
Abstract
Bread wheat is an important food source worldwide, contributing ~20% of the caloric intake per person worldwide. Due to a domestication bottleneck and highly selective breeding for key traits, modern wheat cultivars have a narrow genetic base. Wheat production faces several challenges due to both abiotic and biotic stresses as well as changing climatic conditions and genetic improvement of wheat is generally considered to be the most sustainable approach to develop climate resilient cultivars with improved yield and end-use traits. Since wheat cultivars and landraces have been explored extensively to identify novel genes and alleles, one way to overcome these pitfalls is by looking into the proverbial treasure trove of genomic diversity that is present in wheat’s wild relatives. These wild relatives hold reservoirs of genes that can confer broad-spectrum resistance to pathogens, increase yield, provide additional nutrition, and improve dough quality. Genetic approaches and techniques have existed to introgress wild chromatin to bread wheat, as well as trace introgressions present in the germplasm for over 7 decades. However with the availability of NGS technologies, it is now easier to detect and efficiently integrate the genetic diversity that lies within wheat’s gene pools into breeding programs and research. This chapter provides a concise explanation of current technologies that have allowed for the progression of genomic research into wheat’s primary, secondary, and tertiary gene pools, as well as past technologies that are still in use today. Furthermore, we explore resources that are publicly available that allow for insight into genes and genomes of wheat and its wild relatives, and the application and execution of these genes in research and breeding. This chapter will give an up-to-date summary of information related with genomic resources and reference assemblies available for wheat’s wild relatives and their applications in wheat breeding and genetics.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Wheat
- Gene pool
- Wild wheat relatives
- Reference assemblies
- Resistance genes
- Exome
- Yield related traits
- NGS
- Comparative genomics
- Alien introgressions
12.1 Introduction
Bread wheat is one of the most important staple crops and provides over 1/5th of the calories consumed by the world’s population (FAOSTAT 2020). Global wheat production needs to be increased in light of the growing human population and changing climatic conditions (Hickey et al. 2019; Ray et al. 2012, 2013; Tilman et al. 2011). To cope with the numerous challenges that wheat faces, such as heat, drought, and diseases, it is important to find useful sources of genes and alleles for its improvement, and at the same time, develop approaches for efficient transfer of this useful genetic variability to cultivated wheat. Efforts have already been made in this direction, with the major and successful efforts that have been made after the wheat genome reference assembly using T. aestivum cv. CHINESE SPRING became available as a model in the 2018 (Appels et al. 2018). Since then, more and more resources have been added up to speed up breeding activities and the development of markers for important traits. For instance, in the years 2019 and 2020 a wheat pan-genome resource containing an assembly of 10+ wheat genomes including elite cultivars from across the globe and a 1 K exome capture data were generated (He et al. 2019; Walkowiak et al. 2020). In fact, although high-quality reference assembly is available for CHINESE SPRING, it does not capture the complete species-specific variation that can be exploited for variety development. Therefore, the above genomic resources including the pan-genome and exome capture data have proven to be highly useful. These resources have also been exploited for identification of useful wild introgressions in wheat followed by marker development for biotic and abiotic stress tolerance traits. The current pan-genome resource consists of ten genomes with pseudomolecules level assembly and five genomes with assemblies of hexaploid wheat.
One of the major objectives of any breeding program has been to develop resilient wheat varieties against environmental conditions as well as biotic stresses and significant progress has been made in the genetic improvement of wheat, mainly after the green revolution either using conventional or molecular breeding approaches through marker assisted selection. The introduction of dwarfing genes during the green revolution revolutionized wheat variety development and led to dramatic increase in wheat yield across the globe (Ali et al. 1973; Hedden 2003; Pingali 2012). Similarly, important genetic markers have also been identified for the QTL/genes providing resistance against different biotic and abiotic stresses (Saini et al. 2022; Singh et al. 2021). This has certainly led to the enhancement in the breeding populations of wheat; however, at the same time it has also narrowed down the genetic base thus resulting in reduced species variability. This ultimately necessitates the need to explore the wild and related species of wheat which are an important reservoir of useful genetic diversity as well as genes for biotic and abiotic stresses.
Based on the evolutionary distance between the species and the success rate of interspecies hybridization, Harlan and de Wet (1971) introduced the idea of wheat gene pools that included primary, secondary, and tertiary gene pool (Fig. 12.1) (Jiang et al. 1993; Mujeeb-Kazi et al. 2013). While, the genomes of primary and secondary gene pool share some homology with the wheat genome, the species in the tertiary gene pool do not share any homology with the wheat genome and, therefore, are sexually incompatible through homologous recombination. It is also difficult to cross the species of secondary and tertiary gene pool with hexaploid wheat when compared to the species of primary gene pool (Mujeeb-Kazi et al. 2013).

Overview of bread wheat’s gene pools with examples in each category
The species in the primary gene pool include modern wheat cultivars and other T. aestivum landraces, Triticum spelta (AABBDD), tetraploid durum wheat T. turgidum (AABB), diploid wheat species T. urartu (AA), and Aegilops tauschii (DD). Examples of species in the secondary gene pool are tetraploid species T. timopheevii (AAGG), and diploid species T. monococcum (AmAm) and Ae. speltoides (SS). Species in the tertiary gene pool include cultivated species such as rye (RR) and barley (HH) as well as wild relatives of wheat. Importantly, wild relatives of wheat contain a treasure trove of variability that can overcome the genetic bottlenecks found in bread wheat (Tiwari et al. 2015). Examples of these are wild grasses such as diploid Thinopyrum elongantum (EE), tetraploid Ae. geniculata (UUMM), and octoploid Leymus arenarius (XXXXNNNN) (Pour-Aboughadareh et al. 2021; Anamthawat-Jónsson 2001). Due to the absence of pairing at meiosis between the tertiary pool chromosomes and those of wheat, techniques such as radiation induced chromosomal breaks or gene editing must be used to create introgression lines (Benlioğlu and Adak 2019; Jiang et al. 1993; Mujeeb-Kazi et al. 2013).
As mentioned above, the availability of genomic resources in hexaploid bread wheat has driven the development of useful markers leading to stress resilient wheat cultivars. However, looking at the complexity of the wheat genome owing to its large genome size and polyploid nature, it became necessary to develop genomic resources for the above wild relatives of wheat. Considerable progress has already been in this direction. For example, diploid relatives Ae. longissima, Ae. speltoides, and Ae. sharonensis, as well as several accessions of Ae. tauschii all have recently released reference quality assemblies available for BLAST and genome browsing (Avni et al. 2022; Gaurav et al. 2022; Zhou et al. 2021). Further, wild tetraploid species T. turgidum ssp. dicoccoides v. “ZAVITAN” have also recently had a high-quality assembly released with the use of optical maps for more accurate scaffolding.
The present chapter is mainly focused on providing an overview of the available reference assemblies, and genomic resources in wheat’s wild relatives, which have been explored to identify useful introgressions in wheat. Some examples include (i) Fhb7 (from T. elongatum) providing resistance against Fusarium head blight in wheat (Guo et al. 2015); (ii) the well-known 1BL/1RS translocations from rye which has useful genes for improved grain yield and biomass especially under abiotic stress (Lukaszewski 1993), Lr57 and Yr40 from Ae. geniculata providing resistance against rust disease (Kuraparthy et al. 2007a, b). Recent developments in the next generation sequencing technologies have led to the development of low-cost sequencing reactions such as skim sequencing which provides a useful resource for the identification of alien introgressions with even a low coverage of less than 0.1x (Adhikari et al. 2022b). A comparative overview of synthetic relationships between wheat and wild relatives is also discussed. Overall, the present chapter will serve as a useful resource for the students and researchers working in alien wheat genomics and exploring useful alien wheat introgressions in development of wheat cultivars.
12.2 State of Reference Assemblies in Wheat and Its Wild Relatives
Wild and related species in wheat are a reservoir of important genes for different abiotic and biotic stress tolerances. Therefore, the availability of genomic resources for these wild relatives will prove to be an asset for identification of genes/QTLs and their linked markers which may be helpful in simplifying wheat genomics leading to development of elite wheat cultivars which is otherwise difficult due to complex and large wheat genome. Reference genome assemblies are now available for some of the important wild species belonging to all the three wheat gene pools. Reference assemblies for the important wheat relatives are explained in brief below.
12.2.1 Primary Gene Pool Reference Genomes
The first draft of the reference genome of bread wheat first became public in 2014, utilizing survey sequencing of individual chromosomes. Though this is considered a significant breakthrough in the world of wheat genomics, this initial draft sequence only accounted for ~61% of the entire wheat genome (Lukaszewski et al. 2014). Four years later, with the use of additional genetic data, including radiation hybrids, and sequence data, with the advancement of next generation sequencing (NGS) technologies, the fully annotated CHINESE SPRING reference genome was released with pseudomolecule assemblies for all 21 chromosomes (Appels et al. 2018). This reference genome has been continuously updated with the use of new technologies, both with the intent of more accurate contig establishment and scaffolding as well as annotation of genes not initially reported in the V.1.0. (Alonge et al. 2020; Zhu et al. 2021). Extensive comparative data shows that CHINESE SPRING is a genetic outlier when compared to domesticated species of Triticum sp. (Walkowiak et al. 2020).
The development of the pan-genome of wheat has allowed for more precise research and insight into the primary gene pool of wheat, including T. spelta. As of December 2022, 13 cultivars of wheat and one cultivar of T. spelta are available for BLAST as well as genome browsing. Interestingly, with the information gained by the 10+ genome project, alien introgressions were able to be traced using reads derived from T. timopheevii and T. ponticum (JJJJJJsJsJsJsJs) in T. aestivum cv. LANCER, and Ae. ventricosa (NvNvDvDv) in T. aestivum cv. JAGGER in order to get more exact coordinates of these loci.
Tetraploid species of both cultivated (T. durum) and wild emmer (T. dicoccoides) wheat are also a part of the primary gene pool, due to the ability for homologous recombination to occur within the shared sub-genomes (A and B). When compared to hexaploid wheat, only 5% of wheat grown for human consumption is durum, and 95% is hexaploid. This may be attributed to the genome plasticity of hexaploid wheat which allowed for a broader potential for adaptation compared to tetraploid wheat (Mastrangelo and Cattivelli 2021). Also, compared to hexaploid wheat, the elite gene pool of durum wheat has little genetic diversity, and most elite durum wheat cultivars are moderately to highly susceptible to disease resistance breeding (Clarke et al. 2010; Miedaner and Longin 2014). This is also not surprising due to the widely known fact that hexaploid bread wheat actually evolved from an inter-specific hybridization between T. dicoccoides and diploid species Ae. tauschii (Dvorak et al. 2012; Lukaszewski et al. 2014; Mcfadden and Sears 1946). However, it is evident from the published reports that wild emmer introgressions were responsible for significant gains in genetic diversity among the hexaploid lines as shown recently using the 1000 Wheat Exome Project (He et al. 2019). Similarly, the phenotypic variance contributed by several important traits including harvest weight, drought response, and plant height is largely attributed to these wild emmer introgressions (Nigro et al. 2022; Zhu et al. 2019).
