
Abstract
Background
KIAA1549-BRAF fusion is the most common genetic event in pilocytic astrocytoma (PA), and leads to activation of the mitogen activated protein kinase (MAPK) signaling pathway. Fusions of BRAF with other partner genes, as well as other genetic alterations not involving BRAF but also leading to MAPK pathway activation have been described rarely.
Case presentation
We present a new fusion partner in the low-grade glioma of a 10-year-old male, who presented with headaches and recent episodes of seizures. Magnetic resonance imaging (MRI) demonstrated a right temporal lobe tumor. Histological and immunohistochemical evaluation, and a next generation sequencing assay (Oncopanel, Illumina, 500 genes) including breaKmer analysis for chromosomal rearrangements were performed.
Histology was remarkable for a low-grade glioma composed of mildly atypical astrocytes with piloid processes, in a focally microcystic background. Mitoses were not seen; unequivocal Rosenthal fibers or eosinophilic granular bodies were absent. The tumor was positive for OLIG2 and GFAP and negative for BRAF V600E and IDH1 R132H mutant protein immunostains. Oncopanel showed low SOX2 (3q26.33) copy number gain, and no gains at 7q34. There were no significant single nucleotide variants. BreaKmer detected a GIT2-BRAF fusion with loss of BRAF exons 1–8. The integrated diagnosis was low-grade glioma with piloid features, most consistent with pilocytic astrocytoma, WHO grade I.
Conclusion
GIT2-BRAF fusion has not been reported in the literature in any tumor. Given that the BRAF sequence deleted is identical to that seen in other fusion events in PA, it most likely acts as tumor driver by activation of the MAPK pathway.
Similar content being viewed by others


Background
PA is the most common glioma in the pediatric population [1] and it represents 5.4% of all gliomas in children and adults [2]. These tumors can arise anywhere within the neuraxis, however have a predilection for the posterior fossa, most classically the cerebellum [3]. On imaging, PA is usually well-circumscribed, cystic and solid, and demonstrates contrast enhancement [4, 5]. Histologically, PAs are composed of neoplastic astrocytes with mild-to-moderate atypia and bipolar piloid processes. Most PAs demonstrate a biphasic pattern of growth with areas of increased cellularity and a dense fibrillary background alternating with hypocellular areas with microcysts; interspersed Rosenthal fibers and/or eosinophilic granular bodies (EGBs) are seen in classic cases most typically in the cerebellum, but are not a specific diagnostic feature. Areas of infiltration, indistinguishable from those of a diffuse astrocytoma, may be present at the periphery of the tumor [5].
PAs generally have an excellent prognosis and are classified as WHO grade I neoplasms [5]. Surgical excision alone is frequently curative, with a survival rate of greater than 95% at 10-year follow-up [2, 6]. This benign clinical course underscores the importance of accurately distinguishing these tumors from other glial neoplasms. Occasionally, the histological diagnosis can be problematic because PAs can demonstrate mitoses, foci of increased proliferative labeling index by immunohistochemistry for MIB1, microvascular proliferation and sometimes geographic necrosis, all features that are also encountered in high-grade gliomas [5]. At the same time, midline glioma with H3 K27 M mutation, a WHO grade IV tumor with dismal prognosis, can present with low-grade histology, especially on biopsy specimens [7]. Most PAs have activation of mitogen activating protein kinase (MAPK) signaling through numerous described alterations, the most common being a tandem duplication at 7q34 that results in KIAA1459-BRAF fusion, therefore demonstration of a known alteration is helpful in supporting a diagnosis of PA [8,9,10].
In this manuscript we describe a tumor with a novel fusion of BRAF with GIT2, hypothesized to result in activation of MAPK in a similar fashion as in PAs with the canonical KIAA1459-BRAF fusion.
Case presentation
Clinical history
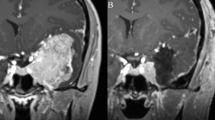
We report the case of a 10-year-old male patient with a 1-year-history of headaches that were thought initially to represent migraines. However, he progressed to having blurred vision and complex partial seizures characterized by jaw and mouth movements, teeth grinding and upper extremity stiffness/shaking. Magnetic resonance imaging without contrast showed a 2.0 × 1.9 × 1.8 cm tumor in the right temporal lobe along the inferolateral margin of the temporal horn of the right ventricle, with mild mass effect (Fig. 1a). The mass was slightly heterogeneous in signal characteristics, slightly hyperintense on T1 sequence (Fig. 1b) and peritumoral edema was not observed (Fig. 1c, FLAIR sequence). The patient underwent surgery and a gross total resection was achieved. A gray-tan, soft, partially gelatinous tumor was received by pathology.

