal tumor with multilayered rosettes (TTYH1–C19MC) and in the newly identified CNS neuroblastoma tumors with FOXR2 activation (e.g., JMJD1C–FOXR2) [2, 109, 134, 161, 216, 253]. Such type of rearrangements are now known as enhancer hijacking events and will not be discussed in this review [161]. A new fusion product occurs when the promotor and 5′-coding region of one gene is fused with the 3′ coding region and UTR of the second gene, resulting in a chimeric transcript that can be translated into a fusion protein. Although many of these fusions have been identified on a RNA level, in this review we will address them as fusion proteins since their functional domains and protein functions are being discussed.
For this review, we present an overview of all chimeric proteins in pediatric CNS neoplasms that are either identified in two or more independent studies or for which additional molecular validation has been presented.
Chimeric proteins in pediatric CNS tumors
Fusion proteins have been reported in 43 different pediatric CNS neoplasms. By totalling all fusions per entity, 171 distinct fusion–entity combinations have been detected (Fig. 1a. Online Resource) [3, 8, 11, 13,14,15, 17, 19, 21,22,23, 27, 28, 30, 33, 34, 36, 39, 40, 42, 44, 46, 47, 50, 58, 59, 62,63,64, 66, 69, 70, 74, 76, 79, 81,82,83,84,85,86,87, 93, 96, 97, 99, 101, 105,106,107, 111, 114,115,116, 119, 121, 127,128,129, 132, 133, 135, 142, 145, 146, 154, 155, 157, 159, 162, 164, 167, 169, 175,176,177, 180,181,182, 184, 189, 190, 192, 198, 201, 202, 204,205,206,207, 210, 214, 216, 217, 221, 222, 225,226,227,228,229, 235, 239, 240, 242, 244,245,246, 248,249,250, 252, 255]. These 171 combinations exist of 110 unique 5’ and 3’ fusion partner gene combinations. The majority (66%; 73/110) of these unique fusions are entity specific (Fig. 1b), and their detection could aid diagnosis. For example, Yes1 associated transcriptional regulator (YAP1)–MAMLD1 fusions are restricted to supratentorial ependymoma (ST-EPN). The remaining 37 (34%) fusion proteins have been detected in multiple (two to six) tumor types (Fig. 1a, b). Additionally, there are genes with multiple fusion partners such as neurotrophic receptor tyrosine kinases (NTRKs) and fibroblast growth factor receptors (FGFRs). However, the fusions with one of the partner genes may still be specific to a tumor type, while other fusion partners occur in multiple tumor types. With the ever-expanding sequencing of tumors, these unique fusion genes might be detected in the future in other tumor types as well. Furthermore, fusions that we have not included here due to their presence in only a single case might in the future be confirmed in other cases adding to the complexity of the fusion network in pediatric CNS tumors.

Characteristics of fusion proteins observed in pediatric brain tumors. a Complete fusion partner network in pediatric CNS neoplasms. Green = N terminal partner, red = C terminal partner. Multiple connecting lines indicate that fusions were identified in multiple tumor types. b The majority (73/110) of fusions have been observed in a single tumor type. 37/110 fusions have been detected in two or more tumor types. The tumor types are annotated as mentioned in the original publication, it might be possible that some tumor types have been wrongly diagnosed or that diagnosis was changed after this publication. This is not considered. c The majority (152/171) of the pediatric CNS tumors with fusions present are glial tumors. d Based on WHO classification there are slightly more pediatric CNS low grade (LG) tumors than high grade (HG) tumors with driving fusion proteins. The WHO classification is based on the original publication and/or the tumor type. e Fusions in pediatric CNS tumors are slightly more often the results of inter-chromosomal rearrangements than intra-chromosomal rearrangements. f Most of the fusion proteins have at least one partner that functions as a kinase. Most other fusions have at least one transcription factor
Strikingly, most of the reported fusion proteins (89%; 152/171) are observed in glial tumors, while only five (3%) fusions are present in sarcomas, two (1%) in embryonal tumors and 12 (7%) fusions occur in other CNS tumor diagnoses (Fig. 1c, Online Resource 1). Most of the well-known fusion proteins, such as KIAA1549–BRAF, are especially prevalent in pediatric low-grade gliomas (LGG). Thus we examined whether the 171 identified fusion–entity pairs represent more low-grade tumors, based on data extracted from the original reports where tumor types were graded according to the WHO grading system [136]. When we divide tumors into low grade (I–II) and high grade (III–IV), low-grade tumors (50%; 85/171) harbor more fusions than high-grade tumors (40%; 69/171) (Fig. 1d). Since prevalence is not taken into account, it might be possible that overall, fusions are even more common in a certain tumor grade. For example, KIAA1549–BRAF has been detected in at least five different entities, however this fusion is reported in 70–80% of the pilocytic astrocytomas, making the fusion the most prevalent in grade I tumors [91, 112]. More than half of the reported unique fusions are inter-chromosomal (54%; 59/110) (Fig. 1e). However, due to the difficulties in detecting intra-chromosomal fusions, their incidence number might be underrepresented and might therefore increase with improved detection algorithms.
For most of the unique fusion proteins (65%; 72/110), one partner is either a tyrosine or a serine/threonine kinase (Fig. 1f). Interestingly, these 72 fusions are comprised of 12 different kinase genes, classified into seven kinase families: (1) NTRK family (NTRK1–3), (2) Raf proto-oncogene, serine/threonine kinase (RAF) family (RAF1/BRAF), (3) FGFR family (FGFR1–3), (4) ALK receptor tyrosine kinase (ALK), (5) ROS proto-oncogene 1, receptor tyrosine kinase (ROS1), (6) MET proto-oncogene, receptor tyrosine kinase (MET), and (7) protein kinase C alpha (PRKCA) (Fig. 2a). PRKCA only accounts for one fusion–entity pair and will thus not be discussed further. In pediatric CNS neoplasms these 72 kinase fusions appear as 118 unique fusion–entity pairs, mainly in glial entities (97%; 114/118), except for some NTRK- (2%; 2/118), ALK- (1%; 1/118) and ROS1- (1%; 1/118) fusions that have been identified in non-glial tumors (Fig. 3a) [133, 145, 190].

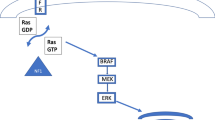
Fusion proteins are most common in the RAS/MAPK pathway. a Schematic representation of the RAS/MAPK, PI3K/AKT/mTOR and JAK/STAT pathway. Kinases implicated in fusions in pediatric CNS tumors are marked with a red dashed circle. Created with BioRender.com b Five common trends in fusion proteins observed in pediatric CNS tumors. I. C-terminal kinase protein fused to a protein with dimerization domains. II. C-terminal kinase protein fused to a protein that is highly expressed in the CNS. III. N-terminal kinase protein fused to a protein with dimerization domains. IV. Transcription factors with a transcription activation domain fused to proteins with a DNA binding domain. V. C-terminal transcription factors with a transcription activation domain and a DNA binding domain fused to a protein with no clear functional domains. c Examples of oncogenic fusions in pediatric CNS tumors that belong to the five different fusions types as mentioned in b. For every protein, the exons that are retained in the fusion protein are specified, as well as the total exons (in between brackets) in the original protein

Characteristics of kinase fusions in pediatric CNS neoplasms a Eight kinase families are responsible for all kinase fusions that are mainly present in glial tumors. b Kinase proteins are not limited to one pediatric CNS tumor type but are present in multiple different tumor types. Graphs made by https://app.rawgraphs.io. AA anaplastic astrocytoma, DA diffuse astrocytoma, DIPG diffuse intrinsic pontine glioma, DLGT diffuse leptomeningeal glioneural tumor, DNET dysembryoplastic neuroepithelial tumor, DOD diffuse oligodendroglioma, EPN ependymoma, FSCN fibroblastic spindle cell neoplasm, GBM glioblastoma, GNT glial neuronal tumor, CNS-GNB central nervous system ganglioneuroblastoma, HGG high grade glioma, IHG interhemiscpheric glioma, IMT inflammatory myofibroblastic tumor nervous system—ganglioneuroblastoma, LGG low grade glioma, MNG meningioma, NBS-HGG non brain stem high grade glioma, NET neural epithelial tumor, OA oligoastrocytoma, OD oligodendroglioma, PA pilocytic astrocytoma, GG ganglioglioma, PGNT papillary glial neuronal tumor, PLNTY polymorphous neuro-epithelial tumor of the young, PXA pleomorphic xanthoastrocytoma
We determined whether the different groups of kinase fusions have a preference towards a certain tumor type. Most are present in multiple different tumor types (Fig. 3b). However, ALK fusions mostly occur in infant hemispheric gliomas (IHG), while pilocytic astrocytomas are mainly characterized by BRAF and RAF1 fusions. Furthermore, no BRAF and RAF1 fusions were found in glioblastoma, which are characterized by fusions with NTRK, FGFR, MET or ROS1. In oligodendrogliomas there are mostly fusions with members of the FGFR family. Nonetheless, it should be kept in mind that there may be discrepancies due to updated nomenclature and differences in diagnostic methods and criteria used.
The 39 fusions lacking a kinase domain, representing 53 fusion–entity pairs, are mainly observed in glial tumors (72%; 38/53) (Fig. 3a). However, they also occur in embryonal tumors (2%; 1/53), sarcomas (9%; 5/53) and other CNS tumor diagnoses (17%; 9/53). They cover a variety of fusion partners, including recurrent partners such as EWS RNA binding protein 1 (EWSR1), MYB proto-oncogene, transcription factor (MYB), and zinc finger translocation associated (ZFTA, formerly C11orf95) [15, 27, 33, 105, 132, 176, 207, 226, 245, 255]. Instead of harboring kinase domains, these fusions are mostly characterized by transcription factors and partners with a DNA binding domain, indicating that besides kinases, these are important partners in oncogenic fusions in pediatric CNS tumors.
Receptor Tyrosine Kinases and the MAPK pathway in CNS tumors
Ten of the 13 kinase families involved in fusion proteins are receptor tyrosine kinases (RTKs), while BRAF, RAF1 and PRKCA serine/threonine kinases (STKs) are part of their downstream pathways (Fig. 2a). Under physiological conditions, RTKs, only activate the downstream pathways upon binding of extracellular growth factors, thereby inducing proliferation, differentiation, and cell survival [20, 95]. In pediatric CNS neoplasms, mutations or fusions cause dimerization and cross-phosphorylation of the intracellular domain independent of this mitogenic signal. Therefore, the RTKs constitutively activate their downstream signaling pathways, including the RAS/MAPK pathway. BRAF, RAF1 and PRKCA are all signaling through this pathway. During development, the RAS/MAPK pathway is important in cortex, midbrain and cerebellum formation [194, 197]. Its role in neurogenesis is especially interesting in regards to glial pathogenesis, considering that the cell-of-origin for gliomas is now proposed to be a neural stem cell or neural precursor instead of a post-mitotic glial cell [96, 130]. The exact mechanism how the RAS/MAPK pathway contributes to brain development is still controversial and potentially also depends on the spatial localization of the cell [25, 68]. Nonetheless, the RAS/MAPK pathway is often deregulated in gliomas [90, 91, 96, 98, 113, 128]. High-grade gliomas as well as low-grade gliomas are defined by aberrations in the pathway, although the aberrations differ between different entities. High-grade gliomas more often have aberrations in the upstream components such as the receptor tyrosine kinase NTRK, while the low-grade gliomas such as pilocytic astrocytoma have more aberrations in the intracellular RAF kinases [90]. Genomic studies showed that almost all of the pilocytic astrocytomas bear an aberration in the RAS/MAPK pathway without additional mutations or alterations [97, 179, 245].
Kinase fusions types
We have distinguished three different types of kinase fusion proteins. In the first type, the kinase domain of the RTK/STK is always retained. The RTK/STK is the C-terminal partner, meaning that the expression of the chimeric protein is driven by the promoter of its N-terminal fusion partner (Fig. 2b I). This may lead to an elevated RTK/STK expression level compared to the endogenous RTK/STK expression, as is seen in fusions with CLIP2, EML4, ETV6, KIF5B, QKI, and TFG. Due to these genes being relatively high expressed in the glial cell lineages, the oncogenic C-terminal RTK/STK expression is elevated. These fusion partners further contribute to the oncogenicity of the fusion protein by facilitating the dimerization of the RTK in absence of its ligands, an example of such fusion is HIP1–ALK (Fig. 2c). In most of these kinase fusions type I (Fig. 2b) the extracellular domain as well as the transmembrane domain of the RTK is lost and therefore the fusion protein localizes to the cytoplasm. Since the RAS signaling is lipid membrane-dependent it remains to be elucidated how oncogenic fusions activate the RAS/MAPK pathway [126]. Some of these chimeric proteins, such as EML4–ALK, can induce lipid membrane-independent RAS signaling by formation of protein-based granules [231]. Further research is necessary to show if more fusion proteins make use of this mechanism.