Looking into the importance of wild emmer introgressions in hexaploid bread wheat, improved reference genomes of both wild emmer and cultivated durum wheat were published in 2019. The improved reference genome of wild emmer wheat cv. ZAVITAN (WEW) utilized optical maps as well as advancements in alignment technologies in order to increase the effective size of the reference genome by ~67 Mb, as well as adding over 2,000 high confidence genes. Additionally, between WEW_v1.0 and WEW_v.2.0, gaps of unknown size dropped from 2,767 to only 471 (Avni et al. 2017; Zhu et al. 2019). Later in 2019, a high-quality reference genome of T. durum cv. SVEVO was published, and by utilizing the WEW data, it was shown that the short-term evolutionary changes showed little change to synteny between WEW and durum. There were, however, lower copy numbers of important gene families such as NLRs in SVEVO in comparison with Zavitan, which implies a reduction of canonical R-genes (Maccaferri et al. 2019).
Diploid progenitor species of bread wheat genomes A (T. urartu) and D (Ae. tauschii), as well as close B genome relative Ae. speltoides (SS) all serve as a less complex system to work with for genomics research than the hexaploid bread wheat (Kerby and Kuspira 1987). Therefore, in recent years, reference genomes for all the three wheat genome donors (A genome; T. urartu, B genome: Ae. speltoides; D genome: Ae. tauschii) have been produced in order to help with wheat improvement. While the donors for A and D genome are included in the primary gene pool, the donors for the B genome are included in secondary gene pool. Therefore, the reference assemblies for the donors of A and D genome are discussed in more detail below, and the reference assemblies for the B genome donor (Ae. speltoides) are discussed in separate sub-heading in the next section involving secondary gene pool.
12.2.2 A Genome
The T. urartu reference genome was first published in 2018 (Ling et al. 2018), four months before the release of the CHINESE SPRING v.1.0 reference genome. In their analysis, done using the 2014 draft wheat genome v. 0.4, strong structural variations were observed between the T. urartu A genome and the bread wheat A genome, proposing evolutionary rearrangements. Within the diverse population of T. urartu accessions used for this study, and using the reference genome, three distinct groups were identified in the Fertile Crescent. The above diverse accessions were screened for powdery mildew resistance, and excitingly, after inoculation with powdery mildew (PM), one group (group 2) showed significant resistance against the pathogen. Further, analysis using the SNP data revealed a single putative candidate gene that was involved in providing resistance against powdery mildew. This resistance was perhaps due to the natural selection for powdery mildew resistance as well adaptation to grow at high altitudes.
12.2.3 D Genome
The D genome progenitor Ae. tauschii is a well of genetic variability in wheat, due to the low level of variation seen within D genome of wheat (Dubcovsky and Dvorak 2007; Voss-Fels et al. 2015). This lack of variation is partially due to the small proportion of diversity that was obtained during polyploidization when hybridization between ancient, domesticated T. turgidum (AABB), and the small population of Ae. tauschii near the Caspian Sea (Dubcovsky and Dvorak 2007; Gaurav et al. 2022; Luo et al. 2017; Voss-Fels et al. 2015). However, due to the ability to develop synthetic wheat by hybridizing tetraploid species with Ae. tauschii, diversity in the D genome can be integrated into the breeding germplasm (Li et al. 2018). The first Ae. tauschii reference genome was released in 2017 in the background of accession AL8/78; the current version (Aet v.5.0) has been improved using optical maps as well as Pac-Bio long-read sequencing (Luo et al. 2017; Wang et al. 2021).
Since the initial release, several strides have been made in Ae. tauschii genomics. For instance, Zhou et al. (2021) developed reference quality genomes of four additional accessions representing four sub-lineages of Ae. tauschii with the intent to trace wild introgressions better in the germplasm. In the same year, the Open Wild Wheat Consortium (OWWC) generated whole genome sequencing (WGS) data for 242 non-redundant accessions of Ae. tauschii, to probe the evolution of bread wheat, determine the variation within the population, and perform genome-wide association studies (GWAS) for important traits using the AL8/78 reference genome (Gaurav et al. 2022). This study was able to show the two major lineages that make up the D genome in wheat, and using the wheat pan-genome, show the physical regions that come from these lineages. Additionally, a third lineage not associated with the evolution of bread wheat was also characterized.
Further, using k-mer-based GWAS, candidate genes for flowering time, stem rust (Sr) resistance, trichome number, spikelet number, PM resistance, and wheat curl mite resistance were also reported. Efforts are currently underway by the OWWC to develop a pan-genome resource for Ae. tauschii which will provide further information pertaining to the diversity prevailing in the genome sequences of diverse Ae. tauschii accessions (openwildwheat.org).
12.2.4 Secondary Gene Pool Reference Genomes
In comparison with the primary gene pool of wheat, genomic resources for members of the secondary gene pool are limited. Therefore, efforts are being made in this direction. For instance, (i) the development of reference assemblies for wild and cultivated T. monococcum accessions are available for public use (Ahmed et al. 2023).
Diploid wheat T. monococcum which is a close relative of T. urartu (A genome donor) is the only species with both domesticated (T. monococcum ssp. monococcum) and wild type (T. monococcum ssp. aegilopoides) accessions. Therefore, the reference assemblies for these species once available will certainly help in simplifying wheat genomics and may be an improvement over the reference assembly available for T. urartu. (ii) Transcriptome data for T. monococcum is also available from an earlier study (Fox et al. 2014). (iii) A core set of wild einkorn as well as domestic einkorn was also recently categorized by Adhikari et al. (2022a). Using GBS data, 145 domesticated einkorn accessions and 584 wild einkorn accessions were divided into α, ß, γ, and monococcum. A set of T. urartu accessions were also a part of this study, and as expected, they clustered together distally from T. monococcum accessions.
When compared to A and D genome, B genome of wheat has been difficult to study in a diploid species due the proposed extinction of the direct progenitor (Riley et al. 1958; Sarkar and Stebbins 1956). Researchers, however, have found a workaround this issue by working with species in the Sitopsis section of Aegilops (S*S*) due to their close relatedness with the B genome (Kerby and Kuspira 1987). In the last decade, reference quality genomes for five Sitopsis species were released to help with additional resources for not only the elucidation of the B genome of wheat, but also as a further resource in researching the D genome of wheat (Li et al. 2022; Sandve et al. 2015; Yamane and Kawahara 2005; Yu et al. 2022). Recently, Avni et al. (2022) also communicated the release of three reference quality genomes in the same section (Sitopsis) which included two new assemblies for Ae. sharonensis (SlSl), and Ae. speltoides and one assembly for Ae. sharonensis which was in fact first communicated by Yu et al. (2022).
Alignments of the above assemblies with the different sub-genomes of wheat revealed a strong linear alignment of Ae. sharonensis and Ae. longissima with the D genome of bread wheat, and that of Ae. speltoides with the B genome which is obvious due to their strong relationship with the respective sub-genomes (Fig. 12.2). This was also further supplemented with the clustering of high confidence gene annotations of Ae. sharonensis and Ae. longissima with bread wheat’s D genome as well as Ae. tauschii, and that of Ae. speltoides with WEW, durum wheat, and bread wheat’s B genome.

Synteny between diploid wheat chromosomes 1A, 1S, 1D and hexaploid bread wheat’s genome
In March 2022, reference assemblies of two additional “S” genomes (Ae. bicornis (SbSb) and Ae. searsii (SbSb)) were communicated, finally completing the Sitopsis section of the Triticeae. Both the above S genome assemblies also clustered with the D genome and D genome progenitors of wheat, in comparative alignments showing their closer association within the ancestry of wheat’s evolution. Interestingly, with this complete information, it was found that the divergence of the D-related Sitopsis clade from the D progenitors was predicted to have happened around 5.23 Mya, whereas Ae. speltoides and the B genomes of both durum and bread wheat happened more recently at 4.44 Mya. With these available genomes, more precise genomic research can now be performed in the B genome of wheat, as well as diving deeper into the evolution of the D genome and its progenitors.
12.2.5 Tertiary Gene Pool Reference Genomes
The tertiary gene pool of wheat is underrepresented in terms of resource availability and research, due to the difficulty in defining these species, as well as limited genomic information (Qi et al. 2007; Schneider et al. 2008; Tiwari et al. 2015). Discussions in the literature have considered the Sitopsis section species as members of the tertiary gene pool, not including Ae. speltoides, but since the recent advancements in their genomic resources, it is more fitting to place them in the secondary genepool. Although some species, such as T. elongantum (EE), have had assemblies and annotations competed for attention with regards gene cloning, no reference genomes for wild grasses in the tertiary gene pool are currently available (Wang et al. 2020).
Two cultivated species, on the other hand, belonging to the tertiary gene pool, barley (Hordeum vulgare; 2n = 2x = 14; HH) and rye (Secale cereale; 2n = 2x = 14; RR), have had reference genomes published in the last ten years. The original barley genome was the first species in the Triticeae tribe to have a reference genome (Melonek and Small 2022; Mochida and Shinozaki 2013; Purugganan and Jackson 2021). Originally sequenced and annotated in 2012, one of the biggest achievements in this assembly was overcoming the size and complexity of cereal genomes, due to the highly repetitive elements (Mayer et al. 2012). Since the release, updates have been made to properly order the chromosomes and create a better physical map, as well as reduce the unanchored sequences from ~250 to 83 Mb (Beier et al. 2017; Mascher et al. 2017; Monat et al. 2019). In a similar fashion to the achievements in wheat, a pan-genome project was also developed in barley, which included the sequencing and assembly of 19 additional barley lines including two highly transformable lines (GOLDEN PROMISE and IGRI) as well as a wild barley genotype (Jayakodi et al. 2020). This resource was, and is, an important milestone in the advancement of cereal crop genomics due to its early elucidation. Rye is an important member of the tertiary gene pool as a contributor of high tolerance for both biotic and abiotic stresses. Additionally, rye has been an important player in wheat breeding due to the importance of the 1BL/1RS and 1AL/1RS which confer resistance to multiple biotic diseases (Zeller and Sears 1973; Jung and Seo 2014). Moreover, synthetic hybrids of rye and wheat, named Triticale, have gained popularity due to their nutritional value as forage (Zhu 2018).