a MRI without contrast, axial section: a T2 hyperintense relatively well defined tumor in the right temporal lobe. b T1 sequence, slightly hyperintense well defined tumor in the right temporal lobe. c FLAIR sequence highlights the tumor; peritumoral edema is not seen
Histological and immunohistochemical stains
Hematoxylin-eosin-stained sections were performed on 4-μm thick formalin-fixed paraffin-embedded sections. Antibodies against glial fibrillary acidic protein (GFAP, Leica Biosystems, Richmond IL, clone GA5, predilute), NeuN (Chemicon, MilliporeSigma, Temecula, CA, clone A60, 1:100), IDH1(R132H) mutant protein (Dianova GmbH, Hamburg, Germany, Clone H09 1:25), OLIG2 (Cell Marque, Sigma Aldrich, Rocklin, CA, clone EP112, predilute), Ki67 (Cell Marque, Sigma Aldrich, Rocklin, CA, clone SP6, predilute) and BRAF V600E (Ventana, Tucson, AZ, clone VE1, predilute) were applied. Coverslips were mounted with Permount (Fisher Scientific). The slides were examined under an Olympus BX41 light microscope. Photographs were taken using an Olympus DP25 camera.
Next generation sequencing
Next generation sequencing was performed using a previously described method [11]. In summary, DNA was analyzed by massively parallel sequencing using a solution-phase Agilent SureSelect hybrid capture kit. Somatic mutations in tumor DNA were detected using the exome-sequencing platform OncoPanel (Illumina HiSeq) in a CLIA-certified laboratory. Common single nucleotide polymorphisms (SNP) were accounted for using the following informatic steps: any SNP present at >0.1% in Exome Variant Server (NHLBI GO Exome Sequencing Project [ESP], Seattle, WA; URL: http://evs.gs.washington.edu/EVS/) or present in dbSNP was filtered. Variants also present in the COSMIC mutation database were rescued for manual review. Structural variants were detected using the BreaKmer algorithm, which identifies rearrangements and nucleotide-level breakpoints by realigning variant contigs generated from assembling all misaligned reads within the targeted NGS data [12].
Results
H&E sections demonstrated a focally infiltrative tumor composed of mildly atypical cells (Fig. 2a) with prominent bipolar piloid processes (Fig. 2b). Areas of increased cellularity alternated with areas less cellular (Fig. 2a) and occasional multinucleated cells with eccentric nuclei (“pennies on a plate”) (Fig. 2c) were present. Some areas demonstrated oligodendroglioma-like morphology (Fig. 2d). Mitoses were difficult to find and there was no necrosis or microvascular proliferation. Unequivocal Rosenthal fibers or eosinophilic granular bodies were not identified. The tumor demonstrated extensive immunostaining with GFAP (Fig. 2e) and OLIG2 (not shown), and was negative for BRAF V600E and for IDH1 R132H mutant protein. A MIB-1 immunostain showed a proliferative labeling index of 1–2% (Fig. 2f). The tumor was provisionally signed out as low-grade astrocytoma with piloid features, W.H.O. grade I or II.

a H&E section showing some peripheral infiltration in this astrocytoma with mild nuclear atypia. b Smear preparation during the intraoperative microscopic examination, showing mildly atypical astrocytes with prominent bipolar piloid processes. c Dense, relatively hypercellular area with mild nuclear atypia and occasional multinucleated cells with peripheral nuclei (“penny on a plate”). d Focal oligodendroglioma-like morphology, with perinuclear clearing and round nuclei with speckled chromatin. e Extensive cytoplasmic GFAP immunostaining. f Low MIB (KI67) proliferation labeling index of 1–2%
Molecular analysis using Oncopanel was performed, and the BreaKmer algorithm demonstrated a fusion involving intron 14 of GIT2 and intron 8 of BRAF, resulting in loss of BRAF exons 1–8 (Fig. 3a-c). Overall, 43 split reads were detected out of 259 total reads demonstrating a translocation at base 110,387,138 of chromosome 12 within intron 14 of GIT2, and 140,494,011 of chromosome 7 within intron 8 of BRAF. 29 discordant reads were also detected (Fig. 3d). There were no copy number changes besides low copy gain of SOX2 (3q26.33). There were no significant single nucleotide variants identified. Given the molecular test results, an integrative diagnosis of “low-grade astrocytoma with piloid feature most consistent with pilocytic astrocytoma, WHO grade I” was rendered.

