
Abstract
Infective diseases have become health threat of a global proportion due to appearance and spread of microorganisms resistant to majority of therapeutics currently used for their treatment. Therefore, there is a constant need for development of new antimicrobial agents, as well as novel therapeutic strategies. Quinolines and quinolones, isolated from plants, animals, and microorganisms, have demonstrated numerous biological activities such as antimicrobial, insecticidal, anti-inflammatory, antiplatelet, and antitumor. For more than two centuries quinoline/quinolone moiety has been used as a scaffold for drug development and even today it represents an inexhaustible inspiration for design and development of novel semi-synthetic or synthetic agents exhibiting broad spectrum of bioactivities. The structural diversity of synthetized compounds provides high and selective activity attained through different mechanisms of action, as well as low toxicity on human cells. This review describes quinoline and quinolone derivatives with antibacterial, antifungal, anti-virulent, antiviral, and anti-parasitic activities with the focus on the last 10 years literature.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Antibiotics
- Antifungals
- Anti-parasitics
- Antivirals
- Anti-virulence activity
- Quinoline/quinolone derivatives
1 Introduction
Antimicrobial drugs, structurally diverse molecules, can be natural products, semi-synthetic derivatives of natural compounds, or chemically synthesized compounds. The development of antimicrobials in general, first of all antibiotics, but also antivirals and antimalarials, revolutionized medicine in many ways, and as such is one of the greatest successes of modern medicine. Unfortunately, time with these drugs is rapidly running out. Occurrence and global spread of resistance of bacteria, fungi, viruses, and protozoan parasites to available antimicrobial medicines threaten to send humanity back to pre-antimicrobial era. Therefore, there is an urgent need not just to develop novel antimicrobials but also to introduce into practice novel therapeutic options to fight against both drug-sensitive and drug-resistant pathogens.
A promising alternative to the classic antibiotic approach has recently been established and is known as anti-virulence therapy. Instead of targeting microbial viability, this alternative strategy aims to target pathogens’ virulence machinery required to cause host damage and disease. Microbial virulence machinery includes plethora of virulence factors, which are diverse in structure, function, and localization. One of the most important virulent characteristics of both bacteria and fungi is their ability to form biofilms. Biofilms are multicellular communities enclosed in self-synthetized polymeric matrices attached to biotic or abiotic surfaces (Hall-Stoodley et al. 2004; Costa-Orlandi et al. 2017). According to the National Institutes of Health of the United States, more than 75% of microbial infections that occur in the human body are promoted by the formation and persistence of biofilms (Miquel et al. 2016). Biofilm confers an extreme capacity for persistence against phagocytosis, oxidative stress, nutrient/oxygen restriction, metabolic waste accumulation, interspecies competition, and most importantly, conventional antimicrobial agents (Moradali et al. 2017). Quorum sensing (QS), a cell-to-cell communication system, is a global regulatory system of virulence factor production and biofilm formation in both bacteria and fungi (Albuquerque and Casadevall 2012; Defoirdt 2018), with no homologous components in humans, thus its inhibition is considered the most attractive strategy for the development of anti-virulence agents.
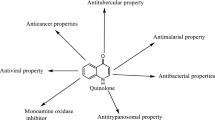
Quinine (Fig. 1), quinoline alkaloid isolated from the bark of the Cinchona tree in 1820, used in the treatment of malaria played a historical role in the development of quinoline alkaloids as therapeutics. These quinoline based compounds have been isolated and identified from natural sources (plants, animals, and microorganisms), and many studies have documented their antitumor, antimalarial, antibacterial, antifungal, antiviral, anti-parasitic and insecticidal, anti-inflammatory, antiplatelet and other activities (Shang et al. 2018). Quinoline and 4-quinolone (Fig. 1) moieties were used as a scaffold for drug development for more than two centuries (Heeb et al. 2011). The most successful drug based on quinoline scaffold is chloroquine (Fig. 1), which was specifically developed as antimalarial agent. Until today, numerous quinoline-based compounds and drugs were developed as antimalarial agents, designed to target all stages of parasite life-cycle.

Quinoline and quinolone scaffolds and their best-known drugs
In fact quinolines still serve as inexhaustible models for design and development of new semi-synthetic or synthetic quinoline/quinolone antimicrobial agents, which are the focus of this review article.
2 Antibacterial Activity
Important group of antibacterial agents are synthetic antibiotics with 4-quinolone as core structure, which are used in the treatment of urinary tract and respiratory infections (Anderson et al. 2012). Today antibacterial 4-quinolones in clinical use include nalidixic acid, that is introduced to medical practice in 1964 (Bisacchi 2015), followed by ciprofloxacin, levofloxacin, norfloxacin, besifloxacin, and moxifloxacin (Fig. 2). Majority of them belong to fluoroquinolone chemotype. The quinolone antibiotics are very potent towards a wide range of Gram negative bacteria, with minimal inhibitory concentrations (MICs) in the ng/ml range, and are reasonably active against many Gram positive bacteria (MICs in the mg/ml range) (Anderson et al. 2012). Their antibacterial activity is based on inhibition of DNA replication through inhibition of DNA gyrase and topoisomerase IV activities to varying extents depending on the pathogen. In order to maintain antibacterial activity, positions C(3) (unsubstituted carboxyl group) and C(4) (keto group) in 4-quinolone ring should not be altered (Gualerzi et al. 2013).

Quinoline and quinolone antibiotics
After more than 40 years, in 2012, a new antibiotic with quinoline as a core pharmacophore developed to fight multidrug-resistant (MDR) mycobacteria, was granted accelerated approval by the United States Food and Drug Administration, process applicable only to the therapeutics that should fill an unmet medical needs (World Health Organization 2013). Unlike other members of quinolone antibiotics, new antibiotic bedaquiline (Fig. 2) imposes its antimycobacterial activity (MIC 0.06 μg/ml), through interaction with proton pump of the ATP synthase of Mycobacterium tuberculosis (Andries et al. 2005).
Using building-block approach in quest for novel antibacterial agents Dolan and colleagues (Dolan et al. 2016) combined structural moieties, such as functional groups bearing fluorine atoms, quinoline bicycles, saturated N-heterocycles, and thioureas, all of them recognized as motifs in antimicrobial agents, and synthetized a series of thiourea-containing compounds. Derivative 1 (Fig. 3) was the most active compound with MIC90 values of 7.90–10.52 μM, 10.52–15.78 μM, and 17.74 μM for Escherichia coli, Staphylococcus aureus, and methicillin resistant S. aureus (MRSA), respectively, but without any effect on Pseudomonas aeruginosa. Its antibacterial activities were in the line with MIC90 values obtained for vancomycin, well-known antibiotic of the last resort often used to treat drug-resistant infections. The derivative 1 was found to be non-toxic to Galleria mellonella larvae at concentrations of up to 1000 μg/ml. Hence, the quinine-thiourea structure, as found in compound 1, has potential as a new class of non-toxic, anti-MRSA agent.

Quinoline derivatives with antibacterial activity
Synergistic effect of quinoline and pyrazole derivatives (also known for numerous bioactivities including antituberculosis, antiviral, and anti-inflammatory ones) on antimicrobial activity of three diverse series was assessed (El Shehry et al. 2018). Majority of derivatives exhibited antibacterial and antifungal activities, and the most promising derivative proved to be compound 2 (Fig. 3). Its MICs ranged from 0.12 to 0.98 μg/ml for Shigella flexneri, Klebsiella pneumonia, Staphylococcus epidermidis, and Proteus vulgaris, were in the range of ampicillin and gentamycin MICs, thus demonstrating that quinoline derivative bearing pyrazole motifs are interesting scaffolds for development of antimicrobial agents.
Similarly, antibacterial and antifungal activities of a novel quinoline series of Schiff bases and hydrazide derivatives containing moiety, synthesized via condensation of aromatic amines or hydrazines with 2-substituted quinoline-3-carbaldehydes was assessed (Hamama et al. 2018). Antimicrobial activities of quinoline derivatives 3 and 4 (Fig. 3; MIC values 30.6–93.7 μg/ml and 62.5–125 μg/ml, respectively) were even better than activity of ampicillin (MIC values 125–187.5 μg/ml) against E. coli, P. aeruginosa, S. aureus and B. subtilis. The same derivative exhibited similar antifungal activity (MIC values 4.5–7.8 μg/ml and 15.6–23 μg/ml) as clotrimazole (5.6–5.8 μg/ml). The activities of quinoline derivatives 3 and 4 are most likely due to their combination with another quinoline and thiazole moieties, respectively.
Several studies were focused on synthesis of different halogenated quinolines (HQ) and the assessment of bioactivities of the obtained derivatives, namely antibacterial and biofilm eradicating ability. In the first study, the HQ scaffolds or esters of halogenated 8-hydroxyquinoline derivatives were synthetized (Abouelhassan et al. 2014). Five brominated quinolines demonstrated more potent antibacterial activity (MICs 0.2–1.56 μM) against S. aureus and S. epidermidis strains in comparison to nitroxoline (MIC = 12.5–25 μM). In biofilm dispersion assay against MRSA isolate seven quinoline derivatives were very efficient (concentration at which 50% of pre-formed biofilms is disrupted; BDIC50 ≤ 5 μM), with derivative 5 as the most potent (Fig. 4; BDIC50 = 2.06 μM). HQ compounds were less potent against S. epidermidis, although derivatives 6 and 7 exhibited excellent biofilm eradication potential (Fig. 4; BDIC50 = 3.26 μM and BDIC50 = 5.56 μM, respectively). Nitroxoline BDIC50 values were 10.5 μM and 14.2 μM for MRSA and S. epidermidis biofilms, receptively. In the abovementioned study it has been revealed that the C(2)-position of the HQs is of crucial importance for their antibacterial profile against different bacterial species and their potential to eradicate preformed biofilms. Therefore a series of C(2)-substituted analogues of the HQs were synthetized through reductive amination and tested for bioactivity (Basak et al. 2015). These derivatives demonstrated equipotent or slightly reduced antibacterial activities as parent compound 7 (Fig. 4) against clinical isolate MRSA, while products 8 (aniline derived) and 9 (4-bromoaniline derived) were the most potent analogues against methicillin-resistant S. epidermidis (MRSE), fivefold more potent than vancomycin (MIC = 0.78 μM) (Fig. 4). Derivatives 8 through 11 proved to be three- to six-fold more potent (MIC = 0.39–0.78 μM) against vancomycin-resistant Enterococcus faecium (VRE) than 7 (MIC = 2.35 μM) or linezolid (MIC = 3.13 μM). The best potency for MRSA biofilm eradication activities demonstrated 9 with minimum biofilm eradication concentration (MBEC) of 125 μM, being at least 16-fold more potent than vancomycin, daptamycin, or linezolid antibiotics (MBECs>2000 μM). Derivatives 8 (MBEC = 3.0 μM) and 10 (MBEC = 5.9 μM) were the most potent MRSE biofilm eradicators in this series, while 7 and 8 through 11 demonstrated potent biofilm eradication activities against VRE biofilms (MBEC = 1.0–1.5 μM) and as such were equipotent to linezolid, which is used to treat VRE infections. Haemolytic activity of the compounds has varied between 21.3% and less than 4%, as for 8 and 9, indicating that analogues do not eradicate biofilms through the destruction of bacterial membranes, but through other mechanism. In addition, the authors have concluded that the HQs antibacterial activity is realised through a metal(II)-dependent mode of action possibly through the targeting of a metalloprotein critical to bacterial biofilm viability (Basak et al. 2015).

