
Abstract
Ixodes holocyclus is the paralysis tick commonly found in Australia. I. holocyclus does not cause paralysis in the primary host – bandicoots, but markedly affects secondary hosts such as companion animals, livestock and humans. Holocyclotoxins are the neurotoxin molecules in I. holocyclus responsible for paralysis symptoms. There is a limited understanding of holocyclotoxins due to the difficulties in purifying and expressing these toxins in vitro. Next-generation sequencing technologies were utilised for the first time to generate transcriptome data from two cDNA samples –salivary glands samples collected from female adult ticks engorged on paralysed companion animals and on bandicoots. Contig-encoded proteins in each library were annotated according to their best BLAST match against several databases and functionally assigned into six protein categories: housekeeping, transposable elements, pathogen-related, hypothetical, secreted and novel. The “secreted protein” category is comprised of ten protein families: enzymes, protease inhibitors, antigens, mucins, immunity-related, lipocalins, glycine-rich, putative secreted, salivary and toxin-like. Comparisons of contig representation between the two libraries reveal the differential expression of tick proteins collected from different hosts. This study provides a preliminary description of the I. holocyclus tick salivary gland transcriptome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
I. Introduction
A. Ixodes holocyclus
Ticks belong to the Class Arachnida, which includes other arthropods such as spiders and scorpions. Ticks are members of the Subclass Acari that consists of three families: Argasidae, Ixodidae and Nutalliellidae [1]. Ixodes holocyclus is a tick species categorized in the family Ixodidae comprised of hard-bodied ticks. In addition, they are referred to as one of the most ancient terrestrial arachnids and possibly the earliest organisms to have evolved with blood-feeding capabilities [2].
I. holocyclus is a native and unique paralysis tick species of Australia that is commonly found in areas of high humidity and moderate temperature along the east coast of Australia [3, 4]. The primary host of I. holocyclus are native marsupials such as bandicoots, whereas livestock, companion animals and humans are considered as secondary hosts. Figure 1 describes the lifecycles of the three-host tick, I. holocyclus, which is conformed by four lifecycle stages from egg, larva, nymph to adult. As for other Ixodidae (hard ticks), I. holocyclus can feed on hosts for a minimum few days up to several weeks [4, 5].
I. holocyclus is also a vector of various diseases, for example Rickettsial Spotted Fever, and is able to transmit pathogens to hosts during infestation [6, 7]. I. holocyclus is capable of inducing paralysis in hosts upon engorgement, which eventually leads to death if left untreated [8]. In addition, I. holocyclus is recognised as the most severe and harmful form of tick paralysis compared to its counterparts such as Dermacenor andersoni and Dermacentor variabilis in North America and Rhipicephalus evertsi evertsi in Africa [6]. In contrast, paralysis symptoms caused by I. holocyclus infestations are often irreversible and deaths occur regardless of the immediate treatment given [8]. Between 1900 and 1945, there were approximately 30 fatal human cases reported caused by I. holocyclus, victims were mostly under 4 years of age [5, 9]. Domestic animals are the major tick paralysis victims with an estimated number of 100,000 animals affected by the Australian tick paralysis each year [4, 10]. Five to ten percent of affected canines presenting to veterinary clinics reported fatal regardless of tick removal and antivenom administration [11, 12].
The development of paralysis syndrome is dependent on the duration of infestation and the composition of the salivary gland (SG). Generally, it takes 3-5 days for clinical symptoms to appear in victims of I. holocyclus. The clinical presentation includes loss of appetite, voice and coordination. Other severe indications are ascending flaccid paralysis, excessive salivation, asymmetric pupillary dilation and respiratory distress [13].

The lifecycle of the three-host tick Ixodes holocyclus [5].
B. Tick saliva and sialome studies
The saliva of blood-feeding ticks play an essential role for effective and successful feeding. A large array of saliva components are known to facilitate tick-host interactions and counteract the hosts’ defences [14, 15]. To provide a comprehensive understanding of tick saliva, next-generation sequencing, which generates high-throughput sequencing data and deep coverage of transcriptomes [16], has been used to generate the salivary gland transcriptome (sialome) of several genera within ticks [17-20]. Sialome studies revealed that different tick species have a set of novel salivary proteins or protein families, which are unique and beneficial to their own hematophagy (blood-feeding behaviour) [21, 22]. These novel salivary proteins are for example anti-haemostatic, anti-inflammatory and immune-modulatory components, which facilitate the tick hematophagy [15]. Previous studies on Ixodes scapularis and Rhipicephalus appendiculatus suggested that the variation among tick sialomes is influenced by tick feeding conditions such as the vertebrate host(s) and/or developmental stages [23, 24]. However only 3 sialomes, out of the 243 tick species in the genus of Ixodes, have been described to date including I. scapularis [20, 24], Ixodes pacificus [25] and Ixodes ricinus [17, 26].