a Diagram of chromosomes 7 and 12 illustrating chromosomal breakpoints at location of BRAF (q34) and GIT2 (q24.11), respectively. b Schematic of BRAF gene, including autoregulatory domain (CR1) and kinase domain (CR3), and GIT2 gene. Breakpoints at intron 8 of BRAF and intron 14 of GIT2 are diagramed. c Resulting GIT2-BRAF fusion gene, with deletion of autoregulatory domain of BRAF. The kinase domain (CR3) is hypothesized to be constitutively activated in the resulting fusion protein (Additional file 1: Figure S1)
Follow-up
Given that the patient underwent a gross total resection, the patient is followed periodically with imaging studies and neuro-oncology consultation; radiotherapy or chemotherapy was not used. At nine months from surgery, the patient is stable, without evidence of recurrence.
Discussion
PAs demonstrate MAPK signaling pathway activation through different genetic alterations. The first described and most common genetic event, KIAA1549-BRAF fusion, was described in 2008 by Bar et al. [8], who observed frequent gains at 7q34 and increased signaling in BRAF-MEK-ERK in PAs. More recent studies [9, 13, 14] describe numerous other genetic events that result in MAPK pathway activation: BRAF V600E mutations, BRAF intragenic deletions and insertions, NTRK2 fusions, FGFR1 and KRAS point mutations. Recently described BRAF fusion partners are FAM131B, RNF130, CLCN6, MKRN1, and GNAII. We report a novel inframe GIT2-BRAF fusion with breakpoints at intron 14 of GIT2 (band q24.11) and intron 8 of BRAF (band q34), with retention of exon 9 and higher in BRAF. There were no significant copy number changes observed, and no single copy gain at 7q34, therefore this fusion is the result of a deletion, and not tandem duplication; the same mechanism of MAPK activation has been described in pilocytic astrocytomas with FAM131B-BRAF fusions. Because the deleted region of BRAF is identical to the one affected in other described fusions, resulting in deletion of the inhibitory domain of the BRAF and constitutive activation, most likely this fusion activates the MAPK signaling pathway.
GIT2 encodes the ARF GTPase-activating protein GIT2, which plays a role in the regulation of cell-to-cell adhesion and cell motility. While overexpression of GIT2 has been described in the literature in breast and lung carcinomas [15, 16], this fusion with BRAF is novel in tumorigenesis and has not been described in any other type of tumor, hence it is difficult to predict the effect that the deletion will have on the GIT2 gene. Although the glioma described in this case report had focal infiltration, the histologic features were widely within the range seen in other pilocytic astrocytomas reviewed at our institution and reported in the literature; there were no specific histologic features that could be directly associated to BRAF-GIT2 fusion.
In the daily practice of neuropathology, it may be difficult to decide if the case presented is histologically WHO grade I or II due to its focally infiltrative nature. While areas of focal infiltration can be concerning for a histologic grade II neoplasm, many pediatric low-grade astrocytomas and glioneuronal tumors that are WHO grade I, including PA, have areas of infiltration. This is a feature seen in practice and mentioned in the literature, including in the revised WHO Classification of the Tumors of the Central Nervous System [5]. Such difficulty in precise histologic grading underscores the value of molecular findings in helping to predict clinical behavior. While there are infiltrative gliomas with MAPK pathway activation that may demonstrate progression over time, or have a first presentation with high-grade histology, these tumors usually have MAPK pathway activation through structural alterations in FGFR or NTRK family genes, or have pathogenic variants of NF1 or BRAF [17,18,19,20,21,22]. Alternatively, structural rearrangements including fusions, duplications, and intragenic deletions involving the inhibitory domain of BRAF and thought to result in constitutive activation of the kinase domain, as seen in this case report, are very common in PA and possibly even specific to this neoplasm. They are associated with the increased survival expected in pilocytic astrocytomas and other WHO grade I pediatric gliomas, and therefore such a molecular finding may suggests a diagnosis of PA in the appropriate histologic context [23]. Although a body of literature suggests that a small percentage of diffuse astrocytomas, WHO grade II, have BRAF structural rearrangements, the progression to high-grade gliomas that is expected in diffuse astrocytomas is not expected in tumors with BRAF fusions [24].
Conclusion
We are expanding the knowledge of genetic events that activate the MAPK signaling pathway in low-grade gliomas by describing a novel GIT2-BRAF fusion in PA. Given the evolving targeted therapeutic options for patients with recurrent or inoperable PAs, complete molecular characterization of these tumors is more important than ever.