Halogenated quinoline derivatives with antibacterial and antibiofilm activities
In the following study Basak and colleagues (Basak et al. 2016) derivatised broxyquinoline (12, Fig. 4) through multi-step synthetic routes to achieve highly diverse HQ analogues alkylated and aminated at the C(2)-position. Among 39 synthetized derivatives, 13 (Fig. 4) emerged as a highly potent anti-planktonic compound and eradicating agent against MRSA (MIC = 0.39 μM, MBEC = 7.8–93.8 μM), MRSE (MIC = 0.39 μM, MBEC = 5.9 μM) and VRE (MIC = 0.78 μM, MBEC = 1 μM) biofilms, when compared to vancomycin (MRSA: MIC = 0.59 μM, MBEC>2000 μM; MRSE: MIC = 0.78 μM, MBEC>2000 μM; VRE: MIC>100 μM, MBEC = 150 μM). With haemolytic activity <5% and low cytotoxicity, this compound certainly represent a promising lead for further development of useful treatments against persistent infection caused by Gram positive bacteria (Basak et al. 2016).
Abouelhassan and colleagues (Abouelhassan et al. 2015) tested whether phytochemicals, typically considered as a safe, could potentiate antibacterial activity of halogenated derivatives of 8-hydroxyquinoline (14, Fig. 4). They have shown that gallic acid, which itself does not act as antibacterial agent, potentiate several derivatives from authors’ libraries described in previous studies against different S. aureus strains, by lowering their MICs at least fourfold and, in some cases, up to 11,800-fold. On the other hand, gallic acid in combination with antibiotics of different classes, including quinolone, has not potentiated their antibacterial activity, thus indicating that antibacterial mechanisms of the HQs differs from those of conventional antibiotics. It was also demonstrated that gallic acid in combination with compound 7 potentiate MRSA biofilm eradication fourfold.
For further development of 8-hydroxyquinolines derivatives microwave-enhanced Friedländer synthesis protocol was used in order to synthesize the HQ compounds with C(2)- or C(2)- and C(3)- substituted positions (Garrison et al. 2017). Several derivatives exhibited significant antibacterial activity. Compounds 15 and 16 (Fig. 4) with MICs against MRSA isolate of 0.39 μM and 0.59 μM, respectively, were similar to vancomycin (MIC = 0.59 μM) or better than daptamycin (MIC = 4.69 μM) and linezolid (MIC = 3.13 μM). Compounds 15, 17, and 18 exhibited MICs of 0.10 μM, 0.15 μM, and 0.10 μM, respectively, against MRSE, and as such were more efficient than vancomycin (MIC = 0.78 μM), daptamycin (MIC = 12.5 μM) and linezolid (MIC = 3.13 μM). Among all, derivative 18 demonstrated the highest potency against VRE (MIC = 0.3 μM). When tested for eradication potency, compound 16 proved to be one of the most potent biofilm eradicators ever reported against both MRSA (MBEC = 3.9–23.5 μM) and MRSE (MBEC = 1.0 μM), while compounds 15 and 18 exhibited promising biofilm eradication activities against MRSA (MBEC = 31.3 μM) and VRE (MBEC = 1.5 μM), respectively. Since negligible haemolytic activity was observed for those most promising derivatives (<8% at 200 μM) it seems that the HQs could be a promising class of compounds capable of treating biofilm associated infections.
These studies demonstrated great potentials of numerous HQ derivatives as antibacterial agents and biofilm eradicators, so further investigations have been focused on development of the HQ analogues with improved water solubility while maintaining potent biofilm eradication properties against major human pathogens (Basak et al. 2018; Huigens 2018). Conducting diverse synthetic modification at the C(2)-position of the HQ scaffold in order to enhanced water solubility, a new compound that had lower ClogP value (3.44) than parent compound 7; ClogP = 4.19) was developed. This derivative, with morpholine moiety at the C(2)-position 19 (Fig. 4), was the most effective eradicator of MRSE biofilm (MBEC = 2.35 μM) but showed no activity on MRSA biofilms. Therefore, although HQ could play a critical role in the development of next-generation antibacterial therapeutics, at the moment it is still work in progress.
Massoud and colleagues (Massoud et al. 2013) described antibacterial activity of six new Ag(I) compounds with quinoline-derived ligands tested against 15 different MDR bacteria isolated from diabetic foot ulcers and compared them to antibacterial activity of silver sulfadiazine used clinically to prevent infections in burns and wounds. Compound [Ag(8-nitroquinoline)2]NO3 × H2O showed activity similar to topical antibiotic against clinical isolates, being active against all strains and having slightly better average silver efficiency than silver sulfadiazine (5 vs. 6 μg Ag/ml).
Promising candidates for development of new drug are dimeric molecules since they exhibit some unique properties in comparison to corresponding monomer, such as enhanced biological activity. In the last three decades, numerous quinoline and quinolone dimers were assessed for their biological activities, including antimicrobial, and quite recently very detailed review article has been published on the subject (Chu et al. 2019); hence, quinoline/quinolone dimeric molecules are not reviewed here.
It is worth noting that bacterial responses to antibiotics (not just 4-quinolone) are concentration-dependent. At high concentrations, antibiotics exhibit their antimicrobial activities on susceptible cells, but at subinhibitory concentrations they can induce diverse biological responses in bacteria. At these non-lethal concentrations, bacteria may perceive antibiotics as extracellular signals and thus trigger different cellular responses, which may include an altered antibiotic resistance/tolerance profile (Bernier and Surette 2013). Bacterial responses to subinhibitory concentrations of antibiotics and mechanisms of their responses vary depending on antibiotic and species, thus we advise readers for literature search on this topic.
3 Antifungal Activity
Fungal infections have become an everyday problem, but also a serious threat to human health due to the development of resistant strains causing weak and unsatisfactory therapeutic response to known antifungals. Although the collection of antifungal drugs is broad, the most commonly used agents have major drawbacks such as side effects and high level of toxicity. Together with the emerging resistance, these drawbacks restrict the number of medicines which can be used to treat such infections. Thus, there is a clear need for development of novel more effective antifungal agents with a broad-spectrum activity, better pharmacokinetic profile and low toxicity. As eukaryotic organisms, fungi share numerous conserved pathways with their human hosts, therefore only a few drug targets can be exploited to selectively kill these pathogens. Enzymes involved in the synthesis of cell wall polysaccharides are one of the most popular targets for development of antifungal drugs.
A quinoline scaffold can be found in many classes of biologically active compounds which are used as antifungals (Musiol et al. 2010). The unmodified quinoline exhibits relatively high activity against some fungal strains at nontoxic concentrations, which is a clear advantage in the context of designing of novel antifungal drugs. The fungistatic activity of 8-hydroxyquinoline (14, Fig. 4) and its metal complexes has been known since the early 1920s and these compounds are still broadly used in healthcare. Relatively simple quinoline modifications have been widely investigated in order to obtain better antifungal activity and some of these compounds are still in use. Previous efforts in the development of quinoline-based antifungals have been extensively reviewed earlier (Musiol et al. 2010), thus here we focus on the achievements from last 10 years.
A series of small HQ molecules exhibiting a potent antifungal activity against Candida albicans and Cryptococcus neoformans through intracellular mode of action have been synthetized (Zuo et al. 2016). HQ analogues with bromine and chlorine halogens (7, 20) (Figs. 4 and 5) inhibited C. albicans growth with MIC of 100 nM which is four to eight times more potent than the best-known antifungal agents amphotericin B and itraconazole, while several analogues inhibited C. neoformans at MICs of 50–780 nM. Importantly, the HQ analogues could eradicate mature C. albicans and C. neoformans biofilms with MBEC of 6.25–62.5 μM. The same HQ analogues also showed a range of antibacterial activities inhibiting their planktonic and biofilm forms, suggesting that these compounds could be a promising scaffold for the development of therapeutics against mixed-species and/or biofilm-associated infections. The biological activity of the HQs depended on the nature of the groups attached to the C(5)- and C(7)-positions of quinoline ring. The presence of chlorine, bromine or iodine atoms in the C(5)- or C(7)- positions of the compounds was related to strong antifungal activity, which was not observed with the fluorinated compounds. Although the observed antifungal activity can be attributed to the presence of halogens, those substituents can also cause higher toxicity of the compounds. Bromoquinol (broxyquinoline; 12, Fig. 4) is identified as one of the most potent compounds with antifungal activity during screening of 40,000 drug-like molecules from diverse chemical compound libraries against Aspergillus fumigatus (Ben Yaakov et al. 2017). Its activity was blocked by iron, copper or zinc supplementation, which suggested that it interferes with the utilization of these metals. It was demonstrated that bromoquinol induces oxidative stress and apoptosis in A. fumigatus. Bromoquinol significantly reduced mortality rates of Galleria mellonella infected with A. fumigatus, but was ineffective in a murine model of infection.

Quinoline derivatives with antifungal activity
Clioquinol (21, Fig. 5) is a dihalogenated 8-hydroxyquinoline and antiseptic drug effective against multidrug resistant Candida (Pippi et al. 2018). It blocks hyphal development thus preventing biofilm formation. Clioquinol also reduces the metabolic activity of sessile Candida but the susceptibility was lower compared to planktonic cells (0.031–0.5 μg/ml required to inhibit 50% planktonic cells and 4–16 μg/ml to inhibit 50% preformed biofilms). Clioquinol, as well as other 8-hydroxyquinoline derivatives such as 8-hydroxy-5-quinolinesulfonic acid and 8-hydroxy-7-iodo-5-quinolinesulfonic acid effectively inhibited growth of Candida spp., Microsporum spp. and Trichophyton spp. (Pippi et al. 2017) with MIC values ranging from 0.031–2 μg/ml, 1–512 μg/ml, and 2–1024 μg/ml, respectively.
Most of the known quinoline drugs have a side chain on the C(4)- or C(8)-position of the quinoline scaffold. However, other positions also provide opportunities for the design of novel bioactive compounds. Two subseries of 2,6-disubstituted quinolines, consisting of 6-amide and 6-urea derivatives exhibit fungicidal activity against C. albicans with minimal fungicidal concentration (MFC) values lower than 15 μM (Delattin et al. 2012). The 6-amide derivatives displayed the highest fungicidal activity against C. albicans and a bit lower activity against C. glabrata (MFC < 50 μM) (22 as the most potent, Fig. 5). Some of the 6-amide derivatives and the 6-urea derivatives could also eradicate C. albicans biofilms by inducing accumulation of endogenous reactive oxygen species.
Styrylquinolines (23–26, Fig. 5) are a novel group of quinoline drugs that have p53-independent antiproliferative activity and antiviral properties. They were also found to have antifungal activity and to decrease the activity of ABC multidrug transporters in C. albicans. They also show synergistic activity with first-line drug fluconazole (Szczepaniak et al. 2017).
Novel phenylcarbamate derivatives with tetrahydroquinoline were designed as inhibitors of HIV-1 reverse transcriptase (RT) also showed antifungal activity against C. albicans and A. niger (Chander et al. 2016). The most active derivative (27, Fig. 5) displayed antifungal activity against C. albicans almost comparable to fluconazole with MIC value of 8 μg/ml.
The 7-chloroquinoline moiety was extensively studied mainly because of its antimalarial properties. Duval and co-workers synthetized 7-chloroquinolin-4-yl arylhydrazone derivatives and addressed their antifungal activity against several Candida species and two yeasts species of Rhodotorula (Duval et al. 2011). The active derivatives showed MIC and MFC values in the range of 25 μg/ml and 50 μg/ml, which was the activity comparable with the fluconazole. Four of these hydrazones potently inhibited enzymatic processes in C. albicans at sub-antifungal concentrations, showing enzymatic repression of phospholipase and aspartyl proteases, which are the most frequent enzymes produced by C. albicans (de Azambuja Carvalho et al. 2016). Notably, the compounds exhibited low cytotoxicity against mouse fibroblasts (NIH/3 T3 cell line) at sub-antifungal concentrations.
On the way to N-functionalized 3-, 5-, 6- and 8-aminoquinolines, were obtained and their antifungal activity was assessed against C. albicans, Rhodotorula bogoriensis and A. flavus (Vandekerckhove et al. 2015). Several compounds displayed antifungal activity against all three microorganisms tested, while derivative 28 (Fig. 5) was selected as the most potent one, exhibiting activity toward the A. flavus strain comparable to amphotericin B.
Evaluation of antifungal activity of N-alkyl tetra and decahydroisoquinolines showed that the activity of these compounds depends on the length of the alkyl chain, with an optimum of about 10–12 carbon atoms, whereas longer or shorter alkyl chains lead to a decrease or complete loss of activity (Krauss et al. 2014). The trans-decahydroisoquinolines showed high antifungal activity, comparable to the reference antifungal drug clotrimazole with MIC values against C. glabrata between 2.4 and 25 μg/ml. The decahydroisoquinoline 29 (Fig. 5) was found to inhibit the enzyme D14-reductase in C. glabrata, while an additional inhibition of the downstream enzyme D8,7-isomerase was not excluded. The same group of researchers synthetized a series of (±)-trans-N-alkylperhydroquinolines, which also showed high antifungal activity with MIC values against C. glabrata between 5 and 50 μg/ml (Krauss et al. 2015). The activity of the most potent derivative was comparable to drug clotrimazole. The maximum of activity was found with the derivatives having C10 or C12 alkyl chains, while shorter alkyl chains led to a decrease in activity, as already found for other N-alkyl heterocycles. In comparison to N-alkyl perhydroisoquinolines, perhydroquinoline compounds showed similar antifungal activity, but higher cytotoxicity against a human cell line. Both perhydroquinolines and perhydroisoquinolines targeted the same enzyme in ergosterol biosynthesis (Δ8,7-isomerase), but the latter chemotype showed higher selectivity.
The lipid-like choline and colamine analogues based on 1,2,3,4- tetrahydro(iso)quinoline have been developed as a part of compound libraries of lipid-like systems that combine two fragments: hydrophilic – 1,2,3,4-tetrahydro(iso)quinolinium pharmacophore system, and lipophilic – long chain alkyl substituent (30, Fig. 5), that helps the molecule in its passive transport across lipophilic barriers and plasma membranes in vivo (Zablotskaya et al. 2017). Tetrahydroisoquinoline derivatives and compounds possessing substituents with chain length of 10 or 11 carbon atoms showed the strongest antifungal activities with MIC values between 8 and 64 μg/ml. Tetrahydroisoquinolinium heptyl and decyl derivatives 30 exhibited high antifungal action as compared with corresponding tetrahydroquinolinium analogues. These compounds exhibited stronger antifungal activity than fluconazole.
Quinoline based 1,2,3 triazoles emerged as a group of very potent antifungal compounds with IC50 values (half maximal inhibitory concentration) for the most active derivative 31 (Fig. 5) of 0.044 μg/ml against C. albicans, 12.02 μg/ml against C. glabrata, and 3.60 μg/ml against C. tropicalis (Irfan et al. 2015; Irfan et al. 2017). Moreover, these antifungal concentrations were not cytotoxic. The presence of these compounds affected the secretion of extracellular hydrolytic enzymes (proteinases and phospholipases). The antifungal target for these triazoles was plasma membrane as suggested by altered cell membrane, reduced plasma membrane H+ATPase activity, and significant inhibition in ergosterol biosynthesis in the presence of these compounds.
A series of 2,5 and 1,5-regioisomers of tetrazole with quinoline and benzylthio substituents, were synthesized (Shaikh et al. 2017). The designed compounds consist of a quinoline nucleus, which serves as pharmacophore and the non-heterocyclic part such as phenyl group separated by a tetrazole bridge. The antifungal activity of synthesized compounds was tested against A. fumigatus and C. albicans strains and the obtained MIC values for the most active compounds 32 and 33 (the 2,5- and 1,5-isomer with fluoro substituent at the C(6)-position) (Fig. 5) were between 2.5 and 25 μg/ml. All the derivatives having halogens as substituents showed better activity than other derivatives. The active compounds came to be inhibitors of dihydrofolate reductase and N-myristoyl transferase, and exhibited no cytotoxic effects.
New hybrid analogues containing 7-chloro-4-aminoquinoline and 2-pyrazoline N-heterocyclic fragments were synthesized and evaluated for antifungal activity against C. albicans and C. neoformans (Montoya et al. 2016). The compounds displayed stronger activity against C. neoformans with the most active derivative showing MIC50 values of 15.5 and < 3.9 μg/ml against C. albicans and C. neoformans, respectively.
In an attempt for development of novel antimicrobial agents, three series of quinoline derivatives bearing pyrazole moiety were synthesized (El Shehry et al. 2018). The most active compound 2 (Fig. 3) showed fourfold potency of amphotericin B in inhibiting the growth of A. clavatus (MIC 0.49 μg/ml) and C. albicans (MIC 0.12 μg/ml), respectively. The same compound showed equipotent activity to amphotericin B in inhibiting the growth of A. fumigatus (MIC 0.98 μg/ml).
New quinoline derivatives (Khan et al. 2019) bearing vinyl benzylidene imine with substituted aniline at the C(2)-position showed antibiofilm and antifungal activity. The antibiofilm activity of compounds 34 and 35 (Fig. 5) against C. albicans (IC50 values 66.2 and 51.2 μM, respectively) were similar to activity of fluconazole (IC50 = 40.0 μM). Compound 34 exhibited slightly lower antifungal activity (MIC = 94.2 μg/ml) than fluconazole (MIC = 50.0 μg/ml).
Benzimidazole fused pyrrolo[3,4-b] quinoline (Villa et al. 2019) compound 36 (Fig. 5) exhibited potent antifungal activity against C. albicans, C. parapsilosis, C. tropicalis, C. glabrata, and C. neoformans with the MIC values ranging from 0.0625 to 2 μg/ml. Fungal strains that are resistant to fluconazole were also inhibited by 36 with the MIC ranging from 0.25 to 0.5 μg/ml. These compounds also showed antibiofilm activities without apparent toxicity to mammalian cells. A fluorescent benzimidazo[1,2-α] quinoline was identified within a set of nine benzimidazole derivatives as bifunctional Candida spp. biofilm detector and eradicator (de Souza et al. 2016). Compound 37 (Fig. 5) was the most active derivative showing antifungal activity against different Candida strains including C. tropicalis, C. albicans, and C. parapsilosis with MIC values in the range of 4 μg/ml against C. albicans and 32 μg/ml against the other isolates. Spraying of 37 over Candida sp. biofilm contaminated surface enabled detection of the biofilms under UV light. At the same time, 37 showed the potential to eradicate the detected biofilms. Benzimidazole 37 thus has a good potential to enable the usage of disinfected medical and surgical instruments in clinical and surgical procedures contributing to increased safety for patients.
As described, quinoline scaffold can be considered a valuable pharmacophore to be used for a simple synthesis and development of novel antifungal agents with the superior efficacy to commonly used antifungal drugs. The structural diversity of synthetized compounds can provide high and selective activity achieved through different modes of action, as well as low cytotoxicity on human cells.
4 Anti-virulence Activity
P. aeruginosa produces more than 50 different alkylquinolines (Deziel et al. 2004) of which 2-heptyl-3-hydroxy-4-quinolone (PQS) and its direct precursor 2-heptyl-4-hydroxyquinoline (HHQ) are PQS pathway autoinducers regulating production of several virulence factors such as pyocyanin and hydrogen cyanide, as well as biofilm formation. Alkylquinolines signalling has so far been detected in P. aeruginosa and certain Burkholderia and Alteromonas species (Diggle et al. 2006; Vial et al. 2008).
The majority of bacterial QS inhibitors (QSIs) have been designed based on the core structures of autoinducers. Quinolone-based compounds are expected to exhibit species-specific QSI activity against Pseudomonas, some Burkholderia and Alteromonas species.
Following a ligand-based drug design approach, a set of HHQ and PQS analogs were synthesized by variyng the side chain and introducing substituents into the benzene moiety of the quinolone molecule. PQS analogues with hydroxy group showed PqsR agonistic activity in P. aeruginosa, and this effect was circumvented by synthetizing a series of HHQ analogs (Lu et al. 2012). Highly potent PQS inhibitors have been synthetized by introducing modifications in benzene ring and 3-alkyl substituents achieving almost complete PqsR inhibition at 5–10 μM concentrations (38, Fig. 6) and reduction of virulence factor pyocyanin production by 74% at 3 μM. Introduction of polar groups at the C(3)-position of derivative 39 resulted in novel compounds with enhanced anti-virulence activity, with the most active derivative 40 inhibiting pyocyanin production at IC50 of 3.8 μM (Lu et al. 2014a). The QSI activities of these compounds were confirmed in in vivo experiments (Lu et al. 2014b).

