
Abstract
Serratia plymuthica are plant-associated, plant beneficial species belonging to the family Enterobacteriaceae. The members of the genus Serratia are ubiquitous in nature and their life style varies from endophytic to free-living. S. plymuthica AS9 is of special interest for its ability to inhibit fungal pathogens of rapeseed and to promote plant growth. The genome of S. plymuthica AS9 comprises a 5,442,880 bp long circular chromosome that consists of 4,952 protein-coding genes, 87 tRNA genes and 7 rRNA operons. This genome is part of the project entitled “Genomics of four rapeseed plant growth promoting bacteria with antagonistic effect on plant pathogens” awarded through the 2010 DOE-JGI Community Sequencing Program (CSP2010).
Similar content being viewed by others


Introduction
The genus Serratia belongs to a group of Gammaproteobacteria, commonly found in soil, water, plants, insects and humans [1]. The genus includes antagonists of soil borne pathogens of different plant species, plant growth promoters and insect pathogens, as well as opportunistic human pathogens. The most common human pathogen in this genus is Serratia marcescens which causes nosocomial infections in humans, while other species are harmless. In agriculture, S. plymuthica is successfully used for control of many soil borne fungal pathogens of different crops (e.g. strawberry, rapeseed) [2,3], while S. proteamaculans promotes the growth of poplar trees [4].
S.plymuthica AS9 (= CCUG 61396) was isolated from field samples of rapeseed roots in Uppsala, Sweden. Our interest in S. plymuthica AS9 is attributed to its ability to stimulate rapeseed plant growth, to inhibit soil borne fungal pathogens and to increase oilseed production. Here we present a description of the complete genome sequencing of S. plymuthica AS9 and its annotation.
Classification and features
The bacterial strain AS9 was previously considered a member of the family Enterobacteriaceae [5]. Recently, comparison of 16S rRNA gene sequences with the most recent databases from GenBank using NCBI BLAST [6] under default settings showed that S. plymuthica AS9 shares 99% similarity with many Serratia species including S. plymuthica (AJ233433) and Serratiaproteamaculans (CP000826.1). When considering high-scoring segment pairs (HSPs) from the best 250 hits, the most frequent matches were with various Serratia species (17.2% with maximum identity of 97–100%) with S. plymuthica (5.2% with maximum identity of 97–99%), S. proteamaculans (4.8% with maximum identity of 97–99%), S. marcescens (4.8% with maximum identity of 96–97%) and various Rahnella species. (7% with maximum identity of 97–98%).
Figure 1 shows the phylogenetic relationship of S. plymuthica AS9 with other species within the genus Serratia in a 16S rRNA based tree. The tree shows its close relationship with the type strain of S. plymuthica, which was confirmed by digital DNA-DNA hybridization values [11] above 70% with the (unpublished) draft genome sequence of the S. plymuthica type strain Breed K-7T from a DSM4540 culture using the GGDC web server [12].

Phylogenetic tree highlighting the position of S. plymuthica AS9 in relation to other species within the genus Serratia, which is based on 1,479 characters of the 16S rRNA gene sequence aligned in ClustalW2 [7]. The tree was inferred under the maximum likelihood criterion [MEGA5, 8] and rooted with Yersinia pseudotuberculosis (a member of the family Enterobacteriaceae). The branches are scaled in terms of the expected number of substitutions per site. The numbers above branches are support values from 1,000 bootstrap replicates if larger than 60% [9]. Lineages with type strain genome sequences registered in GOLD [10] are shown in blue.
S. plymuthica AS9 is a Gram-negative, rod shaped, motile bacterium, 1–2 µm long and 0.5–0.7 µm wide (Figure 2 and Table 1). It forms red to pink colored colonies 1–2 mm in diameter on tryptic soy agar and potato dextrose agar. The color of the bacterium is the result of its production of the red pigment, prodigiosin, but the colony color or production of pigment depends on the ingredients, pH of the medium and the incubation temperature [26–28]. S. plymuthica is a facultative anaerobe, grows between 4 °C and 40 °C and within the pH range 4–10. It can utilize a wide range of carbon sources and also has chitinolytic, proteolytic, cellulolytic, and phospholytic activity [5].

Scanning electron micrograph of S. plymuthica AS9
Chemotaxonomy
The whole cell lipid pattern of S. plymuthica AS9 contains a mixture of saturated and unsaturated fatty acids. The main fatty acids in AS9 strain comprise C16:0 (24.13%), C16:1ω7c (19.41%), C18:1ω7c (18.76%), C14:0 (5.24%) along with other minor fatty acid components. Previously it has been shown that Serratia spp. contain a mixture of C14:0, C16:0, C16:1 and C18:1+2 fatty acids of which 50–80% of the total was C14:0 and other were less than 3% each [29]. This is consistent with the fact that the C14:0 3OH is characteristic of the family Enterobacteriaceae.
