
Abstract
Coraliomargarita akajimensis Yoon et al. 2007 is the type species of the genus Coraliomargarita. C. akajimensis is an obligately aerobic, Gram-negative, non-spore-forming, non-motile, spherical bacterium that was isolated from seawater surrounding the hard coral Galaxea fascicularis. C. akajimensis is of special interest because of its phylogenetic position in a genomically under-studied area of the bacterial diversity. Here we describe the features of this organism, together with the complete genome sequence, and annotation. This is the first complete genome sequence of a member of the family Puniceicoccaceae. The 3,750,771 bp long genome with its 3,137 protein-coding and 55 RNA genes is a part of the Genomic Encyclopedia of Bacteria and Archaea project.
Similar content being viewed by others


Introduction
Strain 04OKA010-24T (DSM 45221 = JCM 23193 = KCTC 12865) is the type strain of the species Coraliomargarita akajimensis and was first described in 2007 by Yoon et al. [1]. Strain 04OKA010-24T was isolated from seawater surrounding the hard coral Galaxea fascicularis L., collected at Majanohama, Akajima, Okinawa, Japan. Yoon et al. considered strain C. akajimensis 04OKA010-24T to represent a novel species in a new genus belonging to subdivision 4 of the phylum Verrucomicrobia. Based on 16S rRNA the phylum Verrucomicrobia has been divided into five subdivisions [2]. In the second edition of Bergey’s Manual of Systematic Bacteriology three subdivisions were included at the rank of family: ‘Verrucomicrobiaceae’ (subdivision 1), ‘Xiphinematobacteriaceae’ (subdivision 2) and ‘Opitutaceae’ (subdivision 4) [3]. There were three identified species in subdivision 4, Opitutus terrae [4–6] isolated from soil and the marine bacteria ‘Fucophilus fucoidanolyticus’ [7], isolated from a sea cucumber and Alterococcus agarolyticus [8], isolated from a hot spring that was originally misclassified as a member of the Gammaproteobacteria.
In 2007, coincident to the description of C. akajimensis, the class Opitutae, which comprises two orders: the order (Puniceicoccales containing the family Puniceicoccaceae and the order Opitutales containing the family Opitutaceae) was proposed for the classification of species belonging to subdivision 4 of the phylum ‘Verrucomicrobia’ [9]. Besides the genus Coraliomargarita [1] the genera Cerasicoccus [10], Pelagicoccus [11], Puniceicoccus [9] belong into the family Puniceicoccaceae. Here we present a summary classification and a set of features for C. akajimensis 04OKA010-24T, together with the description of the complete genomic sequencing and annotation.
Classification and features
Within the class Opitutae, strain C. akajimensis 04OKA010-24T shares the highest degree of 16S rRNA gene sequence similarity with Puniceicoccus vermicola (88.3%), isolated from the digestive tract of a marine clamworm [5], and Pelagicoccus croceus (87.6%) [12], whereas the other members of the class share 84.1 to 87.2% sequence similarity [13]. ‘Lentimonas marisflavi’ and ‘Fucophilus fucoidanolyticus’ are the closest related cultivable strains (94.0% sequence similarity), whose names are not yet validly published. ‘Fucophilus fucoidanolyticus’ was isolated from sea cucumbers (Sticopus japonicus) and is able to degrade fucoin [14]. GenBank contains also a large number of 16S rRNA sequences with reasonably high sequence similarity from phylotypes (uncultured bacteria) reflecting the problem of efficient culturing of bacteria from the class Opitutae. However, only few sequences from genomic and marine metagenomic surveys surpass 90% sequence similarity, indicating that members of the genus Coraliomargarita are not widely distributed globally in the habitats screened thus far (status April 2010).
Figure 1 shows the phylogenetic neighborhood of C. akajimensis 04OKA010-24T in a 16S rRNA based tree. The two copies of the 16S rRNA gene in the genome are identical with the previously published sequence generated from DSM 45221 (AB266750).

Phylogenetic tree highlighting the position of C. akajimensis 04OKA010-24T relative to the other type strains within the phylum Verrucomicrobia. The tree was inferred from 1,373 aligned characters [15,16] of the 16S rRNA gene sequence under the maximum likelihood criterion [17] and rooted in accordance with the current taxonomy [18]. The branches are scaled in terms of the expected number of substitutions per site. Numbers above branches are support values from 300 bootstrap replicates [19] if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [20] are shown in blue (Akkermansia muciniphila CP001071, Opitutus terrae CP001032), published genomes in bold.