Quinoline and quinolone derivatives with anti-virulence activity
A 4-quinolone isostere quinazoline was designed as highly potent competitive inhibitor of PqsR with derivatives 41–43 being the most active ones (Fig. 6) (Ilangovan et al. 2013). A simple isostere replacement (OH for NH2) changed quinolone isostere quinazoline activity from potent agonistic to potent antagonistic. Derivative 41 inhibited pyocyanin production, virulence genes expression, aminoquinoline biosynthesis, and biofilm development.
A series of 4-aminoquinoline derivatives have been described as dual P. falciparum and botulinum neurotoxin inhibitors (Opsenica et al. 2012; Videnovic et al. 2014; Solaja et al. 2008) as well as ligands in complexes with anticancer activities (Nikolić et al. 2015). Following the drug repurposing approach several 4-aminoquinoline-based molecules with QSI activities have been further developed. Within a series of 31 derivatives of antiprotozoal 4-aminoquinolines compounds with PqsR antagonistic activity were identified inhibiting receptor activity by 70–85% at 10 μM (Soukarieh et al. 2018). Derivatives 44 and 45 inhibited pyocyanin production, while compound 44, inhibited also HHQ, PQS, and 2-heptyl-4-hydroxyquinoline N-oxide (HQNO) production, and biofilm formation (Fig. 6). The efficacy of these compounds depended on the strains of P. aeruginosa (P. aeruginosa PA14 vs. P. aeruginosa PAO1).
Recently, PQS inhibitory activity of long-chain 4-aminoquinoline derivatives (Fig. 6) has been reported (Aleksić et al. 2017). Within this series, derivatives 46 and 47 with C12 alkyl chain at C(4)-position and chlorine at C(7)-position were the most active anti-virulence compounds inhibiting biofilm formation in P. aeruginosa with IC50 values 63 μM and 69 μM, respectively, and pyocyanin production with IC50 values of 40 and 2.5 μM, respectively. The same compounds also inhibited biofilm formation in Serratia marcescens with the same efficacy.
PQS, HHQ and some of the 4-quinolone derivatives can modulate interspecies and interkingdom interactions (Fernández-Piñar et al. 2011; Reen et al. 2011, 2012, 2015), which makes this chemotype even more attractive for further development of QSI.
Long chain amide derivatives of 2-amino-4-quinolone showed ability to inhibit biofilm formation in P. aeruginosa and S. aureus (Espinosa-Valdes et al. 2019). The most active compounds were those with an alkyl chain of more than 12 carbon atoms and in general they were more active against biofilm formation in P. aeruginosa than in S. aureus. The most active compounds 48 and 49 (Fig. 7) also inhibited pyocyanin production between 62.6 and 68.2% at concentration of 20 μM. None of the tested compounds affected bacterial growth suggesting that observed activity of the derivatives could be through modulation of QS.

Quinoline and quinolone derivatives with antibiofilm activity
A library of quinoline amino alcohol derivatives have been evaluated as biofilm inhibitors against the Gram-negative pathogen Vibrio cholerae (Leon et al. 2015). The most potent compound was meta-substitued pentafluorosulfonyl derivative 50 (Fig. 7) with BIFC50 of 4.4 μM and BDIC50 value of 7.4 μM. The antibiofilm activity of this compound was observed at concentration that did not affect bacterial growth (MIC = 78.1 μM) suggesting that mode of antibiofilm action was different from its bactericidal activity. This compound also exhibited cytotoxicity to HeLa cells with IC50 value of 7.27 μM, thus it can be considered as active component for functionalization of materials for medical use rather than for systemic application.
Chloroquine (CQ, Fig. 1) has been tested in combination with antifungal drugs fluconazole, voriconazole, amphotericin B, and caspofungin for ability to inhibit C. albicans planktonic forms and biofilms (Shinde et al. 2013). No synergistic activity of CQ and antifungals has been observed against planktonic growth. Alone fluconazole and voriconazole didn’t affect formation of C. albicans biofilms even at high concentrations, but in a combination with 250 μg/ml CQ, antibiofilm activity was observed at fluconazole concentration of 4 μg/ml and voriconazole concentration of 0.25 μg/ml. Combinations of CQ with amphotericin B, or CQ with caspofungin showed no synergistic activities against biofilm formation. Antifungal drugs fluconazole and voriconazole showed no effect on mature C. albicans biofilms, while amphotericin B and caspofungin inhibited mature biofilms but high concentrations were required (0.5 and 1 μg/ml, respectively). In the presence of 250 μg/ml CQ, mature biofilms were disrupted as a result of synergistic activity with fluconazole and voriconazole, while combination with amphotericin B and caspofungin showed low effect against mature biofilms. CQ can be developed as a promising partner molecule of antifungal drugs for combination therapy against C. albicans biofilm.
5 Antiviral Activity
Viral infections have concerning effects on the health of human populations worldwide. The successful treatments of these infections require highly effective antiviral drugs, which together with the growing spread of resistance require new therapeutic agents. A lot of efforts has been done on antiviral properties of quinolines and quinolones and their structural analogues against human immunodeficiency virus (HIV), but their antiviral activity was also demonstrated against human cytomegalovirus (HCMV), SARS corona virus, Zika virus, Chikungunya virus, hepatitis C virus (HCV), and Ebola virus (Luthra et al. 2018; Plantone and Koudriavtseva 2018; Al-Bari 2015; Delvecchio et al. 2016; Barbosa-Lima et al. 2017; Loregian et al. 2010). Chloroquine shows antiviral effects by inhibiting pH-dependent steps of the replication of several viruses including members of the flaviviruses, retroviruses, and coronaviruses (Savarino et al. 2003).
Human immunodeficiency virus type 1 (HIV-1) integrase (IN), HIV-1 RT and HIV-1 protease are the essential enzymes for retroviral replication, and represent the important targets for interrupting the viral replication cycle and thus development of novel antiviral therapeutics. Reverse transcriptase and protease inhibitors, which have been used in retroviral therapy, cannot achieve complete suppression and there is a risk for resistance development. The best-studied effects of CQ are those against HIV replication, and its analogues have been used in clinical trials as investigational anti-retroviral agents in humans with HIV-1/AIDS (Al-Bari 2015). Approved 51 (Fig. 8), later called elvitegravir, is the first quinolone-based anti-HIV drug, exhibiting potent inhibitory activity against integrase-catalyzed DNA strand transfer (Sato et al. 2006). Recently, a series of quinolone-3-carboxylic acids have been synthetized as HIV-1 integrase inhibitors featuring a fluorine atom at C(5) position (He et al. 2013). The most active compound 52 (Fig. 8) exhibited activity against both wild-type and the mutant virus, with an EC50 value of 0.032 and 0.082 μΜ, respectively. Another series of quinolone-3-carboxylic acids have been synthesized by introducing different hydrophobic groups at N(1), C(2), C(7), and C(8) positions (Hajimahdi et al. 2016). Most of the compounds of this group showed anti-HIV activity without cytotoxicity at concentration of 100 μM. The most active compound 53 (Fig. 8) showed anti-HIV activity with an inhibition rate of 84%. Docking study revealed that the anti-HIV activity of this group of compounds might involve a metal chelating mechanism. Several other quinoline analogues have been synthetized, which exhibit low micromolar inhibitory potency against HIV-1 IN as recently reviewed by Wadhwa et al. (Wadhwa et al. 2018). A series of multi-substituted quinolines, with the focus on the substitution pattern of the 4-phenyl moiety and incorporation of heteroaromatic or polycyclic substituents at the C(4)-position, were prepared and examined for their ability to trigger HIV-1 IN multimerisation via binding to an allosteric site (Jentsch et al. 2018). 4-(4-Chlorophenyl)quinoline and 2,3-benzo[b][1,4]dioxine showed highest potency with EC50 values (half maximal eradication concentration) of 0.1 and 0.08 μM, respectively. A series of isoquinoline analogues with allosteric IN inhibitory activity has been recently reported (Wilson et al. 2019). Compound 54 (Fig. 8) was highly potent with EC50 of 1.1 μM.

Quinoline and quinolone derivatives with anti-HIV activities
Protein kinase CK2 plays a role in the stimulation of RT and protease. Substituted pyrimido[4,5-c]quinoline ATP competitive inhibitors of protein kinase CK2 showed a good antiviral activity with the compound 55 (Fig. 8) having IC50 values as low as 80 nM against HIV-1 viruses with an excellent therapeutic index (Pierre et al. 2011).
Using the ligand based drug design approach the 1-(4-chlorophenyl)-2-(3,4-dihydroquinolin-1(2H)-yl)ethyl phenylcarbamate derivatives were designed as inhibitors of HIV-1 RT (Chander et al. 2016). The most active compounds 56 and 57 (Fig. 8) inhibited the RT activity with IC50 8.12 and 5.42 μM, respectively. Cytotoxicity and anti-HIV activity of these compounds were evaluated on T lymphocytes. Both compounds exhibited potent anti-HIV activity with EC50 values 11.10 and 2.76 μg/ml, for 56 and 57 respectively, while showing a very good safety index.
A molecular-hybrid approach is a powerful tool in the design of new molecules with improved affinity and efficacy and as such was applied for development of novel antiviral agents involving quinoline scaffold. The fluoroquinolone-isatin hybrids 58 and 59 (Fig. 8) showed inhibition on the replication of HIV-1 in human cells with EC50 of 11.3 and 13.9 μg/ml, and the selectivity index values were > 5. The promising antiviral properties of isatin and fluoroquinolone hybrids have been extensively discussed recently (Xu et al. 2019). Further, a series of diarylpyrimidine–quinolone hybrids was synthesized and evaluated against both wt HIV-1 and mutant viral strains (Mao et al. 2015). The most active hybrid 60 (Fig. 8) displayed an EC50 value of 0.28 ± 0.07 μM against HIV-1 by targeting RT.
HIV infects multiple cells in human body, but its replication starts after infecting CD4 lymphocytes (T-cell or CD4- cell). The treatment of proliferating CD4 T-cells with antiproliferative activity agents reduces the ability of these cells to support HIV replication. Combination therapy involving cytostatic compounds such as hydroxychloroquine or hydroxyurea with proven anti-retroviral drugs as didanosine decreases viral replication and increases the CD4 count in antiretroviral-naive HIV patients thus helping to control the infection. A novel di-gold(I) complex of ferrocene–quinoline (61, Fig. 8) was investigated for cytostatic behaviour as well as antiviral activity (Gama et al. 2016). Di-gold quinoline derivative showed inhibition of virus infectivity by 83% at concentration of 10 μg/ml and cytostatic activity with significant S and G2/M phase cell arrest, which make it a good candidate for use in HIV-1 infection as a virostatic agent.
Although quinoline/quinolone derivatives showed promising antiviral effects in in vitro studies, published clinical studies evaluating the effects of chloroquine/hydroxychloroquine administration, alone or in combination with other drugs, in HIV infected subjects reported different effects in terms of immune activation, viral load, and CD4 counts (Savarino and Shytaj 2015). The outcome of the studies depended on applied doses, duration of the treatments or the drug exposure, or on the individual differences in drug metabolism and distribution.
6-Aminoquinolones with amino group at the C(6) position of the bicyclic quinolone ring system, specifically inhibit HIV replication (Cecchetti et al. 2000). On the other side, 62 (Fig. 9), which is characterized by a cyclopropyl group at the N(1) position and a 4-(2-pyridyl)-1-piperazine moiety at the C(7) position showed specific activity against HCMV by inhibiting transactivation activity of immediate-early 2 (IE2), a multifunctional factor essential for viral replication (Loregian et al. 2010; Mercorelli et al. 2014). The target of this compound is different from that of the anti-HCMV drugs currently approved for clinical use. 62 demonstrated activity against laboratory strains of HCMV but also against clinical isolates and virus strains resistant to clinically relevant anti-HCMV agents. Importantly, this compound does not show cross-resistance with ganciclovir, acyclovir, cidofovir, and foscarnet, which is important advantage for its potential clinical use, since drug-resistant HCMV strains often emerge after long-term treatment with antiherpetic drugs.

