
Abstract
The restoration of the immune system prompted by antiretroviral therapy (ART) has allowed drastically reducing the mortality and morbidity of HIV infection. However, one main source of clinical concern is the persistence of immune hyperactivation in individuals under ART. Chronically enhanced levels of T-cell activation are associated with several deleterious effects which lead to faster disease progression and slower CD4+ T-cell recovery during ART. In this article, we discuss the rationale, and review the results, of the use of antimalarial quinolines, such as chloroquine and its derivative hydroxychloroquine, to counteract immune activation in HIV infection. Despite the promising results of several pilot trials, the most recent clinical data indicate that antimalarial quinolines are unlikely to exert a marked beneficial effect on immune activation. Alternative approaches will likely be required to reproducibly decrease immune activation in the setting of HIV infection. If the quinoline-based strategies should nevertheless be pursued in future studies, particular care must be devoted to the dosage selection, in order to maximize the chances to obtain effective in vivo drug concentrations.
Similar content being viewed by others
Background
The quest for clinical candidates to counteract immune activation has become a “hot topic” in AIDS research, because HIV infection is characterized by malignant immune hyperactivation which correlates with disease progression and poor response to antiretroviral therapy (ART) [1–5]. Moreover, immune hyperactivation is also regarded as a major obstacle to a cure for AIDS [6].
In the beginning of the millennium, an article authored by one of us launched chloroquine as a tool to inhibit viral replication and the related malignant immune activation associated with some viral diseases [7]. This article sparked a new wave of studies, in that it extended a theory, previously designed for HIV/AIDS [8], to other viral diseases characterized by excessive immune activation. As will be discussed below, by accumulating in the acidic organelles, chloroquine exerts both direct antiviral effects on enveloped viruses and decreases activation of several cell types involved in the immune response. Chloroquine has since shown promise in preclinical studies (both in vitro and in vivo), as a therapeutic agent against emerging viruses such as MERS CoV [9]. Of note, chloroquine has been indicated as a promising candidate for filovirus treatment [10], especially during the latest Ebola epidemic [11, 12]. In two studies out of three, chloroquine showed antiviral activity in mice at the maximum tolerated dose [10, 13, 14], thus rendering this drug an interesting agent for further testing of combination anti-Ebola therapies. However, the effects of chloroquine and its hydroxyl analogue hydroxychloroquine, on HIV infection, i.e. the initial target for the repurposing of these drugs, have remained controversial. On the one hand, based on the results of some earlier clinical trials, chloroquine/hydroxychloroquine has been recently re-suggested as a promising candidate to restrict the HIV-related immune activation [15, 16]. On the other hand, the results from the latest clinical trials indicate that chloroquine/hydroxychloroquine has no beneficial effect on immune activation [17, 18].
We here provide a state of the art of the studies investigating the use of chloroquine/hydroxychloroquine as a therapeutic tool for HIV/AIDS and suggest the possible biological grounds for the clinical results obtained. Moreover, we describe the reasons why our group decided to proceed further with strategies based on another drug, i.e. auranofin, which shares with chloroquine an anti-rheumatic effect [19].
Immune activation in HIV/AIDS
Several reviews have recently been published on immune activation in HIV infection [6, 16, 20, 21]. Briefly, immune hyperactivation, commonly measured as the expression levels on peripheral blood lymphocytes of markers such as HLA-DR, CD38, or CD69 correlates with, and also predicts, disease progression (reviewed in [22, 23]). Immune activation gradually decreases following therapy initiation [24] and is maintained high in immunological non-responders, who are individuals maintaining low CD4 counts despite prolonged exposure to ART [3, 4]. While the initial studies were focused on the relationship between disease progression and activation of CD8+ T-cells [1], later studies better concluded that there is a broader relationship between disease progression and immune hyperactivation, involving also CD4+ T-cells [5, 25] and innate immunity [26].
Immune activation and viral replication are believed to be mutually enhanced in a vicious circle. The virus, recognized by the immune system as non-self, induces immune activation, which, in turn, fuels viral replication by furnishing to the virus material to synthesize the different viral components. For example, lymphocyte activation increases the cytoplasmic levels of deoxyribonucleotides necessary for viral DNA synthesis by reverse transcriptase [27]. This vicious circle may still persist in anatomical compartments incompletely penetrated by ART.
