
Abstract
Here, we report an inorganic hexagonally ordered mesoporous fibre-like carrier made of silica as an effective drug delivery system with mineralisation potential. Fibre-like SBA-15 has been modified by employing a simple surface activation (rehydroxylation) procedure. The surface-rehydroxylated fibre-like SBA-15 (SBA-15-R) was used to investigate the possible mechanism of hydroxyapatite (HA) nucleation and deposition onto silica’s surface after immersion in simulated body fluid (SBF). Amorphous calcium phosphate, Ca-deficient HA and bone-like HA deposits were observed on SBA-15-R surface consecutively after 7, 14 and 21 days of immersion in SBF. Accordingly, our low-angle XRD, STEM and N2 adsorption/desorption results indicated that deposited ions were mostly located at the silica’s surface and could modify the size of the mesopores. The SBA-15-R was studied in vitro as the potential bioactive drug delivery system using doxorubicin (DOX) as a model water-soluble and anticancer drug. The adsorbed DOX molecules were mostly located at the pore walls and pore openings, likely together with the silanol groups. The DOX release was diffusion-controlled and relatively slower in SBF (pH = 7.4) than in phosphate-buffered solution (pH = 5.0), most probably due to both the stronger electrostatic interactions occurring between the DOX and the SBA-15-R and the simultaneous deposition of calcium and phosphates ions from SBF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The development of new local drug carriers for bone diseases focuses on enhanced bioactivity—mineralisation potential, higher delivery efficiency, lower toxicity and controlled drug release. Hexagonally ordered mesoporous silicas (HOMS), e.g. SBA-15 or MCM-41, obtained in sol–gel process, have received considerable attention as a drug delivery system due to their large surface area (600–1200 m2/g), uniform mesopore size (4–10 nm), high surface area-to-volume ratio and modifiable particle shape (sphere to rod-like and fibre-like microparticles). The potential role of HOMS in formulating local drug carriers for bone diseases is emphasised in several studies (1,2,3). The bone bonding property of HOMS can be demonstrated in vitro by the hydroxyapatite layer (HA) formation on the silica’s surface in the simulated body fluid (SBF) (4,5,6,7). A similar HA layer was also observed in vivo at the bone/implant interface (8). The SBF solution biomimics the blood plasma in both ionic composition and concentration and is kept under the physiological temperature and pH (9). Importantly, the apatite layer that formed on a surface of the silica immersed in SBF usually features low to medium crystallinity, similar to that of the bone tissue apatite. This phenomenon has been recently explored as a potential way of generating in situ biomimetic HA coatings onto the implantable drug carriers (10).
The mineralisation potential of HOMS depends on their chemical and physical properties such as pore size, specific surface area, bulk composition and surface chemistry (6). It has been found that high surface area of the HOMS is related to their higher mineralisation potential. Moreover, both a negative surface charge and porous surface of the HOMS are important properties for the efficient nucleation, growth and spread of the HA crystals. The mineralisation potential is directly associated with the presence of polar silanol groups (Si–OH) on the HOMS’ surface. It appears that the surface silanols induce the heterogeneous nucleation of HA through strong intermolecular bonding to the HA precursors which are present in SBF, i.e. ions of Ca2+, PO43− and OH−. After being formed and immobilised on the HOMS’ surface, the apatite nuclei grow spontaneously into crystalline particles, further consuming the calcium and phosphate ions from the SBF (5,6,11). However, HOMS is synthesised/calcinated at a temperature of 500°C or more. At these conditions, silanols tend to condense into siloxane bridges; thus, after the thermal treatment only, the residual hydroxyl groups are present on the surface. This results in delayed HA formation on the HOMS’ surface. The typical induction times for HA nucleation on the surface of HOMS are in the range of 30–60 days (5,6,12). However, this HA formation rate is still much slower if compared with an amorphous, hydroxyl-rich silica synthesised at low temperatures. For such type of silica, the HA deposition was reported after 7–10 days (13,14). Therefore, it is a great challenge to design HOMS drug carriers with the enhanced bioactivity and controlled drug release property.
In this paper, we focused on the evaluation of the HA nucleation and deposition onto the rehydroxylated surface of the fibre-like SBA-15 (SBA-15-R). To the best of our knowledge, such study has not been carried out before. The mineralisation potential of SBA-15-R was studied under biomimetic conditions. Scanning electron microscopy (SEM) is the most common method for morphological analysis of apatite formation on the surface of bioactive material. However, due to SEM’s limited resolution, the shape of a small HA nanocrystals can be only resolved with scanning transmission electron microscope (STEM) (15). The compositional distribution can be studied in detail using STEM microscope combined with field emission scanning electron microscopy mapping (FESEM). FESEM is widely used to observe the surface and cross sections, as well as to analyse the elemental composition by the EDX (energy dispersive X-ray) analysis. It has been demonstrated that a dedicated STEM microscope working in a high-angle annular dark-field mode (HAADF) is capable of taking an image with an atomic resolution (16). Therefore, the detailed insight into the HA formation on the SBA-15-R surface was investigated by HAADF/STEM with FESEM mapping and EDX analysis, and also by SEM imaging, X-ray scattering and N2 adsorption/desorption analysis. In addition, the SBA-15-R was studied in vitro as a potential bioactive drug delivery system using doxorubicin (DOX) as a model water-soluble and anticancer drug, which is characterised as amphipathic weak base consisting of an anthracycline moiety and an amino sugar (17). DOX is the most common drug used to treat primary bone cancer (18,19). In case of water-soluble drug such as DOX, the electrostatic attraction between positively charged DOX and the negatively charged SBA-15-R surface may cause the adsorption of the solute onto the silica’s surface. The 1 mg DOX load per 200 mg of SBA-15-R was designed to deliver a low dose of DOX such as the DOX-loaded hydroxyapatite that was tested in the animal tumour models (18). The authors found that the drug exhibited physiological activity at very low concentrations; the IC50 value for osteosarcoma was about 0.35 μg/mL. They also observed minimal premature release in the extracellular region, which could decrease the drug side effects. In this paper, we additionally studied the differences in the DOX in vitro release using release media with various pH (phosphate-buffered solution, pH = 5.0 and SBF, pH = 7.4). In both DOX adsorption and release studies, the non-rehydroxylated SBA-15 was used as a control.
MATERIALS AND METHODS
Materials
Pluronic P123 triblock copolymer ((EO)20(PO)70(EO)20, Mav = 5800) tetraethyl orthosilicate (TEOS), benzylamine, doxorubicin hydrochloride (DOX), ethanol and tris(hydroxymethyl)aminomethane (TRIS) were all obtained from Sigma-Aldrich.