To better understand the underlying genetics behind the important aspects of rye, two reference quality genomes of wheat were released simultaneously in 2021. In the article by Rabanus-Wallace et al. (2021), a chromosome scale assembly was developed in the background of cv LO7, showing similar genomic makeup as other members of in Triticeae, and strong collinearity with the barley genome. Using this assembly, the researchers were able to determine a translocated region conferring frost tolerance in a 5A/5RL translocation line, first denoted using chromosome labeling and confirmed using read depth analysis on bread wheat’s 5A chromosome and rye’s 5R chromosome. In another article by Li et al. (2021), an additional genotype of rye, cv. WEINING, had a reference assembly created, which provided further support for the strong collinearity between the tertiary gene pool genomes. In their study, utilizing 2,517 single-copy orthologous genes, Li et al. (2021) developed a phylogenetic tree depicting 12 grasses and their evolutionary divergence. Although it is not necessarily new information, with the rye genome sequenced, the authors were able to compare rye with the other 11 sequenced genomes to deduce that rye had diverged from wheat ~5 Mya after barley and wheat’s divergence, giving further evidence of rye’s closer relationship with bread wheat and its progenitors. For a summary of the state of reference genomes in Triticeae from the past five years (see Fig. 12.3).

Timeline of reference genomes in Triticeae from 2017 to 2022
12.3 Alien Introgressions and Comparative Genomics
As described above, wild wheat relatives play an important role in the production of high performing wheat cultivars. Modern breeding techniques have reduced the genetic diversity in the breeding germplasm to select for higher yield (Keilwagen et al. 2022; Sansaloni et al. 2020; Schneider et al. 2008). Utilizing DNA segments from wild relatives that have been integrated into bread wheat’s genome is a method to overcome this reduction in genetic diversity (Fig. 12.4); however, methods for detecting these introgressions are a must to properly trace these segments in breeding programs (Hao et al. 2020; Molnár-Láng et al. 2015). In this section, we will describe the methods, both old and new, that researchers utilize to detect and trace these introgressions, describe the important genes that come from these introgressions, as well as show the usefulness of modern technologies for comparative genomic analysis.

Methods for developing introgression lines from wild relatives coming from a the primary gene pool and b the secondary and tertiary gene pools
12.3.1 Methods for Detecting Alien Introgressions
Different methods for detecting the alien introgressions can be broadly classified into cytological/cytogenetic, PCR-based markers and Recent Next Generation Sequencing (NGS)-based methods including skim sequencing.
12.3.2 Cytological Methods
Cytological methods for detecting chromosomal morphological differences have been used for almost 100 years (Gill and Friebe 1996). A popular method for observing different sizes and compositions of chromosomes was achieved by using centromeric heterochromatin staining, or C-banding, which allows for visualization of chromosomes and/or karyotypes of different species on a conserved scale (Endo and Gill 1996; Gill et al. 1991). This method was used for detecting rye/wheat hybrid pairing as far back as 1977 as well as determining T. timopheevii introgressions in T. timopheevii × T. aestivum hybrids (Badaeva et al. 1991; Dhaliwal et al. 1977). The C-banding method used alongside genomic in situ hybridization (GISH) also allowed for the detection of introgressions from Ae. umbullata (UU), Ae. speltoides, Ae. comosa (MM), Ae. longissima, and T. timopheevii as well as several others as far back as the early 90s (Friebe et al. 1996).
More recently, regions of Leymus racemosus DNA containing important Fusarium Head Blight (FHB) resistance gene Fhb3 introgressed into bread wheat were traced using GISH and C-banding (Qi et al. 2008). Another, still popular, method of visualizing introgressions in wheat is the use of fluorescence in situ hybridization or FISH, which utilizes fluorescent-labeled DNA probes to detect important regions of chromosomes, such as introgressions (Campos-Galindo 2020; Jiang and Gill 2006). This method is still much in use today to provide further evidence of translocations in wheat, including the previously mentioned frost tolerance associated region in rye introgressed into wheat background (Rabanus-Wallace et al. 2021). This method has also been used to dissect introgressions coming from T. elongantum, Ae. columnaris (UcUcXcXc), Ae. caudata (CC), T. timopheevii, as well as many more not noted here (Badaeva et al. 2017; Devi et al. 2019; Grewal et al. 2020; Guo et al. 2022). Another use of this method was described in 2021, where FISH and GISH markers were utilized to visualize the recombination patterns of susceptible vs resistant genotypes of Ae. geniculata (UgUgMgMg) introgression lines in F3 families (Steadham et al. 2021).
12.3.3 PCR-Based Markers
Another method to detect alien introgressions is by using PCR-based markers that are polymorphic between bread wheat and the wild species. The use of PCR-based markers for identifying alien introgressions in bread wheat dates back to early 90s when Rogowsky et al. (1993) designed PCR and RFLP markers to detect famous 1AS.1RL, 1BS.1RL, and 1DS.1RL rye introgressions in wheat background. Since then, PCR-based markers are continuously being implemented for identifying introgressions. More recently Li et al. (2019) designed markers to detect Thinopyrum intermedium ssp. trichophorum (JJJsJsStSt) introgressions in wheat that provide significant stripe rust resistance. To illustrate the importance of old and new technologies, these researchers utilized GISH, FISH, and C-banding in order to validate the effectiveness of the PCR markers, which now can be utilized in marker assisted breeding (MAS) to incorporate these genes into the breeding germplasm. Further, polymorphic SSR markers were also developed recently to detect introgressions from synthetic amphidiploid species T. kiharae (AtAtGGDD) which holds a reservoir of genes that have the potential to improve resistance to many diseases as well as increase the quality of flour production (Orlovskaya et al. 2020).
12.3.4 NGS Technology
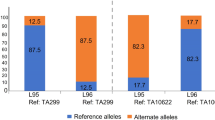
With the advent of cost-effective NGS methods, researchers now have the ability to obtain sequence data coming from the transcriptome, exome, as well as the whole genome. This data can be generated from any species that the researchers are interested in, including the wild relatives of wheat. Examples of this have been mentioned in Sect. 12.2 of this chapter, in regard to whole genome assembly; however, data for wild relatives is constantly being generated for purposes of gene mapping and cloning, as well as diving deeper into wild relatives. One such example comes from Tiwari et al. (2015), where the 5 Mg chromosome of Ae. geniculata was sorted, sequenced, and assembled to gain insight into this important species. This information helped with the fine mapping of Lr57 and Yr40 in translocation wheat lines (Steadham et al. 2021).
In the past 5 years, NGS data has been utilized to detect introgressions in Triticeae species without the additional step of SNP calling, which can create artifacts as well as require more computational resources (Li and Wren 2014). Genotyping by sequencing (GBS) data provides short and low coverage genomic data, usually for the purpose of creating VCF files in order to genotype a population with relatively low computational and storage requirements (Perea et al. 2016). This data has now been shown to be able to discern introgressions in both wheat and barley. In a study by Keilwagen et al. (2019), they were able to detect putative introgressions from wild relatives in wheat, including the 1BL/1RS translocation. Interestingly, in the panel of 209 elite European winter wheat varieties in which GBS data was generated, many of the regions where introgressions were detected, these overlapped with important genes used in breeding programs such as Yr17 from Ae. ventricosa (NvNvDvDv) and Lr19 from T. ponticum as well as genes not yet known to be from wild relative introgressions such as Glu-D1 and Ppo-D1. Due to the decrease in the cost of WGS data generation, one group set out to see the benefit of using resequencing data from multiple wild relatives to detect introgressions, utilizing the 10+ genomes described above. Keilwagen et al. (2022) used wild relative WGS data from both public repositories as well data generated from their own experiments to determine the regions of wild introgressions in 10 genotypes, gathered from the 10+ wheat genome project. In doing so, 9 introgressions coming from wild relatives Ae. ventricosa, Ae. markgrafii (CC), Ae. speltoides, T. timopheevii, Ae. umbullata, Ae. uniaristata (NN), and T. ponticum were found to be present on chromosomes 2A, 2B, 2D, 3D, and 4A. The researchers determined that within introgressions found on 2AS (from either Ae. ventricosa or Ae. markgrafii), 2B (from T. timopheevii) and 2DL (from Ae. markgrafii or, Ae. umbullata) contained genes that shared >90% amino acid similarity with genes coding for leaf rust and stripe rust genes, respectively. Fascinatingly, when checking the two introgressions that were present in all 10 genotypes, on 2A and 4AL coming from Ae. speltoides, in relatives of bread wheat, these introgressions were found in T. urartu, T. boeoticum, and T. monococcum, but not in T. dicoccoides or T. spelta. The studies also determined that these introgressions were able to be detected using only 1% of the total data.
To further save on computational cost, researchers have shown that skim sequencing of genomes can be used at a coverage as low as 0.025x, to determine introgressions, as described by Adhikari et al. (2022a, b). These authors used this method to determine barley introgressions on chromosomes 7A, 7B, and 7D in a population of 384 wheat–barley introgression lines. Additionally, they screened T. intermedium-durum wheat amphiploid lines to find not only lines where there were possible introgressions, but also certain lines containing whole wheat chromosomes. Due to the efficacy and precision of this method of detecting introgressions, this method is more than likely to define what the future of alien introgression mapping procedures looks like for researchers not only in wheat, but in all important crops.
12.3.5 Agronomically Important Genes Coming from Alien Introgressions
One of the most important alien introgressions in wheat is the 1BL/1RS translocation, in which the short arm of chromosome 1R in rye has replaced the short arm of 1B of wheat. This introgression has been used in wheat breeding not only for the disease resistance that is associated with this introgression, which has since become obsolete, but also because of the increased root biomass that has a positive effect on yield (Zeller and Hsam 1983; Sharma et al. 2011; Villareal et al. 1998). Despite the negative effects of this translocation on bread making quality, ~30% of modern cultivars contain the 1BL/1RS segment (Wang et al. 2017; Zeller et al. 1982). For a list of varieties containing 1R translocations visit http://www.rye-gene-map.de/rye-introgression/index.html (see also Ru et al. 2020). In recent years new 1BL/1RS lines have been developed to overcome some of the shortcomings of older introgressed lines, in which resistance against stripe rust, as well as drought tolerance was observed (Ren et al. 2022; Sharma et al. 2022; Gabay et al. 2020).