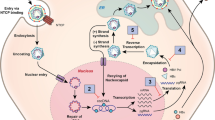
HIV-induced immune activation is not limited to specific immunity, but exerts its effects on innate immunity as well. HIV-1 was shown to activate plasmacytoid dendritic cells (pDCs), which, differently from myeloid dendritic cells (the most potent antigen-presenting cells in the body), induce innate antimicrobial immunity by producing type I interferons (Figure 1) [26]. pDCs internalize HIV-1 through viral envelope/CD4 interactions, and the internalized virus activates these cells mainly through toll-like receptor 7 (TLR-7) signaling (Figure 1). Comparative pathology corroborates the hypothesis that over-stimulation of this pathway may be associated with deleterious effects. Sooty mangabeys (Cercocebus atys), which can be infected by a simian homolog of HIV (i.e. simian immunodeficiency virus, SIV) but do not develop AIDS, display weak IFN-α production upon stimulation with TLR-7 antagonists [28]. On the contrary, rhesus macaques (Macaca mulatta), which do progress to AIDS, produce high amounts of IFN-α when their pDCs are subjected to the same stimuli [28]. Moreover, another species displaying nonpathogenic SIV infection, i.e. the African green monkey (Chlorocebus aethiops), is characterized by an efficient control of IFN-α production following acute infection [29].

Mechanistic model of HIV-induced persistent immune-activation. a HIV enters CD4-expressing plasmacytoid dendritic cells (pDCs); b the virus is endocytosed, decapsided and its RNA is recognized by toll-like receptor 7 (TLR-7); c stimulation of TLR-7 prompts a signaling cascade inducing IFN-α transcription in the nucleus; d production of IFN-α favors activation of several cell subsets such as T, B and natural killer (NK) lymphocytes. Chloroquine (CQ) is postulated to reduce the efficiency of this mechanism by accumulating in endosomes and decreasing HIV-mediated TLR-7 signaling [44].
pDCs decrease in peripheral blood during progression to AIDS, because, upon activation, they migrate to the lymphoid tissue [30]. As a huge number of cells reside in the gut-associated lymphoid tissue (GALT), according to the microbial translocation theory, the intestinal mucosa damaged by the consequent inflammation may become permeable to products of the gut microbiome which further enhance HIV-related immune hyperactivation [31, 32].
Finally, immune activation is one primary driver of both generation and maintenance of the viral reservoir, which is mainly constituted by latently infected, central and transitional memory CD4+ T-cells (henceforth TCM and TTM, respectively) [33]. Also in this case, comparative pathology has provided clues for understanding this phenomenon. It was shown that CD4+ TCM cells from sooty mangabeys express, upon activation, low levels of CCR5, the main coreceptor for virus entry into cells, thus limiting infection of this important cellular compartment [34]. Instead, activated TCM cells from AIDS-developing species, such as humans and rhesus macaques, up-regulate the levels of CCR5 to a higher extent than cells from sooty mangabeys, thus facilitating the generation of a consistent viral reservoir [34]. After these cells become quiescent, viral replication switches off, and latently infected, HIV-reservoir cells proliferate through low-level antigenic stimulation (TCM) or IL-7-driven homeostatic proliferation (TTM) [33]. Both processes are enhanced by generalized immune activation.
Mechanisms of action of chloroquine
Multiple in vitro effects of chloroquine could support its possible use as a modulator of immune activation in HIV/AIDS:
-
1.
Chloroquine and its hydroxyl analogue hydroxychloroquine were shown in several studies to inhibit HIV-1 replication (reviewed in: [7]). The effects of these quinolines, mainly due to the induction of a defect in the maturation of the viral envelope glycoprotein gp120 [35, 36], might mimic the effects of broadly neutralizing antibodies directed against the viral envelope, although the effects of these antibodies are weaker than those directed against the CD4-binding site [37]. These effects are additive to those of non-nucleosidic reverse transcriptase inhibitors (NNRTIs) and synergistic to those of protease inhibitors (PIs) [38]. As quinoline drugs accumulate in lymphoid tissues [39], they might decrease ongoing viral replication during ART in anatomical sanctuaries and, consequently switch off one of the main drivers of immune activation. Chloroquine is also an inhibitor of P-glycoprotein (P-gp) and multidrug resistance proteins (MRPs) [40, 41], cell surface glycoproteins which extrude several antiretroviral drugs to the extracellular medium. In line with this evidence, chloroquine was shown to increase the intracellular levels of PIs [38]. The effects of chloroquine in combination with NRTIs are instead controversial: some reported an additive effect [42], while others did not detect it [43]. The combined effects of chloroquine and integrase inhibitors are as yet unknown.
-
2.
Chloroquine accumulates in phagosomes of pDCs and inhibits their HIV-induced activation [44]. It might therefore impact on innate immunity-induced immune hyperactivation.
-
3.