Quinoline and quinolone derivatives with anti-HCMV and anti-HCV activities
Most of the current clinical compounds and approved drugs against HCV target non-structural (NS) proteins NS3, NS5A, and NS5B. 3-Heterocyclyl quinolones have been described as a series of allosteric-site (NNI-2) inhibitors of the HCV NS5B polymerase (Kumar et al. 2012). The most potent compounds in this series were 63 and 64 (Fig. 9), with EC50 below 250 nM. Derivatives of 1H -quinazolin-4-one were reported as allosteric HCV NS5B thumb pocket 2 (TP-2) inhibitors (Hucke et al. 2014) with picomolar antiviral potency in genotype (gt) 1a and 1b (EC50 = 120 and 110 pM, respectively) and with EC50 ≤ 80 nM against gt 2−6.
A group of 6-aminoquinolone derivatives demonstrated inhibitory activity against NS5B polymerase (Manfroni et al. 2014). The most active compound of this group was 6-amino-7-[4-(2-pyridinyl)-1-piperazinyl]quinolone derivative 65 (Fig. 9) with an IC50 value of 0.069 μM. Its selective antiviral effect was EC50 3.03 μM, while cytotoxicity was not observed.
Derivatives of 2-oxadiazoloquinoline were designed to inhibit HCV NS4B, which is integral membrane protein believed to act primarily as an endoplasmic reticulum-localized scaffold for the assembly of the replicase complexes needed for HCV RNA replication (Phillips et al. 2014). Compound 66 (Fig. 9) from this series showed exceptional activity against different replicons with EC50 values ranging between 0.08 nM and 3.1 nM.
The compound 67 (RG7109, Fig. 9), was synthetized as a result of an effort to develop a combination therapy against HCV consisting of only direct-antiviral agents. The standard therapy of HCV included pegylated interferon α (Peg-IFN) and ribavirin (RBV), however this therapy was effective against gt 2 or 3, but gt 1 did not respond well, with SVR (absence of detectable HCV RNA in blood serum for 24 weeks after treatment withdrawal) rates of <50% even after 48 weeks long therapy. Upon FDA approval of HCV protease inhibitors the treatment of gt 1 included Peg-IFN and RBV and a protease inhibitor, which improved the SVR rates up to 90% with gt 1 naïve patients. Interferon-associated side effects limit treatment success in a large number of patients. With the aim to develop a combination therapy consisting of only direct-antiviral agents, Talamas and co-workers identified a bicyclic template with potent activity against the NS5B polymerase containing 3,5,6,8-tetrasubstituted quinoline core 67, which was selected for advancement to clinical development (Talamas et al. 2014).
Isoquionoline-containing compound called asunaprevir (68) (Fig. 9), a tripeptidic acylsulfonamide inhibitor of the NS3/4A enzyme developed by Scola and co-workers (Scola et al. 2014a; b) went through phase III clinical trials for efficient treatment of HCV infection.
6 Derivatives with Anti-parasitic Activity
Parasitic diseases caused by protozoan species represent one of the major health challenges worldwide, affecting millions of people of all ages and social classes. The most threatened are children, pregnant women, immunocompromised individuals, and people living in developing countries. Consequences of illness burden not only patients and their families, their financial status, but also local and global economy. Taking into account the limitations of currently available drugs, the only proven strategy to fight against these diseases is development of the new therapeutics under clearly defined criteria (Nwaka and Hudson 2006).
6.1 Antimalarial Activity
According to WHO annual report, in 2017 approximately 219 million cases of malaria occurred worldwide, resulting in 435,000 deaths (World Health Organisation 2019). Most cases were registered in African regions, followed by South-East Asia and Eastern Mediterranean regions. Malaria is caused by five Plasmodium species with Plasmodium falciparum as the most dangerous one, since it causes cerebral malaria and is the major cause of death.
Although many different pharmacophores have been developed during past decades, quinoline derivatives (Figs. 1 and 10) are still a backbone for development of novel antimalarial drugs (Barnett and Guy 2014). Actually, one of the most successful antimalarial therapeutics, a well-known quinoline derivative chloroquine, has been used for decades to threat this illness. The main mechanisms of action of CQ and its congeners (Figs. 1 and 10) are inhibition of hemozoin polymerization and release of a free toxic haem (Egan and Marques 1999). Among diverse antimalarials used in clinics CQ, considered as safe even for children and pregnant women, is the easiest to synthesize. Unfortunately, malaria parasite has developed resistance to most of antimalarial drugs including CQ. Structural modifications of this chemotype, which would include altering the nature of the quinoline core and side chain at C(4)-position, or synthesis of organic (Boudhar et al. 2016) or organo-metallic hybrids (Salas et al. 2013) present a good direction for obtaining novel derivatives with better activity against CQ resistant strains (Manohar et al. 2013).

The most-successful quinoline derived antimalarial drugs developed in the twentieth century
Introducing of steroidal structure fragment, various thiophen, benzothiophen and adamantyl groups, numerous derivatives of 4-aminoquinoline have been synthetized and tested for their antimalarial activity. Steroidal compounds reported by (Videnovic et al. 2014), which were developed based on the results of the previous study (Solaja et al. 2008), showed low nanomolar IC50 (50% inhibition of β-haematin formation) activities against CQ resistant and MDR P. falciparum strains. Among them derivatives 69–71 were the most active in the series (Fig. 11), being more active than mefloquine (Fig. 10) against MDR strains. High potency of these derivatives was demonstrated with three times better inhibition of hemozoin production than CQ. Derivatives bearing electron-rich thiophen or benzothiophen groups were designed to enhance activity against CQ resistant and MDR strains of P. falciparum, by additional π-interactions with hem (Opsenica et al. 2015). 4-Fluoro derivatives, 72 and 73, were the most active in in vivo experiments showing curative activity (Fig. 11). Addition of aromatic ring, elongation of diaminoalkyl linker, and replacement of F-atom with CN-group (74–76, Fig. 11) enhance active against CQ resistant strain (Konstantinovic et al. 2017). The most active benzothiophene derivate 77 cured mice infected with P. berghei when dosed orally (Fig. 11). Derivatives with adamantyl-substituent, 78–81 (Fig. 11) (Terzic et al. 2016) were designed to increase lipophilicity of quinoline core and to provide amphiphilic structure (Wanka et al. 2013) and metabolic stability. These compounds demonstrated in vitro antimalarial activity at low nanomolar concentrations against CQ resistant and MDR strains, presenting the highest potency up today. This modification increased microsomal metabolic stability over 60 min. Introducing the F-atom at C(3)-position attenuated in vitro activity against erythrocyte form of P. falciparum. However, five F-C(3) derivatives showed excellent in vitro and in vivo activity against liver stage of P. berghei, with 78 as the most active derivative with IC50 = 0.1 μM.

Quinoline derivatives with antimalarial activity
Changing the character of 4-amino group, or replacing it with different functional group, alters electronic properties of quinoline core. One of the such example is derivative 82 (Fig. 12) who contained carbonyl group attached to C(4) (Baragana et al. 2015). Compound 82 showed potent activity against multiple stages of the Plasmodium parasite life-cycle and exhibited excellent pharmacokinetic properties and an acceptable safety profile. This molecule having EC90 = 2.4 nM against MDR P. falciparum strain reduced parasitemia for 90% in mice infected with P. berghei after a single oral dose. Translation elongation factor 2, a protein responsible for the GTP-dependent translocation of the ribosome along mRNA and as such required for protein synthesis at all stages of the parasite life cycle was identified as the target. Therefore, with its potent activity against multiple stages of the parasite life cycle, high metabolically stability, novel mode of action, and excellent drug-like properties, 82 meets the key criteria for new antimalarial drug.

Keto- and amidino-quinolines with antimalarial activity
Few derivatives of yet another series of 4-amidinoquinolines, tested against CQ sensitive and CQ resistant strains, exhibited IC50 values less than 10 nM (Korotchenko et al. 2015). Introduction of the third substituent on the amidine-group provided both good activity and enhanced metabolic stability. Derivatives 83 and 84 (Fig. 12) showed activity with IC50 = 1.9–34 nM. However, those promising results were diminished with high resistant index (activity against resistant vs. activity against sensitive strains) ratio.
Amodiaquine (Fig. 10), well known and intensely studied drug, could replace CQ in antimalarial therapy (Olliaro and Mussano 2003). It is successful in treatment of CQ resistant strains but unfortunately when used prophylactically toxic metabolites are produced (Schlitzer 2007). Therefore, a series of N-pyridyl, N-1H-benzo[d]imidazole, N-benzo[d]oxazole, and N-benzo[d]tiazole were tested (Ongarora et al. 2015). Three derivatives (85–87, Fig. 13) showed in vivo efficacies in dose-depend manner with 99.5% parasitemia reduction among which derivative 85 was the most successful. 8-Aminoquinolines primaquine and tefenoquine (Fig. 10) are also known and promising antimalarial compounds (Waters 2011). Shared common disadvantage of these compounds is biotransformation of terminal amino-groups into aldehydes, which are toxic. To prevent such transformation and to maintain good antimalarial activity a new derivatives that contained aminoxy and oxime-groups were synthetized (Leven et al. 2019). The compounds were evaluated in vitro against asexual blood stages, liver stages, and sexual stages of P. falciparum. 8-Aminoquinolines bearing a 2-alkoxy and a 5-phenoxy substituent (88–92, Fig. 13) were the most active compounds, with the IC50 values in the range 0.95–1.3 μM, comparable to primaquine and tefenoquine. Derivatives 93 and 94 showed significant activity against asexual blood stage of parasites (Fig. 13). However, identification and toxic profile of potential metabolites remained inexplicable.

4-Amino and 8-aminoquinolines with antimalarial activity
Synthesis of hybrid compounds that contained two or more pharmacophore is a new promising approach in development of more successful antimalarials. It was expected that those hybrid-compounds cover distinct mechanism of actions, or affect different phase of plasmodium life cycle; hence, synergistic effect was anticipated.
1,2,4,5-Tetraoxane-8-aminoquinoline hybrids with various aryl or heteroaryl group as substituents at the metabolically labile C(5)-position of the 8-aminoquinoline moiety were tested in vitro as dual-stage antimalarial agents against P. falciparum CQ resistant strain (blood stage) and P. berghei (liver stage) (Capela et al. 2018). Derivatives 95–98 (Fig. 14) have improved metabolic stability, and proved to be efficient as dual-stage antiplasmodial agents with good metabolic stability, inhibiting the development of intra-erythrocyte forms of P. falciparum (EC50 = 15–17 nM) and P. berghei liver stage (EC50 = 1.11 μM).

Quinoline hybrides with antimalarial activity
Hybrid compound 99 (Fig. 14) containing a ruthenocene, 4-aminoquinoline, and a 1,2,4-trioxane motifs exhibited IC50 = 65.33 nM and IC50 = 62.0 nM against CQ resistant and MDR strain of P. falciparum, respectively (Martínez et al. 2017). As such derivative 99 was better than CQ, and the parent metal-free and quinoline-free trioxane, indicating possible route for designing new derivatives than could overcome resistance problem.
Hybrid derivative 100 and 101 (Fig. 14), contained 4-aminoquinoline and a quinolizidine (Sparatore et al. 2005) fragments displayed a very attractive bioactivity profile (Basilico et al. 2017). Both racemic (1R/1S)-AM1 and enantiomers exhibited an antimalarial activity in low nanomolar range against sensitive and CQ resistant strains, as well as against MDR strain of P. falciparum. Racemic (1R/1S)-mixture showed remarkable antimalarial activity against ten different isolates of Plasmodium vivax with three times higher potency than CQ. Enantiomer (1S)-100 displayed an additive effect when applied in combination with dihydroartemisinin. All forms, racemate and enantiomers, were in vivo active with no significant differences in potency among them.
Enantiopure chiral 4-aminoquinoline derivatives that contain piperazine were synthesized (Dola et al. 2017) among which derivatives 102–104 (Fig. 14) showed activities in the range of 5.03–39.39 nM against P. falciparum strains. Compound 102, showed the best activity in vivo, and successfully cured mice infected with CQ resistant P. yoelii strain. This derivative has been identified as a preclinical candidate molecule, due to excellent physicochemical properties, an acceptable pharmacokinetic profile, and moderate metabolic clearance in liver microsomes.
Large number of examined quinoline derivatives has provided necessary information on structure-activity relationship and revealed new potential targets for the treatment of malarial parasite. However, although those results are highly valuable, new derivatives still have not brought much needed breakthrough in the therapy. Currently, in accordance with WHO recommendation, the most successful clinical treatments are combination therapies, which include established antimalarial drugs, such as artemisinin (Chico and Chandramohan 2011).
6.2 Antileishmanial Activity
Leishmaniasis is caused by Leishmania parasites and is transmitted to humans by the bite of infected female phlebotomine sandflies. The disease may occur in humans and animals, including those from the closest human surrounding. About 70 animal species and humans can be a reservoir of Leishmania and depending on the source it can be classified as zoonotic or anthroponotic. More than 20 different Leishmania species that infect mammals can cause infection in humans. There are four main forms of the disease: visceral leishmaniasis (also known as kala-azar), post-kala-azar dermal leishmaniasis, cutaneous leishmaniasis, and mucocutaneous leishmaniasis (World Health Organisation 2018). While cutaneous leishmaniasis is the most common form of the disease, visceral leishmaniasis is the most serious and almost always fatal if untreated. Based on the assumption that similar chemotypes could be active against related pathogens, broad series of quinoline-like structures (Woodring et al. 2015), and quinolines (Baragana et al. 2015; Devine et al. 2015; Konstantinovic et al. 2017; Solaja et al. 2008) were examined simultaneously as antimalarial, antileishmaniasis and antitrypanosomal agents, including application in combination therapy (Wijnant et al. 2017).
Ten out of thirty tested quinoline derivatives that contained adamantine or benzothiophen moiety exhibited IC50 values less than 1 μM against Leishmania infantum and Leishmania tropica promastigote stage (Konstantinovic et al. 2018). Two derivatives, 105 and 106 (Fig. 15), exhibited dose-dependent in vivo activity, and a high level of mice survival and reduction of parasites level – up to 99% after 50 mg/kg/day and 5 days treatment. Those two compounds induced production of nitric oxide by IFNγ-primed macrophages, but only when highest doses were used. Also, both derivatives induced a persistent increase of reactive oxygen species (ROS) at all tested doses.