Ae. geniculata is also a genetic goldmine due to the strong disease resistance genes that are present in some accessions. The line TA10437, in which the 5 Mg chromosome was sequenced in 2015, contains important resistance genes against nefarious pathogens such as stripe rust and leaf rust (Tiwari et al. 2015). Recently, leaf and stripe rust resistance genes, Lr57 and Yr40 respectively, have been fine mapped in Ae. geniculata translocation lines utilizing mapping populations derived from a cross between resistant TA10437 derived introgression lines and susceptible disomic 5 Mg addition lines in the background of CHINESE SPRING (Steadham et al. 2021). In this study, Lr57 and Yr40 were not only fine mapped to a 1.5 Mb region of the introgressed Ae. geniculata 5 Mg segment, but through phenotyping of the mapping population and the donor parent of the 5 Mg segment, Lr57 was shown to provide further evidence of its broad-spectrum resistance, confirming the results of an earlier study (Kuraparthy et al. 2007a, b). Moreover, this study showed that recombination is achievable in alien introgressions by crossing introgression lines with disomic lines containing homologous chromosomes of the alien species.
Sources of biotic disease resistance coming from wild relatives are unequivocally important for the sustenance and improvement of wheat; however, due to the associated linkage drag, their utilization in modern cultivars by durum and bread wheat breeders is limited for integrating “exotic” resistance genes from wild or cultivated relatives into their elite material (Hafeez et al. 2021; Steiner et al. 2019). But with the ever-increasing knowledge of wild wheat relatives, new genes that confer resistance are being integrated into the germplasm without a yield penalty. Powdery mildew and stripe rust resistance genes, Pm5V and Yr5V respectively, transferred from the annual diploid wheat relative D. villosum (VV) via amphiploid generation (T. turgidum × D. villosum, AABBVV) (Zhang et al. 2022). In order to integrate these genes into the germplasm, subsequent crossing with elite D. villosum introgression lines was performed and yielded lines with comparable yield to that of elite bread wheat lines. However, due to grain softness that is also associated with the 5 V chromosome, chemical mutagenesis was performed to knockout this undesirable trait, resulting in comparable yielding, hard grained genotypes for utilization in wheat breeding. A summary of disease resistance genes coming from wild relatives is described in Table 12.1.
Outside of resistance, genes controlling yield-related and end-use traits coming from wild relatives have also been utilized by researchers to further address the benefits of these species. Wild tetraploid wheat Agropyron cristatum (PPPP) has been used as a donor for abiotic and biotic disease resistance, as well as for yield-related traits for over 30 years (Chen et al. 1992; Zhang et al. 2015). In a study by Zhang et al. (2018), researchers found that Pubing260, a T3BL.3BS/6PL translocation line containing a small terminal introgression from Ag. cristatum had increased grains per spike, spikelets per spike, thousand kernel weight, and flag leaf width in comparison with elite bread wheat genotypes without this segment. Additionally, in 2022, a high molecular weight glutenin subunit (HMW-GS) gene coming from Ae. tauschii was directly introduced into bread wheat, and although the dough quality was reduced slightly, the quality of Chinese steamed bread increased (Bo et al. 2022).
12.4 Available Resources for Sequence Data and Plant Material
Availability and accessibility of resources is paramount for the development of higher yielding, disease resistant cultivars of wheat. Fortunately, there exists web-based databases for the extraction of genomic and transcriptomic information regarding wheat and its relatives. Furthermore, there are avenues available for requesting seed material for many of the species mentioned above. In this section, we will provide an overview of the publicly available sites that can be utilized to not only browse and obtain genomic data from bread wheat and wheat’s wild relatives but also where to request seeds from repositories across the world.
12.4.1 Web-Based Databases for Sequence Data
The National Center for Biotechnology Information is a resource for genetic research for almost any species that has had any type of sequence information generated (Sayers et al. 2022). Their user-friendly website allows for easy search for any topic, giving results for all 35 of their databases. A simple search for the term “Triticum” on December 12, 2022, yielded results in 26 of the 35 available databases. Over 4 million hits from this search go to their nucleotide database, whereas ~3 million hits come from the protein database. Moreover, NCBI’s sequence read archive (SRA) is a significant repository of sequencing data coming from NGS reads from researchers across the globe. These SRAs are mostly publicly available and include genome and transcriptome data that is BLASTable. A search for T. intermedium in the SRA database yields over 4 thousand results, 184 of which are from reads coming from genome sequencing. Suffice to say, NCBI’s website is a significant source of information, especially for those who may not have access to funding their own NGS studies. However, due to the abundance of avenues in which data is deposited into their databases, curated navigation for specific species may be overwhelming. Specifically, when BLASTing against their database, many of the hits received may be outdated, or repeats of similar information.
Ensembl overcomes some of the pitfalls of NCBI by allowing users to select specific organisms to browse (Cunningham et al. 2022). Moreover, Ensembl plant removes species coming from Animalia, Fungi, and prokaryotes are removed to deconvolute searches for specific species. Although their database is not as robust as NCBI, the navigation of certain aspects is made much easier. Their biomart and downloads tabs allow for easy access to nucleotide and protein data for the species hosted by the website, which can be downloaded from a single web page. Ensembl plant stays up to date with current versions of reference genomes, including the newest versions of T. aestivum, Ae. tauschii, and H. vulgare, although old versions are still available. Another significant feature of Ensembl is their variation track that is available for some species. This feature allows for users to find variants of specific genomic regions, either found naturally or induced via chemical mutagenesis. By clicking on this feature, users are able to browse either the effects of these variations, or in some cases such as in T. aestivum, find accession numbers for mutant genotypes. This is very important for researchers who are looking for variants of candidate genes in gene cloning projects, making it easy to find knockouts and/or missense mutations in candidate regions. Unfortunately, very few relatives of wheat are available for BLAST, genome browsing, or data acquisition. Currently, diploid species T. urartu, Ae. tauschii, rye, and barley are the only diploid relatives of wheat that are accessible using this website.
For researchers who work specifically in small grains, GrainGenes is a curated database that has many features that are useful (Yao et al. 2022). Genome browsers are easy to find and available for several wild relatives of wheat, including the five accessions of Ae. tauschii, and three of the genomes in the Sitopsis section mentioned above. Additionally, BLASTing is robust, being able to select from many wild relatives, including all members of the Sitopsis section. Additionally, GrainGenes has an easy-to-use search for markers and probes found in literature. There also are some useful tools that are found in GrainGenes, including genome specific primer (GSP) design. The website, however, has become more cumbersome over the years as more and more data is being added to the site, though currently a more user-friendly interface is being developed.
A wheat specific database also exists in the form of URGI (Alaux et al. 2018; see also Chap. 2). This site allows for wheat-curated research in the form of BLASTs that can be performed on specific chromosomes for all available versions. This is important because many of the times, in the literature, different versions of reference genomes are used for research. This site, although not as user friendly as the previously mentioned databases, contains a significant amount of sequence data for wheat.
12.4.2 Germplasm Acquisition Resources
Researchers across the globe are willing to share material with one another for the greater good of assuring food security. Specifically in wheat research, seed requests can be performed from multiple sources. One such example is the Wheat Genetics Resource Center, hosted by Kansas State University. This site gives direct access to alien species coming from the aforementioned Sitopsis section, as well as multiple other species coming from Aegilops, such as Ae. geniculata. Along with this, there is access to Triticum species including diploid monococcum and urartu. WGRC also contains 95 unique accessions of Dasypyrum villosum coming from several different countries. Alien translocation lines with transfers coming from Aegilops, Dasypyrum, Triticum, Secale, and Agropyron species are directly accessible from this resource as well. This site links to other important germplasms and seed distributors such as CIMMYT and the USDA.
CIMMYT (The International Maize and Wheat Improvement Center) and the USDA utilize the Germplasm Resource Information Network (GRIN) or GRIN-global to give international institutions access to germplasms of several different species of plants, including wheat and some of its wild relatives. Although this resource is not specifically catered to wheat researchers, wild species belonging to Aegilops and Triticum are available. Similarly, Genesys is a resource for multiple different crop systems, but their user-friendly interface allows for easy search for species in Triticum. This site contains over 12 thousand accessions coming from Aegilops alone, and they are designated by subsets, including Aegilops core sets.
The OWWC, mentioned in Sect. 12.2.1, has their panel available through the Germplasm Resource Unit (GRU) hosted by the John Innes Centre. This resource has similar resources as the aforementioned sites; however, they have a core collection of Titiceae wild relatives that include Dasypyrum, Aegilops, Triticum, and Eremopyron. This site also contains seed resources for mutant, DH, and other mapping populations in wheat, as well as historical landraces.
12.5 Concluding Remarks
The ever-increasing breadth of knowledge coming from wheat and its relatives have large implications for improving the overall quality of cultivars in the coming years. This chapter gives an up-to-date overview of recent advances in genomic resources within wheat, highlighting the importance of wild relatives, and alien introgressions within the germplasm. The availability of the wheat pan-genome has allowed for researchers to trace introgressions that are present within cultivars across the world, some of these alien introgressions were found within the entire pan-genome, giving further evidence of the importance of the genetic diversities (Keilwagen et al. 2022). As more of these wild genomes get reference quality assemblies associated with them, the more we can learn about the important genes that lie within these species. On the OWWC website, a pan-genome of Ae. tauschii is currently underway, allowing researchers to get a more in depth understanding of the diversities that are present in these progenitor species. High-quality reference genomes are still required for some important species, such as T. elongatum, D. villosum, and Agrypyron species. Researchers would also benefit from pan-genomes representing other important wild relatives that have been mentioned in this chapter, such as that of wild diploid Triticum species, tetraploid Aegilops species.
Extensive resources for obtaining both genomic data as well as seed material for these species are available for public use, making further novel research possible across the globe. It is an exciting time to work in the field of wheat research with the ability to obtain diverse populations of not only bread wheat and its primary gene pool, but also members of secondary and tertiary gene pools from collaborators in different countries. The web-based resources that exist now make it possible for quick turnaround for not only basic scientific knowledge but also for the integration of this diversity into local breeding programs. A future prospect that could make this process even more efficient is a localized database where these independent seed and data repositories can be accessed. CIMMYT and the USDA make it easy to find material from either establishment by utilizing systems like GRIN and GRIN-global, which share germplasm requests; however, many other institutions do not utilize this as a means for requests and distribution, and further many of these are not necessarily catered toward wheat-based research. A similar central database would be beneficial for the amount of sequence data that is becoming available in Triticeae. A system to search for data pertaining to specific gene pools could prove to be beneficial for future research, especially as more genomes are being sequenced. The examples and information provided here will hopefully make it easier for researchers, students, and curious minds alike to find information pertaining to wheat and the many species that make up its gene pools.