Two cDNA libraries were constructed in this study with large numbers of transcripts yielded from feeding female adult I. holocyclus ticks engorged on different hosts. This research paper aimed to provide a preliminary description of the transcriptomes of the Australian paralysis tick, I. holocyclus by annotating the contigs obtained from the two cDNA libraries and to understand the differential expression of I. holocyclus protein families when engorged on different hosts.
II. Materials and methods
A. Two cDNA samples
Two pooled RNA samples were prepared from feeding female adult ticks for cDNA library construction and sequencing. Fully engorged female adult I. holocyclus were collected from 2 groups of hosts – (i) paralysed cats and dogs, and (ii) bandicoots. The first sample (SG_FA_CD) and second sample (SG_FA_BA) were SG RNA obtained from female adult ticks collected from cats and dogs with paralysis symptoms and bandicoots, respectively.
B. Next generation sequencing
The cDNA libraries were sent to the Australian Genome Research Facility Ltd (AGRF) for next generation sequencing. The SG_FA_CD cDNA sample was sequenced using Illumina HiSeq. The Illumina sequenced reads were then assessed with FastQC [27] to avoid inherent biases associated with sequencing and the starting libraries. In order to standardize the sequencing reads and to eliminate sequencing errors prior to de novo assembly, the processed Illumina reads were subjected to Content Dependent Read Trimmer (ConDeTri) [28], which removes redundant and low scoring reads. Assemblies of Illumina sequences were performed using Velvet short read assembler [29] for each odd kmer ranged between 59-79, then merged using Velvet’s Oases [30]. The second SG_FA_BA sample was sequenced using 454 FLX Roche technologies. The 454 pyrosequences were assembled with GS Assembler (Newbler) v2.5.3 [31], and resulting final contig assemblies were examined in this study.
C. Contig annotation and contig representation in each library
The contigs of the assembled ESTs (Expressed Sequence Tag) in the four individual libraries were blast using different BLAST searches (blastx, balstn, or rpblast searches) against various databases, including non-redundant protein database (NR) of the National Centre for Biotechnology Information (NCBI) [32], the Swiss-Prot database [33], gene ontology (GO) fasta subset [34], the NCBI conserved domains database [35], KOG [36], KEGG [37], PFAM [38] and SMART [39] motifs for contig annotation. All BLAST searches were undertaken using Yabi [40] with the default parameters.
Transcripts, which have no significant hit with any records in different databases, were submitted to the SignalP server [41] to identify possible secreted proteins and the TMHMM server [42] to detect possible trans-membrane helices. Finally, all transcripts in each individual library were functionally classified by the key words provided from the BLAST match, considering their significant hits from different search engines. All annotated contigs for each individual library are compiled in a hyperlinked Excel spreadsheet. The number of contigs per functional group of each library was calculated for comparison of contig representation between the two I. holocyclus libraries.
III. Results
A. Next generation sequencing and sequencing assembly
A total of 130,071,262 reads were generated for SG_FA_CD using Illumina technology. To improve the read quality, Illumina read quality was filtered using FastQC and ConDenTri which filtered out 19,968,632 poor quality reads and 58,020,916 redundant reads, resulting in 26,040,857 paired end reads (52,081,714 reads) for the SG_FA_CD sample. The assembly of Illumina paired-end reads yielded 41,797 contigs with an average consensus size of 830bp for the SG_FA_CD library. Alternatively, 454 pyrosequencing generated 286,047 reads with an average read length of 310bp for SG_FA_BA. SG_FA_CD 454 pyrosequencing reads were assembled into 3,772 with an average consensus size of 663bp.
B. Functional annotation
All contigs from the two libraries were annotated and can be viewed in Supplementary files 1 and 2. Column A contains a specific identification code for each contig. The descriptions returned from different BLAST searches against various databases are summarized in column D. Columns E and F describe hits returned from NR and the NR score. Columns G-J contain hits returned with Uniprot and the Uniprot alignment score, Expected-value E, and sequence similarity. While columns K-P describe Enzyme commission number, KOG function, KOG general categories, KEGG pathway ID, KEGG pathways, KEGG general category, followed by hits returned from SMART, PFAM ISU, SSU, SignalP and TMHMM.
Based on the best matches, each protein-encoded contig, for each I. holocyclus library, was functionally annotated in the Excel spreadsheet using the classification of Schwarz et al [33]. All contigs were functionally classified into 15 main protein families (column C) describing their predicted functions – housekeeping, hypothetical, pathogen-related, transposable elements, novel, putative secreted, saliva, mucin, glycine rich, enzyme, lipocalin, protease inhibitor, toxin like, immunity related and antigen. The nineteen protein families were conceptually grouped into 6 categories (column D), which are housekeeping products, hypothetical proteins, pathogen-related sequences, transposable elements, novel products and secreted proteins.