Preparation of Simulated Body Fluid
SBF was prepared according to the Kokubo’s procedure (9). To prepare 1 L of SBF, 7.996 g of NaCl, 0.350 g of NaHCO3, 0.224 g of KCl, 0.228 g of K2HPO4·3H2O, and 0.305 g of MgCl2·6H2O were dissolved in 750 mL of deionised water. Then, 39.0 mL of hydrochloric acid (1 M) was added to the solution; 0.278 g of CaCl2, 0.071 g of Na2SO4 and 6.057 g of TRIS were sequentially dissolved in the solution. Finally, the solution was diluted to 1 L and adjusted to pH = 7.4 with 1 M hydrochloric acid.
Synthesis of Fibre-Like SBA-15
The synthesis of hexagonally ordered mesoporous silica, fibre-like SBA-15, was carried out according to the modified procedure proposed by Zhao et al. (20) using Pluronic P123 triblock copolymer as a structure-directing agent and tetraethyl orthosilicate (TEOS) as silica source. Of Pluronic P123, 4.0 g was dissolved in the mixture of 2 M HCl (120.0 g) and purified water (30.0 g) at 35°C using a magnetic stirrer (300 rpm). Then, 8.0 g of TEOS was added drop-wise, and the reaction solution was continuously stirred to completion for 20 h. The mixture containing precipitated solid products was aged at 80°C for 24 h without stirring and then filtrated under reduced pressure. Solid products were washed with 100 mL of purified water and 30 mL of absolute ethanol and dried at 40°C for 1 h in air. Finally, the products were calcinated at 500°C for 6 h in air (with heating rate of 1°C/min) in muffle furnace (M-525, II series). The fibre-like SBA-15 material with particle size of 10 to 20 μm was obtained by micronisation (Fritsch, Mortar Grinder Pulverisette 2). The micronisation lasted 20 min at 80 rpm.
Surface Activation (Rehydroxylation) of SBA-15
The calcinated micrometre-sized (10–20 μm) fibre-like SBA-15 was subject to post-synthetic surface activation. During the surface activation process, the SBA-15 is rehydroxylated in order to increase the number of silanols (Si–OH) (21), thus to increase the overall surface polarity (Fig. 1). Rehydroxylation of calcinated samples was performed by immersing 1.0 g of SBA-15 in 150 mL HCl (18 wt%), followed by stirring for 24 h (400 rpm) under reflux. The surface-rehydroxylated SBA-15 was subsequently placed under a vacuum for 15 min in order to remove entrapped air bubbles. Finally, the samples were rinsed with deionised water three times to remove any residual HCl. The pH of the last rinsing solution was checked to confirm it was 7.0.

Schematic representation of the surface activation/rehydroxylation process of hexagonally ordered fibre-like SBA-15 leading to increased surface polarity: acid-catalysed cleavage of a surface siloxane bridge (Si–O–Si) and subsequent generation of two siloxanes (Si–OH)
In order to confirm the surface activation of fibre-like SBA-15 after the rehydroxylation procedure (which will further be referred as SBA-15-R), the SBA-15 and SBA-15-R samples were compared using Fourier transform infrared spectroscopy (FTIR, Jasco model 4700), KBr tablet technique with spectra normalisation to maximum absorption of a dominant peak at ca. 1080 cm−1. The accessible surface concentration of silanols in both SBA-15 and SBA-15-R materials was calculated using semi-quantitative benzylamine (0.015 M) adsorption method from non-polar solvent, cyclohexane (22). The amount of adsorbed benzylamine on silicas’ surface at equilibrium state was determined spectrophotometrically (Shimadzu, model UV-1800) at 256 nm. For both SBA-15 and SBA-15-R materials, the benzylamine adsorption tests were repeated five times and the mean values with standard deviations were calculated.
In Vitro Mineralisation Assay of SBA-15-R
The HA nucleation and deposition onto rehydroxylated silica’s surface was investigated during the mineralisation assay. For this purpose, the SBA-15-R was chosen because the higher concentration of accessible silanols seems to promote the HA formation on silica’s surface (23,24). Briefly, the micrometre-sized (10–20 μm) fibre-like SBA-15-R was dispersed in SBF at a concentration of 1 mg/mL and gently stirred at 37°C for 7, 14 and 21 days, respectively. As recommended by Rámila and Vallet-Regı́ (7), SBF was exchanged after each 24 h of immersion. The samples were then rinsed in deionised water twice and stored in a vacuum until further characterisation.
Drug Loading
The immersion loading method was applied to the fibre-like SBA-15-R loading experiments as previously reported (1). In brief, the loading experiments were carried out as follows: 200.0 mg of micrometre-sized (10–20 μm) fibre-like SBA-15-R was added to a 5 mL doxorubicin hydrochloride (DOX) solution 250 μg/mL in distilled deionised water (DDI), pH close to 6.6. The mixture was stirred at room temperature (25 ± 0.5°C) under light-sealed conditions for 24 h to reach the equilibrium state. To evaluate the DOX loading efficacy, the supernatant was centrifuged at 16,500×g for 10 min using a MPW-350 fixed angle rotor centrifuge (MPW Med. Instruments, Poland). The supernatant was collected, and the residual DOX content was determined using the calibration curve of DOX standard solutions in DDI by the UV-Vis measurement at 480 nm. Drug-loading efficiency (E) was calculated using Eq. 1:
The theoretical drug loading was based on mass fraction of drug and SBA-15-R used to prepare samples. The same loading procedure was performed for the non-rehydroxylated SBA-15 as a control. For both SBA-15 and SBA-15-R, the loading experiments were repeated five times. The results are expressed as mean values ± standard deviations.
In Vitro Dissolution Study
Dissolution studies were performed for the micrometre-sized (10–20 μm) fibre-like DOX-loaded SBA-15-R at different pH values. The phosphate buffer (pH = 5.0) was used to mimic a physiological condition at the targeted tumour site (19), and the above-mentioned simulated body fluid (SBF, pH = 7.4) was used for comparative purposes. The release studies were performed on a ERWEKA USP Apparatus 4 dissolution system (closed loop) (Erweka DFZ-720, Germany). In each experiment, the DOX-loaded SBA-15-R samples with an approximate mass of DOX, equal 1000 μg, were placed on glass beads in a 22.6-mm flow-through cell. The glass beads were used to produce laminar flow conditions and to prevent powders from descending into the inlet tubing (25). The flow rate was 4 mL/min in attempt to mimic in vivo conditions (26). The eluate was filtered upon leaving the cell and was collected in fractions. At certain time intervals, a portion of the eluate (2 mL) was taken out to measure the released drug concentration and then fresh buffer (2 mL) was added to keep the volume constant. Sink conditions, where DOX concentrations in the release medium are below 10% of its aqueous solubility (2.1 mg/mL), were maintained during the studies. The buffer solution was changed for a fresh one every 24 h. DOX was stable in the media during the sampling intervals. DOX was analysed quantitatively using UV/Vis spectroscopy at λ max = 480 nm (Shimadzu, model UV-1800). Analytical studies were conducted in accordance with the requirements for quantitative analyses, calibrating the detector with a standard solution of the DOX in specific buffer. The calibration curve was linear in the tested range from 1.0 to 40.0 μg/mL with a regression coefficient of 0.999, regardless of buffer used. The DOX detection limit was 0.9 μg/mL. Drug elution data were plotted as the cumulative percent of DOX released (Q) as a function of time (t). The results were obtained from data groups of n ≥ 3 and are expressed as mean values ± standard deviations. The same dissolution studies were performed for non-rehydroxylated SBA-15 as a control.