References
Adhikari L, Raupp J, Wu S, Wilson D, Evers B, Koo DH, Singh N, Friebe B, Poland J (2022a) Genetic characterization and curation of diploid A-genome wheat species. Plant Physiol 188(4):2101–2114. https://doi.org/10.1093/plphys/kiac006
Adhikari L, Shrestha S, Wu S, Crain J, Gao L, Evers B, Wilson D, Ju Y, Koo DH, Hucl P, Pozniak C, Walkowiak S, Wang X, Wu J, Glaubitz JC, DeHaan L, Friebe B, Poland J (2022b) A high-throughput skim-sequencing approach for genotyping, dosage estimation and identifying translocations. Sci Rep 12(1). https://doi.org/10.1038/s41598-022-19858-2
Ahmed HI, Heuberger M, Schoen A, Koo DH, Quiroz-Chavez J, Adhikari L, Raupp J, Cauet S, Rodde N, Cravero C, Callot C (2023) Einkorn genomics sheds light on history of the oldest domesticated wheat. Nature. https://doi.org/10.1038/s41586-023-06389-7
Alaux M, Rogers J, Letellier T, Flores R, Alfama F, Pommier C, Mohellibi N, Durand S, Kimmel E, Michotey C, Guerche C, Loaec M, Lainé M, Steinbach D, Choulet F, Rimbert H, Leroy P, Guilhot N, Salse J, Quesneville H et al (2018). Linking the international wheat genome sequencing consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol 19(1). https://doi.org/10.1186/s13059-018-1491-4
Ali CH, Khan A, Choudhari HA (1973) Income impact of the green revolution author. Pak Econ Soc Rev 11(1)
Alonge M, Shumate A, Puiu D, Zimin AV, Salzberg SL (2020) Chromosome-scale assembly of the bread wheat genome reveals thousands of additional gene copies. Genetics 216(2):599–608. https://doi.org/10.1534/genetics.120.303501
Anamthawat-Jónsson, K (2001) Molecular cytogenetics of introgressive hybridization in plants. Methods Cell Sci 23:141–150. https://doi.org/10.1023/A:1013182724179
Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ, Choulet F, Distelfeld A, Poland J, Ronen G, Barad O, Baruch K, Keeble-Gagnère G, Mascher M, Ben-Zvi G, Josselin AA, Himmelbach A, Balfourier F, Wang L et al (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361(6403). https://doi.org/10.1126/science.aar7191
Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K, Jordan KW, Golan G, Deek J, Ben-Zvi B, Ben-Zvi G, Himmelbach A, MacLachlan RP, Sharpe AG, Fritz A, Distelfeld A et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357(6346):93–97. https://doi.org/10.1126/science.aan0032
Avni R, Lux T, Minz-Dub A, Millet E, Sela H, Distelfeld A, Deek J, Yu G, Steuernagel B, Pozniak C, Ens J, Gundlach H, Mayer KFX, Himmelbach A, Stein N, Mascher M, Spannagl M, Wulff BBH, Sharon A (2022) Genome sequences of three Aegilops species of the section Sitopsis reveal phylogenetic relationships and provide resources for wheat improvement. Plant J 110(1):179–192. https://doi.org/10.1111/tpj.15664
Badaeva ED, Budashkina EB, Badaev NS, Kalinina NP, Shkutina FM (1991) General features of chromosome substitutions in Triticum aestivum × T. timopheevii hybrids. Theor Appl Genet 82(2):227–232. https://doi.org/10.1007/BF00226218
Badaeva ED, Ruban AS, Aliyeva-Schnorr L, Municio C, Hesse S, Houben A (2017) In situ hybridization to plant chromosomes, pp 477–494. https://doi.org/10.1007/978-3-662-52959-1_49
Beier S, Himmelbach A, Colmsee C, Zhang X-Q, Barrero RA, Zhang Q, Li L, Bayer M, Bolser D, Taudien S, Groth M, Felder M, Hastie A, Šimková H, Staňková H, Vrána J, Chan S, Muñoz-Amatriaín M, Ounit R, Mascher M et al (2017) Construction of a map-based reference genome sequence for barley, Hordeum vulgare L. Sci Data 4(1):170044. https://doi.org/10.1038/sdata.2017.44
Benlioğlu B, Adak MS (2019) Importance of crop wild relatives and landraces genetic resources in plant breeding programmes. J Exp Agric Int 1–8. https://doi.org/10.9734/jeai/2019/v37i330268
Bo C, Fan Z, Ma X, Li A, Wang H, Kong L, Wang X (2022) Identification and Introgression of a Novel HMW-GS Gene from Aegilops tauschii. Agronomy 12(11):2709. https://doi.org/10.3390/agronomy12112709
Campos-Galindo I (2020) Cytogenetics techniques. In: Human reproductive genetics. Elsevier, pp 33–48. https://doi.org/10.1016/B978-0-12-816561-4.00003-X
Clarke JM, Clarke FR, Pozniak CJ (2010) Forty-six years of genetic improvement in Canadian durum wheat cultivars. Can J Plant Sci 90(6):791–801. https://doi.org/10.4141/cjps10091
Chen Q, Jahier J, Cauderon Y (1992) Production and cytogenetic analysis of BC1, BC2, and BC3 progenies of an intergeneric hybrid between Triticum aestivum (L.) Thell. and tetraploid Agropyron cristatum (L.) Gaertn. Theoret Appl Genet 84:698–703. https://doi.org/10.1007/BF00224171
Cox TS, Raupp WJ, Gill BS (1994) Leaf rust-resistance genes Lr41, Lr42, and Lr43 transferred from Triticum tauschii to common wheat. Crop Sci 34(2):339–343
Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov AG, Barnes I, Bennett R, Berry A, Bhai J, Bignell A, Billis K, Boddu S, Brooks L, Charkhchi M, Cummins C, da Rin Fioretto L, Flicek P et al (2022) Ensembl 2022. Nucleic Acids Res 50(D1):D988–D995. https://doi.org/10.1093/nar/gkab1049
Delibes A, Romero D, Aguaded S, Duce A, Mena M, Lopez-Brana I, Andrés MF, Martin-Sanchez JA, García-Olmedo F (1993) Resistance to the cereal cyst nematode (Heteroder aavenae Woll.) transferred from the wild grass Aegilops ventricosa to hexaploid wheat by a ‘stepping–stone’ procedure. Theor Appl Genet 87(3):402–408
Devi U, Grewal S, Yang CY, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge A, King IP, King J (2019) Development and characterisation of interspecific hybrid lines with genome-wide introgressions from Triticum timopheevii in a hexaploid wheat background. BMC Plant Biol 19(1). https://doi.org/10.1186/s12870-019-1785-z
Dhaliwal HS, Gill BS, Waines JG, Egulation R (1977) Analysis of induced homoeologous pairing in a ph mutant wheat X rye hybrid. J Heredity 68. https://academic.oup.com/jhered/article/68/4/207/802618
Dubcovsky J, Dvorak J (2007) Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316(5833):1862–1866. https://doi.org/10.1126/science.1143986
Dvorak J, Deal KR, Luo MC, You FM, von Borstel K, Dehghani H (2012) The origin of spelt and free-threshing hexaploid wheat. J Hered 103(3):426–441. https://doi.org/10.1093/jhered/esr152
Endo TR, Gill BS (1996) The deletion stocks of common wheat. J Heredity 87. https://academic.oup.com/jhered/article/87/4/295/2186527
Food and Agriculture Organization of the United Nations (2020) FAOSTAT statistical database
Fox SE, Geniza M, Hanumappa M, Naithani S, Sullivan C, Preece J, Tiwari VK, Elser J, Leonard JM, Sage A, Gresham C, Kerhornou A, Bolser D, McCarthy F, Kersey P, Lazo GR, Jaiswal P (2014) De novo transcriptome assembly and analyses of gene expression during photomorphogenesis in diploid wheat Triticum monococcum. PLoS ONE 9(5). https://doi.org/10.1371/journal.pone.0096855
Friebe B, Badaeva ED, Kammer K, Gill BS (1996) Standard karyotypes of Aegilops uniaristata, Ae. mutica, Ae. comosa subspeciescomosa andheldreichii (Poaceae). Plant Syst Evol 202(3–4):199–210. https://doi.org/10.1007/BF00983382
Gabay G, Zhang J, Burguener GF, Howell T, Wang H, Fahima T, Lukaszewski A, Moriconi JI, Santa Maria GE, Dubcovsky J (2020) Structural rearrangements in wheat (1BS)–rye (1RS) recombinant chromosomes affect gene dosage and root length. Plant Genome. https://doi.org/10.1002/tpg2.20079
Gaurav K, Arora S, Silva P, Sánchez-Martín J, Horsnell R, Gao L, Brar GS, Widrig V, John Raupp W, Singh N, Wu S, Kale SM, Chinoy C, Nicholson P, Quiroz-Chávez J, Simmonds J, Hayta S, Smedley MA, Harwood W, Wulff BBH et al (2022) Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat Biotechnol 40(3):422–431. https://doi.org/10.1038/s41587-021-01058-4
Gill BS, Friebe B (1996) Plant cytogenetics at the dawn of the 21st century. Curr Opin Plant Biol 1:109–124. http://biomednet.com/elecref/1369526600100109
Gill BS, Friebe B, Endo TR (1991) Standard karyotype and nomenclature system for description of chromosome bands and structural aberrations in wheat (Triticum aestivum). Genome 34(5):830–839. https://doi.org/10.1139/g91-128
Grewal S, Othmeni M, Walker J, Hubbart-Edwards S, Yang CY, Scholefield D, Ashling S, Isaac P, King IP, King J (2020) Development of wheat-aegilops caudata introgression lines and their characterization using genome-specific KASP markers. Front Plant Sci 11. https://doi.org/10.3389/fpls.2020.00606
Guo J, Zhang X, Hou Y, Cai J, Shen X, Zhou T, Xu H, Ohm HW, Wang H, Li A, Han F, Wang H, Kong L (2015) High-density mapping of the major FHB resistance gene Fhb7 derived from Thinopyrum ponticum and its pyramiding with Fhb1 by marker-assisted selection. Theor Appl Genet 128(11):2301–2316. https://doi.org/10.1007/s00122-015-2586-x