BRAF fusions belong to the second type of fusions, where the kinase domain is retained in the C-terminal partner but there is no common oncogenic contribution within the N-terminal partners (Fig. 2b II, c II). In FGFR fusions, who belong to the third type of fusions, FGFR retains its kinase domain and is always the N-terminal partner. The C-terminal partner almost always contains a dimerization domain, as also seen with TACC1 (Fig. 2c III). Since FGFR is the N-terminal partner it does not depend on the expression level of their partner (Fig. 2b III). A reason for this could be that tumors with FGFR fusions arise during brain development at a stage that FGFR genes are highly expressed in progenitors, or alternatively, because the regulatory domains in the 3′ UTRs of FGFR genes that negatively regulate FGFR expression are lost in these fusions [38].
Fusions involving kinases in pediatric CNS tumors
RAF fusions
BRAF and RAF1 are both RAF kinases that occur as fusion proteins with various and different partners. BRAF is seen as an oncogenic driver in a wide variety of solid and hematological malignancies. Most of the aberrations found in BRAF are mutations, and by far the most common mutation occurs within the kinase domain at amino acid V600 (V600E). While the V600E mutation in melanoma is approved for therapy with BRAF inhibitors dabrafenib and vemurafenib, these same inhibitors are controversial in other malignancies and they seem not able to inhibit tumorigenesis in pediatric astrocytomas harboring BRAF fusions and may even lead to tumor progression [103]. However, the MEK inhibitor selumetinib has recently been tested in pediatric patients with LGG harboring the well-known KIAA1549–BRAF fusion and has shown to be effective in a phase I and II trial [16, 55, 56]. Many new BRAF fusions have been identified over the last couple of years, with one study identifying 29 different BRAF fusions across seven different tumor types [189]. In pediatric CNS tumors, 14 different BRAF fusions have been described (Online Resource 2). In these fusions, the C-terminal part of BRAF is fused to the N-terminal part of its partner. The most common partner is KIAA1549 but other N-terminal partners like CLCN6, GNAI1, GTF2I, GIT2, or FAM131B, have also been reported [76, 97, 184, 192, 225, 245]. The BRAF kinase domain (encoded by exon 11–18) is retained in all pediatric CNS fusions. Most fusion breakpoints occur at the 9th exon of BRAF and in all fusions the inhibitory regulatory domain that is located within the first six exons is cut off by the fusion. So far there has been no evidence that the N-terminal partner is of specific importance other than the removal of the regulatory domain [110, 208, 238]. Six of the 14 fusion partners have either no significantly important domain in the fusion part or a domain of unknown function. The other fusion partners have domains that vary from E3 ligase, zinc-finger to coiled-coil, thus showing no clear trend in the N-terminal fusion partners.
RAF1 is the second member of the RAF kinase family that is implicated in fusions and functions alongside BRAF in the RAS/MAPK pathway. RAF1 fusions are identified in pediatric LGG (p-LGG) as well as several adult malignancies such as prostate cancer, breast cancer, pancreatic cancer and thyroid cancer [171, 215, 243]. As with BRAF fusions, due to the limited number of discovered fusion events, their prevalence and oncogenic potential as well as the effectivity of inhibitory compounds is still being elucidated. While second generation RAF inhibitors are effective for BRAF fusions, they are not effective for RAF1 fusions. Vemurafenib has even been found ineffective targeting RAF1 fusions in pediatric astrocytoma due to a paradoxical activation of the RAS/MAPK pathway [208]. RAF1 has at least six different fusion partners in pediatric CNS tumors (Online Resource 3). It is hypothesized that the N-terminal fusion partners in RAF1 fusions are important for the oncogenic potential of the fusion proteins in contrast to the partners of BRAF. Moreover, several partners such as QKI and SGRAP have already been implicated in other malignancies as well [15, 37, 118, 123]. Furthermore, all RAF1 fusion proteins identified in pediatric CNS tumors have N-terminal partners that possess a coiled-coil or other dimerization domain, meaning they belong to type I of kinase fusions (Fig. 2b). This might indicate that the dimerization of these fusion partners is necessary for the oncogenic mechanism of the fusion protein. Subsequently, RAF inhibitors may not be able to disrupt these oncogenic dimers and are thus unable to inhibit downstream signaling. For further research, combination therapies should be considered that combine a RAF inhibitor together with molecules that block the dimerization of N-terminal fusion partners.
ALK fusions
ALK belongs to the insulin receptor superfamily of RTKs. Under physiological conditions, the gene translates into a membrane bound receptor in nerve cells where it activates next to MAPK also the PI3K/AKT/mTOR and JAK/STAT pathways [75]. ALK rearrangements are common in all types of adult and pediatric cancers. Over 30 fusions have been described in various tumor types [35]. In pediatric CNS tumors, 13 different partners have been observed (Online Resource 4). These fusions are mainly found in IHGs and include HIP1–ALK, EML4–ALK and PPP1CB–ALK as well as the more recently identified ZC3H7A–ALK, MAD1L1–ALK and MSI2–ALK [41]. All ALK fusions contain the complete kinase domain of ALK at the C-terminal end, while the N-terminal partners retain variable domains in the chimeric protein, although most of the N-terminal partners have a coiled-coil or dimerization domain (69%; 9/13). ALK fusions are therefore part of the type I kinase fusions (Fig. 2b). The extent of oligomerization that is endorsed by these domains differs per partner, leading to a diversity in the oncogenic potential of the different ALK fusions [212, 213]. Interestingly, due to these variables, different ALK-fusion positive tumors have varying sensitivity to ALK inhibitors such as crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib. Not only the expression, dimerization and stability of the chimeric protein plays a role in this, but also the signaling pathway that is activated, should be considered when treating patients with ALK inhibitors. Fusion proteins that induce a > 0.5 ratio phosphorylated ALK/total ALK can activate the MAPK pathway [35]. Other fusions potentially activate different signaling pathways and this influences the oncogenic mechanism and its sensitivity to the different ALK inhibitors. In vitro and in vivo tests with ALK inhibitors showed pre-clinical evidence for tumor reduction in a PPP1CB–ALK positive tumor as a response to ALK targeted therapy with lorlatinib [40]. However, the effect on tumors with different ALK fusions remains unknown.
ROS1 fusions
ROS1 closely resembles ALK both in sequence and structure. Both RTKs have an extracellular domain, a transmembrane domain and an intracellular kinase domain and both receptors signal via the RAS/MAPK as well as the JAK/STAT and PI3K/AKT/mTOR pathways [1, 160, 172, 254]. ROS1 fusions are identified in several tumor types and are relatively common in non-small cell lung cancer (NSCLC), spitzoid neoplasms and inflammatory myofibroblastic tumors [170, 232]. In pediatric CNS tumors six different fusions have been described (Online Resource 5). In contrast to ALK, the most common ROS1 fusion, GOPC–ROS1, induces oncogenic signaling by translocating to the Golgi apparatus rather than by dimerization [29]. However, more recent research shows that dimerization and kinase activation is also a key step in the constitutive activation of ROS1 in fusion proteins [31]. The exact oncogenic mechanism of the chimeric protein is also determined by the fusion partner [100]. Although the breakpoints might be slightly different, all fusions retain the kinase domain of ROS1 encoded by exons 36–41 [220, 233]. Furthermore, all fusions lose the transmembrane domain of ROS1, leading to a cytoplasmic location of the fusion protein. Additionally, all the fusion partners contain a coiled-coil domain and sometimes an additional leucine zipper domain that leads to the dimerization of the fusion protein and hence the ligand independent activation of the ROS1 kinase. Although this type I kinase fusion activation is the most credible reason for the oncogenic signaling to date, it is possible that the loss of N-terminal regulatory domains as in type II fusions also play a role in enhanced signaling of ROS1 (Fig. 2b). More research is required to determine whether other mechanisms also play a role in ROS1 kinase activation [232]. Specific ROS1 inhibitors do not exist but there is evidence that next generation tyrosine kinase inhibitors (TKI) like entrectinib are potent against ROS1 fusions [53]. Furthermore, entrectinib unlike other TKI can sustain prolonged CNS exposure making it a suitable drug for treating ROS1 positive primary CNS tumors [60]. Current research is focusing on whether entrectinib is also suitable for pediatric CNS tumors [188].
NTRK fusions
Members of the NTRK family of RTKs are especially highly expressed in neural tissue [6, 185]. These receptors participate in the development and proper functioning of the CNS. The NTRK family exists of three members, NTRK1, NTRK2, and NTRK3, which besides the RAS/MAPK pathway can also signal via the PI3K/AKT/mTOR and the PLCγ/PLK pathways, depending on which docking protein binds to the kinase domain. Via these pathways, the signal transduction leads to proliferation, prevention of neuron degeneration, development, synaptic plasticity, sensory neuron maintenance and neuronal differentiation [108, 131, 156, 211]. These receptors and their signaling cascade are also implicated in neoplastic cells [156].
Whether NTRK fusions signal via the same preferred pathways as their full-length counterparts is still unknown. Experiments with the ETV6–NTRK3 fusion showed that this fusion protein signals mainly through RAS/MAPK but also activates PI3K/AKT/mTOR. Activating both pathways might induce the oncogenic potential since it activates proliferation and inhibits apoptosis [152, 223].
Although mutations and alternative splicing occur, fusions are the most common aberrations of NTRK in tumors. The most common alteration is a fusion between an NTRK gene and another N-terminal partner [71, 143, 219, 224, 234]. All these aberrations result in the constitutive activation of the kinase, due to loss of the extracellular domain. Of the 80 different N-terminal partners observed in tumors [80], 22 occur in pediatric CNS neoplasms (Online Resource 6). NTRK fusions have been identified in several pediatric gliomas such as pilocytic astrocytoma, high-grade glioma and glioblastoma (Fig. 3b) [19, 40, 93, 97, 116, 135, 177, 182, 184, 192, 240, 245]. In contrast to other tumor types, NTRK2 is the most common fusion partner of the NTRK family in pediatric brain tumors [215, 229, 234]. NTRK fusions belong to the type I kinase fusions as in 16/22 (73%) NTRK fusions the N-terminal partner has at least one coiled-coil or other type of dimerization domain (Fig. 2b). This probably leads to a constitutively active kinase, continuous downstream signaling and thus proliferation and cell survival. However, N-terminal partners such as CHTOP and VCL do not have a dimerization domain or another oncogenic functional domain. For these fusions it might be possible that the loss of the regulatory domain in the N-terminus of NTRK is enough to drive oncogenesis or there might be another yet undiscovered mechanism.
While the percentage of NTRK fusion driven tumors is quite low, the incidence in pediatric HGG and diffuse infiltrating pontine glioma is around 5% and even 40% in infants with non-brainstem HGG [163, 240]. NTRK inhibitors could be a potential effective targeted therapy in these tumors. Furthermore, these NTRK inhibitors have already shown high efficacy in several case reports of NTRK fusion driven tumors [49, 52, 57, 120, 153, 195, 209, 252]. Recent phase I and II trials have confirmed the effectivity of larotrectinib and entrectinib in pediatric brain tumors. Moreover, larotrectinib is now approved for NTRK fusion positive tumors in pediatric patients [45, 51, 117, 122, 173].
FGFR fusions
The FGFR family exists of four transmembrane tyrosine kinase receptors (FGFR1–4). The FGFR family plays an important role in embryonal CNS development as well as in tumorigenesis, regulating angiogenesis, proliferation, differentiation, migration, and survival. Aberrant signaling of the FGFR family is seen in many different cancers as a result of SNVs, overexpression or rearrangements (reviewed in [230]). FGFR fusions have been identified as oncogenic drivers in brain, bladder, lung and breast tumors. In pediatric CNS tumors nine different FGFR fusions have been identified (Online Resource 7).
All FGFR fusion proteins retain the kinase domain of FGFR and almost all C-terminal partners contain a coiled-coil domain. This means FGFR fusions belong to type III kinase fusions (Fig. 2b). Other oncogenic mechanisms are also proposed, for example, the FGFR3–TACC3 fusion displaces FGFR3 to the mitotic spindle leading to aneuploidy and tumorigenesis [178, 210]. Simultaneously, the constitutively active signal of FGFR3 also transduces via the RAS/MAPK pathway [158]. In contrast to other RTK fusion proteins, FGFR fusions depend on their own promoter for the expression of the chimeric protein. FGFR3 is very lowly expressed in normal brain and fusion negative adult glioblastoma but is highly expressed in fusion positive glioblastoma, which is likely due to the loss of microRNA regulation [174, 199]. The 3′ UTR of the FGFR3 gene is negatively controlled by microRNAs in the normal brain. However, in the fusion gene, this region is lost and FGFR3 can thus no longer be controlled by mir-99a [174]. The sequence of the 3′ UTR regions in FGFR genes is quite diverse but is lost in all the fusion genes. Additionally, computational analysis has shown that these UTRs are presumably regulated by different miRNAs. Therefore, it is likely that the loss of the 3′ UTR of FGFR in fusion genes leads to the enhanced expression of the chimera [94]. To date, no fusions have been detected with family member FGFR4 in pediatric CNS neoplasms. Furthermore, fusions with FGFR1 and FGFR3 often occur due to a small deletion on the chromosome, fusing them to partners in close proximity. In contrast, FGFR2 often translocates to partners on other chromosomes [175].