Abbreviations
- CLIA:
-
Clinical Laboratory Improvement Amendments
- EGB:
-
Eosinophilic granular body
- MAPK:
-
Mitogen activated protein kinase signaling pathway
- PA:
-
Pilocytic astrocytoma.
- SNP:
-
Single-nucleotide polymorphism.
- WHO:
-
World Health Organization.
References
Ostrom OT, Gittleman H, Xu J, Kromer C, Wollinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, Oct 1;18(suppl_5):1–75.
Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005 Jun;64(6):479–89.
Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001 Sep;17(9):503.
Brandao LA, Poussaint TY. Pediatric brain tumors. Neuroimaging Clin N Am. 2013 Aug;23(3):499–525.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D, Perry A, Reifenberger G, von Deimling A. WHO classification of tumors of the central nervous system. Chapter 2. 2016. pp. 80–8.
Burkhard C, Di Patre PL, Schuler D, Schuler G, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003 Jun;98(6):1170–4.
Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ, Perry A. Diffuse midline gliomas with histone H3K27M mutation: a study of 47 cases assessing the spectrum of morphologic variations and associated genetic alterations. Brain Pathol. 2016 Sep;26(5):569–80.
Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008 Sep;67(9):878–87.
DTW J, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang D, Fontebasso AM, Stutz AM, Hutter S, Zuckermann M, Sturm D, Gronych J, Lasitschka B, Schmidt S, Cin HS, Witt H, Sultan M, Ralser M, Northcott P, Hovestadt V, Bender S, Pfaff E, Stark S, Faury D, Schwartzentruber J, Makewski J, Weber UD, Zapatka M, Raeder B, Schlesner M, Worth CL, Bartholomae CC, Kalle C, Umbusch CD, Radomski S, Lawerenz C, van Sluis P, Koster J, Volckmann R, Versteeg R, Lehrach H, Monoranu C, Winkler B, Uterberg A, Mende C, Milde T, Kulozik AE, Ebinger M, Schuhmann M, Cho YJ, Pomeroy SL, von Deimling A, Witt O, Taylor MD, Wolf S, Karajannis M, Eberhart C, Scheurlen W, Hasselblatt M, Ligon K, Kieran MW, Korbel JO, Yaspo ML, Brors B, Lesberg J, Reifenberger G, Collins VP, Jabado N, Eils R, Lichter P, Pfister S. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–32.
Pfister S, Janzarik WG, REmke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, KOrshunov A, Scheurlen W, Omran H, Lichter P. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008 May;118(5):1739–49.
MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4:e7887.
Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015;43:e19. https://doi.org/10.1093/nar/gku1211.
Pathak P, Kumar A, Jha P, Purkait S, Faruq M, Suri A, Suri V, Sharma MC, Sarkar C. Genetic alterations related to BRAF-FGFR genes and dysregulated MAPK/ERK/mTOR signaling in adult pilocytic astrocytoma. Brain Pathol. 2016 Sep 8; https://doi.org/10.1111/bpa.12444. [Epub ahead of print]
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, Tang B, Haupfear K, Punchihewa C, Easton J, Mulder H, Boggs K, Shao Y, Rusch M, Becksfort J, Gupta P, Wang S, Lee RP, Brat D, Collins V, Dahiya S, George D, Konomos W, Kurian KM, McFadden K, Serafini LN, Nickols H, Perry A, Shurtleff S, Gajjar A, Boop FA, Kilmo PD, Mardis ER, Wilson RK, Baker SJ, Zhang J, Wo G, Downing JR, Tatevossian RG, Ellison DW. Genetic alterations I uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016 Jun;131(6):833–45.
Duan B, Cui J, Sun S, Zheng J, Zhang Y, Ye B, Chen Y, Deng W, Du J, Zhu Y, Chen Y, Gu L. EGF-stimulated activation of Rab35 regulates RUSC2-GIT2 complex formation to stabilize GIT2 during directional lung cancer cell migration. Cancer Lett. 2016 Aug 28;379(1):70–83.
Zhou W, Cao MG, Xu J, Fang ZY, Wang XY, Guo ZP, Li SS, Zhou ZH. Effect of GTPase activating protein Git2 on metastasis in breast cancer. Zhonghua Zhong Liu Za Zhi. 2016 Jul;38(7):492–8.
Lassaletta A, Zapotocky M, Bouffet E, Hawkins C, Tabori U. An integrative molecular and genomic analysis of pediatric hemispheric low-grade gliomas: an update. Childs Nerf Syst. 2016 Oct;32(10):1789–97.