Characterisation
The mesostructure ordering of the investigated samples was characterised by low-angle X-ray diffraction (XRD D/max2600, Rigaku, Japan) using the CuKα radiation. XRD patterns were collected in the 2Θ range between 0.5° and 9°, with a step width of 0.02° and at a scanning rate of 0.5°/min. The crystallinity of the investigated samples was examined by wide-angle X-ray diffraction (XRD). XRD data was recorded on Empyrean PANalytical diffractometer using CuKα radiation (40 kV and 25 mA) at a scanning rate of 4°/min with a step width of 0.0263. The peak positions of the measured samples were referenced to those of the JCPDS database.
The morphology of the investigated samples was analysed using Ultrospec (FE-TEM), EDX and elemental mapping with a JEM-2100F (JEOL) instrument operated at 200 kV. Cationic compositions were determined on selected areas of the samples using EDX equipped with a retractable detector with an ultrathin window. The high-angle annular dark field (HAADF) images were obtained in a VG HB501UX scanning TEM. Samples for TEM were sprinkled on a carbon film with holes, supported by a copper gird.
The parameters characterising the porosity of the investigated samples were determined by the measurements of low-temperature nitrogen adsorption–desorption at − 196°C using a volumetric adsorption analyser ASAP 2405 (Micromeritics, Norcros, GA). Prior to the experiment, all of the samples were dried overnight at 80°C under vacuum. The specific surface areas were calculated using the standard Brunauer–Emmett–Teller (BET) equation for nitrogen adsorption data acquired in the range of relative pressure p/p0 in the range from 0.05 to 0.25. The total pore volumes (Vp) were estimated from a single point adsorption at 0.993 p/p0. The average pore diameter (Dp) was determined from the desorption branch of the nitrogen isotherm using the Barrett–Joyner–Halenda (BJH) procedure. The unit cell parameter (a) was calculated using the formula a = 2d100/\( \sqrt{3} \), where d100 presents the d-spacing value of the (100) diffraction peak in XRD patterns (27). The pore wall thickness (b) was calculated using formula b = a − Dp where (a) represents the unit cell parameter and Dp represents the pore diameter obtained from BJH calculations (27).
Scanning electron microscopy (Quanta 3D FEG) at an acceleration voltage 10–20 kV was used to show the morphology of the investigated samples. Samples for SEM were fixed on carbon tape and coated with Pd/Au for 2 min.
The zeta potential was used to analyse the charge densities of SBA-15-R under the conditions of both DOX adsorption and release studies. Zeta potential testing was carried out using an Otasuka Electronics zeta potential analyser (ELS-8000, Ireland). The samples were dispersed in deionised water at concentration of 1 mg/mL, under sonication for 5 min. The zeta potential and isoelectric point (IEP) were measured as a function of pH by titrating with 0.1 M HCl and 0.1 M NaOH at room temperature. The same measurements for the SBA-15 were performed as a control.
RESULTS
Surface Activation of SBA-15
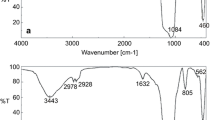
The FTIR spectra of the starting material SBA-15 and surface-activated SBA-15-R are shown in Fig. 2. The most intense bands at ca. 1080 cm−1 with the shoulder at 1150 cm−1 are characteristic for (Si–O–Si) transversal optical mode and (Si–O–Si) longitudinal optical mode asymmetric stretching vibrations, respectively (28,29). The band at 960 cm−1 represents the Si–OH stretching vibrations of silanol groups. Other silica network bands are located at 801 and 455 cm−1 and are signatures of (Si–O–Si) symmetric stretching vibrations and (O–Si–O) bending vibrations, respectively. The band at 1635 cm−1 results from (H–O–H) bending vibrations of physisorbed molecular water. The higher absorbance at 960 cm−1 (upwards arrow) indicates the higher surface concentration of isolated silanols after the rehydroxylation procedure (29). In addition, the frequency shift of the transversal optical peak (leftwards arrow) from 1081 to 1075 cm−1 can be connected with an increase in the Si–O bond length, resulting from hydrolysis of Si–O–Si bridges into Si–OH groups via rehydroxylation procedure (28). This observed frequency shift might be also related to decrease of bridging angle based on central force network model (28,29). The higher concentration of surface silanol groups for SBA-15-R material was also confirmed by higher benzylamine adsorption as compared with the non-rehydroxylated SBA-15 material. The maximum amounts of adsorbed benzylamine at equilibrium state were 1.52 ± 0.2 and 3.43 ± 0.7 mmol per g of silica, for SBA-15 and SBA-15-R, respectively. The corresponding values calculated per surface area (765 m2/g for SBA-15 and 680 m2/g for SBA-15-R) were 2.0 ± 0.3 and 5.0 ± 1.0 μmol/m2 for SBA-15 and SBA-15-R, respectively, showing that after rehydroxylation, the accessible surface concentration of silanols increased by factor of 2.5. As reported elsewhere (22,30) when employing this rehydroxylation procedure, the concentration of surface silanol groups should exceed 4.0 μmol/m2.

The FTIR spectra of SBA-15 (blue line) and SBA-15-R (red line) samples with calculated surface silanol concentration obtained from the benzylamine adsorption studies
Nanostructure of SBA-15-R
Figure 3 shows a microscopic characterisation of the SBA-15-R nanostructure side-view STEM image and corresponding electron energy loss spectroscopy (EELS) mapping with EDX profile and SEM image. Table I lists the unit cell parameter and pore wall thickness obtained from XRD and the N2 adsorption/desorption data of the SBA-15-R. The microscopic analysis (Fig. 3a) indicated that the nanostructure of the SBA-15-R consists of well-ordered nanochannels. Based on XRD and BJH calculations (Table I), the nanochannels were characterised by the average pore diameter of 6.5 nm and pore wall thickness between nanochannels of 4.5 nm. The EDX analysis confirmed that the material’s surface is composed of oxygen and silicon elements. As shown in the SEM image (Fig. 3c), the synthesised SBA-15-R material forms fibrous, rod-like domains aggregated into bundle-like macrostructures, the same as observed for non-rehydroxylated SBA-15 (data not shown).