A recent study showed that hydroxychloroquine selectively induces apoptosis in the memory T-cell compartment (CD45RA− CD45RO+) [45]. As, upon activation, naïve T-cells (CD45RA+ CD45RO−) acquire a CD45RA− CD45RO+ phenotype, the “antimemory” effect should limit immune activation (Figure 2) [46]. There is growing consensus that induction of apoptosis in the memory T-cell compartment might have a detrimental effect on the viral reservoir [47–49]. In this light, chloroquine/hydroxychloroquine should have an anti-reservoir potential. This view is supported by another recent study which shows that chloroquine sensitizes to apoptosis the latently infected cells upon viral reactivation, likely by removing the anti-apoptotic effect of the virus structural gag gene products [50]. These effects are potentially interesting, since it has been well demonstrated that viral reactivation from latency does not necessarily result in cell death [51].
Figure 2 
Comparison of the susceptibility to chloroquine/hydroxychloroquine and auranofin of the cellular subsets involved in HIV production and persistence. Shown in the figure is a schematic depiction of a activation and b differentiation stages of CD4+ T-lymphocytes and their correlation with viral production, latency and viral reactivation. Both chloroquine/hydroxychloroquine and auranofin can influence these transitions by exerting a pro-apoptotic effect, the efficacy of which is graphically exemplified by the intensity of the blue color in the corresponding rectangles. Efficacy gradients are based on data derived from Refs. [45, 48, 50].

In vivo effects of chloroquine/hydroxychloroquine: preclinical models
The macaque AIDS model is an important tool for preclinical assessment of strategies aimed at treating HIV/AIDS [52]. To our knowledge, chloroquine has been tested in this model on two occasions.
In a first study, chloroquine (25 mg every other day for 30 days, i.e. a cumulative dosage comparable to that administered to humans with rheumatioid arthritis) was administered to three Chinese rhesus macaques infected with the simian HIV-homologue, SIVmac251 [53]. Although a decrease in activated pDCs was shown, no effects were seen on viral load and CD4+ and CD8+ T-cell activation (measured as CD38 expression) [53].
As the immune activation set point is established during acute infection [4], Vaccari et al. [54] treated with chloroquine (18.7 mg/day for 112 consecutive days) seven SIVmac251-infected rhesus macaques during the viral load peak that characterizes acute infection. Apart from an unexpected, although transient, increase in the expression of interferon-regulated genes (perhaps not population-relevant as possibly driven by only one animal), no significant differences were reported in viral load and T-cell activation and proliferation (measured as expression of CD69 and Ki67, respectively) [54]. A trend was however noticed for maintenance of decreased levels of Ki67, CD69 and CCR5 in the gut of the chloroquine-treated animals, although the differences with values from the control group did not reach statistical significance. The effect of chloroquine in this simian model in the presence of ART is still unknown.
In vivo effects of chloroquine/hydroxychloroquine: clinical trials
Chloroquine and hydroxychloroquine have so far been tested in several HIV clinical trials. The results summarized in Figure 3 support the hypothesis that the chloroquine/hydroxychloroquine dosage may be an important driver of at least partial clinical success.
Suppressive effects on immune activation by chloroquine were shown in the trial conducted by Murray et al. [55]. However, in this trial, the dosage administered was not the same for all individuals, some of them receiving 500 mg/die instead of 250 mg/die. It is thus possible that the statistical significance of the effects reported in this study was driven by the higher dosage of the drug. This view is supported by a later study which tested chloroquine at 250 mg/die and failed to show any effect of the drug [18].

Published clinical studies evaluating the effects of chloroquine/hydroxychloroquine administration, alone or in combination with other drugs, in HIV infected subjects. Highlighted in blue, red or white are the studies that have reported a positive, negative, or neutral outcome of the therapy respectively. CQ chloroquine, HCQ hydroxychloroquine.
In two clinical trials conducted in the 1990s, Sperber et al. reported suppressive effects on immune activation (measured at that time as IL-6 production) and viral load in individuals treated with 800 mg of hydroxychloroquine/day (bioequivalent to 500 mg/day of chloroquine) [56, 57]. The other clinical trials testing hydroxychloroquine at a lower dosage (i.e. 400 mg/day) led to conflicting results. Earlier studies [58, 59] and the more recent study of Piconi et al. [60] reported significant effects on viral load [58], CD4 counts [59], and immune activation. [60]. Instead, a more recent clinical trial, randomized and double blind, showed disappointing results, even hinting at possibly deleterious effects of hydroxychloroquine on viral load and CD4 counts [17]. This trial was conducted in the absence of ART, and this might explain differences between this study and the study of Piconi et al., which was conducted on individuals under ART [60]. Another trial in ART-treated patients is currently ongoing and will provide more information on the effects of hydroxychloroquine (ClinicalTrials.gov identifier: NCT01232660).