Guo X, Shi Q, Yuan J, Zhang J, Wang M, Wang J, Wang C, Fu S, Su H, Liu Y, Huang Y, Liu C, Liu Q, Sun Y, Wang L, Wang K, Jing D, Zhang P, Li J, Han F (2022) Alien chromatin other than the GST-encoding Fhb7 candidate confers Fusarium head blight resistance in wheat breeding. https://doi.org/10.1101/2021.02.03.429547 (BioRxiv)
Hafeez AN, Arora S, Ghosh S, Gilbert D, Bowden RL, Wulff BBH (2021) Creation and judicious application of a wheat resistance gene atlas. Mol Plant 14(7):1053–1070. Cell Press. https://doi.org/10.1016/j.molp.2021.05.014
Hao M, Zhang L, Ning S, Huang L, Yuan Z, Wu B, Yan Z, Dai S, Jiang B, Zheng Y, Liu D (2020) The resurgence of introgression breeding, as exemplified in wheat improvement. In: Frontiers in plant science, vol 11. Frontiers Media S.A. https://doi.org/10.3389/fpls.2020.00252
Harlan JR, Wet JMJ (1971) Toward A rational classification of cultivated plants. Taxon 20(4):509–517. https://doi.org/10.2307/1218252
He F, Pasam R, Shi F, Kant S, Keeble-Gagnere G, Kay P, Forrest K, Fritz A, Hucl P, Wiebe K, Knox R, Cuthbert R, Pozniak C, Akhunova A, Morrell PL, Davies JP, Webb SR, Spangenberg G, Hayes B, Akhunov E et al (2019) Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat Genet 51(5):896–904. https://doi.org/10.1038/s41588-019-0382-2
Hedden P (2003) The genes of the green revolution. Trends Genet 19(1):5–9. https://doi.org/10.1016/S0168-9525(02)00009-4
Hickey LTN, Hafeez A, Robinson H, Jackson SA, Leal-Bertioli SCM, Tester M, Gao C, Godwin ID, Hayes BJ, Wulff BBH (2019) Breeding crops to feed 10 billion. In: Nature biotechnology, vol 37, issue 7. Nature Research, pp 744–754. https://doi.org/10.1038/s41587-019-0152-9
Hsam SLK, Huang X, Ernst F, Hartl L, Zeller F (1998) Chromosomal location of genes for resistance to powdery mildew in common wheat (Triticum aestivum L. em Thell.). 5. Alleles at the Pm1 locus. Theor Appl Genet 96:1129–1134
Jahier J, Tanguy AM, Abelard P, Rivoal R (1996) Utilization of deletions to localize a gene for resistance to the cereal cyst nematode, Heterodera avenae, on an Aegilops ventricosa chromosome. Plant Breed 115:282–284
Jayakodi M, Padmarasu S, Haberer G, Bonthala VS, Gundlach H, Monat C, Lux T, Kamal N, Lang, D, Himmelbach A, Ens J, Zhang XQ, Angessa TT, Zhou G, Tan C, Hill C, Wang P, Schreiber M, Boston LB, Stein N et al (2020) The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 588(7837):284–289. https://doi.org/10.1038/s41586-020-2947-8
Jiang J, Gill BS (2006) Current status and the future of fluorescence in situ hybridization (FISH) in plant genome research. Genome 49(9):1057–1068. https://doi.org/10.1139/G06-076
Jiang J, Friebe B, Gill BS (1993) Recent advances in alien gene transfer in wheat. Euphytica 73(3):199–212. https://doi.org/10.1007/BF00036700
Jung WJ, Seo YW (2014) Employment of wheat-rye translocation in wheat improvement and broadening its genetic basis. J Crop Sci Biotechnol 17(4):305–313. Korean Society of Crop Science. https://doi.org/10.1007/s12892-014-0086-1
Keilwagen J, Lehnert H, Berner T, Beier S, Scholz U, Himmelbach A, Stein N, Badaeva ED, Lang D, Kilian B, Hackauf B, Perovic D (2019) Detecting large chromosomal modifications using short read data from genotyping-by-sequencing. Front Plant Sci 10. https://doi.org/10.3389/fpls.2019.01133
Keilwagen J, Lehnert H, Berner T, Badaeva E, Himmelbach A, Börner A, Kilian B (2022) Detecting major introgressions in wheat and their putative origins using coverage analysis. Sci Rep 12(1). https://doi.org/10.1038/s41598-022-05865-w
Kerber (1987) Resistance to leaf rust in hexaploid wheat: Lr32 A third gene derived from Triticum tauschii. Crop Sci 27(2):204–206
Kerby K, Kuspira J (1987) The phylogeny of the polyploid wheats Triticum aestivum (bread wheat) and Triticum turgidum (macaroni wheat). Genome 29(5):722–737. https://doi.org/10.1139/g87-124
Kuraparthy V, Chhuneja P, Dhaliwal HS, Kaur S, Bowden RL, Gill BS (2007a) Characterization and mapping of cryptic alien introgression from Aegilops geniculata with new leaf rust and stripe rust resistance genes Lr57 and Yr40 in wheat. Theor Appl Genet 114(8):1379–1389. https://doi.org/10.1007/s00122-007-0524-2
Kuraparthy V, Sood S, Chhuneja P, Dhaliwal HS, Kaur S, Bowden RL, Gill BS (2007b) A cryptic wheat-Aegilops triuncialis translocation with leaf rust resistance gene Lr58. Crop Sci 47(5):1995–2003
Li H, Wren J (2014) Toward better understanding of artifacts in variant calling from high-coverage samples. In: Bioinformatics, vol 30, issue 20. Oxford University Press, pp 2843–2851. https://doi.org/10.1093/bioinformatics/btu356
Li A, Liu D, Yang W, Kishii M, Mao L (2018) Synthetic hexaploid wheat: yesterday, today, and tomorrow. Engineering 4(4):552–558. https://doi.org/10.1016/j.eng.2018.07.001
Li J, Chen Q, Zhang P, Lang T, Hoxha S, Li G, Yang Z (2019) Comparative FISH and molecular identification of new stripe rust resistant wheat-Thinopyrum intermedium ssp. trichophorum introgression lines. Crop J 7(6):819–829. https://doi.org/10.1016/j.cj.2019.06.001
Li G, Wang L, Yang J, He H, Jin H, Li X, Ren T, Ren Z, Li F, Han X, Zhao X, Dong L, Li Y, Song Z, Yan Z, Zheng N, Shi C, Wang Z, Yang S, Wang D et al (2021) A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat Genet 53(4):574–584. https://doi.org/10.1038/s41588-021-00808-z
Li LF, Zhang Z, Wang ZH, Li N, Sha Y, Wang XF, Ding N, Li Y, Zhao J, Wu Y, Gong L, Mafessoni F, Levy AA, Liu B (2022) Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Mol Plant 15(3):488–503. https://doi.org/10.1016/j.molp.2021.12.019
Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y, Yu Y, Du H, Qi M, Li Y, Lu H, Yu H, Cui Y, Wang N, Chen C, Liang C et al (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557(7705):424–428. https://doi.org/10.1038/s41586-018-0108-0
Liu Z, Sun Q, Ni Z et al (2002) Molecular characterization of a novel powdery mildew resistance gene Pm30 in wheat originating from wild emmer. Euphytica 123:21–29
Liu W, Danilova TV, Rouse MN, Bowden RL, Friebe B, Gill BS, Pumphrey MO (2013) Development and characterization of a compensating wheat-Thinopyrum intermedium Robertsonian translocation with Sr44 resistance to stem rust (Ug99). Theor Appl Genet 126(5):1167–1177
Lukaszewski AJ (1993) Reconstruction in wheat of complete chromosomes 1B and 1R from the 1RS.1BL translocation of “Kavkaz” origin. Genome 36(5):821–824. https://doi.org/10.1139/g93-109
Lukaszewski AJ, Alberti A, Sharpe A, Kilian A, Stanca AM, Keller B, Clavijo BJ, Friebe B, Gill B, Wulff B, Chapman B, Steuernagel B, Feuillet C, Viseux C, Pozniak C, Rokhsar DS, Klassen D, Edwards D, Akhunov E, Dubská Z et al (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science, 345(6194). https://doi.org/10.1126/science.1251788
Luo M-C, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo N, Zhu T, Wang L, Wang Y, McGuire PE, Liu S, Long, H, Ramasamy RK, Rodriguez JC, Van SL, Yuan L, Wang Z, Xia Z, Dvořák J et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551(7681):498–502. https://doi.org/10.1038/nature24486
Maccaferri M, Harris NS, Twardziok SO, Pasam RK, Gundlach H, Spannagl M, Ormanbekova D, Lux T, Prade VM, Milner SG, Himmelbach A, Mascher M, Bagnaresi P, Faccioli P, Cozzi P, Lauria M, Lazzari B, Stella A, Manconi A, Cattivelli L et al (2019) Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51(5):885–895. https://doi.org/10.1038/s41588-019-0381-3
Mago R, Zhang P, Vautrin S, Simkova H, Bansal U, Luo MC, Rouse M, Karaoglu H, Periyannan S, Kolmer J, Jin Y, Ayliffe MA, Bariana H, Park RF, McIntosh R, Dolezel J, Berges H, Spielmeyer W, Lagudah ES, Ellis JG, Dodds PN (2015) The wheat Sr50 gene reveals rich diversity at a cereal disease resistance locus. Nat Plants 1:15186
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang XQ, Zhang Q, Barrero RA, Li L, Taudien S, Stein N et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544(7651):427–433. https://doi.org/10.1038/nature22043
Mastrangelo AM, Cattivelli L (2021) What makes bread and durum wheat different? In: Trends in plant science, vol 26, issue 7. Elsevier Ltd., pp 677–684. https://doi.org/10.1016/j.tplants.2021.01.004
Mayer KFX, Waugh R, Langridge P, Close TJ, Wise RP, Graner A, Matsumoto T, Sato K, Schulman A, Ariyadasa R, Schulte D, Poursarebani N, Zhou R, Steuernagel B, Mascher M, Scholz U, Shi B, Madishetty K, Svensson JT, Stein N et al (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491(7426):711–716. https://doi.org/10.1038/nature11543
Mcfadden ES, Sears ER (1946) The origin of Triticum spelta and its free-threshing hexaploid relatives. J Hered 37(3):81–89. https://doi.org/10.1093/oxfordjournals.jhered.a105590