Housekeeping contigs were proteins involved in organismal systems, environmental/ cellular processes and signaling, information storage and processing and metabolism. Hypothetical proteins are proteins labeled hypothetical and putative in NCBI databases, which are predicted proteins that have not been well characterized. All ticks used for this transcriptome study were collected from the field (including from hosts) and may carry pathogens and/or symbionts. Pathogen-related are associated with several contigs originating from pathogens found in the two I. holocyclus libraries. A minor number of contigs were reported to be transposable elements, including both class I (retrotransposons) and class II (transposons) transposable elements, reverse transcriptases and several transposases. Contigs, which did not have significant similarities with protein or nucleotide sequences on current databases were classified under novel products and are predicted to be unique proteins in I. holocyclus. The remaining 10 protein families are classified under the category of “Secreted”. Descriptions of secreted protein families, as described in previous studies, are summarized in Table 5. Figures 5 and 6 depict the classification of the categories and protein families in the two I. holocyclus libraries.
B. 1. Functional annotations for Salivary Gland collected from Female Adults collected from paralysed Cats and Dogs (SG_FA_CD)
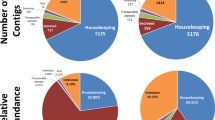
For the SG_FA_CD library, a total number of 21,982 contigs (52.85%) were predicted to encode for novel proteins (Figure 2). Hypothetical proteins were 34.84% (n=14,657) while pathogen-related proteins and transposable elements represent 0.23% (n=98) and 0.24% (n=106) respectively from all the SG_FA_CD contigs. Housekeeping products comprised of 8.69% (n=3,746) of the contigs. There were 1208 contigs (2.87%) encoding for secreted proteins. Most of the secreted proteins (26.82%) were enzymes.

Functional classification of contig-encoded Ixodes holocyclus proteins in SG_FA_CD library. The overall contig proportion of different protein families/categories is shown, and all contigs categorized under “Secreted” were further divided into different subfamilies.

Functional classification of contig-encoded Ixodes holocyclus proteins in SG_FA_BA library. The overall contig proportion of different protein families/categories is shown, and all contigs categorized under “Secreted” were further divided into different subfamilies.
B. 2. Functional annotations for Salivary Gland collected from Female Adults collected from BAndicoots (SG_FA_BA).
Unlike contigs assembled from Illumina reads generated from female adult ticks engorged on companion animals with paralysis symptoms, the largest group of contigs in SG_FA_BA encoded for hypothetical proteins, which is comprised of 36.66% (n=1,383) of the all the contigs for the SG_FA_BA library (Figure 3). In the SG_FA_BA library, novel transcripts are the second most abundant group of contigs and is made up of 26.43% (n=997) of the total contigs, followed by housekeeping proteins (20.44%, n=771), secreted proteins (16.41%, n=619), and pathogen-related proteins (0.05%, n=2). Putative secreted proteins, which are predicted secreted proteins previously identified in other organisms, are the major groups of transcripts under secreted proteins (23.26%, n=144) in the SG_FA_BA library.
B.3 Contig representation in the two Ixodes holocyclus next-generation libraries
Comparison was made to study the expression of different protein families from the salivary gland of I. holocyclus collected from different hosts: salivary glands collected from companion animals with paralysis symptoms (SG_FA_CD) and bandicoots (SG_FA_BA). In order to visualize the contig representation in each library and the comparisons, percentages of the six protein family categories and secreted protein families were calculated to generate comparative graphs (Figure 4).

Comparing the abundance of six categories of protein families between salivary gland samples collected from adult female ticks engorged on paralysed companion animals (SG_FA_CD) and bandicoots (SG_FA_BA). Red bars represent the SG_FA_CD library while green bars represent the SG_FA_BA library.

Comparing the abundance of secreted protein families between salivary gland samples collected from adult female ticks engorged on paralysed companion animals (SG_FA_CD) and bandicoots (SG_FA_BA). Red bars represent the SG_FA_CD library while green bars represent the SG_FA_BA library.
B.4 Comparison of Salivary Gland collected from Female Adults collected from paralysed Cats and Dogs (SG_FA_CD) and Salivary Gland collected from Female Adults collected from BAndicoots (SG_FA_BA) libraries
The SG_FA_CD and SG_FA_BA libraries show significant differences in the secreted and novel categories (Figure 4). For the contigs representing secreted proteins, SG_FA_CD yielded only 2.87%, which is approximately eight times less that SG_CD_BA, which yielded 16.41% of secreted proteins. In contrast, SG_FA_CD yielded 52.85% of contigs coding for novel products, which is two times more than SG_FA_BA, which yielded 26.43% novel proteins. Figure 5 further demonstrated the differences in the yields of different secreted protein families, significantly for salivary, enzyme, lipocalin, protease inhibitor and immunity-related secreted proteins.