The hydroxychloroquine levels show high inter-subject variability and, although individuals receiving the higher hydroxychloroquine dosages (800 and 1,200 mg/day) also showed significantly higher blood levels of the drug than those receiving 400 mg/die, the range of the blood concentrations was in part overlapping in the different dosage groups [61]. Chloroquine has similar pharmacokinetics [62]; therefore, not only the dosage but also individual differences in drug metabolism and distribution may explain the different conclusions of the aforementioned studies. A large clinical trial has recently been completed (ClinicalTrials.gov Identifier: NCT00819390) and its results can help to better represent the response of a population, thus abolishing the bias due to limited sample size. In this trial, however, chloroquine has been tested at 250 mg/day in the absence of ART; thus, in light of the results of the aforementioned clinical trials and considerations derived from basic science (see next paragraph), it is not surprising that the preliminary results released so far for this trial (https://clinicaltrials.gov/ct2/show/NCT00819390) do not show any significant effect of chloroquine on immune activation, viral load and CD4 counts.
Lessons learnt from chloroquine/hydroxychloroquine use in HIV infection
Chloroquine/hydroxychloroquine-treated individuals display blood concentrations that are highly variable and only rarely exceed 10 or 20 µM, respectively [61, 62]. Therefore, at the steady state levels, these blood concentrations only in part overlap those at which a therapeutic effect is expected. For example, the EC50 of chloroquine on PBMC proliferation upon activation is, in general, ≥10 µM [63], and this value can explain the varying results obtained in the different clinical trials, with clearer effects associated with the higher drug dosages. Similarly, the pro-apoptotic effect of hydroxychloroquine on the memory T-cells is only moderate at the concentrations reachable in blood, especially in the lower range [45, 61]. The pro-apoptotic effect of chloroquine described by Li et al. on latently infected cells upon viral reactivation is instead more marked, although still partial, at the upper range of clinically achievable blood concentrations (5–10 µM) [50]. This effect could therefore be visible in vivo in terms of viral reservoir reduction, but only treating with high chloroquine dosages in the presence of suppressive ART. Moreover, to maximize the chances to obtain viral reservoir reduction in vivo, chloroquine treatment should be prolonged, as the events of virus reactivation from latency are rather rare (estimated as one event of transition from latency to productive infection every 10 mL of blood each day) [64].
The effect of chloroquine on pDC activation (see Figure 1) was initially observed in vitro by pre-incubating pDCs with 100 µM of chloroquine for 1 h [44]. This treatment results in intracellular concentrations comparable to those observed during chronic in vivo administration [65]. In this case, the in vitro effect is in line with the results of two in vivo studies [53, 60]. The use of chloroquine-related compounds with increased potency is yielding promising results in vitro [66], and it will be interesting to test the best-performing candidates in the simian AIDS model.
The effects of chloroquine/hydroxychloroquine on viral replication have been repeatedly shown in vitro at lower drug levels than those inducing the cellular effects [35, 36, 63, 65]. The blood concentration/EC50 ratio is however much narrower than those shown by antiretroviral drugs [63]. The antiretroviral effects of chloroquine/hydroxychloroquine may though become visible in anatomical sanctuaries of those individuals treated with PI-containing antiretroviral regimens. In any case, we recommend that chloroquine/hydroxychloroquine be tested at the highest recommended dosages in future HIV clinical trials.
Alternative/complementary interpretations of the results so far obtained are possible. For example, the effectiveness of the ART regimen employed may play a role in determining the magnitude of the effects (if any) observed following chloroquine/hydroxichloroquine addition. The study of Piconi et al. [60], showing some benefit in immunological non responders, may indicate that the effects of chloroquine may be visible only in some subsets of individuals with peculiar immunological characteristics, and that these effects can be hindered when immunologically non homogeneous cohorts are studied. In this regard, larger studies, with cohorts stratified according to immunological responsiveness to ART, could provide further information on the effects of chloroquine/hydroxychloroquine.
Another open question remains the influence of the duration of drug exposure, as it has been shown that chloroquine/hydroxychloroquine has cumulative effects [67]. As a proportion of HIV-infected patients in Africa may already be on chloroquine medication to prevent malaria, it might be worth examining the long-term effects of this treatment. In this regard, an ongoing phase III clinical trial will assess the long-term effects of chloroquine and trimethoprim-sulfamethoxazole phrophylaxis on survival and disease control in HIV-infected individuals with suppressed viral load and good clinical response to ART [68].
Current and future directions: another approach based on antirheumatic therapy
Given the aforementioned problems in the pharmacokinetics of chloroquine/hydroxychloroquine, our group chose to follow a different, yet partly similar, approach to corroborate treatment of HIV/AIDS. Based on the feedback received from basic science studies and clinical trials that have been published throughout the years, we decided to use drugs the desired effects of which be striking in vitro at concentrations lower than the trough plasma concentrations in vivo. We also decided to re-direct our research on the basis of the plasma concentrations rather than on whole-blood concentrations (widely used for chloroquine/hydroxychloroquine), because we thought that the former might better mimic the tissue culture concentrations. The drug that we selected is the gold-based compound auranofin, the pharmacodynamics and pharmacokinetics of which are well known, due to its decade-long employment for treatment of rheumatoid arthritis [69].