Since new aberrations in the FGFR pathway are detected in a variety of tumor types, there is an interest in FGFR pathway inhibitors. Clinical trials with FGFR inhibitors in brain tumors are being conducted [48, 92]. The FGFR inhibitor ponatinib has an improved therapeutic activity of temozolide in in vitro patient derived DIPG cells [200]. Further research will show the efficacy of the FGFR inhibitors in the different FGFR fusion positive pediatric tumors.
MET fusions
The least common RTK fusions in pediatric CNS neoplasms are fusions involving MET. In the three different MET fusions in pediatric CNS tumors, CLIP2-MET, TFG-MET, and PTPRZ1-MET, the kinase domain of MET is retained within the fusion (Online Resource 8). In CLIP2-MET and TFG-MET, only the C-terminal part of the protein containing the kinase domain is retained and therefore lacks its autoregulatory domain leading to a constitutive active MET [19, 141]. MET fusions can thus be classified as type II kinase fusions (Fig. 2b). Additionally, TFG has also been described as a partner for RTKs in chimeric proteins in other neoplasms [72, 77]. The PTPRZ1–MET fusion has been described in adult glioblastomas and entails the full length MET protein fused to the first exons of PTPRZ1 and probably uses its promoter to overexpress MET [17]. Additional to the MET fusions, all patients also harbored TP53 mutations or CDKN2A and CDKN2B deletions, indicating that the tumorigenesis due to MET fusions is probably dependent on additional aberrations in the cell cycle regulation [19]. It is shown that in a preclinical setting MET inhibition with RTK inhibitors is effective and MET fusion positive pediatric glioblastoma respond positively before relapse [19]. This initial data promotes further research with MET inhibitors in pediatric glioblastomas.
Fusions involving transcription regulators in pediatric CNS tumors
There are 38 fusions reported in 53 fusion–entity pairs, that do not have a kinase domain. Most of these (94%; 50/53), however contain a transcription activation domain, indicating that transcription regulators also play an important role in oncogenic fusions in pediatric CNS tumors. There are three (6%) fusion–entity pairs with two fusions that do not belong to this group and only have uncharacterized domains: RNF213–SLC26A11, and SETD2–ROBO1. Nine fusion partners are responsible for the majority of fusions (86%; 43/50) driven by transcriptional regulators (Fig. 4a). These nine transcription regulators occur with multiple partners, while the remaining transcription regulators have only one fusion partner and account for the remaining seven fusion–entity pairs: NAB2–STAT6, ATXN–NUTM1, PAX3–NCOA1 and PLAGL1–FOXG1, these will not be discussed further (Online resource 1). Transcription regulator fusions mostly occur in the glial tumor types (69%; 37/53) (Fig. 4a). The different fusions are not exclusively present in certain tumor types (Fig. 4b).

Characteristics of transcription regulator fusions in pediatric CNS neoplasms a Nine transcription factors are responsible for most transcription factor fusions that are mainly present in glial tumors. b Kinase proteins are not limited to one pediatric CNS tumor type but are present in multiple different tumor types. Graph made by https://app.rawgraphs.io. AB astroblastoma, AFH angiomatoid fibrous histiocytoma, AG angiocentric glioma, APA anaplastic pleomorphic astrocytoma, AO anaplastic oligodendroglioma, CNS EFT–CIC central nervous system Ewing Sarcoma Family Tumor with CIC alteration, CNS—ET central nervous system embryonal tumor, CNS-HGNET MN1 central nervous system high grade neuroepithelial tumor with MN1 alteration, DA diffuse astrocytoma, DSRCT desmoplastic small round cell tumor, ELTMD ependymoma like tumor with mesenchymal differentiation, EPN ependymoma, GBM glioblastoma, GG ganglioglioma, GNT glial neuronal tumor, HPC hemangiopericytoma, IHG interhemiscpheric glioma, IMMT intracranial myxoid mesenchymal tumor, IRMS intracranial rhabdomyosarcoma, MNG meningioma, NF neurofibroma, OD oligodendroglioma, PA pilocytic astrocytoma, PESS primary epidural spinal sarcoma, PNET primitive neuroectodermal tumor, SFT solitary fibrous tumor
Overall, we have identified two different types of transcription regulator fusions. Most of these fusions contain at least one partner that functions as a transcription factor and retains its transcription activation domain, while the other partner retains its nuclear localization signal (NLS) as well as, sometimes, a DNA binding domain (Fig. 2b, IV). An example of this kind of fusion is ZFTA–RELA (Fig. 2c, IV). In fusion type V, the transcription activation and DNA binding domain as well as the NLS are retained within one fusion partner, while the other partner influences the regulation of the chimeric protein. This is also seen in MYB–PCDHGA1, where PCDHGA1 is responsible for the truncation of the negative regulation domain in MYB (Fig. 2b V, c V). For BCOR fusions the mechanism of the chimeric proteins is unknown.
Furthermore, many of these fusions make use of epigenetic mechanisms to activate transcription of the alternative target genes to induce tumorigenesis. Hence, more research in epigenetic mechanisms could aid the overall understanding of these tumors and the identification of potential therapeutic treatments. As these fusions partners themselves are often difficult to target, it is worth identifying targetable downstream factors.
ETV1 fusions
ETS variant transcription factor 1 (ETV1) belongs to the ETS family of transcription factors. This family regulates genes that are responsible for processes such as cell growth, angiogenesis, proliferation and differentiation [102]. Furthermore, these transcription factors are known oncogenes in Ewing sarcoma, melanoma and prostate cancer [12, 88, 89]. It is unclear what the exact oncogenic mechanism is of DGKB–ETV1 fusions in glioblastoma and PTPRZ1–ETV1 in glioblastoma, pilocytic astrocytoma and anaplastic oligodendroglioma (Fig. 4b, Online Resource 9). However, as elevated expression levels of ETV1 are detected in multiple tumor types and can lead to increased invasiveness of tumor cells, this might also be the oncogenic mechanisms of these fusions [93, 146]. The promoters of PTPRZ1 and DGKB are highly active in the CNS and the DNA binding domain of ETV1 is retained in the fusion, possibly leading to an enhanced activation of ETV1 target genes [19]. As ETV1 itself retains its NLS, DNA binding domain and its transactivation domain, it belongs to the type V transcriptional regulator fusions.
EWSR1 fusions
EWSR1 is an RNA binding protein that plays an important role in transcription initiation. The protein is vital for cell survival in the CNS and regulates genomic integrity and RNA maturation processes [146]. Translocations with different partners are seen in several cancers, mainly soft tissue sarcomas. EWSR1 most often fuses with transcription factors. The new chimeric proteins alter pathways that are important for cell growth, differentiation, and proliferation, which ultimately leads to tumorigenesis [147]. In pediatric CNS tumors five different EWSR1 fusions have been detected, such as EWSR1–PLAGL1 and EWSR1–CREB1 (Online Resource 10). In all these fusions the transcription activation domain of EWSR1 is retained as well as the DNA binding domain (zinc finger, leucine zipper) and the NLS of its partners, making the fusions part of the type IV transcription regulator fusions. Additionally, to these type IV fusions, there is one EWSR1–SMARCA5 fusion in a Ewing sarcoma/primitive neuroectodermal tumor [205, 217]. Apart from its DNA binding domain (SANT) and its NLS, SMARCA5 also retains its helicase domain, which might implicate an epigenetic function in tumorigenesis for this fusion [24]. At the moment due to the difficulties in targeting transcription regulators, there are no targeted therapies available for EWSR1 fusions [187].
ZFTA fusions
ZFTA–RELA is the most recurrent fusion in ST-EPN. Around 70% of the ST-EPNs are driven by this fusion and EPNs with this fusion belong to the subtype ST-EPN–ZFTA. While ZFTA is a poorly characterized transcription factor, RELA (p65) is important in the canonical NFκB pathway. Initially, oncogenicity of the fusion was attributed to NFκB activation by RELA. However, due to identification of five additional fusion partners for ZFTA (MAML2–3, NCOA1–2 and YAP1), the poorly characterized ZFTA seems to be the driving fusion partner (Online Resource 11) [105, 115, 176, 226, 249, 255]. All these C-terminal partners are transcription factors or co-activators like RELA. This leads to the hypothesis that the zinc finger domains as well as the NLS in ZFTA are important for the oncogenic action of the fusion, as is seen in type IV transcription regulator fusions. This zinc finger domain and its interactions might alter the trafficking, degradation or target specificity of the chimeric fusion partner and thereby altering the transcription of their targets [176, 251]. Recent research has shown that the zinc finger domain in ZFTA is indeed essential for tumorigenesis [249]. More specifically, the zinc finger is responsible for nuclear translocation of the fusion protein, the binding to chromatin and the recruitment of chromatin remodeling complexes. This way ZFTA binds its fusion partners across the whole genome, modifies the chromatin state and facilitates transcriptional co-activators to promote expression of oncogenic genes [9, 115]. All ZFTA fusion partners retain their transcription activation domain, thus being able to enhance the activation of these oncogenic genes. In utero electroporations with ZFTA fusions in mice have confirmed that induction of the fusion alone is sufficient to drive ependymomas in vivo [249]. These studies also show that drugs can potentially be directed to genes in the downstream signaling cascade of ZFTA-fusions. Further research is needed to see if ZFTA positive tumors can be therapeutically targeted in this way. Additionally, therapeutics should be investigated that can cause a fast degradation of ZFTA (-fusions) [9, 115]. Recently, ZFTA fusions have also been discovered in tumors that although morphologically and genetically resemble ependymomas, do not cluster together with ST-EPN based on their DNA methylation profile [226]. Hence, for now they are represented as a separate type called ependymoma-like tumors with mesenchymal differentiation (ELTMDs). This data, together with the expression of ZFTA fusions in chondroid lipomas shows that ZFTA fusions might be oncogenic drivers in multiple diseases, also outside of the CNS [61, 226].
YAP1 fusions
YAP1 fusions are also mostly detected in ST-EPN, although less frequently than ZFTA–RELA. YAP1 is a regulator of the Hippo pathway and can fuse to mastermind-like proteins MAMLD1 and MAML2 or a thus far uncharacterized protein called FAM118B (Online Resource 12). No other recurrent aberrations have been identified in addition to these fusions, making the fusion the likely oncogenic driver. Indeed, in vivo experiments have demonstrated that ectopic expression of the YAP1–MAMLD1 fusion in fetal mouse brain is sufficient to induce tumors [168]. YAP1 and the Hippo pathway are responsible for the limitation of organ growth and can promote tumorigenesis. Under normal conditions YAP1 is retained in the cytosol until it is activated by the Hippo pathway and translocated to the nucleus, where YAP1 with its TEAD domain acts as a transcriptional activator. In fusions, YAP1 is retained in the nucleus and therefore has an oncogenic potential [151, 247]. The NLS in MAMLD1 is necessary for the retention of YAP1 and its transactivation domain in the nucleus. YAP1–FAM118B is also retained in the nucleus, although the exact mechanism here is unclear since the NLS of FAM118B fused to YAP1 is not enough to drive ependymoma [168]. MAMLD1 thus might have an additional function that drives oncogenesis apart from the nuclear localization. A YAP1 fusion with MAML2 has been detected in meningiomas. Where the TEAD domain of YAP1 is also retained as well as the transcriptional activation domain of MAML2, leading to the co-activation of the Hippo pathway [206, 241]. As most of the YAP1 fusions make use of the NLS of the fusion partner, these fusions are part of the type IV transcription regulator fusions.
CIC fusions
Capicua transcriptional repressor (CIC) is a transcription factor and in pediatric brain tumors a fusion was initially reported between CIC and NUTM1 in CNS Ewing sarcoma family of tumors with CIC alterations (CNS EFT–CIC) [216]. Additionally, fusions between CIC and LEUTX have been detected in an anaplastic pleomorphic astrocytoma and a CNS embryonal tumor (Fig. 4b, Online Resource 13). Recently, a CIC fusion with DUX4 was detected in a single case of primary epidural spinal sarcoma [50]. Although, this is a known fusion in Ewing sarcomas, it has never been detected in the CNS and in contrast to the CNS EFT–CIC tumors, the tumor was located in the spinal cord and not in the cerebrum. It remains to be investigated whether all CIC fusions belong to the same CNS entity or whether they may represent distinct entities or specific subtypes.