Microscopic characterisation of parent SBA-15-R. a STEM image along the nanochannels. b Enlarged STEM image of the blue square region with corresponding EELS mapping of oxygen (O-orange) and silicon (Si-yellow). c SEM micrograph. d EDX profile
In Vitro Mineralisation of SBA-15-R
The representative STEM images of the SBA-15-R nanostructure after 7 days of immersion in SBF are shown in Figs. 4 (side view) and 5 (top view), along with the corresponding EELS mapping and EDX profile. As shown in Figs. 4 and 5, the hexagonal pore arrangement is less ordered as compared with the parent SBA-15-R (Fig. 3a) and the nanometre-sized clusters (bright spots in Figs. 4a and 5a) are present and spread throughout the entire surface. The corresponding EDX analysis revealed that the new observed deposit consists of calcium and phosphorous elements with a Ca/P atomic ratio of 1.82 (Fig. 4c). This is close to the Ca/P ratio in Ca-rich amorphous calcium phosphate (ACP; Ca/P = 1.8) (31). The EDX data has also shown other elements such as Na and Mg, however at much lower concentration. As shown in top-view STEM (Fig. 5), the relatively thick layer of the Ca and P deposit was formed on the SBA-15-R surface. These results suggest that, after immersing in SBF for 7 days, a Ca-rich amorphous calcium phosphate (ACP) forms on the SBA-15-R surface.

The SBA-15-R after 7 days of immersion in SBF. a Side-view STEM-HAADF image along the nanochannels (bright spots: nanoclusters of CaP precipitates). b FESEM mapping of calcium (Ca-green), phosphorous (P-red) and oxygen (O-blue) for the enlarged image of the blue square region shown in a. c EDX profile: Ca/P = 1.82

The SBA-15-R after 7 days of immersion in SBF. a Top-view STEM-HAADF image. b EELS mapping of calcium (Ca-yellow), phosphorous (P-red) and silicon (Si-green) for the enlarged image of the blue square region shown in a Bottom: elemental maps of Si, Ca and P
Figure 6 shows the cross-sectional view STEM image with the corresponding EELS mapping and the EDX profile of SBA-15-R after immersion for 14 days in the SBF. After increasing the time of immersion in SBF to 14 days, we have noticed additional changes in the morphology of SBA-15-R, as shown in the cross-sectional view STEM image (Fig. 6a). The presence of plate-shaped crystals, 10–20 nm wide and 100 nm long, in the silica network was observed. This was accompanied by the almost uniform distribution of Ca and P ions in the bulk of the silica network. The corresponding EDX data (Fig. 6d) confirmed a microstructural evolution of amorphous calcium phosphate to nanocrystalline apatite, and the observed Ca/P atomic ratio of 1.54 was close to the value of a Ca-deficient apatite phase (31).

The SBA-15-R after 14 days of immersion in SBF. a Cross-sectional STEM image showing rectangular plate-shaped individual nanocrystals. For the enlarged image of the blue square region shown in a: b FESEM mapping of calcium (Ca-green) with phosphorous (P-red) and silicon (Si-yellow) and c FESEM mapping of oxygen (O-blue) with phosphorous (P-red). d EDX profile: Ca/P = 1.54. Bottom: elemental maps of Si, P, Ca and O
Figure 7 shows the STEM image with the corresponding EDX profile and SEM image after immersion of SBA-15-R in SBF for 21 days. After 21 days of assay, both the STEM and SEM images of the SBA-15-R showed accordingly the precipitation of the biomimetic coating. The elongated fibre-like domains of SBA-15-R have been covered with spherulites of around 5–10 μm, consisting of plate-like–shaped primary crystals. The EDX measurement (Fig. 7b) showed that in comparison to the samples immersed for 14 days in SBF, the corresponding atomic ratio of Ca/P increased to 1.62, which is close to the Ca/P ~ 1.75 value observed in human bone (32).

The SBA-15-R after 21 days of immersion in SBF. a Cross-sectional STEM image: plate-shaped nanocrystals forming spherulites. b EDX profile: Ca/P = 1.62. c, d SEM micrographs at different magnification (arrow: plate-like domains aggregated into spherical-shaped particles made of apatite crystals with corresponding rod-like domains aggregated into bundle-like macrostructure of SBA-15-R)
Figure 8 presents a low-angle XRD pattern of the SBA-15-R both non-immersed and immersed in SBF for 7 and 14 days. The low-angle XRD pattern of parent SBA-15-R confirmed the presence of (100), (110), (200) and (210) planes indicating the highly ordered SBA-15-type 2D hexagonal nanostructure featuring p6mm symmetry (20)—no disorganisation of ordered pore structure was observed after the rehydroxylation procedure. We observed that after 7 days of immersion in SBF, the intensity of the (100) peak decreased significantly and (110), (200) and (210) peaks almost vanished. Noteworthy, after 14 days in SBF, all the characteristic low-angle XRD peaks of SBA-15-R vanished, indicating that the hexagonal pores in the silica nanostructure were covered by the layer of amorphous calcium phosphate which scattering power is similar to that of silica (33).

Low-angle XRD pattern of the parent SBA-15-R and immersed in SBF for various periods
Figure 9 shows a wide-angle XRD pattern of the SBA-15-R immersed in SBF for various periods. After 7 days in SBF, the presence of a broad and diffuse peak with maxima at 25° 2Θ was observed, which appears to be a combined amorphous phase of inorganic deposit, with Ca and P as main components, along with silica from SBA-15-R. After 14 days in SBF, the XRD pattern indicated the presence of low-crystallinity hydroxyapatite-like calcium phosphate (CaP) phase. The appearance of the HA crystalline phase after 14 days is confirmed by the STEM observation of rectangular plate–shaped nanocrystals (Fig. 6a). Finally, after 21 days in SBF, the wide-angle XRD analysis confirmed that the observed spherulites with plate-like domains (Fig. 7) are composed of hydroxyapatite featuring crystallinity similar to that of hydroxyapatite present in bone tissue (32).