IV. Discussion
Classical Sanger sequencing was utilized for most previous tick sialome studies [17, 24, 25, 43-46] and only a few have used next-generation sequencing [26, 47]. A comparison between I. ricinus transcriptomes generated using Sanger and next-generation sequencing technologies discovered a higher and more comprehensive transcriptome coverage with next-generation sequencing methodologies, which is beneficial in providing in-depth data towards understanding SG transcript dynamics [17, 26]. The high coverage of next-generation transcriptome sequencing demonstrated a more accurate distribution of sequence reads, which reflects the patterns of gene expression during tick feeding. In addition, even in the absence of a reference genome, next generation sequencing has been proven to produce sufficient numbers of sequence reads and thus a reliable and robust transcriptome assembly for genome studies [48]. In this study, the sialome from I. holocyclus was studied for the first time in the absence of a reference genome. We employed both 454 and Illumina next-generation sequencing methodologies in this study. This is also the first paralysis tick sialome study, which provides a preliminary resource and pioneering step towards a comprehensive understanding of paralysis tick saliva composition.
In order to counteract the host defense system and achieve successful feeding, ticks have developed unique saliva compositions, which has been revealed in previous salivary gland transcriptome (sialome) studies [49, 50]. The transcriptome analysis conducted in this study corroborated previous reports through the identification of ten secreted protein families (Table 5) reported in the two I. holocyclus cDNA libraries. These protein families were classified as:
Antigen
Two types of antigen families were identified in I. holocyclus – antigen 5 and antigen P23. Members of antigen 5 protein families are found in the venom of Vespid wasps and snakes and were identified previously in many hematophagous tick transcriptomes [51, 52]. However, most of the antigen 5 proteins have no known function. Antigen P23 is protein identified in I. scapularis and has demonstrated anti-coagulation activity [53].
Enzymes
Some of the enzymes identified in the saliva of hematophagous arthropods were shown to assist with blood feeding. For example 5’ nucleotidase, which is also called apyrase, hydrolyses ATP and ADP released by damaged cells to prevent activation of platelets and neutrophils [15]. Metalloproteases are another large group of hematophagy-assisting enzymes, which inhibits the production of fibrin and fibrinogen [54]. Endonuclease has been found in the saliva of the mosquito Culex pipiens quinquefasciatus. Endonucleases are predicted to have the ability to lower the viscosity of the skin matrix and allow diffusion of pharmacological components through the host dermis [22]. Chitinases may be involved in anti-fungal activity or housekeeping functions associated with the cuticular structure [49]. Serine proteases may interrupt the fibrinolysis pathways in the host or exert a prophenoloxidase anti-pathogen activity [49]. Other minor enzymes identified in the I. holocyclus adult female SGs include: inositol polyphosphate, phosphatase, carboxypeptidase, sulfotransferase, dipeptidylpeptidase, leukotriene hydrolase and phospholipase.
Glycine rich
Glycine rich proteins have been identified in sialome studies of ticks and some were predicted as salivary cement proteins that are utilized during tick-host attachment [49]. Another group of glycine rich proteins are collagen-like proteins, which function as part of the extracellular matrix of the salivary glands and as part of the tracheolar system [49]. GGY peptides, which are rich in glycine and tyrosine, also belong to glycine rich protein family. GGY peptides are predicted to have an antimicrobial function [55].
Immunity related
Immunity-related proteins are often present in the saliva of hematophagous insects and ticks [49]. There are two major groups of immunity-related peptides - antimicrobial peptides and pattern recognition proteins. Antimicrobial peptides prevent contamination in the feeding cavity and microbial growth in the ingested blood. Examples of antimicrobial peptides are lysozyme, defensin, microplusin and histidine-rich peptides [49]. Ficolin, ixoderin, peptidoglycan recognition proteins and ML domain containing proteins are examples of pattern recognition proteins. Ficolin and ixoderins activate the invertebrate complement system [56] while peptidoglycan recognition proteins are important for the activation of anti-microbial enzymes [49]. ML domains enable the recognition of lipids important in innate immunity and lipid metabolism [49].
Lipocalin
Lipocalins have a highly conserved barrel structure, which enables them to bind and carry hydrophobic ligands. Lipocalins are ubiquitously distributed and abundantly expressed in the salivary glands of ticks [24, 25, 57]. In ticks, lipocalins are described to be involved in histamine and serotonin binding [58], anti-haemostasis[59], anti-complement activity [60], immunoglobulin binding [61] and toxicity [62]. An extensive family of lipocalins is predicted to have more than 20 members and has a tendency of increasing in number with diverse functions being discovered in other ticks [62].
Mucin
Mucins are putative glycoproteins with O-galactosylation sites and a chitin-binding domain [63]. This group of proteins is predicted to serve as mechanical protection against pathogen invasion of ticks, by coating the chitinous feeding mouthparts or the feeding lesion [49].