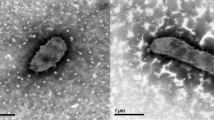
Cells of C. akajimensis 04OKA010-24T are Gram-negative, obligately aerobic cocci with a diameter of 0.5–1.2 µm (Figure 2 and Table 1) [1]. The cells are non-motile and spores are not formed. On half strength R2A agar medium with 75% artificial seawater C. akajimensis forms circular, convex, white colonies. The optimum temperature for growth ranges from 20 to 30°C. No growth was observed at 4 or 45°C. The pH range for growth is 7.0–9.0. NaCl concentrations up to 5% (w/v) are tolerated [1].

Scanning electron micrograph of C. akajimensis 04OKA010-24T
Strain 04OKA010-24T produces acid from glycerol, galactose, fructose, mannose, mannitol, sorbitol, trehalose, D-turanose, D-lyxose, D-tagatose, D-fucose, L-fucose, D-arabitol, and 5-ketogluconate [1]. C. akajimensis is able to hydrolyze urea and DNA, but cannot hydrolyze agar, casein, aesculin, starch and gelatin [1]. Nitrate is not reduced to nitrite. C. akajimensis is catalase negative, oxidase positive [1] and is resistant to ampicillin and penicillin G [10].
Chemotaxonomy
The fatty acid profile of strain C. akajimensis 04OKA010-24T revealed straight chain acids C14:0 (24.2%), C18:1ω9c (23.5%) and C18:0 (15.6%) as the major fatty acids and iso-C14:0 (8.2%), anteiso-C15:0 (2.9%), C16:0 (3.3%) C19:0 (2.8%) and C21:0 (6.9%) in minor amounts [1]. MK-7 is the predominant menaquinone [1]. Muramic acid and diaminopimelic acid are absent, indicating that the cell wall does not contain peptidoglycan [1].
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [27], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [28]. The genome project is deposited in the Genome OnLine Database [20] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2.
Growth conditions and DNA isolation
C. akajimensis 04OKA010-24T, DSM 45221, was grown in DSMZ medium 514 (bacto marine growth medium) [29] at 25°C. DNA was isolated from 0.5–1 g of cell paste using a MasterPure Gram Positive DNA purification kit (Epicentre MGP04100), adding 5 µl mutanolysin to the standard lysis solution for 40 min at 37°C and a final 35 min incubation on ice after the MPC-step.
Genome sequencing and assembly
The genome of C. akajimensis was sequenced using a combination of Illumina and 454 technologies. An Illumina GAii shotgun library with reads of 714 Mb, a 454 Titanium draft library with average read length of 282 +/− 187.7 bases, and a paired end 454 library with average insert size of 24.632 +/− 6.158 kb were generated for this genome. All general aspects of library construction and sequencing can be found at http://www.jgi.doe.gov/. Draft assembly was based on 3.8 Mb 454 standard and 454 paired end data (498,215 reads). Newbler (Roch, version 2.0.0-PostRelease-10/28/2008) parameters are -consed -a 50 -l 350 -g -m -ml 20. The initial Newbler assembly was converted into a phrap assembly by making fake reads from the consensus and collecting the read pairs in the 454 paired end library. Illumina sequencing data was assembled with Velvet [30], and the consensus sequences were shredded into 1.5 kb overlapped fake reads and assembled together with the 454 data. The Phred/Phrap/Consed software package (www.phrap.com) was used for sequence assembly and quality assessment in the following finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution (http://www.jgi.doe.gov/), Dupfinisher, or sequencing cloned bridging PCR fragments with subcloning or transposon bombing [31]. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J-F. Cheng, unpublished). A total of 297 additional Sanger reactions were necessary to close gaps and to raise the quality of the finished sequence. Illumina reads were also used to improve the final consensus quality using Polisher [32]. The error rate of the completed genome sequence is less than 1 in 100,000.
Genome annotation
Genes were identified using Prodigal [33] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [34]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [35].
Genome properties
The genome is 3,750,771 bp long and comprises one main circular chromosome with a 53.6% GC content (Table 3 and Figure 3). Of the 3,192 genes predicted, 3,137 were protein-coding genes, and 55 RNAs. Seventeen pseudogenes were also identified. The majority of the protein-coding genes (63.6%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.