As also seen in CIC–NUTM1 and CIC–LEUTX fusion proteins, the chimeric CIC–DUX4 protein retains most of the functional regions of CIC, including the DNA-binding high-mobility group (HMG) box and the MAPK phosphorylation sites, while the partners retain their TEAD domain [50, 216]. This is seen in type IV transcription regulator fusions and might indicate that these domains are important for the oncogenic potential of the fusion. CIC is a transcriptional repressor that prevents activation of genes downstream of RTK signaling. In oligodendrogliomas, mutations in this gene are correlated with a poor outcome. These mutations lead to a loss-of-function and thus the activation of downstream RTK signaling. It is hypothesized that in the CIC fusions the downstream signaling is also activated by the recruitment of chromatin modifiers such as histone acetyl transferases to the transcription activation domains of the fusion partners [65, 216].
MN1 fusions
MN1 proto-oncogene, transcriptional regulator (MN1) is a transcriptional coregulator that has rearrangements in meningioma and leukemia [78, 125]. The first MN1 fusions located in the CNS were described in an entity named central nervous system high-grade neuroepithelial tumor with MN1 alterations (CNS HGNET–MN1) [216]. Recently, 73 CNS HGNET–MN1 tumors have been analyzed [32]. Most of these tumors have an oncogenic fusion between MN1 and BEND2 (Online Resource 14). However, the partner CXXC5 is also commonly present and additionally an MN1–GTSE1 fusion has been detected. In the MN1–BEND2 fusion, the transactivating domain of MN1 is retained as well as the BEN domains of BEND2. These BEN domains are thought to play a role in DNA binding, chromatin organization and neural transcriptional regulation [43, 196, 216]. The transactivation domain of MN1 is thought to recruit transcription activators, although, the exact oncogenic mechanism is unknown [186]. For MN1–CXXC5 and MN1–GTSE1 no research has been done into the chimeric fusion structure and the oncogenic mechanism. It is known that CXXC5 retains its NLS and DNA binding domain as also seen in BEND2, and GTSE1 retains its NLS, making MN1 fusions part of the type IV transcription regulator fusions. Recently, in the new 5th edition of WHO classification of CNS tumors, the HGNET–MN1 tumors have been renamed into astroblastoma, MN1 altered, as most of these tumors show an astroblastoma morphology [124, 137, 138]. It remains to be investigated whether tumors with MN1–BEND2 or MN1–CXXC5 fusions indeed all belong to this same entity or to what extent they may differ from each other molecularly and/or clinically.
PATZ1 fusions
PATZ1 (POZ/BTB and AT hook containing zinc finger 1) is a transcription factor and is important in maintaining pluripotency and hindering differentiation in stem cells [166]. Additionally, at different expression levels of PATZ1, the protein can act as either a transcriptional repressor or activator influencing senescence and proliferation, respectively [140]. Two different PATZ1 partners, EWSR1 and MN1 have been identified in several different pediatric CNS neoplasms (Online Resource 15). As described above, both EWSR1 and MN1 are implicated in other tumor entities as well and positively influence transcription regulation [10, 125]. Although the MN1–PATZ1 tumors also contain MN1 fusions, DNA methylation data of these tumors do not cluster together with the other above mentioned astroblastoma, MN1 altered tumors [22]. While the exact oncogenic mechanism of the PATZ1 fusion proteins is still unknown, it is known that the transactivation domain of MN1/EWSR1 is retained as well as the zinc finger domain of PATZ1. PATZ1 fusions thus belong to type IV transcription regulator fusions. It is hypothesized that this leads to enhanced activation of genes near the DNA binding site from PATZ1, via the recruitment of the transcription activators to the transactivation domain. Additionally, PATZ1 might benefit from the elevated expression levels by using the promoter of EWSR1 and MN1 [5]. However, a PATZ1–MN1 fusion was also identified, indicating that the aberrant expression is not the only oncogenic mechanism [255].
BCOR fusions
BCL6 corepressor (BCOR) epigenetically silences different genomic regions and is important in embryonal development. Several aberrations in BCOR have been implicated in different tumor types, including internal tandem duplications in CNS high-grade neuroepithelial tumors with BCOR alterations (CNS HGNET–BCOR) [216]. In the last years, two different fusions with BCOR have been identified in pediatric brain tumors (Online Resource 16). Interestingly, the two fusion partners are paralogues of each other. EP300–BCOR and BCOR–CREBBP have been described as potential oncogenic drivers in gliomas [181, 228]. EP300 and CREBBP are both histone acetyltransferases, which are important in proliferation, differentiation and may even have tumor suppressor functions. For EP300–BCOR several different fusion sites have been identified. In two of the three known fusions, the transactivation domain is truncated, and the acetyltransferase domain is retained, in the third fusion all domains are retained [228]. In the BCOR–CREBBP fusion the acetyltransferase domain is lost, and it probably creates a premature stop codon in CREBBP. The reciprocal CREBPP–BCOR fusion was not detected [181]. Since the order of the partners within the chimeric protein is different and the retention of the domains varies between the different fusions, it is difficult to predict the fusions’ oncogenic mechanism.
MYB fusions
MYB alterations and fusions have been identified in p-LGG for the first time in 2010 [222]. This transcription factor plays a role in the proliferation and differentiation of hematopoietic and other progenitor cells. An oncogenic effect has already been described in both leukemia as well as solid tumors. Ten percent of p-LGG harbor MYB alterations, with the most common alteration being a MYB–QKI fusion [15]. DNA methylation data of pediatric gliomas with a MYB or MYBL alteration cluster together as one entity [33], which is now classified as a new subtype of diffuse gliomas: diffuse astrocytoma—MYB altered [54, 137, 138]. The mechanism behind the MYB–QKI fusion has been well studied and is proposed to be a tripartite mechanism: MYB is overexpressed, the regulatory domain of MYB is truncated and the tumor repressor function of QKI is lost. There are two different fusion sites for the MYB protein in MYB–QKI, one in which the regulatory domain is truncated and one in which it is retained. Other fusion partners of MYB in pediatric gliomas have also been identified: ESR1, MAML2, MMP16 and PCDHGA1 (Online Resource 17). However, not much is known about these fusions. The hypothesis is that these fusions have an oncogenic potential due to the truncation of the C-terminal end of the MYB protein, meaning they belong to type V transcription regulator fusions, but there might also be other mechanisms involved. As seen in MYB–ESR1, where the negative regulation domain of MYB is retained as well as the ESR1 co-activator domain. Currently, there are no small molecular inhibitors for MYB and their development is rather challenging. However, transcriptional targets of MYB fusion proteins could be targeted [15]. Further research is necessary to determine whether similar MYB target genes are activated by the different chimeric proteins.
Validation of fusion proteins as oncogenic drivers
Over the coming years, a variety of fusions will likely be detected by NGS, as this detection mode is becoming standard care in diagnostics. Additionally, pan-cancer studies combining sequencing data from all over the world, can identify many new fusion genes in different tumor types. Moreover, for the fusions that have already been detected, there is still a lack of experimental evidence for the oncogenic potential. In vitro and in vivo, only a few of these fusions have been validated, some of which only in models that are not pediatric brain tumors but do give an indication of the oncogenic potential (Table 1). Only 30 of the 110 fusion proteins presented in this review have been validated in an in vitro or in vivo model.
Fusion genes in diagnostics
With the recent 5th edition of the WHO classification for tumors of the CNS, in which they advance the role of molecular diagnostics for CNS tumors, the detection of fusion genes has become an important diagnostic marker in pediatric CNS neoplasms [137]. Routinely, targeted assays were being used for diagnostic purposes to detect these fusions. Although these assays are cost-effective, they have clear limitations. As only specific fusions and breakpoints are included within these assays, alternative breakpoints, additional fusions or alternative fusion partners are likely to remain undetected. Additionally, targeted assays are unable to discover completely novel oncogenic fusions [193]. More recently, RNA-sequencing (RNA-seq) is being implemented in the diagnostic setting to detect oncogenic gene fusions, using both fresh frozen and formalin fixed paraffin embedded material [18, 119]. RNA-seq is a robust way to pick up expressed fusion RNAs, but calling oncogenic fusions remains challenging. First, there are currently no standardized methods with multiple different algorithms being used.
Second, next to true oncogenic fusions, fusion calling algorithms pick up a lot of false positives such as read-throughs and non-malignant fusions. Determining the cut off in fusion calling algorithms is a manual task that can introduce a bias. The difficulty here is to detect all the real fusion drivers, while ignoring biological and algorithm artefacts [26, 148]. Fusion panels can make detection of fusions less complex in RNA-seq by limiting the analysis to key genes associated with aberrations in pediatric CNS neoplasms. These panels can aid the molecular characterization of the tumors as well as contribute to the therapeutic decision making [119]. While these panels can detect fusions with different breakpoints or partner genes and could thus be used for diagnostics, they are limited to the number of genes in the panel and will thus neglect completely novel fusions with two yet unknown partners. An alternative to distinguish real fusions from artefacts is inclusion of genomic data, such as whole genome or exome sequencing to pinpoint underlying structural variations. This multi-omics approach can aid the assessment of the potential pathogenicity of the fusion and the clinical decision making, especially for lowly expressed oncogenic fusions [18].
Discussion
Fusion proteins as oncogenic drivers are emerging in pediatric CNS neoplasms. Since the implication of NGS more of these drivers have been described in case studies as well as in big multi-center sequencing studies. However, the variety of these fusions, the range of tumor entities in which they can occur, and their molecular mechanism are for a large part still unknown. Literature reveals that most fusions have an active kinase domain, and these kinase fusions are driven by a few main partner genes that are responsible for the oncogenicity of the fusion. We observed that there is a growth in detection of the variety of partners for these main oncogenic partners and we expect that in the next years this variety will only further increase. In line with this, the amount and diversity of non-kinase fusion proteins has also advanced. Transcription regulators now comprise the second biggest group of fusion proteins. These fusions are mainly seen in tumor entities that have been recently discovered or reclassified. With the discovery of these transcriptional regulator fusions probably more brain tumor entities can be reclassified based on their molecular mechanism. Although, this review did not identify the prevalence of these different fusions, the reclassification of the tumor entities based on the fusion that is present, as seen in many of the transcription regulator fusions, shows that the fusions play an important part in these entities. Future research should focus on the mechanism behind these fusions to identify targeted therapeutic options for the distinct tumor entities.