Protease Inhibitor
There are a few protease inhibitor protein families that can be characterized by their protease inhibitor domains such as the Kunitz domain, serpin domain and TIL (Trypsin Inhibitor Like) domain. Kunitz domain containing protease inhibitors are one of the largest protein families in tick saliva and they target proteases of the S1 family and enzymes complexes such as Xase and prothrombinase [49, 64]. Several members of Kunitz protease inhibitors are involved in anti-clotting [49], anti-thrombin [65] and anti-platelet [66] activities. Serpins control the clotting systems of vertebrates by regulating serine protease cascades. Serpins of I. ricinus, which are called IRIS, are found to have immunosuppressive properties in cellular assays [67] and weak anti-hemostatic function [68]. Another serpin, RMS-3, found in R. microplus is predicted to counteract immune responses during the host-parasite interaction [69]. The only characterized TIL domain containing protease inhibitor is ixodidin, which has antimicrobial, anti-trypsin and anti-elastase properties [70]. Cystatins (cysteine proteinase inhibitors) are also a protease inhibitors, which mainly inhibit activities of peptidases of families C1 (papain family) and C13 (legumain family) [71]. Thyropins are another example of protease inhibitors. These members are found to contain Thyroglobulin type I repeats that may inactivate cysteine proteases. However, the function of the thyropin family of inhibitors is still unknown [49].
Putative secreted protein
A significant number of contigs found in the I. holocyclus cDNA libraries matched previously described tick proteins, mainly also deducted from tick sialome studies. These putative secreted proteins were found to have an open reading frame in the previous studies but were not functionally classified. The consistent presence of these groups of proteins in tick saliva suggests that they have tick salivary gland specific functions yet to be determined.
Salivary proteins
Salp15 is an example of salivary protein, which was previously identified in I. scapularis. Salp15 was shown to inhibit CD4 T cells and to be essential for Lyme disease pathogen transmission [24]. Other salivary proteins are described as 8.9-kDa proteins, 14-kDa proteins, 18.7-kDa proteins and 22.5kDa proteins, which were previously identified in other Ixodes ticks. These proteins have an open reading frame and a signal peptide, however their significance and functions remain unknown.
Toxin like
Some proteins, with two to six cysteine residues in the mature peptides, are reported to be toxin-like. Some of these cysteine-rich proteins in I. holocyclus were previously identified in the I. scapularis sialome. Proteins, which produced significant matches with HT-1 [72], are also labeled as toxin-like.
The abundance of the above ten secreted protein families were inconsistent in both of the I. holocyclus transcriptome libraries, reinforcing the significant role of tick salivary dynamics in tick blood-feeding behavior as previously reported in other tick sialome studies [14]. Most of transcripts (52.85% from SG_FA_CD and 26.43% from SG_FA_BA), are novel proteins, which had no significant match with any domain or nucleotide and/or protein sequences. This result is consistent with previous sialome studies from several hard and soft ticks, including I. ricinus [17, 26], I. scapularis [24], I. pacificus [25], Rhipicephalus sanguineus [73], Amblyomma maculatum [47], Amblyomma variegatum [19] and Antricola delacruzi [44], identifying a high abundance of proteins and protein families of unknown functions. These sialome studies suggest an intensive evolution in the salivary proteins as a consequence of host immune pressure. Hence, a large quantity of unique protein families and salivary protein were identified in most of tick sialomes that have been analyzed [2, 21].
V. Conclusions
This report describes for the first time the transcriptome data of the Australian paralysis tick, I. holocyclus infesting different hosts, which were resistant and susceptible respectively. The protein families identified are an important contribution in understanding the I. holocyclus interactions within its hosts. The differential contig representations of the two transcriptome libraries indicate that I. holocyclus feeding on different host regulate gene expression differently.
The contig representation of the contig counts utilized in this study would be an underestimation of the real read coverage. Further analyses such as qPCR are needed to validate the abundances reported here. This transcriptomic study could be further improved by repeating the analysis of the samples with both 454 and Illumina technologies to provide replicate and more robust contig data. Currently, two samples were either processed using 454 or Illumina technologies. The differences observed may be attributed to the inherent differences between the two technologies [74] and therefore reduce the credibility of the results in this study.
References
Cupp E: Biology of ticks. The Veterinary Clinics of North America Small Animal Practice 1991, 21(1):1-26.
Mans BJ, Neitz AWH: Adaptation of ticks to a blood-feeding environment: evolution from a functional perspective. In., vol. 34. England: Pergamon-Elsevier Science LTD; 2004: 1-17.
Bagnall BG, Doube BM: The Australian paralysis tick Ixodes Holocyclus Australian Veterinary Journal 1975, 51(3):159-160.
Bowman DD: Feline clinical parasitology. Ames: Iowa State University Press; 2002.
Underhill D, Sutherland SK: Australia‘s Dangerous Creatures: Reader‘s Digest; 1987.
Estrada-Peña A, Jongejan F: Ticks feeding on humans: a review of records on human-biting Ixodoidea with special reference to pathogen transmission. Experimental and Applied Acarology 1999, 23(9):685-715.
Gothe R, Kunze K, Hoogstraal H: The mechanisms of pathogenicity in the tick paralyses. Journal of Medical Entomology 1979, 16(5):357-369.
Hall-Mendelin S, Craig SB, Hall RA, O‘Donoghue P, Atwell RB, Tulsiani SM, Graham GC: Tick paralysis in Australia caused by Ixodes holocyclus Neumann. Annals of Tropical Medicine and Parasitology 2011, 105(2):95-106.