Genome sequencing information
S. plymuthica AS9, one of the strains isolated from rapeseed roots and rhizosphere soils was selected for sequencing on the basis of its ability to promote rapeseed growth and inhibit soil borne fungal pathogens. The genome project is deposited in the Genomes On Line Databases [10] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI). A summary of the project information is shown in Table 2 and its association with MIGS identifiers.
Growth conditions and DNA isolation
S. plymuthica AS9 was grown in Luria Broth (LB) medium at 28°C for 12 hours (cells were in the early stationary phase) and the DNA was isolated using a standard CTAB protocol for bacterial genomic DNA isolation which is available at JGI [30].
Genome sequencing and assembly
The genome of strain AS9 was sequenced using a combination of Illumina [31] and 454 sequencing platforms [32]. The details of library construction and sequencing are available at the JGI website [30]. The sequence data from Illumina GAii (1,790.7 Mb) were assembled with Velvet [33] and the consensus sequence computationally shredded into 1.5 kb overlapping fake reads. The sequencing data from 454 pyrosequencing (102.2 Mb) were assembled with Newbler (Roche). The initial draft assembly contained 41 contigs in one scaffold and consensus sequences were computationally shredded into 2 kb overlapping fake reads. The 454 Newbler consensus reads, the Illumina velvet consensus reads and the read pairs in the 454 paired end library were integrated using a software phrap (High Performance Software, LLC) [34]. Possible mis-assemblies were corrected with gapResolution [30], Dupfinisher [35], or by sequencing cloned bridging PCR fragments with subcloning or transposon bombing (Epicentre Biotechnologies, Madison, WI). The gaps between contigs were closed by editing in the software Consed [36–38], by PCR and by Bubble PCR (J.-F. Chang, unpublished) primer walks. Thirty seven additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The sequence reads from Illumina were used to correct potential base errors and increase consensus quality using the software Polisher, developed at JGI [39]. The final assembly is based on 47.3 Mb of 454 draft data which provides an average 8.8× coverage of the genome and 1,746.8 Mb of Illumina draft data which provides an average 323.5× coverage of the genome.
Genome annotation
Genes were identified using Prodigal [40] as part of the genome annotation pipeline at Oak Ridge National Laboratory (ORNL), Oak Ridge, TN, USA, followed by a round of manual curation using the JGI GenPRIMP pipeline [41]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) non-redundant database, Uniport, TIGR-Fam, Pfam, PRIAM, KEGG, COG and InterPro databases. The tRNAScanSE tool [42] was used to find tRNA genes. Additional gene prediction analysis and functional annotation were performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [43].
Genome properties
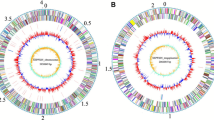
The S. plymuthica AS9 genome includes a single circular chromosome of 5,442,880 bp with 55.96% GC content. The genome had 5,139 predicted genes of which 4,952 were assigned as protein-coding genes, 113 RNA genes and 75 pseudogenes [Figure 3]. The majority of protein coding genes (87.42%) was assigned as a putative function while those remaining were annotated as hypothetical proteins [Table 3]. The distribution into COG functional categories is presented in Table 4.

Graphical circular map of the chromosome. From outside to the center: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew.
References
Grimont F, Grimont PAD. The genus Serratia. In: The Prokaryotes, Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds), New York: Springer. 1992;2822–2848.
Müller H, Berg G. Impact of formulation procedures on the effect of the biocontrol agent Serratia plymuthica HRO-C48 on Verticillium wilt in oilseed rape. BioControl 2008; 53:905–916. http://dx.doi.org/10.1007/s10526-007-9111-3
Kalbe C, Marten P, Berg G. Strains of genus Serratia as beneficial rhizobacteria of oilseed rape with antifungal properties. Microbiol Res 1996; 151:433–439. PubMed http://dx.doi.org/10.1016/S0944-5013(96)80014-0
Taghavi S, Garafola C, Monchy S, Newman L, Hoffman A, Weyens N, Barac T, Vangronsveld J, van der Lelie D. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl Environ Microbiol 2009; 75:748–757. PubMed http://dx.doi.org/10.1128/AEM.02239-08
Alström S. Characteristics of bacteria from oilseed rape in relation to their biocontrol activity against Verticillium dahliae. J Phytopathol 2001; 149:57–64. http://dx.doi.org/10.1046/j.1439-0434.2001.00585.x
Altschul SF, Thomas LS, Alejandro AS, Jingui Z, Webb M, David JL. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997; 25:3389–3402. PubMed http://dx.doi.org/10.1093/nar/25.17.3389
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23:2947–2948. PubMed http://dx.doi.org/10.1093/bioinformatics/btm404
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 2011; 28:2731–2739. PubMed http://dx.doi.org/10.1093/molbev/msr121
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184–200. http://dx.doi.org/10.1007/978-3-642-02008-713
Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2010; 38:D346–D354. PubMed http://dx.doi.org/10.1093/nar/gkp848
Auch AF, von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2010; 2:117–134. PubMed http://dx.doi.org/10.4056/sigs.531120
Auch AF, Klenk HP, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2010; 2:142–148. PubMed http://dx.doi.org/10.4056/sigs.541628
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541–547. PubMed http://dx.doi.org/10.1038/nbt1360
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576–4579. PubMed http://dx.doi.org/10.1073/pnas.87.12.4576
Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 2, Part B, Springer, New York, 2005, p. 1.