The main rationale for the use of auranofin in our studies was its ability to target the central/transitional memory CD4+ T-cell compartment (Figure 2) [48, 70], which is known to harbor the main viral reservoir in patients receiving ART [33]. Auranofin is drastically active at sub-micromolar (i.e. ≤250 nM) concentrations, which are below those readily achievable in human plasma [71]. The administration of auranofin ultimately led to a reduction of the viral reservoir in ART-treated SIVmac251-infected macaques [70]. A review on our preclinical studies has recently been published [46] and the reader is addressed to it for further detail. Not surprisingly for a drug effective against an autoimmune disease such as rheumatoid arthritis, auranofin may as well be beneficial in terms of reduction of cell activation. In particular, the downregulation of the CD28 molecule induced by auranofin can disrupt the co-stimulatory signal often crucial for lymphocyte activation [48]. Moreover, apart from memory CD4+ T-cells, auranofin also targets the memory CD8+ T-cell compartment [48], i.e. a cellular subset known to be hyperactivated during HIV infection [2]. Interestingly, as described for hydroxychloroquine [60], auranofin was shown to disrupt in various cell lines the TLR-4 signaling [72], which is activated by bacterial lipopolysaccharides and likely constitutes another source of immune hyperactivation. In vitro data indicate that the impact of auranofin on lymphocyte activation may be mediated, at least in part, by modulation of oxidative stress [48]. Of note, the addition of a potent pro-oxidant drug, such as buthionine sulfoximine (BSO), increases the potency of auranofin, decreasing phytohemagglutinin-induced activation and expression of the α-chain of the IL-2 receptor [73]. This is in line with our preliminary data in SIVmac251-infected macaques, in which a combined regimen of ART, auranofin and BSO induced a functional cure-like condition following suspension of all therapies [74]. These observations provide proof of concept that drastically decreasing immune hyperactivation arrests SIV disease progression and turns the virus/immune system balance in favor of the latter. Clinical trials will be required to assess the potential of auranofin to decrease immune activation in ART-treated subjects.
Finally, other drugs used or proposed for treatment of rheumatoid arthritis might find a place in the treatment of HIV/AIDS. For example, the janus kinase inhibitors tofacitinib and ruxolitinib have shown a promising in vitro activity against HIV replication [75]. The ongoing in vivo studies on these compounds could provide an opportunity to analyze the effects of this treatment on viral replication and immune activation.
Abbreviations
- ART:
-
antiretroviral therapy
- AIDS:
-
acquired immunodeficiency syndrome
- BSO:
-
buthionine sulfoximine
- CCR5:
-
C–C chemokine receptor type 5
- CD:
-
cluster of designation/differentiation
- GALT:
-
gut associated lymphoid tissue
- HIV:
-
human immunodeficiency virus
- IL:
-
interleukin
- MERS CoV:
-
middle east respiratory syndrome coronavirus
- MRP:
-
multidrug resistance protein
- NRTI:
-
nucleosidic reverse transcriptase inhibitor
- NNRTI:
-
non-nucleosidic reverse transcriptase inhibitor
- pDC:
-
plasmacytoid dendritic cell
- P-gp:
-
P-glycoprotein
- PI:
-
protease inhibitor
- SIV:
-
simian immunodeficiency virus
- TLR:
-
toll-like receptor
- TCM :
-
central memory T-cell
- TTM :
-
transitional memory T-cell
References
Giorgi JV, Ho HN, Hirji K, Chou CC, Hultin LE, O’Rourke S et al (1994) CD8+ lymphocyte activation at human immunodeficiency virus type 1 seroconversion: development of HLA-DR+ CD38− CD8+ cells is associated with subsequent stable CD4+ cell levels. The Multicenter AIDS Cohort Study Group. J Infect Dis 170(4):775–781
Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA et al (1996) Increased numbers of primed activated CD8+ CD38+ CD45RO+ T cells predict the decline of CD4+ T cells in HIV-1-infected patients. AIDS 10(8):827–834
Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H et al (2003) T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis 187(10):1534–1543
Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narváez AB et al (2004) Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104(4):942–947
Hunt PW, Brenchley J, Sinclair E, McCune JM, Roland M, Page-Shafer K et al (2008) Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis 197(1):126–133