Storer E, Sheridan AT, Warren L, Wayte J: Ticks in Australia. Australasian Journal of Dermatology 2003, 44(2):83-89.
Eppleston KR, Kelman M, Ward MP: Distribution, seasonality and risk factors for tick paralysis in Australian dogs and cats. Veterinary Parasitology 2013, 196(3-4):460-468.
Atwell RB, Campbell FE, Evans EA: Prospective survey of tick paralysis in dogs. Australian Veterinary Journal 2001, 79(6):412-418.
Campbell FE, Atwell RB: Long QT syndrome in dogs with tick toxicity ( Ixodes holocyclus ). Australian Veterinary Journal 2002, 80(10):611-616.
Sonenshine DE, Roe RM: Biology of ticks, vol. 2: Oxford University Press; 2013, p. 313-332.
Ribeiro JM: Role of saliva in tick/host interactions. Experimental and Applied Acarology 1989, 7(1):15-20.
Ribeiro JMC: Blood-feeding Arthropods - Live Syringes or Invertebrate pharmacologists? Infectious Agents and Disease - Reviews Issues and Commentary 1995, 4(3):143-152.
Pareek CS, Smoczynski R, Tretyn A: Sequencing technologies and genome sequencing. Journal of Applied Genetics 2011, 52(4):413-435.
Chmelar J, Anderson JM, Mu J, Jochim RC, Valenzuela JG, Kopecký J: Insight into the sialome of the castor bean tick, Ixodes ricinus. BMC Genomics 2008, 9(1):233-233.
Francischetti IMB, Mans BJ, Meng Z, Gudderra N, Veenstra TD, Pham VM, Ribeiro JMC: An insight into the sialome of the soft tick, Ornithodorus parkeri. Insect Biochemistry and Molecular Biology 2008, 38(1):1-21.
Ribeiro JM, Anderson JM, Manoukis NC, Meng Z, Francischetti IM: A further insight into the sialome of the tropical bont tick, Amblyomma variegatum. BMC Genomics 2011, 12(1):136-136.
Valenzuela JG, Francischetti IMB, Pham VM, Garfield MK, Mather TN, Ribeiro JMC: Exploring the sialome of the tick Ixodes scapularis. The Journal of Experimental Biology 2002, 205(Pt 18):2843-2864.
Mans B, Francischetti I: Sialomic perspectives on the evolution of blood-feeding behavior in Arthropods: Future therapeutics by natural design. In: Toxins and Hemostasis. Springer; 2011: 21-44.
Calvo E, Pham VM, Marinotti O, Andersen JF, Ribeiro JMC: The salivary gland transcriptome of the neotropical malaria vector Anopheles darlingi reveals accelerated evolution of genes relevant to hematophagy. BMC Genomics 2009, 10(1):57-57.
Wang H, Kaufman WR, Cui WW, Nuttall PA: Molecular individuality and adaptation of the tick Rhipicephalus appendiculatus in changed feeding environments. Medical and Veterinary Entomology 2001, 15(4):403-412.
Ribeiro JMC, Alarcon-Chaidez F, B. Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, Wikel SK: An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochemistry and Molecular Biology 2006, 36(2):111-129.
Francischetti IMB, My Pham V, Mans BJ, Andersen JF, Mather TN, Lane RS, Ribeiro JMC: The transcriptome of the salivary glands of the female western black-legged tick Ixodes pacificus (Acari: Ixodidae). Insect Biochemistry and Molecular Biology 2005, 35(10):1142-1161.
Schwarz A, von Reumont BM, Erhart J, Chagas AC, Ribeiro JMC, Kotsyfakis M: De novo Ixodes ricinus salivary gland transcriptome analysis using two next-generation sequencing methodologies. Federation of American Societies for Experimental Biology 2013, 27(12):4745.
Andrews S: FastQC: A quality control tool for high throughput sequence data. 2010.
Smeds L, Künstner A, Evolutionsbiologi, Biologiska s, Uppsala u, Institutionen för ekologi och g, Teknisk-naturvetenskapliga v: ConDeTri–a content dependent read trimmer for Illumina data. PloS One 2011, 6(10):e26314.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 2008, 18(5):821-829.
Schulz MH, Zerbino DR, Vingron M, Birney E: Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28(8):1086-1092.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z: Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437(7057):376-380.
Wheeler DL, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL et al: Database resources of the National Center for Biotechnology Information. Nucleic Acids Research 2008, 36(suppl_1):D13-D21.
Apweiler R, Bower L, Roechert B, Bursteinas B, Chan WM, Chavali G, Da Silva A, Dimmer E, Eberhardt R, Fazzini F et al: Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Research 2012, 40(D1):D71–D75.
Gene Ontology C: Gene Ontology: Tool for the unification of biology. Nature Genetics 2000, 25(1):25-29.
Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH: CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Research 2002, 30(1):281-283.
Tatusov RL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA, Fedorova ND et al: The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003, 4(1):41-41.
Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 2000, 28(1):27-30.