4-Aminoquinolines with antileishmanial activity
During the search for new scaffolds that would be able to inhibit three protozoan pathogens, quinoline derivatives 107–109 (Fig. 15) tested for inhibitory activity against two life stages (promastigote and amastigotes) of Leishmania major (Devine et al. 2015). While derivatives 107–109 showed high activities against promastigote life stage, exhibiting EC50 values in the range 0.2–0.4 μM, only derivative 109 also showed activity against amastigotes life stage (EC50 = 0.89 μM).
Two derivatives of 4-aryloxy-7-chloroquinolines showed a strong inhibition of Leishmania donovani promastigote (IC50 = 11.43 μM and IC50 = 7.9 μM) and amastigote proliferation (IC50 = 1.01 μM and IC50 = 1.02 μM) (Valdivieso et al. 2018). Combinations of 110 or 111 (Fig. 16) with amphotericin B showed a synergistic anti-promastigote effect, while combinations of either 110 or 111 with miltefosine showed a synergistic anti-amastigote effect. Compounds showed deleterious effect on the mitochondrial electrochemical potential in IC50 concentrations, suggesting that rapid collapse of the parasite’s mitochondrial membrane potential could be a mechanism of action.

Quinoline derivatives with antileishmanial activity
A quinoline derivative 112 (Fig. 16) exhibited a strong effect against Leishmania amazonensis, inhibiting the growth of its promastigotes and intracellular amastigotes forms (Antinarelli et al. 2015; Antinarelli et al. 2018). The compound also affected the proliferation of amastigote-like forms of L. amazonensis, with IC50 = 7.0 μg/ml. Intramacrophage amastigotes treated with 112 showed a low capacity to reverse the effect of the compound in a drug-free medium. The strong effect on all parasite stages and the absence of reversibility of the action of 112 suggest that severe damage has been caused to the targets, and mechanism of its activity was preliminarily associated with mitochondrial dysfunction.
A quinoline derivative clioquinol (Fig. 5) inhibited L. amazonensis and Leishmania infantum promastigotes with EC50 = 2.55 μg/ml and EC50 = 1.44 μg/ml, respectively, and also showed inhibitory activity against axenic amastigotes of L. amazonensis (EC50 = 1.88 μg/ml) and L. infantum (EC50 = 0.98 μg/ml) (Tavares et al. 2018). Significant reductions in the percentage of infected macrophages after treatment and in the pre-treated assay using clioquinol were observed. Clioquinol induces morphological and biochemical alterations in parasites, including reduction in cell volume, loss of mitochondrial membrane potential, increase in the ROS production and rupture of the plasma membrane.
N-Aryl derivatives of 2- and 3-aminoquinoline were evaluated as antiproliferative agents against Leishmania mexicana, the etiological agent of leishmaniasis and Trypanosoma cruzi (Chanquia et al. 2019). Fluorine-containing derivatives 113 and 114 (Fig. 16) were more than two-fold more potent than geneticin against intracellular promastigote form of L. mexicana, both exhibiting IC50 values of 41.9 μM. The IC50 values of derivatives 113–115 (Fig. 16) were of the same order as drug benznidazole against epimastigote form of T. cruzi. The compounds were capable to inhibit the degradation of haem, inducing intracellular oxidative damage, which is not countered by the antioxidative defence system of the parasite.
Series of triazolyl esters of 2-methyl-4-phenylquinoline-3-carboxylic acid have been evaluated in vitro against L. donovani (Upadhyay et al. 2018). Most of the derivatives exhibited significant antileishmanial activity against promastigotes and intracellular amastigotes, with less cytotoxicity in comparison to the sodium stibo-gluconate. Four compounds 116–119 (Fig. 16) proved to be active derivatives, with lower toxicity and better selectivity index and were selected for in vivo evaluation. Compound 118 showed consistent activity up to day 28 post-treatment in a hamster model, which is promising finding as the hamster model of visceral leishmaniasis closely resembles the human condition.
Sixteen derivatives of 6-substituted quinoline analogues 120 (Fig. 16) showed high inhibitory potency against L. major promastigotes with IC50 = 0.04–5.60 μM, and all were more active in comparison with pentamidine (IC50 = 7.02 μM) (Almandil et al. 2019). Presence of either electron donating or electron withdrawing group on phenyl ring plays an important role in inhibition, but electron donating group showed better activity.
Leishmaniasis did not take much attention in R&D programs in comparison to other parasitic disease, like malaria. However, since the disease is continuously increasing in Eastern Mediterranean Region, with 5 times growth in the period 1998–2015 (World Health Organisation 2017), there is an urgent need for changing of such a treatment. Although many quinolines exhibited good antileishmanial activity, their targets are still unknown.
Identification of the targets is prerequisite to direct further structural modifications of quinoline scaffold in order to optimize their therapeutic activity.
6.3 Antitrypanosomal Activity
Trypanosoma is unicellular parasitic protozoa which causes infections in humans in the tropical and sub-tropical areas. Trypanosomes infect a variety of hosts and cause fatal disease, such as sleeping sickness in humans. More than 19 different Trypanosoma species were identified as causative agents of diseases that differ in symptoms, action and main host. Trypanosoma cruzi causes American trypanosomiasis, or Chagas disease, when it infects a wide variety of wild and domestic mammals, rodents, many species of bloodsucking triatomid insects – the usual vectors of disease, and humans. The other members of the genus Trypanosoma that cause disease in humans are two subspecies of African trypanosomes, T. brucei subspecies gambiense and T. brucei subspecies rhodesiense. These organisms cause West African and East African trypanosomiases, respectively. The main limitation of the drugs that are currently in use against Trypanosoma is toxicity and requirement for intravenous dosing, thus novel drugs are needed. One approach is to develop compounds that show activity against molecular targets that are common for related pathogens. For example, T. brucei (human African trypanosomiasis), T. cruzi (Chagas’ disease), Leishmania spp. (leishmaniases), and Plasmodium spp. (malaria) express kinases and phosphodiesterases which are involved in the process of cellular signalling (Gould and de Koning 2011; Parsons et al. 2005). Recently it was found that certain human kinase inhibitors could be also effective against T. brucei with EC50 in the low micromolar range (Katiyar et al. 2013), including tyrosine kinase inhibitors and new compounds developed on that basis (Patel et al. 2013).
Quinoline and quinoline-like compounds were tested for their antiprotozoan activity (Devine et al. 2015). From 4-aminoquinoline scaffold, derivatives 121–123 (Fig. 17), and derivatives 108 and 109 (Fig. 15) showed medium nanomolar activities against Trypanosoma brucei and Trypanosoma cruzi. In general, derivatives showed better activity against T. brucei (ED50 = 79–450 nM), than against T. cruzi (ED50 = 730–950 nM), with exception of derivative 122, which is more active against T. cruzi (EC50 = 90 nM vs. EC50 = 350 nM).

4-Aminoquinolines with antitrypanosomal activity
The TbrPDEB1 enzyme, a member of cyclic nucleotide phosphodiesterases was used as a target for series of 3-carbamido-4-amino quinolines (Ochiana et al. 2015). Derivatives 124–127 (Fig. 18) exhibited the highest activity, inhibiting >80% of TbrPDEB1 and exhibiting IC50 in the range 3.5–6.4 μM.