Graphical circular map of the genome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
Insights from genome sequence
With 94% identity based on 16S rRNA analysis ‘F. fucoidanolyticus’ is one of the closest related, cultivated organism to C. akajimensis. Sakai and colleagues report the existence of intracellular α-L-fucosidases and sulfatases, which enable ‘F. fucoidanolyticus’ to degrade fucoidan [14]. This fucoidan degrading ability could be shared by C. akajimensis, as the annotation of the genome sequence revealed the existence of 49 sulfatases and 12 α-L-fucosidases belonging to glycoside hydrolase family 29. Furthermore 12 β-agarases are encoded in the genome of C. akajimensis, which is not in accordance to Yoon et al., who reported that agar was not hydrolyzed by C. akajimensis [1]. Forty-two genes coding for transcriptional regulators belonging to the AraC-family were found in C. akajimensis. It might be noteworthy that the genes coding for the AraC-family regulators, agarases, sulfatases and α-L-fucosidases are unequally distributed over the genome, with most of them localized in the first third of the genome (bp 33,731-1,412,308). The genes for several fucosidases and sulfatases are clustered and their expression might be under the control of an AraC-family regulator.
In addition to C. akajimensis only two more genomes of members of the Opitutae are sequenced (but not yet published): Opitutus terrae, an obligately anaerobic, motile bacterium isolated from a rice paddy soil microcosms [6] and Opitutaceae bacterium TAV2 isolated from the gut of a wood-feeding termite. Because of the quite distant relatedness of these three sequenced organisms, a comparison of genomes seems to be of limited use. The reported characteristic differences between the Opitutae [1] are partly reflected in the now known genome sequence. In the case of the motile bacterium O. terrae 36 proteins belonging to the COG pathway ‘flagellum structure and biogenesis’ are predicted, whereas in the genome of the non-motile C. akajimensis, no proteins belonging in this category are encoded. Another characteristic feature is the ability to reduce nitrate. In both genomes genes encoding for nitrate reductase (EC: 1.7.99.4: O. terrae Oter_1740, C. akajimensis Caka_0064, Caka_0348) and nitrite reductase are predicted (EC: 1.7.7.1: O. terrae Oter_1737, C. akajimensis Caka_0346; EC: 1.7.2.2: O. terrae Oter_4608, C. akajimensis Caka_2912), but only for O. terrae nitrate reduction is reported [14]. In the case of starch hydrolysis, the genome data match the experimental data previously reported. The O. terrae reported to be starch-hydrolyzing encodes one α-amylase and for three proteins containing α-amylase domains. For C. akajimensis, starch hydrolysis is not reported and in the genome there is only one gene identified that could encode for an α-amylase.
References
Yoon J, Yasumoto-Hirose M, Katsuta A, Sekiguchi H, Matsuda S, Kasai H, Yokota A. Coraliomargarita akajimensis gen. nov., sp. nov., a novel member of the phylum ‘Verrucomicrobia’ isolated from seawater in Japan. Int J Syst Evol Microbiol 2007; 57:959–963. PubMed doi:10.1099/ijs.0.64755-0
Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 1998; 180:4765–4774. PubMed
Garrity G, Bell JA, Lilburn TG. Taxonomic outline of the prokaryotes, Bergey’s Manual of Systematic Bacteriology, 2nd edition, release 4.0, Springer, New York. 2003.
Janssen PH, Schuhmann A, Morschel E, Rainey FA. Novel anaerobic ultramicrobacteria belonging to the Verrucomicrobiales lineage of bacterial descent isolated by dilution culture from anoxic rice paddy soil. Appl Environ Microbiol 1997; 63:1382–1388. PubMed
Chin KJ, Hahn D, Hengstmann U, Liesack W, Janssen PH. Characterization and identification of numerically abundant culturable bacteria from the anoxic bulk soil of rice paddy microcosms. Appl Environ Microbiol 1999; 65:5042–5049. PubMed
Chin KJ, Liesack W, Janssen PH. Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division ‘Verrucomicrobia’ isolated from rice paddy soil. Int J Syst Evol Microbiol 2001; 51:1965–1968. PubMed
Sakai T, Ishizuka K, Shimanaka K, Ikai K, Kato I. Structures of oligosaccharides derived from Cladosiphon okamuranus fucoidan by digestion with marine bacterial enzymes. Mar Biotechnol 2003; 5:536–544. PubMed doi:10.1007/s10126-002-0107-9
Shieh WY, Jean WD. Alterococcus agarolyticus, gen. nov., sp. nov., a halophilic thermophilic bacterium capable of agar degradation. Can J Microbiol 1998; 44:637–645. PubMed doi:10.1139/cjm-44-7-637
Choo YJ, Lee K, Song J, Cho JC. Puniceicoccus vermicola gen. nov., sp. nov., a novel marine bacterium, and description of Puniceicoccaceae fam. nov., Puniceicoccales ord. nov., Opitutaceae fam. nov., Opitutales ord. nov. and Opitutae classis nov. in the phylum ‘Verrucomicrobia’. Int J Syst Evol Microbiol 2007; 57:532–537. PubMed doi:10.1099/ijs.0.64616-0