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Research 2000, 28(1):263-266.
Schultz J, Copley RR, Doerks T, Ponting CP, Bork P: SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Research 2000, 28(1):231-234.
Hunter AA, Macgregor AB, Szabo TO, Wellington CA and Bellgard MI, Yabi: An online research environment for Grid, High Performance and Cloud computing, Source Code for Biology and Medicine 2012, 7:1 doi:10.1186/1751-0473-7-1 Published: 15 February 2012
Petersen TN, Brunak S, von Heijne G, Nielsen H, Stockholms u, Institutionen för biokemi och b, Naturvetenskapliga f: SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature 2011, 8(10):785-786.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL: Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology 2001, 305(3):567-580.
Mans BJ, Andersen JF, Francischetti IMB, Valenzuela JG, Schwan TG, Pham VM, Garfield MK, Hammer CH, Ribeiro JMC: Comparative sialomics between hard and soft ticks: implications for the evolution of blood-feeding behavior. Insect Biochemistry and Molecular Biology 2008, 38(1):42-58.
Ribeiro JMC, Labruna MB, Mans BJ, Maruyama SR, Francischetti IMB, Barizon GC, de Miranda Santos IKF: The sialotranscriptome of Antricola delacruzi female ticks is compatible with non-hematophagous behavior and an alternative source of food. Insect Biochemistry and Molecular Biology 2012, 42(5):332-342.
Anatriello E, Ribeiro JMC, de Miranda-Santos IKF, Brandão LG, Anderson JM, Valenzuela JG, Maruyama SR, Silva JS, Ferreira BR: An insight into the sialotranscriptome of the brown dog tick, Rhipicephalus sanguineus. BMC Genomics 2009, 11(1):450-450.
Batista IFC, Chudzinski-Tavassi AM, Faria F, Simons SM, Barros-Batestti DM, Labruna MB, Leão LI, Ho PL, Junqueira-de-Azevedo ILM: Expressed sequence tags (ESTs) from the salivary glands of the tick Amblyomma cajennense (Acari: Ixodidae). Toxicon 2008, 51(5):823-834.
Karim S, Singh P, Ribeiro JMC: A deep insight into the sialotranscriptome of the gulf coast tick, Amblyomma maculatum. PloS One 2011, 6(12):e28525.
Vijay N, Poelstra JW, Künstner A, Wolf JBW, Science for Life Laboratory S, Evolutionsbiologi, Teknisk-naturvetenskapliga v, Institutionen för ekologi och g, Uppsala u, Biologiska s: Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA‐seq experiments. Molecular Ecology 2013, 22(3):620-634.
Francischetti IMB, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JMC: The role of saliva in tick feeding. Frontiers in Bioscience 2009, 14(6):2051-2088.
Rodriguez-Valle M, Lew-Tabor A, Gondro C, Moolhuijzen P, Vance M, Guerrero FD, Bellgard M, Jorgensen W: Comparative microarray analysis of Rhipicephalus ( Boophilus ) microplus expression profiles of larvae pre-attachment and feeding adult female stages on Bos indicus and Bos taurus cattle. BMC Genomics 2010, 11(1):437-437.
Megraw T, Kaufman TC, Kovalick GE: Sequence and expression of Drosophila Antigen 5-related 2, a new member of the CAP gene family. Gene 1998, 222(2):297-304.
Hoffman DR: Allergens in Hymenoptera venom. XXV: The amino acid sequences of antigen 5 molecules and the structural basis of antigenic cross-reactivity. The Journal of Allergy and Clinical Immunology 1993, 92(5):707-716.
Schuijt TJ, Narasimhan S, Daffre S, DePonte K, Hovius JWR, Van‘t Veer C, van der Poll T, Bakhtiari K, Meijers JCM, Boder ET et al: Identification and characterization of Ixodes scapularis antigens that elicit tick immunity using yeast surface display. PloS One 2011, 6(1):e15926.
Harnnoi T, Sakaguchi T, Nishikawa Y, Xuan X, Fujisaki K: Molecular characterization and comparative study of 6 salivary gland metalloproteases from the hard tick, Haemaphysalis longicornis. Comparative Biochemistry and Physiology 2007, 147(1):93-101.
Couillault C, Pujol N, Reboul J, Sabatier L, Guichou J-F, Kohara Y, Ewbank JJ: TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nature Immunology 2004, 5(5):488-494.
Rego ROM, Hajdušek O, Kovář V, Kopáček P, Grubhoffer L, Hypša V: Molecular cloning and comparative analysis of fibrinogen-related proteins from the soft tick Ornithodoros moubata and the hard tick I xodes ricinus . Insect Biochemistry and Molecular Biology 2005, 35(9):991-1004.
Alarcon-Chaidez FJ, Sun J, Wikel SK: Transcriptome analysis of the salivary glands of Dermacentor andersoni Stiles (Acari: Ixodidae). Insect Biochemistry and Molecular Biology 2007, 37(1):48-71.