Hatano H (2013) Immune activation and HIV persistence: considerations for novel therapeutic interventions. Curr Opin HIV AIDS 8(3):211–216
Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (2003) Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis 3(11):722–727 (Review)
Boelaert JR, Piette J, Sperber K (2001) The potential place of chloroquine in the treatment of HIV-1-infected patients. J Clin Virol 20(3):137–140 (Review)
de Wilde AH, Jochmans D, Posthuma CC, Zevenhoven-Dobbe JC, van Nieuwkoop S, Bestebroer TM et al (2014) Screening of an FDA-approved compound library identifies four small-molecule inhibitors of middle east respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother 58(8):4875–4884
Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC et al (2013) A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One 8(4):e60579
De Clercq E (2015) Ebola virus (EBOV) infection: therapeutic strategies. Biochem Pharmacol 93(1):1–10 (Review)
Long J, Wright E, Molesti E, Temperton N, Barclay W (2015) Antiviral therapies against Ebola and other emerging viral diseases using existing medicines that block virus entry [v1; ref status: awaiting peer review, http://f1000r.es/510]. F1000Research 4:30
Madrid PB, Panchal R, Warren T, Shurtleff A, Endsley A, Green C et al (2015) Evaluation of Ebola virus inhibitors for drug repurposing. ACS Infect Dis (in press)
Falzarano D, Safronetz D, Prescott J, Marzi A, Feldmann F, Feldmann H (2015) Lack of protection against Ebola virus from chloroquine in mice and hamsters. Emerg Infect Dis (in press)
Saez-Cirion A, Jacquelin B, Barré-Sinoussi F, Müller-Trutwin M (2014) Immune responses during spontaneous control of HIV and AIDS: what is the hope for a cure? Philos Trans R Soc Lond B Biol Sci 369(1645):20130436
Rajasuriar R, Khoury G, Kamarulzaman A, French MA, Cameron PU, Lewin SR (2013) Persistent immune activation in chronic HIV infection: do any interventions work? AIDS 27(8):1199–1208
Paton NI, Goodall RL, Dunn DT, Franzen S, Collaco-Moraes Y, Gazzard BG et al (2012) Hydroxychloroquine Trial Team: effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: a randomized controlled trial. JAMA 308(4):353–361
Routy JP, Angel J, Patel M, Kanagaratham C, Radzioch D, Kema I et al (2015) Assessment of chloroquine as a modulator of immune activation to improve CD4 recovery in immune nonresponding HIV-infected patients receiving antiretroviral therapy. HIV Med 16(1):48–56
Rao UR, Naidu MU, Kumar TR, Shobha U, Askar MA, Ahmed N et al (1995) Comparison of phenytoin with auranofin and chloroquine in rheumatoid arthritis—a double blind study. J Rheumatol 22(7):1235–1240
Klatt NR, Chomont N, Douek DC, Deeks SG (2013) Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev 254(1):326–342 (Review)
Paiardini M, Müller-Trutwin M (2013) HIV-associated chronic immune activation. Immunol Rev 254(1):78–101 (Review)
Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW (2013) Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol 119:51–83 (Review)
Lichtfuss GF, Hoy J, Rajasuriar R, Kramski M, Crowe SM, Lewin SR (2011) Biomarkers of immune dysfunction following combination antiretroviral therapy for HIV infection. Biomark Med 5(2):171–186 (Review)
Autran B, Carcelain G, Li TS, Blanc C, Mathez D, Tubiana R et al (1997) Positive effects of combined antiretroviral therapy on CD4+ Tcell homeostasis and function in advanced HIV disease. Science 277(5322):112–116
Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP et al (1999) Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 179(4):859–870
Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG et al (2005) Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest 115(11):3265–3275
Gao WY, Cara A, Gallo RC, Lori F (1993) Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc Natl Acad Sci USA 90(19):8925–8928
Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S et al (2008) Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med 14(10):1077–1087
Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G et al (2009) Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest 119(12):3544–3555
Sachdeva N, Asthana V, Brewer TH, Garcia D, Asthana D (2008) Impaired restoration of plasmacytoid dendritic cells in HIV-1-infected patients with poor CD4 T cell reconstitution is associated with decrease in capacity to produce IFN-alpha but not proinflammatory cytokines. J Immunol 181(4):2887–2897
Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW (2013) Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol 119:51–83