References
Acquaviva J, Wong R, Charest A (2009) The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta Rev Cancer 1795:37–52. https://doi.org/10.1016/j.bbcan.2008.07.006
Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K et al (2014) Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 28:1725–1735. https://doi.org/10.1038/leu.2014.70
Aghajan Y, Levy ML, Malicki DM, Crawford JR (2016) Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep 2016:1–2. https://doi.org/10.1136/bcr-2016-217189
Alejandro Sweet-Cordero E, Biegel JA (2019) The genomic landscape of pediatric cancers: implications for diagnosis and treatment. Science (80-) 363:1170–1175. https://doi.org/10.1126/science.aaw3535
Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D et al (2021) PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol. https://doi.org/10.1007/s00401-021-02354-8
Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S (2019) Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol 30:VIII5–VIII15. https://doi.org/10.1093/annonc/mdz383
Anderson JL, Denny CT, Tap WD, Federman N (2012) Pediatric sarcomas: translating molecular pathogenesis of disease to novel therapeutic possibilities. Pediatr Res 72:112–121. https://doi.org/10.1038/pr.2012.54.Pediatric
Andreiuolo F, Varlet P, Tauziède-Espariat A, Jünger ST, Dörner E, Dreschmann V et al (2019) Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol 29:205–216. https://doi.org/10.1111/bpa.12659
Arabzade A, Zhao Y, Varadharajan S, Chen H-C, Jessa S, Rivas B et al (2021) ZFTA-RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. https://doi.org/10.1158/2159-8290.cd-20-1066
Azuma M, Embree LJ, Sabaawy H, Hickstein DD (2007) Ewing sarcoma protein Ewsr1 maintains mitotic integrity and proneural cell survival in the zebrafish embryo. PLoS ONE 2:1–10. https://doi.org/10.1371/journal.pone.0000979
Badiali M, Gleize V, Paris S, Moi L, Elhouadani S, Arcella A et al (2012) KIAA1549-BRAF fusions and IDH mutations can coexist in diffuse gliomas of adults. Brain Pathol 22:841–847. https://doi.org/10.1111/j.1750-3639.2012.00603.x
Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y et al (2013) ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev 27:683–698. https://doi.org/10.1101/gad.211011.112
Bale TA (2020) FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol Commun 8:21. https://doi.org/10.1186/s40478-020-00898-6
Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, Ligon K et al (2018) Intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol 28:183–191. https://doi.org/10.1111/bpa.12504
Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R et al (2016) MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48:273–282. https://doi.org/10.1038/ng.3500
Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T et al (2017) A phase i trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro Oncol 19:1135–1144. https://doi.org/10.1093/neuonc/now282
Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL et al (2014) RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res 24:1765–1773. https://doi.org/10.1101/gr.165126.113
Van Belzen IAEM, Cai C, Van Tuil M, Badloe S, Strengman E, Janse A et al (2021) Systematic discovery of gene fusions in pediatric cancer by integrating RNA-seq and WGS. bioRxiv. https://doi.org/10.1101/2021.08.31.458342
Bender S, Gronych J, Warnatz HJ, Hutter B, Gröbner S, Ryzhova M et al (2016) Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 22:1314–1320. https://doi.org/10.1038/nm.4204
Di Benedetto B (2008) Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J Comp Neurol 346:339–346. https://doi.org/10.1002/cne
Bridge JA, Liu X-Q, Sumegi J, Nelson M, Reyes C, Bruch LA et al (2013) Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23:121–128. https://doi.org/10.1111/j.1750-3639.2012.00612.x
Burel-vandenbos F, Pierron G, Thomas C, Reynaud S, Gregoire V, Benaze GDDE et al (2020) A polyphenotypic malignant paediatric brain tumour presenting a MN1-PATZ1 fusion, no epigenetic similarities with CNS High-Grade Neuroepithelial Tumour with MN1 Alteration (CNS HGNET-MN1) and related to PATZ1-fused sarcomas. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12626
Burford A, Mackay A, Popov S, Vinci M, Carvalho D, Clarke M et al (2018) The ten-year evolutionary trajectory of a highly recurrent paediatric high grade neuroepithelial tumour with MN1:BEND2 fusion. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-19389-9
Cantile M, Marra L, Franco R, Ascierto P, Liguori G, De Chiara A et al (2013) Molecular detection and targeting of EWSR1 fusion transcripts in soft tissue tumors. Med Oncol. https://doi.org/10.1007/s12032-012-0412-8
Carbonell WS, Mandell JW (2003) Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J Neurotrauma 20:327–336. https://doi.org/10.1089/089771503765172282
Carrara M, Beccuti M, Cavallo F, Donatelli S, Lazzarato F, Cordero F et al (2013) State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues? BMC Bioinform 14:1–11. https://doi.org/10.1186/1471-2105-14-S7-S2
Chadda KR, Holland K, Scoffings D, Dean A, Pickles JC, Behjati S et al (2021) A rare case of paediatric astroblastoma with concomitant MN1-GTSE1 and EWSR1-PATZ1 gene fusions altering management. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12701
Chan E, Bollen AW, Sirohi D, Van Ziffle J, Grenert JP, Kline CN et al (2017) Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134:671–673. https://doi.org/10.1007/s00401-017-1759-x
Charest A, Kheifets V, Park J, Lane K, McMahon K, Nutt CL et al (2003) Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc Natl Acad Sci USA 100:916–921. https://doi.org/10.1073/pnas.242741799
Charest A, Lane K, McMahon K, Park J, Preisinger E, Conroy H et al (2003) Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosom Cancer 37:58–71. https://doi.org/10.1002/gcc.10207
Charest A, Wilker EW, McLaughlin ME, Lane K, Gowda R, Coven S et al (2006) ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res 66:7473–7481. https://doi.org/10.1158/0008-5472.CAN-06-1193
Chen W, Soon YY, Pratiseyo PD, Sutanto R, Hendriansyah L, Kuick CH et al (2020) Central nervous system neuroepithelial tumors with MN1-alteration: an individual patient data meta-analysis of 73 cases. Brain Tumor Pathol. https://doi.org/10.1007/s10014-020-00372-0
Chiang J, Harreld JH, Tinkle CL, Moreira DC, Li X, Acharya S et al (2019) A single-center study of the clinicopathologic correlates of gliomas with a MYB or MYBL1 alteration. Acta Neuropathol 138:1091–1092. https://doi.org/10.1007/s00401-019-02081-1
Chiang JCH, Harreld JH, Orr BA, Sharma S, Ismail A, Segura AD et al (2017) Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134:159–162. https://doi.org/10.1007/s00401-017-1728-4
Childress MA, Himmelberg SM, Chen H, Deng W, Davies MA, Lovly CM (2018) ALK fusion partners impact response to ALK inhibition: Differential effects on sensitivity, cellular phenotypes, and biochemical properties. Mol Cancer Res 16:1724–1736. https://doi.org/10.1158/1541-7786.MCR-18-0171
Chmielecki J, Bailey M, He J, Elvin J, Vergilio JA, Ramkissoon S et al (2017) Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 77:509–519. https://doi.org/10.1158/0008-5472.CAN-16-1106
Chmielecki J, Hutchinson KE, Frampton GM, Chalmers ZR, Johnson A, Shi C et al (2014) Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 4:1398–1405. https://doi.org/10.1158/2159-8290.CD-14-0617
Choubey L, Collette JC, Smith KM (2017) Quantitative assessment of fibroblast growth factor receptor 1 expression in neurons and glia. PeerJ 5:e3173. https://doi.org/10.7717/peerj.3173
Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DTW et al (2011) Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 121:763–774. https://doi.org/10.1007/s00401-011-0817-z
Clarke M, Mackay A, Ismer B, Pickles JC, Ruth G, Newman S et al (2020). Infant high grade gliomas comprise multiple subgroups characterised by novel targetable gene fusions and favourable outcomes. https://doi.org/10.1158/2159-8290.CD-19-1030
Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S et al (2020) Infant high grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. https://doi.org/10.1158/2159-8290.cd-19-1030
Cocce MC, Mardin BR, Bens S, Stutz AM, Kubieniecki F, In V et al (2016) Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes Chromosom Cancer. https://doi.org/10.1002/gcc
Dai Q, Ren A, Westholm JO, Serganov AA, Patel DJ, Lai EC (2013) The BEN domain is a novel sequence-specific DNA-binding domain conserved in neural transcriptional repressors. Genes Dev 27:602–614. https://doi.org/10.1101/gad.213314.113
Davare MA, Henderson JJ, Agarwal A, Wagner JP, Iyer SR, Shah N et al (2018) Rare but recurrent ROS1 fusions resulting from chromosome 6q22 microdeletions are targetable oncogenes in glioma. Clin Cancer Res 24:6471–6482. https://doi.org/10.1158/1078-0432.CCR-18-1052
Demetri GD, Paz-Ares L, Farago AF, Liu SV, Chawla SP, Tosi D et al (2018) Efficacy and safety of entrectinib in patients with NTRK fusion-positive (NTRK-fp) tumors: pooled analysis of STARTRK-2, STARTRK-1 and ALKA-372-001. Annals of oncology. Oxford University Press, Oxford
Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MU et al (2018) Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136:239–253. https://doi.org/10.1007/s00401-018-1865-4
Diamandis P, Ferrer-Luna R, Huang RY, Folkerth RD, Ligon AH, Wen PY et al (2016) Case report: next generation sequencing identifies a NAB2-STAT6 fusion in Glioblastoma. Diagn Pathol 11:13. https://doi.org/10.1186/s13000-016-0455-9
Dieci MV, Arnedos M, Andre F, Soria JC (2013) Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov 3:264–279. https://doi.org/10.1158/2159-8290.CD-12-0362
Doebele RC, Davis LE, Vaishnavi A, Le AT, Estrada-Bernal A, Keysar S et al (2015) An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov 5:1049–1057. https://doi.org/10.1158/2159-8290.CD-15-0443
Donahue JE, Yakirevich E, Zhong S, Treaba DO, Lakis NS, Ali SM et al (2018) Primary spinal epidural CIC-DUX4 undifferentiated sarcoma in a child. Pediatr Dev Pathol 21:411–417. https://doi.org/10.1177/1093526617707856
Drilon A, Laetsch TW, Kummar S, Dubois SG, Lassen UN, Demetri GD et al (2018) Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 378:731–739. https://doi.org/10.1056/NEJMoa1714448
Drilon A, Li G, Dogan S, Gounder M, Shen R, Arcila M et al (2016) What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol 27:920–926. https://doi.org/10.1093/annonc/mdw042
Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT et al (2020) Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol 21:261–270. https://doi.org/10.1016/S1470-2045(19)30690-4
Ellison DW, Hawkins C, Jones DTW, Onar-Thomas A, Pfister SM, Reifenberger G et al (2019) cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF V600E mutation. Acta Neuropathol 137:683–687. https://doi.org/10.1007/s00401-019-01987-0
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N et al (2019) Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol 20:1011–1022. https://doi.org/10.1016/S1470-2045(19)30277-3
Fangusaro JR, Onar-Thomas A, Young-Poussaint T, Wu S, Ligon AH, Lindeman NI et al (2017) A phase II prospective study of selumetinib in children with recurrent or refractory low-grade glioma (LGG): a pediatric brain tumor consortium (PBTC) study. J Clin Oncol 35:10504. https://doi.org/10.1200/JCO.2017.35.15_suppl.10504
Farago AF, Le LP, Zheng Z, Muzikansky A, Drilon A, Patel M et al (2015) Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol 10:1670–1674. https://doi.org/10.1097/01.JTO.0000473485.38553.f0
Faulkner C, Ellis HP, Shaw A, Penman C, Palmer A, Wragg C et al (2015) BRAF fusion analysis in pilocytic astrocytomas: KIAA1549-BRAF 15–9 fusions are more frequent in the midline than within the cerebellum. J Neuropathol Exp Neurol 74:867–872. https://doi.org/10.1097/NEN.0000000000000226
Ferguson SD, Zhou S, Huse JT, de Groot JF, Xiu J, Subramaniam DS et al (2018) Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J Neuropathol Exp Neurol 77:437–442. https://doi.org/10.1093/jnen/nly022
Fischer H, Ullah M, de la Cruz CC, Hunsaker T, Senn C, Wirz T et al (2020) Entrectinib, a TRK/ROS1 inhibitor with anti-CNS tumor activity: differentiation from other inhibitors in its class due to weak interaction with P-glycoprotein. Neuro Oncol 22:819–829. https://doi.org/10.1093/neuonc/noaa052
Flucke U, Tops BBJ, de Saint Aubain Somerhausen N, Bras J, Creytens DH, Küsters B et al (2013) Presence of C11orf95-MKL2 fusion is a consistent finding in chondroid lipomas: a study of eight cases. Histopathology 62:925–930. https://doi.org/10.1111/his.12100
Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO et al (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46:462–466. https://doi.org/10.1038/ng.2950