Sangamnatdej S, Paesen GC, Slovak M, Nuttall PA: A high affinity serotonin- and histamine-binding lipocalin from tick saliva. Insect Molecular Biology 2002, 11(1):79-86.
Rodriguez-Valle M, Moolhuijzen P, Piper EK, Weiss O, Vance M, Bellgard M, Lew-Tabor A: Rhipicephalus microplus lipocalins (LRMs): genomic identification and analysis of the bovine immune response using in silico predicted B and T cell epitopes. International Journal for Parasitology 2013, 43(9):739-752.
Nunn MA, Sharma A, Paesen GC, Adamson S, Lissina O, Willis AC, Nuttall PA: Complement Inhibitor of C5 Activation from the Soft Tick Ornithodoros moubata. The Journal of Immunology 2005, 174(4):2084-2091.
Wang H, Paesen GC, Nuttall PA, Barbour AG: Male ticks help their mates to feed. Nature 1998, 391(6669):753-754.
Mans BJ, Louw AI, Neitz AW: The major tick salivary gland proteins and toxins from the soft tick, Ornithodoros savignyi, are part of the tick Lipocalin family: implications for the origins of tick toxicoses. Molecular Biology Evolution 2003, 20(7):1158-1167.
Hansen JE, Lund O, Tolstrup N, Gooley AA, Williams KL, Brunak S: NetOglyc: prediction of mucin type O-glycosylation sites based on sequence context and surface accessibility. Glycoconjugate Journal 1998, 15(2):115-130.
Ascenzi P, Bocedi A, Bolognesi M, Spallarossa A, Coletta M, De Cristofaro R, Menegatti E: The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor: A milestone protein. Current Protein and Peptide Science 2003, 4(3):231-251.
Mans BJ, Louw AI, Neitz AWH: Amino acid sequence and structure modeling of savignin, a thrombin inhibitor from the tick, Ornithodoros savignyi. Insect Biochemistry and Molecular Biology 2002, 32(7):821-828.
Karczewski J, Endris R, Connolly TM: Disagregin is a fibrinogen receptor antagonist lacking the Arg-Gly-Asp sequence from the tick, Ornithodoros moubata. Journal of Biological Chemistry 1994, 269(9):6702-6708.
Leboulle Gr, Crippa M, Decrem Y, Mejri N, Brossard M, Bollen A, Godfroid E: Characterization of a novel salivary immunosuppressive protein from Ixodes ricinus Ticks. Journal of Biological Chemistry 2002, 277(12):10083-10089.
Prevot P-P, Adam B, Boudjeltia KZ, Brossard M, Lins L, Cauchie P, Brasseur R, Vanhaeverbeek M, Vanhamme L, Godfroid E: Anti-hemostatic effects of a serpin from the saliva of the tick Ixodes ricinus. The Journal of Biological Chemistry 2006, 281(36):26361-26369.
Rodriguez-Valle M, Vance M, Moolhuijzen PM, Tao X, Lew-Tabor AE: Differential recognition by tick-resistant cattle of the recombinantly expressed Rhipicephalus microplus serine protease inhibitor-3 (RMS-3). Ticks and Tick-Borne Diseases 2012, 3(3):159-169.
Fogaça AC, Almeida IC, Eberlin MN, Tanaka AS, Bulet P, Daffre S: Ixodidin, a novel antimicrobial peptide from the hemocytes of the cattle tick Boophilus microplus with inhibitory activity against serine proteinases. Peptides 2006, 27(4):667-674.
Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P: Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Research 2002, 30(1):242-244.
Vink S, Daly NL, Steen N, Craik DJ, Alewood PF: Holocyclotoxin-1, a cystine knot toxin from Ixodes holocyclus. Toxicon 2014.
Otranto D, Dantas-Torres F, Tarallo VD, Ramos RAdN, Stanneck D, Baneth G, de Caprariis D: Apparent tick paralysis by Rhipicephalus sanguineus (Acari: Ixodidae) in dogs. Veterinary parasitology 2012, 188:325-329.
Egekwu N, Sonenshine DE, Bissinger BW, Roe RM: Transcriptome of the female synganglion of the black-legged tick Ixodes scapularis (Acari: Ixodidae) with comparison between Illumina and 454 systems. PloS One 2014, 9(7):e102667.
Author information
Authors and Affiliations
Corresponding authors
Additional information
1The University of Queensland, Queensland Alliance for Agriculture & Food Innovation, St. Lucia, 4072, Queensland Australia; 2Murdoch University, Centre for Comparative Genomics, Murdoch, 6150, WA, Australia
*Corresponding authors: m.rodriguezvalle@uq.edu.au; a.lewtabor@uq.edu.au
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ong, C., Rodriguez-Valle, M., Moolhuijzen, P. et al. Exploring the transcriptomic data of the Australian paralysis tick, Ixodes holocyclus. GSTF J Vet Sci 3, 1 (2016). https://doi.org/10.7603/s40871-016-0001-y
Published:
DOI: https://doi.org/10.7603/s40871-016-0001-y