Wide-angle XRD pattern of the SBA-15-R after 7, 14 and 21 days of immersion in SBF. Inset: reference of hydroxyapatite
Table I lists the N2 adsorption/desorption data of SBA-15-R immersed in SBF for various time periods. It can be observed that after immersion in SBF, the deposition of the Ca and P ions clearly reduced the available pore volume and specific surface area, compared with the parent SBA-15-R (non-immersed in SBF), most probably due to pore blocking caused by layer of CaP precipitate at the mesoporous structure. The comparison of the data indicates that the mean specific surface area of SBA-15-R after 21 days in SBF was reduced by a factor of 3.5. This corresponds to ca. twofold increase in the mean pore diameter, from 6.50 nm (parent SBA-15-R) to 15.0 nm (after 21 days of immersion in SBF).
Doxorubicin Loading and In Vitro Release
Doxorubicin loading onto SBA-15 was 5.4 ± 0.09 mg/g, whereas for SBA-15-R, it was 5.6 ± 0.14 mg/g, corresponding to adsorption efficiency of 87 ± 7% and 90 ± 9%, respectively (calculated using Eq. 1), under the conditions of the loading experiment. The representative STEM images of a selected region of DOX-loaded SBA-15-R are shown in Fig. 10a (side view), b (top view), along with the corresponding EELS mapping. STEM images of the DOX-loaded SBA-15-R reveal hexagonal arrays of uniform channels (Fig. 10), which closely resemble the SBA-15-R (Fig. 3). The corresponding EELS data revealed that the specific organic atom (carbon corresponding to DOX) was observed within the SBA-15-R cross section (Fig. 10a) and on the surface (Fig. 10b). The SBA-15-R nanochannels exhibit a gradient of the C content, which increases from the interior (C poor) to the surface (C rich), implying that most of drug molecules are located on the surface, together with the silanol groups. Figure 11 shows both the possible ionic attraction, during the loading process, between positively charged DOX (R-NH3+) and negatively charged SBA-15-R surface (≡SiO−) and the co-existed hydrogen bonds.

Microscopic characterisation of DOX-loaded SBA-15-R. a Side-view STEM-HAADF image along the nanochannels with corresponding EELS mapping of carbon (C-red). b Top-view STEM-HAADF image with corresponding EELS mapping of carbon (C-red)

The possible interaction between positively charged DOX and SBA-15-R surfaces during the loading process—illustrative visualisation model (plan view). Atom colour: silicon (Si-gold), oxygen (O-red), hydrogen (H-white), carbon (C-grey), nitrogen (N-blue)
The drug release behaviour of both DOX-loaded SBA-15 and SBA-15-R, as shown in Fig. 12, was studied in buffers at pH values of 7.4 (SBF) and 5.0 (phosphate buffer) at 37°C to mimic the different physiological conditions (19). For both materials, the rate of the DOX release was pH-dependent and increases with the pH decrease. The DOX release was characterised by the biphasic release profiles with an initial burst due to the dissolution of DOX from the outer surface of the fibre-like microparticles followed by a subsequent prolonged release. The highest burst release occurs at the pH = 5.0, where almost 56 ± 2% and 46 ± 3% of the DOX were released within the first 24 h for SBA-15 and SBA-15-R, respectively. At the same time, at pH = 7.4, 36 ± 1% and 32% ± 3% of the DOX were released from SBA-15 and SBA-15-R. The cumulative percent of released drug after 120 h for the SBA-15 was 98 ± 2% and 79 ± 3% (corresponding to 980 ± 20 μg and 790 ± 30 μg of released DOX) at the pH values of 5.0 and 7.4, respectively. For comparison, the corresponding cumulative percent of released drug for SBA-15-R was almost the same at pH = 5.0 (97 ± 3%, 970 ± 30 μg of released DOX), but relatively lower at pH = 7.4 (72 ± 4%, 720 ± 40 μg of released DOX). For the SBA-15, the total drug release at pH = 7.4 was found after 144 h (6 days), whereas for SBA-15-R, it was 26 h longer and was found after 170 h (7 days).

Mean DOX release profiles (±SD, n = 3) of DOX-loaded SBA-15 and SBA-15-R formulations at different dissolution media (phosphate buffer solution, pH = 5.0 and SBF, pH = 7.4)
The measured zeta potential values for SBA-15 and SBA-15-R at pHs of both DOX adsorption (pH = 6.6) and release (pH = 5.0 or pH = 7.4) with IEP are presented in Table II. The IEP for SBA-15 and SBA-15-R was found to be at pH of 2.4 and 3.1, respectively. Thus, the surface of both materials was negatively charged during the adsorption and release studies. However, the rehydroxylated material SBA-15-R was characterised by lower values of zeta potential at each pH compared with non-rehydroxylated SBA-15.
DISCUSSION
This study is part of our ongoing effort to evaluate the suitability of hexagonally ordered mesoporous silica, SBA-15, for two biomedical applications: local anticancer drug delivery and bone tissue regeneration. In this work, we prepared the rehydroxylated fibre-like mesoporous SBA-15 (SBA-15-R) by a soft-template method and by employing a simple surface activation (rehydroxylation) procedure. The SBA-15-R was immersed in the refreshed (every 24 h) SBF to study both the mineralisation potential and the changes in the silica nanostructure, including alteration of pore volume and size, caused by the deposition of calcium phosphate with different stoichiometry and particle size.
It is well known that mesoporous silica can adsorb water, other polar solvents and ions, through creation of hydrogen bonding between surface silanols and any neighbouring solvent or ions (34). Thus, the extra silanol groups generated upon rehydroxylation at the surface of SBA-15-R (cf. Figs. 1 and 2) are expected to induce stronger adsorption of ions which are present in SBF solution. It has been recently demonstrated that the non-modified nanostructured SBA-15 materials induced heterogeneous nucleation of the hydroxyapatite mineral after immersion in SBF for 30 days (5,6). Our results may suggest that the rehydroxylation procedure could reduce the time required for nucleation and subsequent deposition of HA onto the SBA-15 material. We observe that the rehydroxylated, thus more polar and silanol-reach SBA-15-R material induces deposition of an amorphous CaP layer after 7 days, with subsequent crystallisation into Ca-deficient and bone-like hydroxyapatite after 14 and 21 days, respectively. For comparison, we have reported the layer of bone-like apatite on non-rehydroxylated SBA-15 material after 60 days of immersion in SBF (12), whereas Vallet-Regí and co-workers observed the similar HA layer after 30 days (5,6). Considering the significant role of silanols in mechanism of HA formation (23,24), their increased surface concentration seems to promote the deposition of biomimetic apatite layer on SBA-15.