Yoon J, Matsuo Y, Matsuda S, Adachi K, Kasai H, Yokota A. Cerasicoccus arenae gen. nov., sp. nov., a carotenoid-producing marine representative of the family Puniceicoccaceae within the phylum ‘Verrucomicrobia’, isolated from marine sand. Int J Syst Evol Microbiol 2007; 57:2067–2072. PubMed doi:10.1099/ijs.0.65102-0
Yoon J, Yasumoto-Hirose S, Matsuda S, Nozawa M, Matsuda S, Kasai H, Yokota A. Pelagicoccus mobilis gen. nov., sp. nov., Pelagicoccus albus sp. nov. and Pelagicoccus litoralis sp. nov., three novel members of subdivision 4 within the phylum ‘Verrucomicrobia’, isolated from seawater by in situ cultivation. Int J Syst Evol Microbiol 2007; 57:1377–1385. PubMed doi:10.1099/ijs.0.64970-0
Yoon J, Oku N, Matsuda S, Kasai H, Yokota A. Pelagicoccus croceus sp. nov., a novel marine member of the family Puniceicoccaceae within the phylum ‘Verrucomicrobia’ isolated from sea-grass. Int J Syst Evol Microbiol 2007; 57:2874–2880. PubMed doi:10.1099/ijs.0.65286-0
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, Lim YW. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 2007; 57:2259–2261. PubMed doi:10.1099/ijs.0.64915-0
Sakai T, Ishizuka K, Kato I. Isolation and characterization of fucoidan-degrading marine bacterium. Mar Biotechnol 2003; 5:409–416. PubMed doi:10.1007/s10126-002-0118-6
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452–464. PubMed doi:10.1093/bioinformatics/18.3.452
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540–552. PubMed
Stamatakis A, Hoover P, Rougemont J. A Rapid bootstrap algorithm for the RAxML web servers. Syst Biol 2008; 57:758–771. PubMed doi:10.1080/10635150802429642
Euzeby JP. List of Bacterial Names with Standing in Nomenclature: a folder on the Internet. Syst Appl Bacteriol 1997; 47:590–592. doi:10.1099/00207713-47-2-590
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. doi:10.1007/978-3-642-02008-713
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed doi:10.1093/nar/gkp848
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed doi:10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms. Proposal for the domains Archaea and Bacteria. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed doi:10.1073/pnas.87.12.4576
Hedlund BP, Gosink JJ, Staley JT. Verrucomicrobia div. nov., a new division of the bacteria containing three new species of Prosthecobacter. Antonie Van Leeuwenhoek 1997; 72:29–38. PubMed doi:10.1023/A:1000348616863
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119–169.
Classification of bacteria and archaea in risk groups. www.baua.de TRBA 466, supplement 2010; in press.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed doi:10.1038/75556
Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175–182. PubMed doi:10.1016/j.syapm.2010.03.003
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056–1060. PubMed doi:10.1038/nature08656
List of growth media used at DSMZ: http://www.dsmz.de/microorganisms/media_list.php
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs [REMOVED HYPERLINK FIELD]. Genome Res 2008; 18:821–829. PubMed doi:10.1101/gr.074492.107
Sims D, Brettin T, Detter J, Han C, Lapidus A, Copeland A, Glavina Del Rio T, Nolan M, Chen F, Lucas S, et al. Complete genome sequence of Kytococcus sedentarius type strain (541T). Stand Genomic Sci 2009; 1:12–20. doi:10.4056/sigs.761
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008.
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal Prokaryotic Dynamic Programming Genefinding Algorithm. BMC Bioinformatics 2010; 11:119. PubMed doi:10.1186/1471-2105-11-119
Pati A, Ivanova N, Mikhailova N, Ovchinikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A Gene Prediction Improvement Pipeline for microbial genomes. Nat Methods 2010; 7:455–457. PubMed doi:10.1038/nmeth.1457
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed doi:10.1093/bioinformatics/btp393
Acknowledgements
We would like to gratefully acknowledge the help of Marlen Jando (DSMZ) for growing C. akajimensis cultures. This work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, and Oak Ridge National Laboratory under contract DE-AC05-00OR22725, as well as German Research Foundation (DFG) INST 599/1-1.
Author information
Authors and Affiliations
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Mavromatis, K., Abt, B., Brambilla, E. et al. Complete genome sequence of Coraliomargarita akajimensis type strain (04OKA010-24T). Stand in Genomic Sci 2, 290–299 (2010). https://doi.org/10.4056/sigs.952166
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.952166