Melonek J, Small I (2022) Triticeae genome sequences reveal huge expansions of gene families implicated in fertility restoration. In: Current opinion in plant biology, vol 66. Elsevier Ltd. https://doi.org/10.1016/j.pbi.2021.102166
Miedaner T, Longin CFH (2014) Genetic variation for resistance to Fusarium head blight in winter durum material. Crop Pasture Sci 65(1):46. https://doi.org/10.1071/CP13170
Miranda LM, Murphy JP, Marshall D, Cowger C, Leath S (2007) Chromosomal location of Pm35, a novel Aegilops tauschii derived powdery mildew resistance gene introgressed into common wheat (Triticum aestivum L.). Theor Appl Genet 114(8):1451–1456
Mochida K, Shinozaki K (2013) Unlocking triticeae genomics to sustainably feed the future. Plant Cell Physiol 54(12):1931–1950. https://doi.org/10.1093/pcp/pct163
Molnár-Láng M, Ceoloni C, Doležel J (eds) (2015) Alien introgression in wheat. Springer International Publishing. https://doi.org/10.1007/978-3-319-23494-6
Monat C, Padmarasu S, Lux T, Wicker T, Gundlach H, Himmelbach A, Ens J, Li C, Muehlbauer GJ, Schulman AH, Waugh R, Braumann I, Pozniak C, Scholz U, Mayer KFX, Spannagl M, Stein N, Mascher M (2019) TRITEX: chromosome-scale sequence assembly of Triticeae genomes with open-source tools. Genome Biol 20(1). https://doi.org/10.1186/s13059-019-1899-5
Mujeeb-Kazi A, Kazi AG, Dundas I, Rasheed A, Ogbonnaya F, Kishii M, Bonnett D, Wang RRC, Xu S, Chen P, Mahmood T, Bux H, Farrakh S (2013) Genetic diversity for wheat improvement as a conduit to food security. In: Advances in agronomy, vol 122. Academic Press Inc., pp 179–257. https://doi.org/10.1016/B978-0-12-417187-9.00004-8
Murphy JP, Leath S, Huynh D, Navarro RA, Shi A (1999) Registration of NC96BGTA4, NC96BGTA5, and NC96BGTA6 wheat germplasm. Crop Sci 39:883–884
Narang D, Kaur S, Steuernagel B, Ghosh S, Bansal U, Li J, Zhang P, Bhardwaj S, Uauy C, Wulff BBH, Chhuneja P (2020) Discovery and characterisation of a new leaf rust resistance gene introgressed in wheat from wild wheat Aegilops peregrina. Sci Rep 10(1):7573. https://doi.org/10.1038/s41598-020-64166-2
Nigro D, Blanco A, Piarulli L, Signorile MA, Colasuonno P, Blanco E, Simeone R (2022) Fine mapping and candidate gene analysis of Pm36, a wild emmer-derived powdery mildew resistance locus in durum wheat. Int J Mol Sci 23(21):13659. https://doi.org/10.3390/ijms232113659
Orlovskaya O, Dubovets N, Solovey L, Leonova I (2020) Molecular cytological analysis of alien introgressions in common wheat lines derived from the cross of Triticum aestivum with T. kiharae. BMC Plant Biol 20. https://doi.org/10.1186/s12870-020-02407-2
Perea C, de La Hoz JF, Cruz DF, Lobaton JD, Izquierdo P, Quintero JC, Raatz B, Duitama J (2016) Bioinformatic analysis of genotype by sequencing (GBS) data with NGSEP. BMC Genomics 17. https://doi.org/10.1186/s12864-016-2827-7
Petersen S, Lyerly JH, Worthington ML, Parks WR, Cowger C, Marshall DS, Brown-Guedira G, Murphy JP (2015) Mapping of powdery mildew resistance gene Pm53 introgressed from Aegilops speltoides into soft red winter wheat. Theor Appl Genet 128(2):303–312
Pingali PL (2012) Green revolution: impacts, limits, and the path ahead. Proc Natl Acad Sci USA 109(31):12302–12308. https://doi.org/10.1073/pnas.0912953109
Pour-Aboughadareh A, Sanjani S, Nikkhah-Chamanabad H, Mehrvar MR, Asadi A, Amini A (2021) Identification of salt-tolerant barley genotypes using multiple-traits index and yield performance at the early growth and maturity stages. Bull Natl Res Cent 45:117. https://doi.org/10.1186/s42269-021-00576-0
Purugganan MD, Jackson SA (2021) Advancing crop genomics from lab to field. In: Nature genetics, vol 53, issue 5. Nature Research, pp 595–601. https://doi.org/10.1038/s41588-021-00866-3
Qi L, Friebe B, Zhang P, Gill BS (2007) Homoeologous recombination, chromosome engineering and crop improvement. Chromosome Res 15(1):3–19. https://doi.org/10.1007/s10577-006-1108-8
Qi LL, Pumphrey MO, Friebe B, Chen PD, Gill BS (2008) Molecular cytogenetic characterization of alien introgressions with gene Fhb3 for resistance to Fusarium head blight disease of wheat. Theor Appl Genet 117(7):1155–1166. https://doi.org/10.1007/s00122-008-0853-9
Rabanus-Wallace MT, Hackauf B, Mascher M, Lux T, Wicker T, Gundlach H, Baez M, Houben A, Mayer KFX, Guo L, Poland J, Pozniak CJ, Walkowiak S, Melonek J, Praz CR, Schreiber M, Budak H, Heuberger M, Steuernagel B, Stein N et al (2021) Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential. Nat Genet 53(4):564–573. https://doi.org/10.1038/s41588-021-00807-0
Rani K, Raghu BR, Jha SK, Agarwal P, Mallick N, Niranjana M, Sharma JB, Singh AK, Sharma NK, Rajkumar S, Tomar SMS, Vinod (2020) A novel leaf rust resistance gene introgressed from Aegilops markgrafii maps on chromosome arm 2AS of wheat. Theor Appl Genet 133(9):2685–2694
Ray DK, Ramankutty N, Mueller ND, West PC, Foley JA (2012) Recent patterns of crop yield growth and stagnation. Nat Commun 3. https://doi.org/10.1038/ncomms2296
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8(6). https://doi.org/10.1371/journal.pone.0066428
Reader SM, Miller TE (1991) The introduction into bread wheat of a major gene for resistance to powdery mildew from wild emmer wheat. Euphytica 53:57–60
Ren T, Jiang Q, Sun Z, Ren Z, Tan F, Yang W, Li Z (2022) Development and characterization of novel wheat-rye 1RS 1BL translocation lines with high resistance to Puccinia striiformis f. sp. tritici. Phytopathology 112(6):1310–1315. https://doi.org/10.1094/PHYTO-07-21-0313-R
Riar AK, Kaur S, Dhaliwal HS, Singh K, Chhuneja P (2012) Introgression of a leaf rust resistance gene from Aegilops caudata to bread wheat. J Genet 91(2):155–161
Riley R, Unrau J, Chapman V (1958) Evidence on the origin of the B genome of wheat. J Hered 49(3):91–98. https://doi.org/10.1093/oxfordjournals.jhered.a106784
Rogowsky PM, Sorrels ME, Shepherd KW, Langridge P (1993) Characterisation of wheat-rye recombinants with RFLP and PCR probes. In: Theoretical applied genetics, vol 85. Springer
Rong JK, Millet E, Manisterski J, Feldman M (2000) A new powdery mildew resistance gene: introgression from wild emmer into common wheat and RFLP-based mapping. Euphytica 115:121–126
Rowland GG, Kerber ER (1974) Telocentric mapping in hexaploid wheat of genes for leaf rust resistance and other characters derived from Aegilops squarrosa. Can J Genet Cytol 16:137–144
Ru Z, Juhasz A, Li D, Deng P, Zhao J,Gao L, Wang K, Keeble-Gagnere G, Yang Z, Li G, Wang D, Bose U, Colgrave M, Kong C, Zhao G, Zhang X, Liu X, Cui G, Wang Y, Niu Z, Wu L, Cui D, Jia J, Appels R, Kong X (2020) 1RS.1BL molecular resolution provides novel contributions to wheat improvement. https://doi.org/10.1101/2020.09.14.295733 (bioRxiv preprint)
Saini DK, Srivastava P, Pal N, Gupta PK (2022) Meta-QTLs, ortho-meta-QTLs and candidate genes for grain yield and associated traits in wheat (Triticum aestivum L.). Theor Appl Genet 135(3):1049–1081. https://doi.org/10.1007/s00122-021-04018-3
Sandve SR, Marcussen T, Mayer K, Jakobsen KS, Heier L, Steuernagel B, Wulff BBH, Olsen OA (2015) Chloroplast phylogeny of Triticum/Aegilops species is not incongruent with an ancient homoploid hybrid origin of the ancestor of the bread wheat D-genome. New Phytol 208(1):9–10. https://doi.org/10.1111/nph.13487
Sansaloni C, Franco J, Santos B, Percival-Alwyn L, Singh S, Petroli C, Campos J, Dreher K, Payne T, Marshall D, Kilian B, Milne I, Raubach S, Shaw P, Stephen G, Carling J, Pierre CS, Burgueño J, Crosa J, Pixley K (2020) Diversity analysis of 80,000 wheat accessions reveals consequences and opportunities of selection footprints. Nat Commun 11(1):4572. https://doi.org/10.1038/s41467-020-18404-w
Sarkar P, Stebbins GL (1956) Morphological evidence concerning the origin of the B genome in wheat. Am J Bot 43(4):297–304. https://doi.org/10.1002/j.1537-2197.1956.tb10494.x
Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, Connor R, Funk K, Kelly C, Kim S, Madej T, Marchler-Bauer A, Lanczycki C, Lathrop S, Lu Z, Thibaud-Nissen F, Murphy T, Phan L, Skripchenko Y, Sherry ST et al (2022) Database resources of the national center for biotechnology information. Nucleic Acids Res 50(D1):D20–D26. https://doi.org/10.1093/nar/gkab1112
Schneider A, Molnár I, Molnár-Láng M (2008) Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica 163(1):1–19. https://doi.org/10.1007/s10681-007-9624-y
Sears ER (1956) The transfer of leaf rust resistance from Aegilops umbellulata into wheat. Brookhaven Symp Biol 9:1–21