Bhattacharyya et al. (33) found that the low-angle XRD peaks may disappear if the pores have been filled or coated with an amorphous material which scattering power is comparable with that of silica. Our low-angle XRD and STEM results indicate accordingly that the ordered hexagonal channels cannot be clearly distinguished inside the SBA-15-R after 7 and 14 days of immersion in SBF. This suggests the presence of guest ions both onto the SBA-15-R surface and pore opening, most probably by employing diffusion and capillary adhesive forces. This is further confirmed by our N2 adsorption/desorption data (Table I), which shows that at increasing immersion times in SBF, the pore volume decreases. This indicates the clogging of the SBA-15-R mesopores by the in situ–formed CaP precipitate. This observation is particularly relevant to drug delivery applications where drug release rates might be decelerated by the mineral layer deposited either at the surface and/or inside pores of the mesoporous silica.
Our results indicate that the observed nano-sized clusters (bright spots in Figs. 4a and 5a) are amorphous in nature. As revealed by the EDX analysis, they are mainly composed of Ca and P elements, and having Ca/P atomic ratio of 1.82, this precipitate can be considered as Ca-rich amorphous calcium phosphate (Ca-rich ACP) (cf. Fig. 4c). Interestingly, we observed a further evolution of the Ca/P ratio in the deposited layer, along with changes in morphology and sizes of the deposited particles. After 14 days, the amorphous precipitate turned into nanocrystalline particles with well-defined rectangular plate-like shapes (cf. Fig. 6a), while the Ca/P ratio decreased to 1.54 indicating the presence of Ca-deficient hydroxyapatite. In the samples immersed in SBF for 21 days, the Ca/P ratio increased to 1.62, which is close to the ratio of 1.67 in stoichiometric hydroxyapatite. Our XRD results confirmed that after 21 days in SBF, the samples indeed have contained the crystalline phase of hydroxyapatite (cf. Fig. 9). Regarding size and shape, the particles deposited after 21 days were predominantly spherical (spherulite-like), composed of plate-like hydroxyapatite nanocrystals. This spherulite-like particle morphology is commonly observed in hydroxyapatite formed from CaP precipitate in SBF (6,13). A relevant mechanistic description regarding the growth of biological apatite from amorphous calcium phosphate has been provided by Brown et al. (35). The authors proposed that the ACP precursor subsequently transforms to calcium-deficient hydroxyapatite by incorporating phosphate, carbonate, calcium, etc. ions from the surrounding solution. Therefore, the Ca-rich ACP observed after 7 days of immersion in SBF may interact with the phosphate counter-ions in SBF and subsequently form the Ca-deficient low-crystallinity apatite with Ca/P ratio of 1.54 (1,36). As the right stoichiometry is not achieved at this stage, the apatite crystal undergoes further refinement resulting in the Ca/P atomic ratio of 1.62, being now similar in composition to the bone-like apatite (37). Considering our results, the initial stages of the mineralisation process may be summarised in the following steps: (1) diffusion and capillary-induced adhesion of osteogenic ions from the SBF into porous surface of SBA-15-R, (2) the formation of the calcium-silicate interface as a consequence of the electrostatic interactions of negatively charged (deprotonated) surface siloxane groups (≡SiO−) with calcium ions from SBF and (3) the formation of the amorphous calcium phosphate layer (Ca-rich ACP) through interaction of the calcium-terminated silica’s surface with negatively charged phosphate ions from SBF. Feenstra and Bruyn (38) proposed that the transformation mechanisms of ACP to apatite at physiological pH can follow ACP dissolution; then, a transitional octacalcium phosphate (OCP) solid-phase reprecipitation via nucleation and growth, with a final step involving hydrolysis of the OCP phase into the thermodynamically more stable apatite. Therefore, it has been proposed that ACP act as a template for the growth of the apatite crystal.
The wide-angle XRD results (Fig. 9) confirm that the material deposited after 21 days in SBF is semi-crystalline hydroxyapatite. The EDX analysis (Fig. 7b) indicates that the mineral deposit contains other elements such as Na and Mg. These elements are typically observed in the biological apatite of bone tissue (38) and apatite formed on implants in SBF (39).
The effectiveness of surface-rehydroxylated fibre-like SBA-15 (SBA-15-R) as potential bioactive drug delivery systems was further studied in vitro using doxorubicin as a model water-soluble anticancer drug. The topological surface area of DOX molecule is 2.06 nm2, and the hydrodynamic radius for DOX in water is ca. 1.5 nm (40); therefore, the DOX molecule fits into the pores of SBA-15 materials (41,42). This estimate was also confirmed by STEM with the corresponding EELS (cf. Fig. 10). The details on the specific DOX-silica interactions are reported elsewhere (43,44). It has been already well documented that the three following types of interactions between DOX molecules and a carrier can exist: electrostatic, hydrogen bonding and dispersion forces. The steric hindrance can also be a determinant of DOX loading. The pKa of DOX is 8.0 (17); therefore, during the sorption process at pH = 6.6, the DOX features protonated/positively charged amino group (R-NH3+). Based on zeta potential measurements at pH = 6.6 (Table II), the surface of both the SBA-15 and SBA-15-R was negatively charged (− 22 and − 28 mV, respectively), as a consequence of the dissociation of silanol groups (≡SiOH ⇄ SiO− + H+). The relatively lower zeta potential of SBA-15-R surface might be explained as a consequence of the rehydroxylation procedure in which concentration of surface accessible silanols increased by factor 2.5 if compared with non-rehydroxylated SBA-15 (cf. Fig. 2). Thus, increased amount of dissociated silanols caused lower charge of SBA-15-R surface. The positively charged DOX at pH = 6.6 undergoes spontaneous deposition onto the negatively charged SBA-15 materials’ surface and into pores via combination of ionic attraction and hydrogen bonding (cf. Fig. 11). The almost complete DOX loading without significant differences in adsorption efficiency for both materials (87 ± 7% and 90 ± 9% for SBA-15 and SBA-15-R, respectively) might be caused by the chosen adsorption conditions, which were well below the adsorption capacity of used materials (data not shown). Additionally, the electrostatic attraction between silica’s surface and DOX molecules, during the applied immersion method, promotes the drug loading. This result is also in agreement with other reports (45,46).
The DOX release behaviour was studied at the various pHs to mimic the different physiological conditions (47). Apparatus 4 dissolution was chosen as the technique for testing this formulation because the laminar flow through the sample cell correlates well with the likely in vivo conditions, where a constant circulation of body fluids takes place. It is proposed that the rate of DOX release at performed conditions for non-rehydroxylated SBA-15 and SBA-15-R (cf. Fig. 12) is controlled by the diffusion of molecules through the delivery system. The drug release rate decreases with time due to an extended distance the DOX molecules must take to diffuse out of the deeper parts of the nanochannels to the exterior. It is noteworthy that for other mesoporous systems, such as MCM-41, transport via the nanochannels is more important or even dominant (47).