Forshew T, Tatevossian RG, Lawson ARJ, Ma J, Neale G, Ogunkolade B (2009) activation of the ERK/MAPK pathway: a signature genetic defect in posteror fossa pilocytic astrocytomas. https://doi.org/10.1002/path
Frattini V, Pagnotta SM, Tala FJJ, Russo MV, Lee SB, Garofano L et al (2018) A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 553:222–227. https://doi.org/10.1038/nature25171
French C (2014) NUT midline carcinoma. Nat Rev Cancer 14:149–150. https://doi.org/10.1038/nrc3659
Fritchie KJ, Jin L, Rubin BP, Burger PC, Jenkins SM, Barthelmess S et al (2016) NAB2-STAT6 gene fusion in meningeal hemangiopericytoma and solitary fibrous tumor. J Neuropathol Exp Neurol 75:263–271. https://doi.org/10.1093/jnen/nlv026
Fukuoka K, Kanemura Y, Shofuda T, Fukushima S, Yamashita S, Narushima D et al (2018) Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6:134. https://doi.org/10.1186/s40478-018-0630-1
Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH (2011) The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci 31:843–850. https://doi.org/10.1523/JNEUROSCI.3239-10.2011
Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F et al (2020) NTRK fusions in central nervous system tumors: a rare, but worthy target. Int J Mol Sci 21:1–24. https://doi.org/10.3390/ijms21030753
Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S et al (2018) Driver fusions and their implications in the development and treatment of human cancers. Cell Rep 23:227–238.e3. https://doi.org/10.1016/j.celrep.2018.03.050
Geiger TR, Song J-Y, Rosado A, Peeper DS (2011) Functional characterization of human cancer-derived TRKB mutations. PLoS ONE 6:e16871. https://doi.org/10.1371/journal.pone.0016871
Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M et al (1995) The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol 15:6118–6127. https://doi.org/10.1128/mcb.15.11.6118
Grosveld G, Verwoerd T, van Agthoven T, de Klein A, Ramachandran KL, Heisterkamp N et al (1986) The chronic myelocytic cell line K562 contains a breakpoint in bcr and produces a chimeric bcr/c-abl transcript. Mol Cell Biol 6:607–616. https://doi.org/10.1128/mcb.6.2.607
Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C et al (2019) Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10:1–13. https://doi.org/10.1038/s41467-019-12187-5
Hallberg B, Palmer RH (2016) The role of the ALK receptor in cancer biology. Ann Oncol 27:iii4–iii15. https://doi.org/10.1093/annonc/mdw301
Helgager J, Lidov HG, Mahadevan NR, Kieran MW, Ligon KL, Alexandrescu S (2017) A novel GIT2-BRAF fusion in pilocytic astrocytoma. Diagn Pathol 12:1–6. https://doi.org/10.1186/s13000-017-0669-5
Hernández L, Pinyol M, Hernández S, Beà S, Pulford K, Rosenwald A et al (1999) TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 94:3265–3268. https://doi.org/10.1182/blood.V94.9.3265
Heuser M, Argiropoulos B, Kuchenbauer F, Yung E, Piper J, Fung S et al (2007) MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood 110:1639–1647. https://doi.org/10.1182/blood-2007-03-080523
Hsiao SJ, Karajannis MA, Diolaiti D, Mansukhani MM, Bender JG, Kung AL et al (2017) A novel, potentially targetable TMEM106B-BRAF fusion in pleomorphic xanthoastrocytoma. Mol Case Stud 3:a001396. https://doi.org/10.1101/mcs.a001396
Hsiao SJ, Zehir A, Sireci AN, Aisner DL (2019) Detection of tumor NTRK gene fusions to identify patients who may benefit from tyrosine kinase (TRK) inhibitor therapy. J Mol Diagnostics 21:553–571. https://doi.org/10.1016/j.jmoldx.2019.03.008
Hu W, Wang J, Yuan L, Zhang X, Ji Y, Song C et al (2020) Case report: a unique case of pediatric central nervous system embryonal tumor harboring the CIC–LEUTX fusion, germline NBN variant and somatic TSC2 mutation: expanding the spectrum of CIC-rearranged neoplasia. Front Oncol 10:1–6. https://doi.org/10.3389/fonc.2020.598970
Huse JT, Snuderl M, Jones DTW, Brathwaite CD, Altman N, Lavi E et al (2017) Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 133:417–429. https://doi.org/10.1007/s00401-016-1639-9
Ida CM, Lambert SR, Rodriguez FJ, Voss JS, Mc Cann BE, Seys AR et al (2012) BRAF alterations are frequent in cerebellar low-grade astrocytomas with diffuse growth pattern. J Neuropathol Exp Neurol 71:631–639. https://doi.org/10.1097/NEN.0b013e31825c448a
Ishizawa K, Tsukamoto Y, Ikeda S, Suzuki T, Homma T, Mishima K et al (2016) “Papillary” solitary fibrous tumor/hemangiopericytoma with nuclear STAT6 expression and NAB2-STAT6 fusion. Brain Tumor Pathol 33:151–156. https://doi.org/10.1007/s10014-015-0247-z
Ito J, Nakano Y, Shima H, Miwa T, Kogure Y, Isshiki K et al (2020) Central nervous system ganglioneuroblastoma harboring MYO5A-NTRK3 fusion. Brain Tumor Pathol 37:105–110. https://doi.org/10.1007/s10014-020-00371-1
Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB et al (2017) CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 36:6348–6358. https://doi.org/10.1038/onc.2017.276
Jain P, Surrey LF, Straka J, Luo M, Lin F, Harding B et al (2018) Novel FGFR2-INA fusion identified in two low-grade mixed neuronal-glial tumors drives oncogenesis via MAPK and PI3K/mTOR pathway activation. Acta Neuropathol 136:167–169. https://doi.org/10.1007/s00401-018-1864-5
Jané-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner A, Lin WM et al (2011) An oncogenic role for ETV1 in melanoma. Bone 23:1–7. https://doi.org/10.1161/CIRCULATIONAHA.110.956839
Janknecht R (2005) EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene 363:1–14. https://doi.org/10.1016/j.gene.2005.08.007
Jeuken J, van de Broecke C, Gijsen S, Boots-Sprenger S, Wesseling P (2007) RAS/RAF pathway activation in gliomas: the result of copy number gains rather than activating mutations. Acta Neuropathol 114:121–133. https://doi.org/10.1007/s00401-007-0239-0
Jeuken JW, Wesseling P (2010) MAPK pathway activation through BRAF gene fusion in pilocytic astrocytomas; a novel oncogenic fusion gene with diagnostic, prognostic, and therapeutic potential. J Pathol 222:324–328. https://doi.org/10.1002/path.2780
Jimenez-Pascual A, Siebzehnrubl F (2019) Fibroblast growth factor receptor functions in glioblastoma. Cells 8:715. https://doi.org/10.3390/cells8070715
Johnson A, Severson E, Gay L, Vergilio J, Elvin J, Suh J et al (2017) Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22:1478–1490. https://doi.org/10.1634/theoncologist.2017-0242
Johnson DE, Williams LT (1993) Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res 60:1–41. https://doi.org/10.1016/s0065-230x(08)60821-0
Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science (80-) 298:1911–1912. https://doi.org/10.1126/science.1072682
Jones DTW, Gronych J, Lichter P, Witt O, Pfister SM (2012) MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci 69:1799–1811. https://doi.org/10.1007/s00018-011-0898-9
Jones DTW, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932. https://doi.org/10.1038/ng.2682
Jones DTW, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K et al (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677. https://doi.org/10.1158/0008-5472.CAN-08-2097
Jones DTW, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28:2119–2123. https://doi.org/10.1038/onc.2009.73
Jun HJ, Johnson H, Bronson RT, De Feraudy S, White F, Charest A (2012) The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-syt1 phosphorylation. Cancer Res 72:3764–3774. https://doi.org/10.1158/0008-5472.CAN-11-3990
Kao Y-C, Sung Y-S, Zhang L, Chen C-L, Vaiyapuri S, Rosenblum MK et al (2017) EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol 41:482–490. https://doi.org/10.1097/PAS.0000000000000788
Kar A, Gutierrez-Hartmann A (2013) Molecular mechanisms of ETS transcription factor mediated tumoirgenesis. Crit Rev Biochem Mol Biol 48:522–543. https://doi.org/10.1038/jid.2014.371
Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH et al (2014) Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol 16:1408–1416. https://doi.org/10.1093/neuonc/nou059
Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH (2012) Pediatric glioma-associated KIAA1549: BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev 26:2561–2566. https://doi.org/10.1101/gad.200907.112
Keenan C, Graham RT, Harreld JH, Lucas JT, Finkelstein D, Wheeler D et al (2020) Infratentorial C11orf95-fused gliomas share histologic, immunophenotypic, and molecular characteristics of supratentorial RELA-fused ependymoma. Acta Neuropathol. https://doi.org/10.1007/s00401-020-02238-3
Khuong-Quang DA, Brown LM, Wong M, Mayoh C, Sexton-Oates A, Kumar A et al (2020) Recurrent SPECC1L–NTRK fusions in pediatric sarcoma and brain tumors. Cold Spring Harb Mol Case Stud. https://doi.org/10.1101/MCS.A005710
Kiehna EN, Arnush MR, Tamrazi B, Cotter JA, Hawes D, Robison NJ et al (2017) Novel GOPC(FIG)-ROS1 fusion in a pediatric high-grade glioma survivor. J Neurosurg Pediatr 20:51–55. https://doi.org/10.3171/2017.2.PEDS16679
Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL et al (1993) Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75:113–122. https://doi.org/10.1016/S0092-8674(05)80088-1
Kleinman CL, Gerges N, Papillon-Cavanagh S, Sin-Chan P, Pramatarova A, Quang D-AK et al (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46:39–44. https://doi.org/10.1038/ng.2849
Köhler M, Röring M, Schorch B, Heilmann K, Stickel N, Fiala GJ et al (2016) Activation loop phosphorylation regulates B-Raf in vivo and transformation by B-Raf mutants. EMBO J 35:143–161. https://doi.org/10.15252/embj.201592097
Konstantinidis A, Cheesman E, O’Sullivan J, Pavaine J, Avula S, Pizer B et al (2019) Intracranial angiomatoid fibrous histiocytoma with EWSR1-CREB family fusions: a report of 2 pediatric cases. World Neurosurg 126:113–119. https://doi.org/10.1016/j.wneu.2019.02.107
Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H et al (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118:401–405. https://doi.org/10.1007/s00401-009-0550-z
Kumar A, Pathak P, Purkait S, Faruq M, Jha P, Mallick S et al (2015) Oncogenic KIAA1549-BRAF fusion with activation of the MAPK/ERK pathway in pediatric oligodendrogliomas. Cancer Genet 208:91–95. https://doi.org/10.1016/j.cancergen.2015.01.009
Kumar S, Razzaq SK, Vo AD, Gautam M, Li H (2016) Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip Rev RNA 7:811–823. https://doi.org/10.1002/wrna.1382
Kupp R, Ruff L, Terranova S, Nathan E, Ballereau S, Stark R et al (2021) ZFTA-translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discov. https://doi.org/10.1158/2159-8290.cd-20-1052
Kurozumi K, Nakano Y, Ishida J, Tanaka T, Doi M, Hirato J et al (2019) High-grade glioneuronal tumor with an ARHGEF2-NTRK1 fusion gene. Brain Tumor Pathol 36:121–128. https://doi.org/10.1007/s10014-019-00345-y
Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM et al (2018) Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 19:705–714. https://doi.org/10.1016/S1470-2045(18)30119-0
Lahoz A, Hall A (2013) A tumor suppressor role for srGAP3 in mammary epithelial cells. Oncogene 32:4854–4860. https://doi.org/10.1038/onc.2012.489
Lake JA, Donson AM, Prince E, Davies KD, Nellan A, Green AL et al (2020) Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr Blood Cancer 67:1–13. https://doi.org/10.1002/pbc.28028
Landman Y, Ilouze M, Wein S, Neiman V, Yerushalmi R, Yakimov M et al (2018) Rapid response to larotrectinib (LOXO-101) in an adult chemotherapy-naive patients with advanced triple-negative secretory breast cancer expressing ETV6-NTRK3 fusion. Clin Breast Cancer 18:e267–e270. https://doi.org/10.1016/j.clbc.2017.11.017
Lasorella A, Sanson M, Iavarone A (2017) FGFR-TACC gene fusions in human glioma. Neuro Oncol 19:475–483. https://doi.org/10.1093/neuonc/now240
Lassen UN, Albert CM, Kummar S, van Tilburg CM, Dubois SG, Geoerger B et al (2018) Larotrectinib efficacy and safety in TRK fusion cancer: An expanded clinical dataset showing consistency in an age and tumor agnostic approach. Ann Oncol 29:viii133. https://doi.org/10.1093/annonc/mdy279.397
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR et al (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495–501. https://doi.org/10.1038/nature12912
Lehman NL, Usubalieva A, Lin T, Allen SJ, Tran QT, Mobley BC et al (2019) Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7:42. https://doi.org/10.1186/s40478-019-0689-3
Lekanne Deprez RH, Riegman PH, Groen NA, Warringa UL, van Biezen NA, Molijn AC et al (1995) Cloning and characterization of MN1, a gene from chromosome 22q11, which is disrupted by a balanced translocation in a meningioma. Oncogene 10:1521–1528
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134. https://doi.org/10.1016/j.cell.2010.06.011
Libbrecht S, Van Der Meulen J, Mondelaers V, Baert E, Vande Walle C, Van Dorpe J et al (2020) Intracranial myxoid mesenchymal tumor with EWSR1-CREB1 fusion. Pathol Res Pract 216:153239. https://doi.org/10.1016/j.prp.2020.153239
Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D et al (2012) BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549: BRAF fusion variants. J Neuropathol Exp Neurol 71:66–72. https://doi.org/10.1097/NEN.0b013e31823f2cb0
Linzey JR, Marini B, McFadden K, Lorenzana A, Mody R, Robertson PL et al (2017) Identification and targeting of an FGFR fusion in a pediatric thalamic “central oligodendroglioma.” NPJ Precis Oncol. https://doi.org/10.1038/s41698-017-0036-8
Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H et al (2011) Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146:209–221. https://doi.org/10.1016/j.cell.2011.06.014
Loeb DM, Stephens RM, Copeland T, Kaplan DR, Greene LA (1994) A Trk nerve growth factor (NGF) receptor point mutation affecting interaction with phospholipase C-gamma 1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J Biol Chem 269:8901–8910. https://doi.org/10.1016/s0021-9258(17)37053-9
Lopez-Nunez O, Cafferata B, Santi M, Ranganathan S, Pearce TM, Kulich SM et al (2021) The spectrum of rare central nervous system (CNS) tumors with EWSR1-non-ETS fusions: experience from three pediatric institutions with review of the literature. Brain Pathol 31:70–83. https://doi.org/10.1111/bpa.12900
Lopez-Nunez O, John I, Panasiti RN, Ranganathan S, Santoro L, Grélaud D et al (2019) Infantile inflammatory myofibroblastic tumors: clinicopathological and molecular characterization of 12 cases. Mod Pathol. https://doi.org/10.1038/s41379-019-0406-6
López C, Kleinheinz K, Aukema SM, Rohde M, Bernhart SH, Hübschmann D et al (2019) Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun 10:1–19. https://doi.org/10.1038/s41467-019-08578-3
Lopez G, Perry A, Harding B, Li M, Santi M (2019) CDKN2A/B loss is associated with anaplastic transformation in a case of NTRK2 fusion-positive pilocytic astrocytoma. Neuropathol Appl Neurobiol 45:174–178. https://doi.org/10.1016/j.physbeh.2017.03.040
Louis DN, Perry A, Ellison DW, Reifenberger G, Kleihues P, von Deimling A et al (2016) The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. https://doi.org/10.1093/neuonc/noab106
Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA et al (2020) cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 30:844–856. https://doi.org/10.1111/bpa.12832
Lu H, Villafane N, Dogruluk T, Grzeskowiak CL, Kong K, Tsang YH et al (2017) Engineering and functional characterization of fusion genes identifies novel oncogenic drivers of cancer. Cancer Res 77:3502–3512. https://doi.org/10.1158/0008-5472.CAN-16-2745
Ma H, Ow JR, Tan BCP, Goh Z, Feng B, Loh YH et al (2014) The dosage of Patz1 modulates reprogramming process. Sci Rep 4:1–12. https://doi.org/10.1038/srep07519
Mak HHL, Peschard P, Lin T, Naujokas MA, Zuo D, Park M (2007) Oncogenic activation of the Met receptor tyrosine kinase fusion protein, Tpr-Met, involves exclusion from the endocytic degradative pathway. Oncogene 26:7213–7221. https://doi.org/10.1038/sj.onc.1210522
Malgulwar PB, Nambirajan A, Pathak P, Faruq M, Rajeshwari M, Singh M et al (2018) C11orf95-RELA fusions and upregulated NF-KB signalling characterise a subset of aggressive supratentorial ependymomas that express L1CAM and nestin. J Neurooncol 138:29–39. https://doi.org/10.1007/s11060-018-2767-y
Marchetti A, Felicioni L, Pelosi G, Del Grammastro M, Fumagalli C, Sciarrotta M et al (2008) Frequent mutations in the neurotrophic tyrosine receptor kinase gene family in large cell neuroendocrine carcinoma of the lung. Hum Mutat 29:609–616. https://doi.org/10.1002/humu.20707
Marincevic-Zuniga Y, Dahlberg J, Nilsson S, Raine A, Nystedt S, Lindqvist CM et al (2017) Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J Hematol Oncol 10:1–14. https://doi.org/10.1186/s13045-017-0515-y
Maruggi M, Malicki DM, Levy ML, Crawford JR (2018) A novel KIF5B-ALK fusion in a child with an atypical central nervous system inflammatory myofibroblastic tumour. BMJ Case Rep 2018:1–2. https://doi.org/10.1136/bcr-2018-226431
Matjašič A, Zupan A, Boštjančič E, Pižem J, Popović M, Kolenc D (2020) A novel PTPRZ1-ETV1 fusion in gliomas. Brain Pathol 30:226–234. https://doi.org/10.1111/bpa.12776
Mercher T, Schwaller J (2019) Pediatric acute myeloid leukemia (AML): from genes to models toward targeted therapeutic intervention. Front Pediatr. https://doi.org/10.3389/fped.2019.00401
Mertens F, Johansson B, Fioretos T, Mitelman F (2015) The emerging complexity of gene fusions in cancer. Nat Rev Cancer 15:371–381. https://doi.org/10.1038/nrc3947
Mitelman F, Johansson B, Mertens F (2007) The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7:233–245. https://doi.org/10.1038/nrc2091
Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P et al (2015) Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA J Am Med Assoc 314:913–925. https://doi.org/10.1001/jama.2015.10080
Moroishi T, Hansen CG, Guan K-L (2017) The emerging roles of YAP and TAZ in cancer. Physiol Behav 176:139–148. https://doi.org/10.1016/j.physbeh.2017.03.040
Morrison KB, Tognon CE, Garnett MJ, Deal C, Sorensen PHB (2002) ETV6-NTRK3 transformation requires insulin-like growth factor 1 receptor signaling and is associated with constitutive IRS-1 tyrosine phosphorylation. Oncogene 21:5684–5695. https://doi.org/10.1038/sj.onc.1205669
Nagasubramanian R, Wei J, Gordon P, Rastatter JC, Cox MC, Pappo A (2016) Infantile fibrosarcoma with NTRK3–ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr Blood Cancer 63:1468–1470. https://doi.org/10.1002/pbc.26026
Nakada S, Minato H, Nojima T (2016) Clinicopathological differences between variants of the NAB2-STAT6 fusion gene in solitary fibrous tumors of the meninges and extra-central nervous system. Brain Tumor Pathol 33:169–174. https://doi.org/10.1007/s10014-016-0264-6
Nakada S, Minato H, Takegami T, Kurose N, Ikeda H, Kobayashi M et al (2015) NAB2-STAT6 fusion gene analysis in two cases of meningeal solitary fibrous tumor/hemangiopericytoma with late distant metastases. Brain Tumor Pathol 32:268–274. https://doi.org/10.1007/s10014-015-0220-x
Nakagawara A (2001) Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett 169:107–114. https://doi.org/10.1016/S0304-3835(01)00530-4
Nakano Y, Tomiyama A, Kohno T, Yoshida A, Yamasaki K, Ozawa T et al (2019) Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 36:14–19. https://doi.org/10.1007/s10014-018-0330-3
Nelson KN, Meyer AN, Siari A, Campos AR, Motamedchaboki K, Donoghue DJ (2016) Oncogenic gene fusion FGFR3-TACC3 Is regulated by tyrosine phosphorylation. Mol Cancer Res 14:458–469. https://doi.org/10.1158/1541-7786.MCR-15-0497
Ng A, Levy ML, Malicki DM, Crawford JR (2019) Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep. https://doi.org/10.1136/bcr-2018-228248
Nguyen KT, Zong CS, Uttamsingh S, Sachdev P, Bhanot M, Le MT et al (2002) The role of phosphatidylinositol 3-kinase, Rho family GTPases, and STAT3 in Ros-induced cell transformation. J Biol Chem 277:11107–11115. https://doi.org/10.1074/jbc.M108166200
Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D et al (2014) Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 4:428–434. https://doi.org/10.1038/nature13379.Enhancer
Northcott PA, Pfister SM, Jones DTW (2015) Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol 16:e293-302. https://doi.org/10.1016/S1470-2045(14)71206-9
Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R (2018) Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol 8:1–20. https://doi.org/10.1200/po.18.00183
Olsen TK, Panagopoulos I, Meling TR, Micci F, Gorunova L, Thorsen J et al (2015) Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro Oncol 17:1365–1373. https://doi.org/10.1093/neuonc/nov039
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C et al (2019) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol 21:v1–v100. https://doi.org/10.1093/neuonc/noz150
Ow JR, Ma H, Jean A, Goh Z, Lee YH, Chong YM et al (2014) Patz1 regulates embryonic stem cell identity. Stem Cells Dev 23:1062–1073. https://doi.org/10.1089/scd.2013.0430
Pages M, Lacroix L, Tauziede-Espariat A, Castel D, Daudigeos-Dubus E, Ridola V et al (2015) Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3:85. https://doi.org/10.1186/s40478-015-0264-5
Pajtler KW, Wei Y, Okonechnikov K, Silva PBG, Vouri M, Zhang L et al (2019) YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun 10:1–16. https://doi.org/10.1038/s41467-019-11884-5
Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. https://doi.org/10.1016/j.ccell.2015.04.002
Pal P, Khan Z (2017) Ros1. J Clin Pathol 70:1001–1009. https://doi.org/10.1136/jclinpath-2016-204244
Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S et al (2010) Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 16:793–798. https://doi.org/10.1038/nm.2166
Palmer RH, Vernersson E, Grabbe C, Hallberg B (2009) Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 420:345–361. https://doi.org/10.1042/BJ20090387
Papadopoulos KP, Gandhi L, Janne PA, Ou S-HI, Shaw A, Goldberg TR et al (2018) First-in-human study of DS-6051b in patients (pts) with advanced solid tumors (AST) conducted in the US. J Clin Oncol 36:2514. https://doi.org/10.1200/JCO.2018.36.15_suppl.2514
Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P et al (2013) The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest 123:855–865. https://doi.org/10.1172/JCI67144
Parker BC, Engels M, Annala M, Zhang W (2014) Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol 232:4–15. https://doi.org/10.1002/path.4297
Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y et al (2014) C11orf95-RELA fusions drive oncogenic NF- k B signalling in ependymoma. Nature. https://doi.org/10.1038/nature13109
Pehlivan KC, Malicki DM, Levy ML, Crawford JR (2020) TPM3-NTRK1 fusion in a pleomorphic xanthoastrocytoma presenting with haemorrhage in a child. BMJ Case Rep 13:4–6. https://doi.org/10.1136/bcr-2020-234347
Peset I, Vernos I (2008) The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol 18:379–388. https://doi.org/10.1016/j.tcb.2008.06.005
Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N et al (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118:1739–1749. https://doi.org/10.1172/JCI33656
Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin L-W et al (2016) Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p. V600E mutation. Acta Neuropathol 132:757–760
Pisapia DJ, Ohara K, Bareja R, Wilkes DC, Hissong E, Croyle JA et al (2020) Fusions involving BCOR and CREBBP are rare events in infiltrating glioma. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-020-00951-4
Prabhakaran N, Guzman MA, Navalkele P, Chow-Maneval E, Batanian JR (2018) Novel TLE4-NTRK2 fusion in a ganglioglioma identified by array-CGH and confirmed by NGS: potential for a gene targeted therapy. Neuropathology 38:380–386. https://doi.org/10.1111/neup.12458
Pyo KH, Lim SM, Kim HR, Sung YH, Yun MR, Kim SM et al (2017) Establishment of a conditional transgenic mouse model recapitulating EML4-ALK–positive human non-small cell lung cancer. J Thorac Oncol 12:491–500. https://doi.org/10.1016/j.jtho.2016.10.022
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD et al (2016) Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131:833–845. https://doi.org/10.1007/s00401-016-1539-z
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc B Biol Sci 361:1545–1564. https://doi.org/10.1098/rstb.2006.1894
Riedel SS, Lu C, Xie HM, Nestler K, Vermunt MW, Lenard A et al (2021) Intrinsically disordered Meningioma-1 stabilizes the BAF complex to cause AML. Mol Cell 81:2332-2348.e9. https://doi.org/10.1016/j.molcel.2021.04.014
Rizk VT, Walko CM, Brohl AS (2019) Precision medicine approaches for the management of Ewing sarcoma: current perspectives. Pharmgenom Pers Med 12:9–14. https://doi.org/10.2147/PGPM.S170612
Robinson GW, Gajjar AJ, Gauvain KM, Basu EM, Macy ME, Maese LD et al (2019) Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. J Clin Oncol 37:10009. https://doi.org/10.1200/JCO.2019.37.15_suppl.10009
Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J et al (2016) The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 138:881–890. https://doi.org/10.1002/ijc.29825
Rossing M, Yde CW, Sehested A, Ostrup O, Scheie D, Dangouloff-Ros V et al (2017) Genomic diagnostics leading to the identification of a TFG-ROS1 fusion in a child with possible atypical meningioma. Cancer Genet 212–213:32–37. https://doi.org/10.1016/j.cancergen.2017.03.005
Rowley JD (1973) A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature 243:290–293. https://doi.org/10.1038/243290a0
Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J et al (2020) Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell 37:569-583.e5. https://doi.org/10.1016/j.ccell.2020.03.011
Sahm F, Schrimpf D, Jones DTW, Meyer J, Kratz A, Reuss D et al (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. https://doi.org/10.1007/s00401-015-1519-8
Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD et al (2008) Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci 28:6983–6995. https://doi.org/10.1523/JNEUROSCI.0679-08.2008