Marchetti G, Tincati C, Silvestri G (2013) Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev 26(1):2–18 (Review)
Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B et al (2009) HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15(8):893–900
Paiardini M, Cervasi B, Reyes-Aviles E, Micci L, Ortiz AM, Chahroudi A et al (2011) Low levels of SIV infection in sooty mangabey central memory CD4+ T cells are associated with limited CCR5 expression. Nat Med 17(7):830–836
Tsai WP, Nara PL, Kung HF, Oroszlan S (1990) Inhibition of human immunodeficiency virus infectivity by chloroquine. AIDS Res Hum Retroviruses 6(4):481–489
Naarding MA, Baan E, Pollakis G, Paxton WA (2007) Effect of chloroquine on reducing HIV-1 replication in vitro and the DC-SIGN mediated transfer of virus to CD4+ T-lymphocytes. Retrovirology 4:6
Forsman A, Beirnaert E, Aasa-Chapman MM, Hoorelbeke B, Hijazi K, Koh W et al (2008) Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J Virol 82:12069–12081
Savarino A, Lucia MB, ter Heine R, Rastrelli E, Rutella S, Majori G et al (2006) Quinoline antimalarials as investigational drugs for HIV-1/AIDS: in vitro effects on HIV-1 replication, HIV-1 response to antiretroviral drugs, and intracellular antiretroviral drug concentrations. Drug Dev Res 67(10):806–817
Aguirre-Cruz L, Torres KJ, Jung-Cook H, Fortuny C, Sánchez E, Soda-Mehry A et al (2010) Short communication: preferential concentration of hydroxychloroquine in adenoid tissue of HIV-infected subjects. AIDS Res Hum Retroviruses 26(3):339–342
Klohs WD, Steinkampf RW (1988) The effect of lysosomotropic agents and secretory inhibitors on anthracycline retention and activity in multiple drug-resistant cells. Mol Pharmacol 34(2):180–185
Vezmar M, Georges E (2000) Reversal of MRP-mediated doxorubicin resistance with quinoline-based drugs. Biochem Pharmacol 59(10):1245–1252
Boelaert JR, Sperber K (1998) Antiretroviral treatment. Lancet 352(9135):1224–1225
Torres KJ, Reyes-Terán G, Sotelo J, Jung-Cook H, Aguirre-Cruz L (2015) Influence of quinacrine and chloroquine on the in vitro 3′-azido-3′-deoxythymidine antiretroviral effect. AIDS Res Ther 12:7 (eCollection 2015)
Martinson JA, Montoya CJ, Usuga X, Ronquillo R, Landay AL, Desai SN (2010) Chloroquine modulates HIV-1-induced plasmacytoid dendritic cell alpha interferon: implication for T-cell activation. Antimicrob Agents Chemother 54(2):871–881
van Loosdregt J, Spreafico R, Rossetti M, Prakken BJ, Lotz M, Albani S (2013) Hydroxychloroquine preferentially induces apoptosis of CD45RO + effector T cells by inhibiting autophagy: a possible mechanism for therapeutic modulation of T cells. J Allergy Clin Immunol 131(5):1443–6.e1
Shytaj IL, Savarino A (2013) A cure for AIDS: a matter of timing? Retrovirology 10:145
Chomont N, DaFonseca S, Vandergeeten C, Ancuta P, Sékaly RP (2011) Maintenance of CD4+ T-cell memory and HIV persistence: keeping memory, keeping HIV. Curr Opin HIV AIDS 6(1):30–36 (Review)
Chirullo B, Sgarbanti R, Limongi D, Shytaj IL, Alvarez D, Das B et al (2013) A candidate anti-HIV reservoir compound, auranofin, exerts a selective ‘anti-memory’ effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis 4:e944
Zhan XY, Wang N, Liu G, Qin L, Xu W, Zhao S et al (2014) Plasmodium infection reduces the volume of the viral reservoir in SIV-infected rhesus macaques receiving antiretroviral therapy. Retrovirology 11(1):112
Li H, Hatfield G, Pauza CD (2014) Gag-mediated autophagy promotes CD4 T cell survival: a possible mechanism for HIV reservoir persistence. XX international AIDS conference, towards an HIV cure symposium; Melbourne, Australia. PE18 LB
Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC et al (2012) Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36(3):491–501
Evans DT, Silvestri G (2013) Nonhuman primate models in AIDS research. Curr Opin HIV AIDS 8(4):255–261 (Review)
Ma JP, Xia HJ, Zhang GH, Han JB, Zhang LG, Zheng YT (2012) Inhibitory effects of chloroquine on the activation of plasmacytoid dendritic cells in SIVmac239-infected Chinese rhesus macaques. Cell Mol Immunol 9(5):410–416
Vaccari M, Fenizia C, Ma ZM, Hryniewicz A, Boasso A, Doster MN et al (2014) Transient increase of interferon-stimulated genes and no clinical benefit by chloroquine treatment during acute simian immunodeficiency virus infection of macaques. AIDS Res Hum Retroviruses 30(4):355–362
Murray SM, Down CM, Boulware DR, Stauffer WM, Cavert WP, Schacker TW et al (2010) Reduction of immune activation with chloroquine therapy during chronic HIV infection. J Virol 84(22):12082–12086