In order to explain the differences in DOX release rate for SBA-15 and SBA-15-R at different pH conditions, both factors should be considered: the influence of surface silanols and simultaneous deposition of calcium and phosphate ions onto silica’s surface. It has been reported that the surface silanols present in SBA-15 materials are characterised by pKa ca. 2 and 8 for free (Q3, ≡SiOH) and geminal (Q2, =Si(OH)2) silanols, respectively (22). Based on Henderson–Hasselbalch equation (48), free silanols exist in dissociated form (≡SiO−) at both pH values (5.0 and 7.4). In contrast, geminal silanols stay undissociated at pH = 5.0 (=Si(OH)2) and become partially dissociated at pH = 7.4 (=Si(OH)O−). Therefore, the silica surface of both SBA-15 and SBA-15-R is more negatively charged at pH = 7.4 that has been confirmed by zeta potential measurements (cf. Table II). DOX molecules may interact with silanols via both ionic attraction (dissociated silanols) and hydrogen bonds (dissociated and undissociated silanols). Thus, the slower release rate of drug at pH = 7.4 for SBA-15 and SBA-15-R might be explained as a consequence of lower zeta potentials which promoted the electrostatic attractions (both ionic and hydrogen bonds) between positively charged DOX and negatively charged silica’s surface. Moreover, the SBA-15-R was characterised by slower drug release rate compared with SBA-15, especially at pH = 7.4 (cf. Fig. 12), most probably due to higher concentration of surface silanols, as a consequence of the rehydroxylation procedure (cf. Fig. 2). It might be concluded that higher amount of accessible silanols at SBA-15-R surface intensified the electrostatic attractions between silica surface and DOX molecules and consequently slowed down the drug release.
The slower DOX release rate for SBA-15 and SBA-15-R in SBF release medium (pH = 7.4) compared with phosphate buffer (pH = 5.0) might be also connected with simultaneous deposition of calcium on phosphate ions onto silica’s surface in SBF. The surface silanols not occupied by adsorbed DOX molecules act as the nucleation centres for calcium phosphate per se. In case of silanols occupied by adsorbed DOX, they become accessible for calcium ions from SBF after partial release of drug molecules. Considering that most silanols are located in pore walls and at the pore’s entrance (49), the forming calcium phosphate layers may clog the pores and thus impede the release of drug molecules located inside the pores. However, under the biomimetic conditions adapted to drug release, the complete release of DOX was observed for both SBA-15 and SBA-15-R. It means that the formation of hydroxyapatite did not stop the drug diffusion but only slowed it down.
CONCLUSIONS
Fibre-like hexagonally ordered mesoporous silica SBA-15 has been successfully modified by employing a simple surface activation (rehydroxylation) procedure. In result, the surface concentration of silanols (Si–OH) increased from 2 to 5 μmol/m2. The increased surface concentration of silanols seems to enhance the mineralisation potential characterised as the deposition of biomimetic apatite layer onto rehydroxylated SBA-15 silica’s surface. Our findings indicate overall that the mineralisation of bone-like apatite on the rehydroxylated SBA-15 is a multi-stage process, initially involving capillary-induced diffusion and adhesion of ions, and interaction of calcium and phosphate ions with the silanol groups of rehydroxylated SBA-15. The precipitation of amorphous calcium phosphate observed initially on the surface of rehydroxylated SBA-15 appears to be kinetically favourable in SBF. Over time, this metastable amorphous phase transforms into thermodynamically more stable individual nanocrystals of hydroxyapatite, which subsequently form spherulite-like crystalline aggregates with bone-like stoichiometry.
We also studied the features of rehydroxylated fibre-like SBA-15 after immersion in doxorubicin hydrochloride solution by using STEM images with EELS mapping. Our findings indicate that the positively charged drug molecules are located at the surface and inside the nanochannels. We observe that this loading process occurs by spontaneous sorption. Our drug release studies indicate that the rehydroxylated SBA-15 exhibits prolonged and pH-dependent drug release profile. The drug release is diffusion-controlled, and it decreases with time. The concentration of surface silanols and their pH-dependent dissociation cause the negative charge of silica surface and thus influence on drug release rate. The electrostatic interactions (ionic attraction, hydrogen bonds) between negatively charged silica’s surface and positively charged DOX molecules might be a rate-limiting factor influencing the drug release behaviour. The drug release was slower in simulated body fluid (pH = 7.4), if compared with phosphate buffer (pH = 5.0), mostly because of stronger electrostatic interactions and simultaneous deposition of calcium and phosphate ions onto the silica’s surface.
Our results demonstrate that the micrometre-sized, rehydroxylated fibre-like SBA-15 combine the mineralisation properties with the prolonged drug release. The obtained results offer a promising basis for future development of bioactive rehydroxylated mesoporous silica in the targeted drug delivery applications.
References
Prokopowicz M, Czarnobaj K, Szewczyk A, Sawicki W. Preparation and in vitro characterisation of bioactive mesoporous silica microparticles for drug delivery applications. Mater Sci Eng C. 2016;60:7–18.
Shadjou N, Hasanzadeh M. Silica-based mesoporous nanobiomaterials as promoter of bone regeneration process. J Biomed Mater Res Part A. 2015;103:3703–16.
Shi S, Jiang W, Zhao T, Aifantis KE, Wang H, Lin L, et al. The application of nanomaterials in controlled drug delivery for bone regeneration. J Biomed Mater Res Part A. 2015;103:3978–92.
Gomez-Vega JM, Hozumi A, Sugimura H, Takai O. Ordered mesoporous silica coatings that induce apatite formation in vitro. Adv Mater. 2001;13:822–5.
Izquierdo-Barba I, Ruiz-González L, Doadrio JC, González-Calbet JM, Vallet-Regí M. Tissue regeneration: a new property of mesoporous materials. Solid State Sci. 2005;7:983–9.
Vallet-Regí M, Ruiz-González L, Izquierdo-Barba I, González-Calbet JM. Revisiting silica based ordered mesoporous materials: medical applications. J Mater Chem. 2006;16:26–31.
Rámila A, Vallet-Regı́ M. Static and dynamic in vitro study of a sol–gel glass bioactivity. Biomaterials. 2001;22:2301–6.
Díaz A, López T, Manjarrez J, Basaldella E, Martínez-Blanes JM, Odriozola JA. Growth of hydroxyapatite in a biocompatible mesoporous ordered silica. Acta Biomater. 2006;2:173–9.
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27:2907–15.
Shi X, Wang Y, Wei K, Ren L, Lai C. Self-assembly of nanohydroxyapatite in mesoporous silica. J Mater Sci Mater Med. 2008;19:2933–40.