Chamdine O, Gajjar A. Molecular characteristics of pediatric high-grade gliomas. CNS Oncol. 2014 Nov;3(6):433–43.
Wang Z, Zhang C, Sun L, Liang J, Liu X, Li G, Yao K, Zhang W, Juang T. FGFR3, as a receptor tyrosine kinase, is associated with differentiated biological functions and improved survival of glioma patients. Oncotarget. 2016 Dec 20;7(51):84587–93.
Lasorella A, Sanson M, Iavarone A. FGFR-TACC gene fusions in human glioma. Neuro Oncol. 2017 Apr 1;19(4):475–83.
Behling F, Barrabtes-Freer A, Skardelly M, Nieser M, Christians A, Stockhammer F, Rohde V, Tataqiba M, Hartmann C, Stadelmann C, Schittenhelm J. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn Pathol. 2016 Jun 27;11(1):55.
Alexandrescu S, Korshunov A, Lai SH, Dabiri S, Patil S, Li R, Shih CS, Bonnin JM, Baker JA, Du E, Schamhorst DW, Samuel D, Ellison DW, Perry A. Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas – same entity or first cousins. Brain Pathol. 2016 Mar;26(2):215–23.
Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, Alon N, Kahn D, Fried I, Scheinemann K, Tsangaris E, Dirks P, Tressler R, Bouffet E, Jabado N, Tabori U. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011 Jul 15;17(14):4790–8.
Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Alon N, Poke JD, Ray PN, Navickiene V, Managerel J, REmke M, Buczkowicz P, Ramaswamy V, Guerreiro Stucklin A, Li M, Young EJ, Zhang C, Castelo-Branco P, Bakry D, Laughlin S, Schlien A, Chan J, Ligon KL, Rutka JT, Dirks PB, Taylor MD, Greenberg M, Malkin D, Huang A, Bouffet E, Hawkins CE, Tabori U. BRAF mutation and CDKN2A deletion define a clinically distinct subroup of childhood secondary high-grade glioma. J Clin Oncol. 2015 Mar 20;33(9):1015–22.
Acknowledgements
This case report could not have been possible without the dedicated support of the Histology Laboratory at Boston Children’s Hospital and of the Center for Advanced Molecular Diagnostics at Brigham and Women’s Hospital.
Funding
All the data published in this case report is extracted from the electronic medical records. There were no methods that were employed specifically for this manuscript; hence the authors did not need any funding source for the writing of this case report. If the case report will be accepted for publication, the corresponding author will pay the publishing fees from her annual general educational fund that is part of her contract with her employer, The Department of Pathology at Boston Children’s Hospital.
Availability of data and materials
The molecular raw data showing the reads over the breakpoints in BRAF and GIT2 is stored in the clinical records at the Brigham and Women’s Hospital, and it can be provided in a de-identified format upon reasonable request.
Author information
Authors and Affiliations
Contributions
JH – wrote the manuscript and contributed to the molecular interpretation. HL – reviewed the pathology and formulated the diagnosis, and provided edits on the manuscript. NRM – contributed the figure of the BRAF fusion. MWK – is the neur-oncologist treating the patient and contributed the clinical history. He also contributed edits to the manuscript. KLL – is the principal investigator of the DFCI10–417 protocol under which this manuscript was written. He also contributed the interpretation of the oncopanel and edited the manuscript. SA – was the mentor of JH during the write-up of this case report and has overseen all aspects of the manuscript, including the submission. She is also he corresponding author. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The parents of the patient consented to the DFCI10–417 research protocol that was approved by the Institutional Review Board at Dana Farber Cancer Institute on 11/04/2016 and expires on 10/28/2017; one of the sections of the protocol describes the possibility of publication of clinical information and data obtained under the protocol. The DFCI10–417, IRB approval and signed consent are available and can be provided to the editor upon request.
Consent for publication
In addition, the parents signed a separate consent for publication of this case report that includes de-identified clinical data, images and pathology; the consent for publication was signed on 7/13/2017, and can be provided to the editorial office upon request.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Figure S1.
Representative image of BreaKmer interface illustrating GIT2-BRAF translocation. Sequenced contigs corresponding to BRAF are gray, with contiguous rainbow reads corresponding to bases that are part of GIT2. Sequence details of the translocation are shown at the bottom of the schematic (TIFF 25908 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Helgager, J., Lidov, H.G., Mahadevan, N.R. et al. A novel GIT2-BRAF fusion in pilocytic astrocytoma. Diagn Pathol 12, 82 (2017). https://doi.org/10.1186/s13000-017-0669-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-017-0669-5