Quinoline derivatives as antitrypanosomial agents
Another derivatives of chalocone (128; general structure A, Fig. 18) and corresponding 4,5-dihydro-1H-pyrazole derivatives (129; general structure B, Fig. 18) were screened against intracellular amastigotes of T. cruzi and intracellular amastigotes of Leishmania (Vianna) panamensis (Ramirez-Prada et al. 2017). Compound 129 was highly active against T. cruzi (EC50 = 0.70 μg/ml), while the best antileishmanial activity exhibited compound 128, which was active at 0.79 μg/ml.
Novel 7-phenyl-quinolines 130–133 (Fig. 18) were evaluated against bloodstream forms of T. cruzi. Nine quinolines were more effective against amastigotes than benznidazole (EC50 = 2.7 μM) and they showed EC50 values ranging from 0.6 to 0.1 μM (Nefertiti et al. 2018). All examined quinolines were highly active in vitro against African trypanosomes, showing EC50 values ≤0.25 μM. The most potent in vitro candidates 130 were tested in in vivo models of T. b. rhodesiense infection, and showed a more than 98% reduction of the parasitemia and curing a half number of infected mice, after 3 doses of 40 mg/kg intraperitoneal administration However, derivative 131 was even more efficacious, showed complete reduction of the parasitemia and cured a half of infected mice.
A series of sulfone derivatives were tested for their in vitro activity against T. brucei. From 22 examined compounds, derivatives obtained from 8-aminoquinoline 132 and 133 show the best inhibitory activity IC50 = 0.76 μM (Zhang et al. 2018). However, during in vivo evaluation against T. brucei, when infected mice were treated for four consecutive days intraperitoneally, derivative 132 did not shown activity, and the infected mice were positive for parasites 24 h post-treatment. However, second 8-quinoline derivative 133 was able to completely inactivate rhodesain, a cysteine protease essential for parasite survival and infectivity (Ettari et al. 2013), at 20 μM after 1 h of incubation with estimated IC50 value of 800 nM.
Pursuit for efficacious clinical therapy based on quinoline-based compounds still continues. Although there are increasing numbers of reports on trypanosoma growth inhibitors, and identification of potential targets, the mechanism of action(s) remain unknown.
7 Conclusion
As emergence and spread of the resistance to existing therapeutics are inevitable the novel advanced drugs and strategies for treatments of numerous infective diseases, affecting tens of millions of people worldwide, are continuously being demanded. Among large number of various pharmacophores used for development of effective antimicrobials, quinoline/quinolone scaffold can be considered a privileged structure since it displays wide range of bioactivities. The diversity of synthetized quinoline/quinolone derivatives provides high and selective activity through different modes of action and low cytotoxicity on human cells.
The drugs based on quinoline/quinolone structure are well-known and in use for over half a century in the treatment of bacterial, parasitic, and viral infections. Besides, in the last two decades quinoline/quinolone derivatives emerged as efficient anti-virulence agents targeting bacterial and fungal virulence factors, including formation of biofilms, characteristic of chronic infections. Of particular importance is the ability of certain quinoline-based compounds to target both bacteria and fungi, either their planktonic or biofilm life form, suggesting that these compounds are promising chemical matter for the development of therapeutics against mixed-species and/or biofilm-associated infections. Quinoline/quinolone derivatives exhibit strong activity by themselves and can increase efficacy of commonly used drugs when applied together. Recently, a new promising approach was employed in the development of more successful quinoline-derived therapeutics based on the synthesis of hybrid compounds that contain two or more pharmacophores affecting different targets and/or employing distinct mechanisms of action. Hence, there are still plenty of possibilities for development of novel more effective antimicrobials.
The high potency of quinoline/quinolone derivatives is demonstrated by their activities in nanomolar and low micromolar concentrations against wide range of infective agents including resistant strains. The importance of quinoline/quinolone as a scaffold for drugs’ development is emphasized by recently granted accelerated approval for clinical use to a new quinoline-based antibiotic developed to fight MDR mycobacteria.
Therefore, there are more than enough reasons to continue quest for efficacious clinical therapeutics based on quinoline/quinolone structures since they are still proving to be unprecedented source of much needed solutions for infections treatments.
References
Abouelhassan Y, Garrison AT, Burch GM, Wong W, Norwood VM, Huigens RW 3rd (2014) Discovery of quinoline small molecules with potent dispersal activity against methicillin-resistant Staphylococcus aureus and Staphylococcus epidermidis biofilms using a scaffold hopping strategy. Bioorg Med Chem Lett 24(21):5076–5080. https://doi.org/10.1016/j.bmcl.2014.09.009
Abouelhassan Y, Garrison AT, Bai F, Norwood VM, Nguyen MT, Jin S, Huigens RW 3rd (2015) A phytochemical-halogenated Quinoline combination therapy strategy for the treatment of pathogenic bacteria. ChemMedChem 10(7):1157–1162. https://doi.org/10.1002/cmdc.201500179
Al-Bari MA (2015) Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother 70(6):1608–1621. https://doi.org/10.1093/jac/dkv018
Albuquerque P, Casadevall A (2012) Quorum sensing in fungi--a review. Med Mycol 50(4):337–345. https://doi.org/10.3109/13693786.2011.652201
Aleksić I, Šegan S, Andrić F, Zlatović M, Moric I, Opsenica DM, Senerovic L (2017) Long-chain 4-Aminoquinolines as quorum sensing inhibitors in Serratia marcescens and Pseudomonas aeruginosa. ACS Chem Biol 12(5):1425–1434. https://doi.org/10.1021/acschembio.6b01149
Almandil NB, Taha M, Rahim F, Wadood A, Imran S, Alqahtani MA, Bamarouf YA, Ibrahim M, Mosaddik A, Gollapalli M (2019) Synthesis of novel quinoline-based thiadiazole, evaluation of their antileishmanial potential and molecular docking studies. Bioorg Chem 85:109–116. https://doi.org/10.1016/j.bioorg.2018.12.025
Anderson RJ, Groundwater PW, Todd A, Worsley A (2012) Antibacterial agents: chemistry, mode of action, mechanisms of resistance and clinical applications. Wiley, Chichester
Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307(5707):223–227. https://doi.org/10.1126/science.1106753
Antinarelli LM, Dias RM, Souza IO, Lima WP, Gameiro J, da Silva AD, Coimbra ES (2015) 4-Aminoquinoline derivatives as potential antileishmanial agents. Chem Biol Drug Des 86(4):704–714. https://doi.org/10.1111/cbdd.12540
Antinarelli LMR, de Oliveira Souza I, Zabala CPV, Gameiro J, Britta EA, Nakamura CV, Lima WP, da Silva AD, Coimbra ES (2018) Antileishmanial activity of a 4-hydrazinoquinoline derivative: induction of autophagy and apoptosis-related processes and effectiveness in experimental cutaneous leishmaniasis. Exp Parasitol 195:78–86
Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Luksch T, Martinez MS, Luksch T, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, Sinden RE, Winzeler EA, Charman SA, Bebrevska L, Gray DW, Campbell S, Fairlamb AH, Willis PA, Rayner JC, Fidock DA, Read KD, Gilbert IH (2015) A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522(7556):315–320. https://doi.org/10.1038/nature14451
Barbosa-Lima G, Moraes AM, Araujo ADS, da Silva ET, de Freitas CS, Vieira YR, Marttorelli A, Neto JC, Bozza PT, de Souza MVN, Souza TML (2017) 2,8-bis(trifluoromethyl)quinoline analogs show improved anti-Zika virus activity, compared to mefloquine. Eur J Med Chem 127:334–340. https://doi.org/10.1016/j.ejmech.2016.12.058
Barnett DS, Guy RK (2014) Antimalarials in development in 2014. Chem Rev 114(22):11221–11241. https://doi.org/10.1021/cr500543f
Basak A, Abouelhassan Y, Huigens RW 3rd (2015) Halogenated quinolines discovered through reductive amination with potent eradication activities against MRSA, MRSE and VRE biofilms. Org Biomol Chem 13(41):10290–10294. https://doi.org/10.1039/c5ob01883h
Basak A, Abouelhassan Y, Norwood VM, Bai F, Nguyen MT, Jin S, Huigens RW 3rd (2016) Synthetically tuning the 2-position of halogenated Quinolines: optimizing antibacterial and biofilm eradication activities via alkylation and reductive amination pathways. Chemistry 22(27):9181–9189. https://doi.org/10.1002/chem.201600926
Basak A, Abouelhassan Y, Kim YS, Norwood VM, Jin S, Huigens RW 3rd (2018) Halogenated quinolines bearing polar functionality at the 2-position: identification of new antibacterial agents with enhanced activity against Staphylococcus epidermidis. Eur J Med Chem 155:705–713. https://doi.org/10.1016/j.ejmech.2018.06.045
Basilico N, Parapini S, Sparatore A, Romeo S, Misiano P, Vivas L, Yardley V, Croft SL, Habluetzel A, Lucantoni L, Renia L, Russell B, Suwanarusk R, Nosten F, Dondio G, Bigogno C, Jabes D, Taramelli D (2017) In vivo and in vitro activities and ADME-Tox profile of a quinolizidine-modified 4-Aminoquinoline: a potent anti-P. falciparum and anti-P. vivax blood-stage antimalarial. Molecules 22(12). https://doi.org/10.3390/molecules22122102
Ben Yaakov D, Shadkchan Y, Albert N, Kontoyiannis DP, Osherov N (2017) The quinoline bromoquinol exhibits broad-spectrum antifungal activity and induces oxidative stress and apoptosis in aspergillus fumigatus. J Antimicrob Chemother 72(8):2263–2272. https://doi.org/10.1093/jac/dkx117
Bernier SP, Surette MG (2013) Concentration-dependent activity of antibiotics in natural environments. Front Microbiol 4:20. https://doi.org/10.3389/fmicb.2013.00020
Bisacchi GS (2015) Origins of the quinolone class of antibacterials: an expanded “discovery story”. J Med Chem 58(12):4874–4882. https://doi.org/10.1021/jm501881c
Boudhar A, Ng XW, Loh CY, Chia WN, Tan ZM, Nosten F, Dymock BW, Tan KS (2016) Overcoming chloroquine resistance in malaria: design, synthesis, and structure-activity relationships of novel hybrid compounds. Antimicrob Agents Chemother 60(5):3076–3089. https://doi.org/10.1128/AAC.02476-15
Capela R, Magalhaes J, Miranda D, Machado M, Sanches-Vaz M, Albuquerque IS, Sharma M, Gut J, Rosenthal PJ, Frade R, Perry MJ, Moreira R, Prudencio M, Lopes F (2018) Endoperoxide-8-aminoquinoline hybrids as dual-stage antimalarial agents with enhanced metabolic stability. Eur J Med Chem 149:69–78. https://doi.org/10.1016/j.ejmech.2018.02.048
Cecchetti V, Parolin C, Moro S, Pecere T, Filipponi E, Calistri A, Tabarrini O, Gatto B, Palumbo M, Fravolini A, Palu G (2000) 6-Aminoquinolones as new potential anti-HIV agents. J Med Chem 43(20):3799–3802
Chander S, Ashok P, Zheng YT, Wang P, Raja KS, Taneja A, Murugesan S (2016) Design, synthesis and in-vitro evaluation of novel tetrahydroquinoline carbamates as HIV-1 RT inhibitor and their antifungal activity. Bioorg Chem 64:66–73. https://doi.org/10.1016/j.bioorg.2015.12.005
Chanquia SN, Larregui F, Puente V, Labriola C, Lombardo E, Garcia Linares G (2019) Synthesis and biological evaluation of new quinoline derivatives as antileishmanial and antitrypanosomal agents. Bioorg Chem 83:526–534. https://doi.org/10.1016/j.bioorg.2018.10.053
Chico RM, Chandramohan D (2011) Azithromycin plus chloroquine: combination therapy for protection against malaria and sexually transmitted infections in pregnancy. Expert Opin Drug Metab Toxicol 7(9):1153–1167. https://doi.org/10.1517/17425255.2011.598506
Chu XM, Wang C, Liu W, Liang LL, Gong KK, Zhao CY, Sun KL (2019) Quinoline and quinolone dimers and their biological activities: an overview. Eur J Med Chem 161:101–117. https://doi.org/10.1016/j.ejmech.2018.10.035
Costa-Orlandi CB, Sardi JCO, Pitangui NS, de Oliveira HC, Scorzoni L, Galeane MC, Medina-Alarcon KP, Melo W, Marcelino MY, Braz JD, Fusco-Almeida AM, Mendes-Giannini MJS (2017) Fungal biofilms and Polymicrobial diseases. J Fungi 3(2):pii: E22. https://doi.org/10.3390/jof3020022
de Azambuja Carvalho PH, Duval AR, Manzolli Leite FR, Nedel F, Cunico W, Lund RG (2016) (7-Chloroquinolin-4-yl)arylhydrazones: Candida albicans enzymatic repression and cytotoxicity evaluation, part 2. J Enzyme Inhib Med Chem 31(1):126–131. https://doi.org/10.3109/14756366.2015.1010527
de Souza IO, Schrekker CM, Lopes W, Orru RV, Hranjec M, Perin N, Machado M, Oliveira LF, Donato RK, Stefani V, Fuentefria AM, Schrekker HS (2016) Bifunctional fluorescent benzimidazo[1,2-alpha]quinolines for Candida spp. biofilm detection and biocidal activity. J Photochem Photobiol B 163:319–326. https://doi.org/10.1016/j.jphotobiol.2016.08.037
Defoirdt T (2018) Quorum-sensing systems as targets for Antivirulence therapy. Trends Microbiol 26(4):313–328. https://doi.org/10.1016/j.tim.2017.10.005
Delattin N, Bardiot D, Marchand A, Chaltin P, De Brucker K, Cammue BP, Thevissen K (2012) Identification of fungicidal 2,6-disubstituted quinolines with activity against Candida biofilms. Molecules 17(10):12243–12251. https://doi.org/10.3390/molecules171012243
Delvecchio R, Higa LM, Pezzuto P, Valadao AL, Garcez PP, Monteiro FL, Loiola EC, Dias AA, Silva FJ, Aliota MT, Caine EA, Osorio JE, Bellio M, O’Connor DH, Rehen S, de Aguiar RS, Savarino A, Campanati L, Tanuri A (2016) Chloroquine, an endocytosis blocking agent, inhibits Zika virus infection in different cell models. Viruses 8(12). https://doi.org/10.3390/v8120322
Devine W, Woodring JL, Swaminathan U, Amata E, Patel G, Erath J, Roncal NE, Lee PJ, Leed SE, Rodriguez A, Mensa-Wilmot K, Sciotti RJ, Pollastri MP (2015) Protozoan parasite growth inhibitors discovered by cross-screening yield potent scaffolds for Lead discovery. J Med Chem 58(14):5522–5537. https://doi.org/10.1021/acs.jmedchem.5b00515
Deziel E, Lepine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG (2004) Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A 101(5):1339–1344. https://doi.org/10.1073/pnas.0307694100
Diggle SP, Lumjiaktase P, Dipilato F, Winzer K, Kunakorn M, Barrett DA, Chhabra SR, Camara M, Williams P (2006) Functional genetic analysis reveals a 2-Alkyl-4-quinolone signaling system in the human pathogen Burkholderia pseudomallei and related bacteria. Chem Biol 13(7):701–710. https://doi.org/10.1016/j.chembiol.2006.05.006