Sartore-Bianchi A, Ardini E, Bosotti R, Amatu A, Valtorta E, Somaschini A et al (2015) Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. JNCI J Natl Cancer Inst. https://doi.org/10.1093/jnci/djv306
Sathyan KM, Shen Z, Tripathi V, Prasanth KV, Prasanth SG (2011) A BEN-domain-containing protein associates with heterochromatin and represses transcription. J Cell Sci 124:3149–3163. https://doi.org/10.1242/jcs.086603
Sato T, Nakamura H (2004) The Fgf8 signal causes cerebellar differentiation by activating the Ras-ERK signaling pathway. Development 131:4275–4285. https://doi.org/10.1242/dev.01281
Schieffer KM, Agarwal V, Lahaye S, Miller KE, Koboldt DC, Lichtenberg T et al (2021) YAP1-FAM118B fusion defines a rare subset of childhood and young adulthood meningiomas. Am J Surg Pathol 45:329–340. https://doi.org/10.1097/PAS.0000000000001597
Schittenhelm J, Ziegler L, Sperveslage J, Mittelbronn M, Capper D, Burghardt I et al (2020) FGFR3 overexpression is a useful detection tool for FGFR3 fusions and sequence variations in glioma. Neuro-Oncology Pract 8:209–221. https://doi.org/10.1093/nop/npaa075
Schramm K, Iskar M, Statz B, Jäger N, Haag D, Słabicki M et al (2019) DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuse intrinsic pontine gliomas. Neuro Oncol 21:867–877. https://doi.org/10.1093/neuonc/noz057
Setty P, Gessi M, Waha A, Hammes J, El-Maarri O, Pietsch T et al (2011) Sensitive determination of BRAF copy number in clinical samples by pyrosequencing. Diagn Mol Pathol 20:148–157. https://doi.org/10.1097/PDM.0b013e3182143817
Shah N, Lankerovich M, Lee H, Yoon JG, Schroeder B, Foltz G (2013) Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics 14:1–15. https://doi.org/10.1186/1471-2164-14-818
Shtivelman E, Lifshitz B, Gale RP, Canaani E (1985) Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 315:550–554
Siegfried A, Masliah-Planchon J, Roux FE, Larrieu-Ciron D, Pierron G, Nicaise Y et al (2019) Brain tumor with an ATXN1-NUTM1 fusion gene expands the histologic spectrum of NUTM1-rearranged neoplasia. Acta Neuropathol Commun 7:5–7. https://doi.org/10.1186/s40478-019-0870-8
Siegfried A, Rousseau A, Maurage C-A, Pericart S, Nicaise Y, Escudie F et al (2019) EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol 29:53–62. https://doi.org/10.1111/bpa.12619
Sievers P, Chiang J, Schrimpf D, Stichel D, Paramasivam N, Sill M et al (2020) YAP1-fusions in pediatric NF2-wildtype meningioma. Acta Neuropathol 139:215–218. https://doi.org/10.1007/s00401-019-02095-9
Sievers P, Henneken SC, Blume C, Sill M, Schrimpf D, Stichel D et al (2021) Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial ependymoma. Acta Neuropathol 142:827–839. https://doi.org/10.1007/s00401-021-02356-6
Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N et al (2013) Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA 110:5957–5962. https://doi.org/10.1073/pnas.1219232110
Sigal D, Tartar M, Xavier M, Bao F, Foley P, Luo D et al (2017) Activity of entrectinib in a patient with the first reported NTRK fusion in neuroendocrine cancer. J Natl Compr Canc Netw 15:1317–1322
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A et al (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231–1235. https://doi.org/10.1126/science.1220834
Siniscalco D, Giordano C, Rossi F, Maione S, de Novellis V (2011) Role of neurotrophins in neuropathic pain. Curr Neuropharmacol 9:523–529. https://doi.org/10.2174/157015911798376208
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S et al (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566. https://doi.org/10.1038/nature05945
Soda M, Takada S, Takeuchi K, Young LC, Enomoto M, Ueno T et al (2008) A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA 105:19893–19897. https://doi.org/10.1073/pnas.0805381105
Di Stefano AL, Fucci A, Frattini V, Labussiere M, Mokhtari K, Zoppoli P et al (2015) Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin Cancer Res 21:3307–3317. https://doi.org/10.1158/1078-0432.CCR-14-2199
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C (2014) The landscape of kinase fusions in cancer. Nat Commun. https://doi.org/10.1038/ncomms5846
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072. https://doi.org/10.1016/j.cell.2016.01.015
Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA (2011) A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol 24:333–342. https://doi.org/10.1038/modpathol.2010.201
Szulzewsky F, Arora S, Hoellerbauer P, King C, Nathan E, Chan M et al (2020) Comparison of tumor-associated YAP1 fusions identifies a recurrent set of functions critical for oncogenesis. Genes Dev 34:1051–1064. https://doi.org/10.1101/GAD.338681.120
Tacconelli A, Farina AR, Cappabianca L, DeSantis G, Tessitore A, Vetuschi A et al (2004) TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 6:347–360. https://doi.org/10.1016/j.ccr.2004.09.011
Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S et al (2012) RET, ROS1 and ALK fusions in lung cancer. Nat Med 18:378–381. https://doi.org/10.1038/nm.2658
Tan SY, Szymanski LJ, Galliani C, Parham D, Zambrano E (2018) Solitary fibrous tumors in pediatric patients: a rare and potentially overdiagnosed neoplasm, confirmed by STAT6 immunohistochemistry. Pediatr Dev Pathol 21:389–400. https://doi.org/10.1177/1093526617745431
Tatevossian RG, Lawson ARJ, Forshew T, Hindley GFL, Ellison DW, Sheer D (2010) MAPK pathway activation and the origins of pediatric low-grade astrocytomas. J Cell Physiol 222:509–514. https://doi.org/10.1002/jcp.21978
Tognon C, Garnett M, Kenward E, Kay R, Morrison K, Sorensen PH (2001) The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res 61:8909–8916
Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T et al (2008) Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 111:4797–4808. https://doi.org/10.1182/blood-2007-09-113027
Tomić TT, Olausson J, Wilzén A, Sabel M, Truvé K, Sjögren H et al (2017) A new GTF2I-BRAF fusion mediating MAPK pathway activation in pilocytic astrocytoma. PLoS ONE 12:e0184715. https://doi.org/10.1371/journal.pone.0184715
Tomomasa R, Arai Y, Kawabata-Iwakawa R, Fukuoka K, Nakano Y, Hama N et al (2021) Ependymoma-like tumor with mesenchymal differentiation harboring C11orf95-NCOA1/2 or -RELA fusion: a hitherto unclassified tumor related to ependymoma. Brain Pathol. https://doi.org/10.1111/bpa.12943
Torre M, Jessop N, Hornick JL, Alexandrescu S (2018) Expanding the spectrum of pediatric NTRK-rearranged fibroblastic tumors to the central nervous system: a case report with RBPMS-NTRK3 fusion. Neuropathology 38:624–630. https://doi.org/10.1111/neup.12513
Torre M, Meredith DM, Dubuc A, Solomon DA, Perry A, Vasudevaraja V et al (2019) Recurrent EP300-BCOR fusions in pediatric gliomas with distinct clinicopathologic features. J Neuropathol Exp Neurol 78:305–314. https://doi.org/10.1093/jnen/nlz011
Torre M, Vasudevaraja V, Serrano J, Delorenzo M, Malinowski S, Blandin A et al (2020) Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun. https://doi.org/10.1186/s40478-020-00980-z
Touat M, Ileana E, Postel-Vinay S, André F, Soria JC (2015) Targeting FGFR signaling in cancer. Clin Cancer Res 21:2684–2694. https://doi.org/10.1158/1078-0432.CCR-14-2329
Tulpule A, Guan J, Neel DS, Allegakoen HR, Lin YP, Brown D et al (2021) Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 184:2649-2664.e18. https://doi.org/10.1016/j.cell.2021.03.031
Uguen A, De Braekeleer M (2016) ROS1 fusions in cancer: a review. Futur Oncol 12:1911–1928. https://doi.org/10.2217/fon-2016-0050
Uguen A, Marcorelles P, De Braekeleer M (2015) Searching for ROS1 rearrangements in lung cancer by fluorescent in situ hybridization: the importance of probe design. J Thorac Oncol 10:e83–e85
Vaishnavi A, Le AT, Doebele RC (2015) TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 5:25–34. https://doi.org/10.1158/2159-8290.CD-14-0765
Vogels R, Macagno N, Griewank K, Groenen P, Verdijk M, Fonville J et al (2019) Prognostic significance of NAB2–STAT6 fusion variants and TERT promotor mutations in solitary fibrous tumors/hemangiopericytomas of the CNS: not (yet) clear. Acta Neuropathol 137:679–682. https://doi.org/10.1007/s00401-019-01968-3
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013) Cancer genome landscapes. Science (80-) 340:1546–1558. https://doi.org/10.1126/science.1235122
Wai DH, Knezevich SR, Lucas T, Jansen B, Kay RJ, Sorensen PHB (2000) The ETV6-NTRK3 gene fusion encodes a chimeric protein tyrosine kinase that transforms NIH3T3 cells. Oncogene 19:906–915. https://doi.org/10.1038/sj.onc.1203396
Weinberg F, Griffin R, Fröhlich M, Heining C, Braun S, Spohr C et al (2020) Identification and characterization of a BRAF fusion oncoprotein with retained autoinhibitory domains. Oncogene 39:814–832. https://doi.org/10.1038/s41388-019-1021-1
White MD, McDowell MM, Pearce TM, Bukowinski AJ, Greene S (2019) Intracranial myxoid mesenchymal tumor with rare EWSR1-CREM translocation. Pediatr Neurosurg 54:347–353. https://doi.org/10.1159/000501695
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46:444–450. https://doi.org/10.1038/ng.2938
Wu L, Sun T, Kobayashi K, Gao P, Griffin JD (2002) Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol 22:7688–7700. https://doi.org/10.1128/mcb.22.21.7688-7700.2002
Yde CW, Sehested A, Mateu-Regue A, Ostrup O, Scheie D, Nysom K et al (2016) A new NFIA:RAF1 fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer Genet 209:440–444. https://doi.org/10.1016/j.cancergen.2016.09.002
Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H et al (2015) The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34:4845–4854. https://doi.org/10.1038/onc.2014.406
Yuzawa S, Nishihara H, Wang L, Tsuda M, Kimura T, Tanino M et al (2016) Analysis of NAB2-STAT6 gene fusion in 17 cases of meningeal solitary fibrous tumor/hemangiopericytoma: review of the literature. Am J Surg Pathol 40:1031–1040. https://doi.org/10.1097/PAS.0000000000000625
Zhang J, Gang W, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genetics 45:602–612. https://doi.org/10.1038/ng.2611.Whole-genome
Zhang Y, Chen F, Donehower LA, Scheurer ME, Creighton CJ (2021) A pediatric brain tumor atlas of genes deregulated by somatic genomic rearrangement. Nat Commun. https://doi.org/10.1038/s41467-021-21081-y
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761. https://doi.org/10.1101/gad.1602907
Zhao X, Kotch C, Fox E, Surrey LF, Wertheim GB, Baloch ZW et al (2021) NTRK fusions identified in pediatric tumors: the frequency, fusion partners, and clinical outcome. JCO Precis Oncol. https://doi.org/10.1200/po.20.00250
Zheng T, Ghasemi DR, Okonechnikov K, Sill M, Maass KK, Benites P et al (2021) Cross-species genomics reveals oncogenic dependencies in ZFTA / C11orf95 fusion- positive supratentorial ependymomas. Cancer Discov 11:2230–2247. https://doi.org/10.1158/2159-8290.CD-20-0963
Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD et al (2014) Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 20:1479–1484. https://doi.org/10.1038/nm.3729
Zhu JJ, Jillette N, Li XN, Cheng AW, Lau CC (2020) C11orf95-RELA reprograms 3D epigenome in supratentorial ependymoma. Acta Neuropathol. https://doi.org/10.1007/s00401-020-02225-8
Ziegler DS, Wong M, Mayoh C, Kumar A, Tsoli M, Mould E et al (2018) Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br J Cancer 119:693–696. https://doi.org/10.1038/s41416-018-0251-2
Zimmerman MW, Lui Y, He S, Durbin AD, Abraham BJ, Easton J et al (2019) c-MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Physiol Behav 176:139–148. https://doi.org/10.1158/2159-8290.CD-17-0993.c-MYC
Zong CS, Zeng L, Jiang Y, Sadowski HB, Wang LH (1998) Stat3 plays an important role in oncogenic Ros- and insulin-like growth factor I receptor-induced anchorage-independent growth. J Biol Chem 273:28065–28072. https://doi.org/10.1074/jbc.273.43.28065
Zschernack V, Jünger ST, Mynarek M, Rutkowski S, Garre ML, Ebinger M et al (2021) Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141:455–466. https://doi.org/10.1007/s00401-020-02260-5
Acknowledgements
MK was supported by KiKa Fast track grant. Figure 2a is created with Biorender. Figures 3 and 4 are created with https://app.rawgraphs.io. Online resources were generated with and adapted from ProteinPaint https://pecan.stjude.org/proteinpaint/.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MK had the idea for the manuscript. MR performed the literature search and data analysis and wrote the manuscript. ZO and MR made the figures. ZO, JB and MK critically revised the manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roosen, M., Odé, Z., Bunt, J. et al. The oncogenic fusion landscape in pediatric CNS neoplasms. Acta Neuropathol 143, 427–451 (2022). https://doi.org/10.1007/s00401-022-02405-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02405-8