List Editor. Validation of publication of new names and new combinations previously effectively published outside the IJSEM. List no. 106. Int J Syst Evol Microbiol 2005; 55:2235–2238. http://dx.doi.Org/10.1099/ijs.0.64108-0
Garrity GM, Holt JG. Taxonomic Outline of the Archaea and Bacteria. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 155–166.
Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225–420. http://dx.doi.org/10.1099/00207713-30-1-225
Rahn O. New principles for the classification of bacteria. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg 1937; 96:273–286.
Judicial Commission. Conservation of the family name Enterobacteriaceae, of the name of the type genus, and designation of the type species OPINION NO. 15. Int Bull Bacteriol Nomencl Taxon 1958; 8:73–74. http://dx.doi.org/10.1099/0096266X-8-1-73
Bizio B. Lettera di BartolomeoBizio al chiarissimocanonico Angelo Bellani sopra il fenomeno della polenta porporina. Biblioteca Italiana o sia Giornale di Letteratura. [Anno VIII]. Scienze e Arti 1823; 30:275–295.
Sakazaki R. Genus IX. Serratia Bizio 1823, 288. In: Buchanan RE, Gibbons NE (eds), Bergey’s Manual of Determinative Bacteriology, Eighth Edition, The Williams and Wilkins Co., Baltimore, 1974, p. 326–326.
Breed RS, Murray EGD, Hitchens AP. In: Breed RS, Murray EGD, Hitchens AP (eds), Bergey’s Manual of Determinative Bacteriology, Sixth Edition, The Williams and Wilkins Co., Baltimore, 1948, p. 481–482.
BAuA. 2010, Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466, p. 200.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet 2000; 25:25–29. PubMed http://dx.doi.org/10.1038/75556
Bennett JW, Bentley R. Seeing red: the story of prodigiosin. Adv Appl Microbiol 2000; 47:1–32. PubMed http://dx.doi.org/10.1016/S0065-2164(00)47000-0
Alström S, Gerhardson B. Characteristics of a Serratia plymuthica isolate from plant rhizospheres. Plant Soil 1987; 103:185–189. http://dx.doi.org/10.1007/BF02370387
Khanafari A, Assadi MM, Fakhr FA. Review of prodigiosin pigmentation in Serratia marcescens. J Biol Sci 2006; 6:1–13.
Bergan T, Grimont AD, Grimont F. Fatty acids of Serratia determined by gas chromatography. Curr Microbiol 1983; 8:7–11. http://dx.doi.org/10.1007/BF01567306
DOE Joint Genome Institute. http://www.jgi.doe.gov
Bennett S. Solexa Ltd. Pharmacogenomics 2004; 5:433–438. PubMed http://dx.doi.org/10.1517/14622416.5.4.433
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:326–327. PubMed
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821–829. PubMed http://dx.doi.org/10.1101/gr.074492.107
Phrap and Phred for Windows. MacOS, Linux, and Unix. http://www.phrap.com
Han C, Chain P. 2006. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabina HR, Valafar H (eds), CSREA Press. June 26–29, 2006: 141–146.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998; 8:195–202. PubMed
Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. error probabilities. Genome Res 1998; 8:186–194. PubMed
Ewing B, Hillier L, Wendl MC, Green P. Base-Calling of automated sequencer traces using Phred. I. accuracy assessment. Genome Res 1998; 8:175–185. PubMed
Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. PubMed http://dx.doi.org/10.1186/1471-2105-11-119
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455–457. PubMed http://dx.doi.org/10.1038/nmeth.1457
Schattner P, Brooks AN, Lowe TM. The tRNAScanSE, snoscan and snoGPS we servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 2005; 33:W686–W689. PubMed http://dx.doi.org/10.1093/nar/gki366
Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271–2278. PubMed http://dx.doi.org/10.1093/bioinformatics/btp393
Acknowledgements
We would like to gratefully acknowledge the help of Elke Lang for providing cell pastes of reference material and Evelyne-Marie Brambilla for extraction of DNA for digital DNA-DNA hybridizations with the reference strains (both at DSMZ). The work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Neupane, S., Högberg, N., Alström, S. et al. Complete genome sequence of the rapeseed plant-growth promoting Serratia plymuthica strain AS9. Stand in Genomic Sci 6, 54–62 (2012). https://doi.org/10.4056/sigs.2595762
Published:
Issue Date:
DOI: https://doi.org/10.4056/sigs.2595762