Dola VR, Soni A, Agarwal P, Ahmad H, Raju KS, Rashid M, Wahajuddin M, Srivastava K, Haq W, Dwivedi AK, Puri SK, Katti SB (2017) Synthesis and evaluation of Chirally defined side chain variants of 7-Chloro-4-Aminoquinoline to overcome drug resistance in malaria chemotherapy. Antimicrob Agents Chemother 61(3). https://doi.org/10.1128/AAC.01152-16
Dolan N, Gavin DP, Eshwika A, Kavanagh K, McGinley J, Stephens JC (2016) Synthesis, antibacterial and anti-MRSA activity, in vivo toxicity and a structure-activity relationship study of a quinoline thiourea. Bioorg Med Chem Lett 26(2):630–635. https://doi.org/10.1016/j.bmcl.2015.11.058
Duval AR, Carvalho PH, Soares MC, Gouvea DP, Siqueira GM, Lund RG, Cunico W (2011) 7-chloroquinolin-4-yl arylhydrazone derivatives: synthesis and antifungal activity. Sci World J 11:1489–1495. https://doi.org/10.1100/tsw.2011.141
Egan TJ, Marques HM (1999) The role of haem in the activity of chloroquine and related antimalarial drugs. Coord Chem Rev 190–192:493–517
El Shehry MF, Ghorab MM, Abbas SY, Fayed EA, Shedid SA, Ammar YA (2018) Quinoline derivatives bearing pyrazole moiety: synthesis and biological evaluation as possible antibacterial and antifungal agents. Eur J Med Chem 143:1463–1473. https://doi.org/10.1016/j.ejmech.2017.10.046
Espinosa-Valdes MP, Borbolla-Alvarez S, Delgado-Espinosa AE, Sanchez-Tejeda JF, Ceron-Nava A, Quintana-Romero OJ, Ariza-Castolo A, Garcia-Del Rio DF, Loza-Mejia MA (2019) Synthesis, in silico, and in vitro evaluation of long chain alkyl amides from 2-Amino-4-quinolone derivatives as biofilm inhibitors. Molecules 24(2). https://doi.org/10.3390/molecules24020327
Ettari R, Tamborini L, Angelo IC, Micale N, Pinto A, De Micheli C, Conti P (2013) Inhibition of rhodesain as a novel therapeutic modality for human African trypanosomiasis. J Med Chem 56(14):5637–5658. https://doi.org/10.1021/jm301424d
Fernández-Piñar R, Cámara M, Dubern J-F, Ramos JL, Espinosa-Urgel M (2011) The Pseudomonas aeruginosa quinolone quorum sensing signal alters the multicellular behaviour of Pseudomonas putida KT2440. Res Microbiol 162(8):773–781. https://doi.org/10.1016/j.resmic.2011.06.013
Gama N, Kumar K, Ekengard E, Haukka M, Darkwa J, Nordlander E, Meyer D (2016) Gold(I) complex of 1,1′-bis(diphenylphosphino) ferrocene-quinoline conjugate: a virostatic agent against HIV-1. Biometals 29(3):389–397. https://doi.org/10.1007/s10534-016-9921-9
Garrison AT, Abouelhassan Y, Yang H, Yousaf HH, Nguyen TJ, Huigens Iii RW (2017) Microwave-enhanced Friedlander synthesis for the rapid assembly of halogenated quinolines with antibacterial and biofilm eradication activities against drug resistant and tolerant bacteria. Med Chem Commun 8(4):720–724. https://doi.org/10.1039/c6md00381h
Gould MK, de Koning HP (2011) Cyclic-nucleotide signalling in protozoa. FEMS Microbiol Rev 35(3):515–541. https://doi.org/10.1111/j.1574-6976.2010.00262.x
Gualerzi CO, Brandi L, Fabbretti A, Pon CL (2013) Antibiotics: targets, mechanisms and resistance. Wiley-VCH, Weinheim. https://doi.org/10.1002/9783527659685
Hajimahdi Z, Zabihollahi R, Aghasadeghi MR, Hosseini Ashtiani S, Zargh A (2016) Novel quinolone-3-carboxylic acid derivatives as anti-HIV-1 agents: design, synthesis, and biological activities. Med Chem Res 25:1861–1876
Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2(2):95–108
Hamama WS, Ibrahim ME, Gooda AA, Zoorob HH (2018) Efficient synthesis, antimicrobial, antioxidant assessments and geometric optimization calculations of azoles- incorporating Quinoline moiety. J Heterocyclic Chem 55(11):2623–2634
He QQ, Zhang X, Yang LM, Zheng YT, Chen F (2013) Synthesis and biological evaluation of 5-fluoroquinolone-3-carboxylic acids as potential HIV-1 integrase inhibitors. J Enzyme Inhib Med Chem 28(4):671–676. https://doi.org/10.3109/14756366.2012.668540
Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Camara M (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35(2):247–274. https://doi.org/10.1111/j.1574-6976.2010.00247.x
Hucke O, Coulombe R, Bonneau P, Bertrand-Laperle M, Brochu C, Gillard J, Joly MA, Landry S, Lepage O, Llinas-Brunet M, Pesant M, Poirier M, Poirier M, McKercher G, Marquis M, Kukolj G, Beaulieu PL, Stammers TA (2014) Molecular dynamics simulations and structure-based rational design lead to allosteric HCV NS5B polymerase thumb pocket 2 inhibitor with picomolar cellular replicon potency. J Med Chem 57(5):1932–1943. https://doi.org/10.1021/jm4004522
Huigens RW 3rd (2018) The path to new halogenated Quinolines with enhanced activities against Staphylococcus epidermidis. Microbiol Insights 11:1178636118808532. https://doi.org/10.1177/1178636118808532
Ilangovan A, Fletcher M, Rampioni G, Pustelny C, Rumbaugh K, Heeb S, Cámara M, Truman A, Chhabra SR, Emsley J, Williams P (2013) Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR). PLoS Pathog 9(7):e1003508. https://doi.org/10.1371/journal.ppat.1003508
Irfan M, Aneja B, Yadava U, Khan SI, Manzoor N, Daniliuc CG, Abid M (2015) Synthesis, QSAR and anticandidal evaluation of 1,2,3-triazoles derived from naturally bioactive scaffolds. Eur J Med Chem 93:246–254. https://doi.org/10.1016/j.ejmech.2015.02.007
Irfan M, Alam S, Manzoor N, Abid M (2017) Effect of quinoline based 1,2,3-triazole and its structural analogues on growth and virulence attributes of Candida albicans. PLoS One 12(4):e0175710. https://doi.org/10.1371/journal.pone.0175710
Jentsch NG, Hart AP, Hume JD, Sun J, McNeely KA, Lama C, Pigza JA, Donahue MG, Kessl JJ (2018) Synthesis and evaluation of aryl Quinolines as HIV-1 integrase Multimerization inhibitors. ACS Med Chem Lett 9(10):1007–1012. https://doi.org/10.1021/acsmedchemlett.8b00269
Katiyar S, Kufareva I, Behera R, Thomas SM, Ogata Y, Pollastri M, Abagyan R, Mensa-Wilmot K (2013) Lapatinib-binding protein kinases in the African trypanosome: identification of cellular targets for kinase-directed chemical scaffolds. PLoS One 8(2):e56150. https://doi.org/10.1371/journal.pone.0056150
Khan FAK, Kaduskar RN, Patil R, Patil RH, Ansari SA, Alkahtani HM, Almehizia AA, Shinde DB, Sangshetti JN (2019) Synthesis, biological evaluations and computational studies of N-(3-(-2-(7-Chloroquinolin-2-yl)vinyl) benzylidene)anilines as fungal biofilm inhibitors. Bioorg Med Chem Lett 29(4):623–630. https://doi.org/10.1016/j.bmcl.2018.12.046
Konstantinovic J, Videnovic M, Srbljanovic J, Djurkovic-Djakovic O, Bogojevic K, Sciotti R, Solaja B (2017) Antimalarials with benzothiophene moieties as aminoquinoline partners. Molecules 22(3). https://doi.org/10.3390/molecules22030343
Konstantinovic J, Videnovic M, Orsini S, Bogojevic K, D’Alessandro S, Scaccabarozzi D, Terzic Jovanovic N, Gradoni L, Basilico N, Solaja BA (2018) Novel Aminoquinoline derivatives significantly reduce parasite load in Leishmania infantum infected mice. ACS Med Chem Lett 9(7):629–634. https://doi.org/10.1021/acsmedchemlett.8b00053
Korotchenko V, Sathunuru R, Gerena L, Caridha D, Li Q, Kreishman-Deitrick M, Smith PL, Lin AJ (2015) Antimalarial activity of 4-amidinoquinoline and 10-amidinobenzonaphthyridine derivatives. J Med Chem 58(8):3411–3431. https://doi.org/10.1021/jm501809x
Krauss J, Muller C, Kiessling J, Richter S, Staudacher V, Bracher F (2014) Synthesis and biological evaluation of novel N-alkyl tetra- and decahydroisoquinolines: novel antifungals that target ergosterol biosynthesis. Arch Pharm 347(4):283–290. https://doi.org/10.1002/ardp.201300338
Krauss J, Hornacek M, Muller C, Staudacher V, Stadler M, Bracher F (2015) Synthesis and antifungal evaluation of novel N-alkyl tetra- and perhydroquinoline derivatives. Sci Pharm 83(1):1–14. https://doi.org/10.3797/scipharm.1409-13
Kumar DV, Rai R, Brameld KA, Riggs J, Somoza JR, Rajagopalan R, Janc JW, Xia YM, Ton TL, Hu H, Lehoux I, Ho JD, Young WB, Hart B, Green MJ (2012) 3-heterocyclyl quinolone inhibitors of the HCV NS5B polymerase. Bioorg Med Chem Lett 22(1):300–304. https://doi.org/10.1016/j.bmcl.2011.11.013
Leon B, Haeckl FP, Linington RG (2015) Optimized quinoline amino alcohols as disruptors and dispersal agents of Vibrio cholerae biofilms. Org Biomol Chem 13(31):8495–8499. https://doi.org/10.1039/c5ob01134e
Leven M, Held J, Duffy S, Alves Avelar LA, Meister S, Delves M, Plouffe D, Kuna K, Tschan S, Avery VM, Winzeler EA, Mordmuller B, Kurz T (2019) 8-aminoquinolines with an aminoxyalkyl side chain exert in vitro dual-stage antiplasmodial activity. ChemMedChem 14(4):501–511. https://doi.org/10.1002/cmdc.201800691
Loregian A, Mercorelli B, Muratore G, Sinigalia E, Pagni S, Massari S, Gribaudo G, Gatto B, Palumbo M, Tabarrini O, Cecchetti V, Palu G (2010) The 6-aminoquinolone WC5 inhibits human cytomegalovirus replication at an early stage by interfering with the transactivating activity of viral immediate-early 2 protein. Antimicrob Agents Chemother 54(5):1930–1940. https://doi.org/10.1128/AAC.01730-09
Lu C, Kirsch B, Zimmer C, de Jong JC, Henn C, Maurer CK, Musken M, Haussler S, Steinbach A, Hartmann RW (2012) Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem Biol 19(3):381–390. https://doi.org/10.1016/j.chembiol.2012.01.015
Lu C, Kirsch B, Maurer CK, de Jong JC, Braunshausen A, Steinbach A, Hartmann RW (2014a) Optimization of anti-virulence PqsR antagonists regarding aqueous solubility and biological properties resulting in new insights in structure–activity relationships. Eur J Med Chem 79:173–183. https://doi.org/10.1016/j.ejmech.2014.04.016
Lu C, Maurer CK, Kirsch B, Steinbach A, Hartmann RW (2014b) Overcoming the unexpected functional inversion of a PqsR antagonist in Pseudomonas aeruginosa: an in vivo potent antivirulence agent targeting pqs quorum sensing. Angew Chem Int Ed 53(4):1109–1112. https://doi.org/10.1002/anie.201307547
Luthra P, Liang J, Pietzsch CA, Khadka S, Edwards MR, Wei S, De S, Posner B, Bukreyev A, Ready JM, Basler CF (2018) A high throughput screen identifies benzoquinoline compounds as inhibitors of Ebola virus replication. Antivir Res 150:193–201. https://doi.org/10.1016/j.antiviral.2017.12.019
Manfroni G, Cannalire R, Barreca ML, Kaushik-Basu N, Leyssen P, Winquist J, Iraci N, Manvar D, Paeshuyse J, Guhamazumder R, Basu A, Sabatini S, Tabarrini O, Danielson UH, Neyts J, Cecchetti V (2014) The versatile nature of the 6-aminoquinolone scaffold: identification of submicromolar hepatitis C virus NS5B inhibitors. J Med Chem 57(5):1952–1963. https://doi.org/10.1021/jm401362f
Manohar S, Khan SI, Rawat DS (2013) 4-aminoquinoline-triazine-based hybrids with improved in vitro antimalarial activity against CQ-sensitive and CQ-resistant strains of Plasmodium falciparum. Chem Biol Drug Des 81(5):625–630. https://doi.org/10.1111/cbdd.12108
Mao TQ, He QQ, Wan ZY, Chen WX, Chen FE, Tang GF, De Clercq E, Daelemans D, Pannecouque C (2015) Anti-HIV diarylpyrimidine-quinolone hybrids and their mode of action. Bioorg Med Chem 23(13):3860–3868. https://doi.org/10.1016/j.bmc.2015.03.037
Martínez A, Deregnaucourt C, Sinou V, Latour C, Roy D, Schrével J, Sánchez-Delgado RA (2017) Synthesis of an organo-ruthenium aminoquinoline-trioxane hybrid and evaluation of its activity against plasmodium falciparum and its toxicity toward normal mammalian cells. Med Chem Res 26(2):473–483
Massoud AA, Langer V, Gohar YM, Abu-Youssef MA, Janis J, Lindberg G, Hansson K, Ohrstrom L (2013) Effects of different substituents on the crystal structures and antimicrobial activities of six Ag(I) quinoline compounds. Inorg Chem 52(7):4046–4060. https://doi.org/10.1021/ic400081v
Mercorelli B, Luganini A, Muratore G, Massari S, Terlizzi ME, Tabarrini O, Gribaudo G, Palu G, Loregian A (2014) The 6-Aminoquinolone WC5 inhibits different functions of the immediate-early 2 (IE2) protein of human cytomegalovirus that are essential for viral replication. Antimicrob Agents Chemother 58(11):6615–6626. https://doi.org/10.1128/AAC.03309-14
Miquel S, Lagrafeuille R, Souweine B, Forestier C (2016) Anti-biofilm activity as a health issue. Front Microbiol 7:592–592. https://doi.org/10.3389/fmicb.2016.00592
Montoya A, Quiroga J, Abonia R, Derita M, Sortino M, Ornelas A, Zacchino S, Insuasty B (2016) Hybrid molecules containing a 7-Chloro-4-aminoquinoline nucleus and a substituted 2-pyrazoline with antiproliferative and antifungal activity. Molecules 21(8). https://doi.org/10.3390/molecules21080969
Moradali MF, Ghods S, Rehm BH (2017) Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front Cell Infect Microbiol 7:39. https://doi.org/10.3389/fcimb.2017.00039
Musiol R, Serda M, Hensel-Bielowka S, Polanski J (2010) Quinoline-based antifungals. Curr Med Chem 17(18):1960–1973
Nefertiti ASG, Batista MM, Da Silva PB, Batista DGJ, Da Silva CF, Peres RB, Torres-Santos EC, Cunha-Junior EF, Holt E, Boykin DW, Brun R, Wenzler T, Soeiro MNC (2018) In vitro and in vivo studies of the Trypanocidal effect of novel Quinolines. Antimicrob Agents Chemother 62(2). https://doi.org/10.1128/AAC.01936-17
Nikolić S, Opsenica DM, Filipović V, Dojčinović B, Aranđelović S, Radulović S, Grgurić-Šipka S (2015) Strong in vitro cytotoxic potential of new ruthenium–cymene complexes. Organometallics 34(14):3464–3473. https://doi.org/10.1021/acs.organomet.5b00041
Nwaka S, Hudson A (2006) Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov 5(11):941–955. https://doi.org/10.1038/nrd2144
Ochiana SO, Bland ND, Settimo L, Campbell RK, Pollastri MP (2015) Repurposing human PDE4 inhibitors for neglected tropical diseases. Evaluation of analogs of the human PDE4 inhibitor GSK-256066 as inhibitors of PDEB1 of trypanosoma brucei. Chem Biol Drug Des 85(5):549–564. https://doi.org/10.1111/cbdd.12443
Olliaro P, Mussano P (2003) Amodiaquine for treating malaria. Cochrane Database Syst Rev 2:CD000016. https://doi.org/10.1002/14651858.CD000016
Ongarora DS, Strydom N, Wicht K, Njoroge M, Wiesner L, Egan TJ, Wittlin S, Jurva U, Masimirembwa CM, Chibale K (2015) Antimalarial benzoheterocyclic 4-aminoquinolines: structure-activity relationship, in vivo evaluation, mechanistic and bioactivation studies. Bioorg Med Chem 23(17):5419–5432. https://doi.org/10.1016/j.bmc.2015.07.051
Opsenica I, Filipovic V, Nuss JE, Gomba LM, Opsenica D, Burnett JC, Gussio R, Solaja BA, Bavari S (2012) The synthesis of 2,5-bis(4-amidinophenyl)thiophene derivatives providing submicromolar-range inhibition of the botulinum neurotoxin serotype A metalloprotease. Eur J Med Chem 53:374–379. https://doi.org/10.1016/j.ejmech.2012.03.043