Sperber K, Louie M, Kraus T, Proner J, Sapira E, Lin S et al (1995) Hydroxychloroquine treatment of patients with human immunodeficiency virus type 1. Clin Ther 17(4):622–636
Sperber K, Chiang G, Chen H, Ross W, Chusid E, Gonchar M et al (1997) Comparison of hydroxychloroquine with zidovudine in asymptomatic patients infected with human immunodeficiency virus type 1. Clin Ther 19(5):913–923
Paton NI, Aboulhab J, Karim F (2002) Hydroxychloroquine, hydroxycarbamide, and didanosine as economic treatment for HIV-1. Lancet 359(9318):1667–1668
Paton NI, Aboulhab J (2005) Hydroxychloroquine, hydroxyurea and didanosine as initial therapy for HIV-infected patients with low viral load: safety, efficacy and resistance profile after 144 weeks. HIV Med 6(1):13–20
Piconi S, Parisotto S, Rizzardini G, Passerini S, Terzi R, Argenteri B et al (2011) Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy-treated immunologic nonresponders. Blood 118(12):3263–3272
Munster T, Gibbs JP, Shen D, Baethge BA, Botstein GR, Caldwell J et al (2002) Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum 46(6):1460–1469
Augustijns P, Geusens P, Verbeke N (1992) Chloroquine levels in blood during chronic treatment of patients with rheumatoid arthritis. Eur J Clin Pharmacol 42(4):429–433
Savarino A, Gennero L, Chen HC, Serrano D, Malavasi F, Boelaert JR et al (2001) Anti-HIV effects of chloroquine: mechanisms of inhibition and spectrum of activity. AIDS 15(17):2221–2229
Rong L, Perelson AS (2009) Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput Biol 5(10):e1000533
Sperber K, Kalb TH, Stecher VJ, Banerjee R, Mayer L (1993) Inhibition of human immunodeficiency virus type 1 replication by hydroxychloroquine in T cells and monocytes. AIDS Res Hum Retroviruses 9(1):91–98
Royle CM, Tsai MH, Tabarrini O, Massari S, Graham DR, Aquino VN et al (2014) Modulation of HIV-1-induced activation of plasmacytoid dendritic cells by 6-desfluoroquinolones. AIDS Res Hum Retroviruses 30(4):345–354
Tsang AC, Ahmadi Pirshahid S, Virgili G, Gottlieb CC, Hamilton J, Coupland SG (2015) Hydroxychloroquine and chloroquine retinopathy: a systematic review evaluating the multifocal electroretinogram as a screening test. Ophthalmology [Epub ahead of print]
Mehraj V, Jenabian MA, Vyboh K, Routy JP (2014) Immune suppression by myeloid cells in HIV infection: new targets for immunotherapy. Open AIDS J 8:66–78
Suarez-Almazor ME, Spooner CH, Belseck E, Shea B (2000) Auranofin versus placebo in rheumatoid arthritis. Cochrane Database Syst Rev. (2):CD002048 (Review)
Lewis MG, DaFonseca S, Chomont N, Palamara AT, Tardugno M, Mai A et al (2011) Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 25(11):1347–1356
Lewis D, Capell HA, McNeil CJ, Iqbal MS, Brown DH, Smith WE (1983) Gold levels produced by treatment with auranofin and sodium aurothiomalate. Ann Rheum Dis 42(5):566–570
Youn HS, Lee JY, Saitoh SI, Miyake K, Hwang DH (2006) Auranofin, as an anti-rheumatic gold compound, suppresses LPS-induced homodimerization of TLR4. Biochem Biophys Res Commun 350(4):866–871
Vint IA, Chain BM, Foreman JC (1993) The interaction of auranofin and buthionine sulfoximine blocks activation of human peripheral T lymphocytes. Cell Immunol 152(1):152–161
Shytaj IL, Chirullo B, Wagner W, Ferrari MG, Sgarbanti R, Corte AD et al (2013) Investigational treatment suspension and enhanced cell-mediated immunity at rebound followed by drug-free remission of simian AIDS. Retrovirology 10:71
Gavegnano C, Detorio M, Montero C, Bosque A, Planelles V, Schinazi RF (2014) Ruxolitinib and tofacitinib are potent and selective inhibitors of HIV-1 replication and virus reactivation in vitro. Antimicrob Agents Chemother 58(4):1977–1986
Authors’ contributions
AS and ILS contributed to the ideas and to the interpretation of the data presented in the manuscript. AS and ILS contributed to manuscript drafting and preparation of the figures. Both authors read and approved the final manuscript.
Acknowledgements
ILS is supported by a fellowship from the “Sapienza” University (Rome, Italy).
Compliance with ethical guidelines
Competing interests The Istituto Superiore di Sanità holds a US patent on the use of the auranofin for treatment of HIV/AIDS. AS is the leading inventor of the patent.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Savarino, A., Shytaj, I.L. Chloroquine and beyond: exploring anti-rheumatic drugs to reduce immune hyperactivation in HIV/AIDS. Retrovirology 12, 51 (2015). https://doi.org/10.1186/s12977-015-0178-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-015-0178-0