Sharma D, Knott DR (1966) The transfer of leaf rust resistance from Agropyron to Triticum by irradiation. Can J Genet Cytol 8:137–143
Sharma S, Xu S, Ehdaie B, Hoops A, Close TJ, Lukaszewski AJ, Waines JG (2011) Dissection of QTL effects for root traits using a chromosome arm-specific mapping population in bread wheat. Theor Appl Genet 122(4):759–769. https://doi.org/10.1007/s00122-010-1484-5
Sharma P, Chaudhary HK, Kapoor C, Manoj N, Singh K, Sood VK (2022) Molecular cytogenetic analysis of novel wheat-rye translocation lines and their characterization for drought tolerance and yellow rust resistance. Cereal Res Commun 50(4):655–665. https://doi.org/10.1007/s42976-021-00212-7
Singh K, Batra R, Sharma S, Saripalli G, Gautam T, Singh R, Pal S, Malik P, Kumar M, Jan I, Singh S, Kumar D, Pundir S, Chaturvedi D, Verma A, Rani A, Kumar A, Sharma H, Chaudhary J, Gupta PK et al (2021) WheatQTLdb: a QTL database for wheat. Mol Genet Genom 296(5):1051–1056. https://doi.org/10.1007/s00438-021-01796-9
Shi AN, Leath S, Murphy JP (1998) A major gene for powdery mildew resistance transferred to common wheat from wild einkorn wheat. Phytopathology 88(2):144–147
Steadham J, Schulden T, Kalia B, Koo DH, Gill BS, Bowden R, Yadav IS, Chhuneja P, Erwin J, Tiwari V, Rawat N (2021) An approach for high-resolution genetic mapping of distant wild relatives of bread wheat: example of fine mapping of Lr57 and Yr40 genes. Theor Appl Genet 134(8):2671–2686. https://doi.org/10.1007/s00122-021-03851-w
Steiner B, Michel S, Maccaferri M, Lemmens M, Tuberosa R, Buerstmayr H (2019) Exploring and exploiting the genetic variation of Fusarium head blight resistance for genomic-assisted breeding in the elite durum wheat gene pool. Theor Appl Genet 132(4):969–988. https://doi.org/10.1007/s00122-018-3253-9
Tilman D, Balzer C, Hill J, Befort BL (2011) Global food demand and the sustainable intensification of agriculture. Proc Natl Acad Sci USA 108(50):20260–20264. https://doi.org/10.1073/pnas.1116437108
Tiwari VK, Wang S, Danilova T, Koo DH, Vrána J, Kubaláková M, Hribova E, Rawat N, Kalia B, Singh N, Friebe B, Doležel J, Akhunov E, Poland J, Sabir JSM, Gill BS (2015) Exploring the tertiary gene pool of bread wheat: sequence assembly and analysis of chromosome 5Mg of Aegilops geniculata. Plant J 84(4):733–746. https://doi.org/10.1111/tpj.13036
Villareal RL, Bañuelos O, Mujeeb-Kazi A, Rajaram S (1998) Agronomic performance of chromosomes 1B and T1BL.1RS near-isolines in the spring bread wheat Seri M82. Euphytica 103
Voss-Fels K, Frisch M, Qian L, Kontowski S, Friedt W, Gottwald S, Snowdon RJ (2015) Subgenomic diversity patterns caused by directional selection in bread wheat gene pools; subgenomic diversity patterns caused by directional selection in bread wheat gene pools. Plant Genome 8(2). 10.3835/p
Walkowiak S, Gao L, Monat C, Haberer G, Kassa MT, Brinton J, Ramirez-Gonzalez RH, Kolodziej MC, Delorean E, Thambugala D, Klymiuk V, Byrns B, Gundlach H, Bandi V, Siri JN, Nilsen K, Aquino C, Himmelbach A, Copetti D, Pozniak CJ et al (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 588(7837):277–283. https://doi.org/10.1038/s41586-020-2961-x
Wang J, Liu Y, Su H, Guo X, Han F (2017) Centromere structure and function analysis in wheat–rye translocation lines. Plant J 91(2):199–207. https://doi.org/10.1111/tpj.13554
Wang H, Sun S, Ge W, Zhao L, Hou B, Wang K, Lyu Z, Chen L, Xu S, Guo J, Li M, Su P, Li X, Wang G, Bo C, Fang X, Zhuang W, Cheng X, Wu J, Kong L (2020) Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 368(6493). https://doi.org/10.1126/science.aba5435
Wang L, Zhu T, Rodriguez JC, Deal KR, Dubcovsky J, McGuire PE, Lux T, Spannagl M, Mayer KFX, Baldrich P, Meyers BC, Huo N, Gu YQ, Zhou H, Devos KM, Bennetzen JL, Unver T, Budak H, Gulick PJ, Dvorak J (2021). Aegilops tauschii genome assembly Aet v5.0 features greater sequence contiguity and improved annotation. G3 Genes|Genomes|Genetics 11(12). https://doi.org/10.1093/g3journal/jkab325
Xie CJ, Sun QX, Ni ZF, Yang TM, Nevo E, Fahima T (2003) Chromosomal location of a Triticum dicoccoides-derived powdery mildew resistance gene in common wheat by using microsatellite markers. Theor Appl Genet 106:341–345
Yamane K, Kawahara T (2005) Intra- and interspecific phylogenetic relationships among diploid Triticum-aegilops species (Poaceae) based on base-pair substitutions, indels, and microsatellites in chloroplast noncoding sequences. Am J Bot 92(11):1887–1898. https://doi.org/10.3732/ajb.92.11.1887
Yao E, Blake VC, Cooper L, Wight CP, Michel S, Cagirici HB, Lazo GR, Birkett CL, Waring DJ, Jannink JL, Holmes I, Waters AJ, Eickholt DP, Sen TZ (2022) GrainGenes: a data-rich repository for small grains genetics and genomics. Database. https://doi.org/10.1093/database/baac034
Yu G, Matny O, Champouret N, Steuernagel B, Moscou MJ, Hernández-Pinzón I, Green P, Hayta S, Smedley M, Harwood W, Kangara N, Yue Y, Gardener C, Banfield MJ, Olivera PD, Welchin C, Simmons J, Millet E, Minz-Dub A, Wulff BBH (2022) Aegilops sharonensis genome-assisted identification of stem rust resistance gene Sr62. Nat Commun 13(1). https://doi.org/10.1038/s41467-022-29132-8
Zeller FJ, Gunzel G, Fischbeck G, Gerstenkorn P, Weipert D (1982) Alteration of baking properties of wheat-rye chromosome 1B/1R translocation. Getreide Mehl Brot. 36(6):141–143
Zeller FJ, Sears ER (1973) 1B/1R wheat-rye chromosome substitutions and translocations. In: Fourth international wheat genetics symposium, pp 209–221
Zeller FJ, Hsam SL (1983) Broadening the genetic variability of cultivated wheat by utilizing rye chromatin. In Sixth international wheat genetics symposium, pp 161–173
Zhang J, Liu W, Han H, Song L, Bai L, Gao Z, Zhang Y, Yang X, Li X, Gao A, Li L (2015) De novo transcriptome sequencing of Agropyron cristatum to identify available gene resources for the enhancement of wheat. Genomics 106(2):129–136. https://doi.org/10.1016/j.ygeno.2015.04.003
Zhang J, Ma H, Zhang J, Zhou S, Han H, Liu W, Li X, Yang X, Li L (2018) Molecular cytogenetic characterization of an Agropyron cristatum 6PL chromosome segment conferring superior kernel traits in wheat. Euphytica 214(11). https://doi.org/10.1007/s10681-018-2276-2
Zhang J, Hewitt TC, Boshoff WHP, Dundas I, Upadhyaya N, Li J et al (2021a) A recombined Sr26 and Sr61 disease resistance gene stack in wheat encodes unrelated NLR genes. Nat Commun 12:3378
Zhang R, Lu C, Meng X, Fan Y, Du J, Liu R, Feng Y, Xing L, Cápal P, Holušová K, Doležel J, Wang Y, Mu H, Sun B, Hou F, Yao R, Xiong C, Wang Y, Chen P, Cao A (2022) Fine mapping of powdery mildew and stripe rust resistance genes Pm5V/Yr5V transferred from Dasypyrum villosum into wheat without yield penalty. Theor Appl Genet 135(10):3629–3642. https://doi.org/10.1007/s00122-022-04206-9
Zhang Q, Li Y, Li Y, Fahima T, Shen Q, Xie C (2021b) Introgression of the powdery mildew resistance genes Pm60 and Pm60b from Triticum urartu to common wheat using durum as a ‘bridge.’ Pathogens 11(1):25
Zhou Y, Bai S, Li H, Sun G, Zhang D, Ma F, Zhao X, Nie F, Li J, Chen L, Lv L, Zhu L, Fan R, Ge Y, Shaheen A, Guo G, Zhang Z, Ma J, Liang H, Song C-P et al (2021) Introgressing the Aegilops tauschii genome into wheat as a basis for cereal improvement. Nat Plants 7(6):774–786. https://doi.org/10.1038/s41477-021-00934-w
Zhu F (2018) Triticale: nutritional composition and food uses. In: Food chemistry, vol 241. Elsevier Ltd., pp 468–479. https://doi.org/10.1016/j.foodchem.2017.09.009
Zhu T, Wang L, Rodriguez JC, Deal KR, Avni R, Distelfeld A, McGuire PE, Dvorak J, Luo MC (2019) Improved genome sequence of wild emmer wheat Zavitan with the aid of optical maps. G3 Genes, Genomes, Genetics, 9(3):619–624. https://doi.org/10.1534/g3.118.200902
Zhu T, Wang L, Rimbert H, Rodriguez JC, Deal KR, de Oliveira R, Choulet F, Keeble-Gagnère G, Tibbits J, Rogers J, Eversole K, Appels R, Gu YQ, Mascher M, Dvorak J, Luo MC (2021) Optical maps refine the bread wheat Triticum aestivum cv. Chinese spring genome assembly. Plant J 107(1):303–314. https://doi.org/10.1111/tpj.15289
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2024 The Author(s)
About this chapter

Cite this chapter
Schoen, A. et al. (2024). Genome Sequences from Diploids and Wild Relatives of Wheat for Comparative Genomics and Alien Introgressions. In: Appels, R., Eversole, K., Feuillet, C., Gallagher, D. (eds) The Wheat Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-031-38294-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-031-38294-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-38292-5
Online ISBN: 978-3-031-38294-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)