Opsenica IM, Verbic TZ, Tot M, Sciotti RJ, Pybus BS, Djurkovic-Djakovic O, Slavic K, Solaja BA (2015) Investigation into novel thiophene- and furan-based 4-amino-7-chloroquinolines afforded antimalarials that cure mice. Bioorg Med Chem 23(9):2176–2186. https://doi.org/10.1016/j.bmc.2015.02.061
Parsons M, Worthey EA, Ward PN, Mottram JC (2005) Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 6:127. https://doi.org/10.1186/1471-2164-6-127
Patel G, Karver CE, Behera R, Guyett PJ, Sullenberger C, Edwards P, Roncal NE, Mensa-Wilmot K, Pollastri MP (2013) Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis. J Med Chem 56(10):3820–3832. https://doi.org/10.1021/jm400349k
Phillips B, Cai R, Delaney W, Du Z, Ji M, Jin H, Lee J, Li J, Niedziela-Majka A, Mish M, Pyun HJ, Saugier J, Tirunagari N, Wang J, Yang H, Wu Q, Sheng C, Zonte C (2014) Highly potent HCV NS4B inhibitors with activity against multiple genotypes. J Med Chem 57(5):2161–2166. https://doi.org/10.1021/jm401646w
Pierre F, O’Brien SE, Haddach M, Bourbon P, Schwaebe MK, Stefan E, Darjania L, Stansfield R, Ho C, Siddiqui-Jain A, Streiner N, Rice WG, Anderes K, Ryckman DM (2011) Novel potent pyrimido[4,5-c]quinoline inhibitors of protein kinase CK2: SAR and preliminary assessment of their analgesic and anti-viral properties. Bioorg Med Chem Lett 21(6):1687–1691. https://doi.org/10.1016/j.bmcl.2011.01.091
Pippi B, Reginatto P, Machado G, Bergamo VZ, Lana DFD, Teixeira ML, Franco LL, Alves RJ, Andrade SF, Fuentefria AM (2017) Evaluation of 8-hydroxyquinoline derivatives as hits for antifungal drug design. Med Mycol 55(7):763–773. https://doi.org/10.1093/mmy/myx003
Pippi B, Machado G, Bergamo VZ, Alves RJ, Andrade SF, Fuentefria AM (2018) Clioquinol is a promising preventive morphological switching compound in the treatment of Candida infections linked to the use of intrauterine devices. J Med Microbiol 67(11):1655–1663. https://doi.org/10.1099/jmm.0.000850
Plantone D, Koudriavtseva T (2018) Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig 38(8):653–671. https://doi.org/10.1007/s40261-018-0656-y
Ramirez-Prada J, Robledo SM, Velez ID, Crespo MDP, Quiroga J, Abonia R, Montoya A, Svetaz L, Zacchino S, Insuasty B (2017) Synthesis of novel quinoline-based 4,5-dihydro-1H-pyrazoles as potential anticancer, antifungal, antibacterial and antiprotozoal agents. Eur J Med Chem 131:237–254. https://doi.org/10.1016/j.ejmech.2017.03.016
Reen FJ, Mooij MJ, Holcombe LJ, McSweeney CM, McGlacken GP, Morrissey JP, O’Gara F (2011) The Pseudomonas quinolone signal (PQS), and its precursor HHQ, modulate interspecies and interkingdom behaviour. FEMS Microbiol Ecol 77(2):413–428. https://doi.org/10.1111/j.1574-6941.2011.01121.x
Reen FJ, Clarke SL, Legendre C, McSweeney CM, Eccles KS, Lawrence SE, O’Gara F, McGlacken GP (2012) Structure–function analysis of the C-3 position in analogues of microbial behavioural modulators HHQ and PQS. Org Biomol Chem 10(44):8903–8910. https://doi.org/10.1039/C2OB26823J
Reen FJ, Shanahan R, Cano R, O’Gara F, McGlacken GP (2015) A structure activity-relationship study of the bacterial signal molecule HHQ reveals swarming motility inhibition in Bacillus atrophaeus. Org Biomol Chem 13(19):5537–5541. https://doi.org/10.1039/C5OB00315F
Salas PF, Herrmann C, Cawthray JF, Nimphius C, Kenkel A, Chen J, de Kock C, Smith PJ, Patrick BO, Adam MJ, Orvig C (2013) Structural characteristics of chloroquine-bridged ferrocenophane analogues of ferroquine may obviate malaria drug-resistance mechanisms. J Med Chem 56(4):1596–1613. https://doi.org/10.1021/jm301422h
Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H (2006) Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem 49(5):1506–1508. https://doi.org/10.1021/jm0600139
Savarino A, Shytaj IL (2015) Chloroquine and beyond: exploring anti-rheumatic drugs to reduce immune hyperactivation in HIV/AIDS. Retrovirology 12:51. https://doi.org/10.1186/s12977-015-0178-0
Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (2003) Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis 3(11):722–727
Schlitzer M (2007) Malaria chemotherapeutics part I: history of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem 2(7):944–986. https://doi.org/10.1002/cmdc.200600240
Scola PM, Sun LQ, Wang AX, Chen J, Sin N, Venables BL, Sit SY, Chen Y, Cocuzza A, Bilder DM, D’Andrea SV, Zheng B, Hewawasam P, Tu Y, Friborg J, Falk P, Hernandez D, Levine S, Chen C, Yu F, Sheaffer AK, Zhai G, Barry D, Knipe JO, Han YH, Schartman R, Donoso M, Mosure K, Sinz MW, Zvyaga T, Good AC, Rajamani R, Kish K, Tredup J, Klei HE, Gao Q, Mueller L, Colonno RJ, Grasela DM, Adams SP, Loy J, Levesque PC, Sun H, Shi H, Sun L, Warner W, Li D, Zhu J, Meanwell NA, McPhee F (2014a) The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem 57(5):1730–1752. https://doi.org/10.1021/jm500297k
Scola PM, Wang AX, Good AC, Sun LQ, Combrink KD, Campbell JA, Chen J, Tu Y, Sin N, Venables BL, Sit SY, Chen Y, Cocuzza A, Bilder DM, D’Andrea S, Zheng B, Hewawasam P, Ding M, Thuring J, Li J, Hernandez D, Yu F, Falk P, Zhai G, Sheaffer AK, Chen C, Lee MS, Barry D, Knipe JO, Li W, Han YH, Jenkins S, Gesenberg C, Gao Q, Sinz MW, Santone KS, Zvyaga T, Rajamani R, Klei HE, Colonno RJ, Grasela DM, Hughes E, Chien C, Adams S, Levesque PC, Li D, Zhu J, Meanwell NA, McPhee F (2014b) Discovery and early clinical evaluation of BMS-605339, a potent and orally efficacious tripeptidic acylsulfonamide NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem 57(5):1708–1729. https://doi.org/10.1021/jm401840s
Shaikh SKJ, Kamble RR, Somagond SM, Devarajegowda HC, Dixit SR, Joshi SD (2017) Tetrazolylmethyl quinolines: design, docking studies, synthesis, anticancer and antifungal analyses. Eur J Med Chem 128:258–273. https://doi.org/10.1016/j.ejmech.2017.01.043
Shang XF, Morris-Natschke SL, Liu YQ, Guo X, Xu XS, Goto M, Li JC, Yang GZ, Lee KH (2018) Biologically active quinoline and quinazoline alkaloids part I. Med Res Rev 38(3):775–828. https://doi.org/10.1002/med.21466
Shinde RB, Raut JS, Chauhan NM, Karuppayil SM (2013) Chloroquine sensitizes biofilms of Candida albicans to antifungal azoles. Braz J Infect Dis 17(4):395–400. https://doi.org/10.1016/j.bjid.2012.11.002
Solaja BA, Opsenica D, Smith KS, Milhous WK, Terzic N, Opsenica I, Burnett JC, Nuss J, Gussio R, Bavari S (2008) Novel 4-aminoquinolines active against chloroquine-resistant and sensitive P. falciparum strains that also inhibit botulinum serotype A. J Med Chem 51(15):4388–4391. https://doi.org/10.1021/jm800737y
Soukarieh F, Vico Oton E, Dubern J-F, Gomes J, Halliday N, de Pilar CM, Ramírez-Prada J, Insuasty B, Abonia R, Quiroga J, Heeb S, Williams P, Stocks MJ, Cámara M (2018) In silico and in vitro-guided identification of inhibitors of Alkylquinolone-dependent quorum sensing in Pseudomonas aeruginosa. Molecules (Basel, Switzerland) 23(2):257. https://doi.org/10.3390/molecules23020257
Sparatore A, Basilico N, Parapini S, Romeo S, Novelli F, Sparatore F, Taramelli D (2005) 4-Aminoquinoline quinolizidinyl- and quinolizidinylalkyl-derivatives with antimalarial activity. Bioorg Med Chem 13(18):5338–5345. https://doi.org/10.1016/j.bmc.2005.06.047
Szczepaniak J, Cieslik W, Romanowicz A, Musiol R, Krasowska A (2017) Blocking and dislocation of Candida albicans Cdr1p transporter by styrylquinolines. Int J Antimicrob Agents 50(2):171–176. https://doi.org/10.1016/j.ijantimicag.2017.01.044
Talamas FX, Abbot SC, Anand S, Brameld KA, Carter DS, Chen J, Davis D, de Vicente J, Fung AD, Gong L, Harris SF, Inbar P, Labadie SS, Lee EK, Lemoine R, Le Pogam S, Leveque V, Li J, McIntosh J, Najera I, Park J, Railkar A, Rajyaguru S, Sangi M, Schoenfeld RC, Staben LR, Tan Y, Taygerly JP, Villasenor AG, Weller PE (2014) Discovery of N-[4-[6-tert-butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3-quinolyl]pheny l]methanesulfonamide (RG7109), a potent inhibitor of the hepatitis C virus NS5B polymerase. J Med Chem 57(5):1914–1931. https://doi.org/10.1021/jm401329s
Tavares GSV, Mendonca DVC, Lage DP, Granato JDT, Ottoni FM, Ludolf F, Chavez-Fumagalli MA, Duarte MC, Tavares CAP, Alves RJ, Coimbra ES, Coelho EAF (2018) Antileishmanial activity, cytotoxicity and mechanism of action of Clioquinol against leishmania infantum and leishmania amazonensis species. Basic Clin Pharmacol Toxicol 123(3):236–246. https://doi.org/10.1111/bcpt.12990
Terzic N, Konstantinovic J, Tot M, Burojevic J, Djurkovic-Djakovic O, Srbljanovic J, Stajner T, Verbic T, Zlatovic M, Machado M, Albuquerque IS, Prudencio M, Sciotti RJ, Pecic S, D’Alessandro S, Taramelli D, Solaja BA (2016) Reinvestigating old pharmacophores: are 4-aminoquinolines and tetraoxanes potential two-stage antimalarials? J Med Chem 59(1):264–281. https://doi.org/10.1021/acs.jmedchem.5b01374
Upadhyay A, Kushwaha P, Gupta S, Dodda RP, Ramalingam K, Kant R, Goyal N, Sashidhara KV (2018) Synthesis and evaluation of novel triazolyl quinoline derivatives as potential antileishmanial agents. Eur J Med Chem 154:172–181. https://doi.org/10.1016/j.ejmech.2018.05.014
Valdivieso E, Mejias F, Torrealba C, Benaim G, Kouznetsov VV, Sojo F, Rojas-Ruiz FA, Arvelo F, Dagger F (2018) In vitro 4-Aryloxy-7-chloroquinoline derivatives are effective in mono- and combined therapy against Leishmania donovani and induce mitocondrial membrane potential disruption. Acta Trop 183:36–42. https://doi.org/10.1016/j.actatropica.2018.03.023
Vandekerckhove S, Van Herreweghe S, Willems J, Danneels B, Desmet T, de Kock C, Smith PJ, Chibale K, D’Hooghe M (2015) Synthesis of functionalized 3-, 5-, 6- and 8-aminoquinolines via intermediate (3-pyrrolin-1-yl)- and (2-oxopyrrolidin-1-yl)quinolines and evaluation of their antiplasmodial and antifungal activity. Eur J Med Chem 92:91–102. https://doi.org/10.1016/j.ejmech.2014.12.020
Vial L, Lepine F, Milot S, Groleau MC, Dekimpe V, Woods DE, Deziel E (2008) Burkholderia pseudomallei, B. thailandensis, and B. ambifaria produce 4-hydroxy-2-alkylquinoline analogues with a methyl group at the 3 position that is required for quorum-sensing regulation. J Bacteriol 190(15):5339–5352. https://doi.org/10.1128/JB.00400-08
Videnovic M, Opsenica DM, Burnett JC, Gomba L, Nuss JE, Selakovic Z, Konstantinovic J, Krstic M, Segan S, Zlatovic M, Sciotti RJ, Bavari S, Solaja BA (2014) Second generation steroidal 4-aminoquinolines are potent, dual-target inhibitors of the botulinum neurotoxin serotype A metalloprotease and P. falciparum malaria. J Med Chem 57(10):4134–4153. https://doi.org/10.1021/jm500033r
Villa P, Arumugam N, Almansour AI, Suresh Kumar R, Mahalingam SM, Maruoka K, Thangamani S (2019) Benzimidazole tethered pyrrolo[3,4-b]quinoline with broad-spectrum activity against fungal pathogens. Bioorg Med Chem Lett 29(5):729–733. https://doi.org/10.1016/j.bmcl.2019.01.006
Wadhwa P, Jain P, Rudrawar S, Jadhav HRA (2018) Quinoline, coumarin and other heterocyclic analogs based HIV-1 integrase inhibitors. Curr Drug Discov Technol 15(1):2–19. https://doi.org/10.2174/1570163814666170531115452
Wanka L, Iqbal K, Schreiner PR (2013) The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem Rev 113(5):3516–3604. https://doi.org/10.1021/cr100264t
Waters NC, Edstein MD (2011) 8-Aminoquinolines: primaquine and tafenoquine. In: Treatment and prevention of malaria. Springer, Basel, pp 69–94
Wijnant GJ, Van Bocxlaer K, Yardley V, Murdan S, Croft SL (2017) Efficacy of paromomycin-chloroquine combination therapy in experimental cutaneous leishmaniasis. Antimicrob Agents Chemother 61(8):pii: e00358-17
Wilson TA, Koneru PC, Rebensburg SV, Lindenberger JJ, Kobe MJ, Cockroft NT, Adu-Ampratwum D, Larue RC, Kvaratskhelia M, Fuchs JR (2019) An Isoquinoline scaffold as a novel class of allosteric HIV-1 integrase inhibitors. ACS Med Chem Lett 10(2):215–220. https://doi.org/10.1021/acsmedchemlett.8b00633
Woodring JL, Patel G, Erath J, Behera R, Lee PJ, Leed SE, Rodriguez A, Sciotti RJ, Mensa-Wilmot K, Pollastri MP (2015) Evaluation of aromatic 6-substituted Thienopyrimidines as scaffolds against parasites that cause trypanosomiasis, Leishmaniasis, and malaria. Med Chem Commun 6(2):339–346. https://doi.org/10.1039/C4MD00441H
World Health Organization (2013). https://www.who.int/tb/features_archive/bedaquilinelaunch/en/. Accessed 20 Apr 2019
World Health Organisation (2017). https://apps.who.int/iris/bitstream/handle/10665/258973/WER9238.pdf?sequence=1. Accessed 22 Mar 2019
World Health Organisation (2018) Leishmaniasis facts. http://www.who.int/en/news-room/fact-sheets/detail/leishmaniasis. Accessed 22 Mar 2019
World Health Organisation (2019). https://apps.who.int/iris/bitstream/handle/10665/275867/9789241565653-eng.pdf?ua=1. Accessed 21 Mar 2019
Xu Z, Zhao SJ, Lv ZS, Gao F, Wang Y, Zhang F, Bai L, Deng JL (2019) Fluoroquinolone-isatin hybrids and their biological activities. Eur J Med Chem 162:396–406. https://doi.org/10.1016/j.ejmech.2018.11.032
Zablotskaya A, Segal I, Geronikaki A, Shestakova I, Nikolajeva V, Makarenkova G (2017) N-heterocyclic choline analogues based on 1,2,3,4-tetrahydro(iso)quinoline scaffold with anticancer and anti-infective dual action. Pharmacol Rep 69(3):575–581. https://doi.org/10.1016/j.pharep.2017.01.028
Zhang H, Collins J, Nyamwihura R, Ware S, Kaiser M, Ogungbe IV (2018) Discovery of a quinoline-based phenyl sulfone derivative as an antitrypanosomal agent. Bioorg Med Chem Lett 28(9):1647–1651. https://doi.org/10.1016/j.bmcl.2018.03.039
Zuo R, Garrison AT, Basak A, Zhang P, Huigens RW 3rd, Ding Y (2016) In vitro antifungal and antibiofilm activities of halogenated quinoline analogues against Candida albicans and Cryptococcus neoformans. Int J Antimicrob Agents 48(2):208–211. https://doi.org/10.1016/j.ijantimicag.2016.04.019
Acknowledgments
This study has been funded by the Ministry of Education, Science and Technological Development, Republic of Serbia (Grants No. 173048 and No.172008).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Senerovic, L., Opsenica, D., Moric, I., Aleksic, I., Spasić, M., Vasiljevic, B. (2019). Quinolines and Quinolones as Antibacterial, Antifungal, Anti-virulence, Antiviral and Anti-parasitic Agents. In: Donelli, G. (eds) Advances in Microbiology, Infectious Diseases and Public Health. Advances in Experimental Medicine and Biology(), vol 1282. Springer, Cham. https://doi.org/10.1007/5584_2019_428
Download citation
DOI: https://doi.org/10.1007/5584_2019_428
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53646-6
Online ISBN: 978-3-030-53647-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)