Szewczyk A, Prokopowicz M. Amino-modified mesoporous silica SBA-15 as bifunctional drug delivery system for cefazolin: release profile and mineralization potential. Mater Lett. 2018;227:136–40.
Szewczyk A, Prokopowicz M, Sawicki W, Majda D, Walker G. Aminopropyl-functionalized mesoporous silica SBA-15 as drug carrier for cefazolin: adsorption profiles, release studies, and mineralization potential. Microporous Mesoporous Mater. 2019;274:113–26.
Prokopowicz M, Żegliński J, Gandhi A, Sawicki W, Tofail SAM. Bioactive silica-based drug delivery systems containing doxorubicin hydrochloride: in vitro studies. Colloids Surf B. 2012;93:249–59.
Prokopowicz M. Bioactive silica-based nanomaterials for doxorubicin delivery: evaluation of structural properties associated with release rate. Mater Sci Eng C. 2013;33:3942–50.
Dumelié N, Benhayoune H, Rousse-Bertrand C, Bouthors S, Perchet A, Wortham L, et al. Characterization of electrodeposited calcium phosphate coatings by complementary scanning electron microscopy and scanning-transmission electron microscopy associated to X-ray microanalysis. Thin Solid Films. 2005;492:131–9.
Watanabe K, Yamazaki T, Hashimoto I, Shiojiri M. Atomic-resolution annular dark-field STEM image calculations. Phys Rev B. 2001;64:115432.
DrugBank. Doxorubicin hydrochloride. https://www.drugbank.ca/salts/DBSALT000060. Accesed 20 June 2018.
Itokazu M, Kumazawa S, Wada E, Wenyi Y. Sustained release of adriamycin from implanted hydroxyapatite blocks for the treatment of experimental osteogenic sarcoma in mice. Cancer Lett. 1996;107:11–8.
Fu W, Mohd Noor MH, Yusof LM, Ibrahim TAT, Keong YS, Jaji AZ, et al. In vitro evaluation of a novel pH sensitive drug delivery system based cockle shell-derived aragonite nanoparticles against osteosarcoma. J Exp Nanosci. 2017;12:166–87.
Zhao D, Huo Q, Feng J, Chmelka BF, Stucky GD. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J Am Chem Soc. 1998;120:6024–36.
Rosenholm JM, Lindén M. Wet-chemical analysis of surface concentration of accessible groups on different amino-functionalized mesoporous SBA-15 silicas. Chem Mater. 2007;19:5023–34.
Rosenholm JM, Czuryszkiewicz T, Kleitz F, Rosenholm JB, Lindén M. On the nature of the Bronsted acidic groups on native and functionalized mesoporous siliceous SBA-15 as studied by benzylamine adsorption from solution. Langmuir. 2007;23:4315–23.
Izquierdo-Barba I, Arcos D, Sakamoto Y, Terasaki O, López-Noriega A, Vallet-Regí M. High-performance mesoporous bioceramics mimicking bone mineralization. Chem Mater. 2008;20:3191–8.
Salinas AJ, Vallet-Regí M. Bioactive ceramics: from bone grafts to tissue engineering. RSC Adv. 2013;3:11116–31.
Bielen N. Performance of USP calibrator tablets in flow-through cell apparatus. Int J Pharm. 2002;233:123–9.
McCarthy I. The physiology of bone blood flow: a review. J Bone Jt Surg. 2006;88:4–9.
Zhang F, Yan Y, Yang H, Meng Y, Yu C, Bo Tu A, et al. Understanding effect of wall structure on the hydrothermal stability of mesostructured silica SBA-15. J Phys Chem B. 2005;109:8723–32.
Jung HY, Gupta RK, Oh EO, Kim YH, Whang CM. Vibrational spectroscopic studies of sol–gel derived physical and chemical bonded ORMOSILs. J Non-Cryst Solids. 2005;351:372–9.
Prokopowicz M. Synthesis and in vitro characterization of freeze-dried doxorubicin-loaded silica xerogels. J Sol-Gel Sci Technol. 2010;53:525–33.
Kozlova SA, Kirik SD. Post-synthetic activation of silanol covering in the mesostructured silicate materials МСМ-41 and SBA-15. Microporous Mesoporous Mater. 2010;133:124–33.
Wilson RM, Elliott JC, Dowker SEP, Rodriguez-Lorenzo LM. Rietveld refinements and spectroscopic studies of the structure of ca-deficient apatite. Biomaterials. 2005;26:1317–27.
Bonucci E. The mineralization of bone and its analogies with other hard tissues. In: Ferreira SO, editor. Adv top Cryst growth. InTech; 2013:145–184.
Bhattacharyya S, Wang H, Ducheyne P. Polymer-coated mesoporous silica nanoparticles for the controlled release of macromolecules. Acta Biomater. 2012;8:3429–35.
Mardones LE, Legnoverde MS, Simonetti S, Basaldella EI. Theoretical and experimental study of isothiazolinone adsorption onto ordered mesoporous silica. Appl Surf Sci. 2016;389:790–6.
Brown WE, Eidelman N, Tomazic B. Octacalcium phosphate as a precursor in biomineral formation. Adv Dent Res. 1987;1:306–13.
Dorozhkin SV, Epple M. Biological and medical significance of calcium phosphates. Angew Chemie Int Ed. 2002;41:3130–46.
Kolmas J, Krukowski S, Laskus A, Jurkitewicz M. Synthetic hydroxyapatite in pharmaceutical applications. Ceram Int. 2016;42:2472–87.
Feenstra TP, De Bruyn PL. Formation of calcium phosphates in moderately supersaturated solutions. J Phys Chem. 1979;83:475–9.
Li X, Chang J. Preparation of bone-like apatite–collagen nanocomposites by a biomimetic process with phosphorylated collagen. J Biomed Mater Res Part A. 2008;85A:293–300.
IUPHAR/BPS Guide To Pharmacology. Doxorubicin-ligand page. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=biology&ligandId=7069. Accesed 20 June 2018.
Pang J, Zhao L, Zhang L, Li Z, Luan Y. Folate-conjugated hybrid SBA-15 particles for targeted anticancer drug delivery. J Colloid Interface Sci. 2013;395:31–9.
Tian B, Liu S, Wu S, Lu W, Wang D, Jin L, et al. pH-responsive poly (acrylic acid)-gated mesoporous silica and its application in oral colon targeted drug delivery for doxorubicin. Colloids Surf B. 2017;154:287–96.
Prokopowicz M, Przyjazny A. Synthesis of sol-gel mesoporous silica materials providing a slow release of doxorubicin. J Microencapsul. 2007;24:694–713.
Prokopowicz M. Atomic force microscopy technique for the surface characterization of sol–gel derived multi-component silica nanocomposites. Colloids Surf A Physicochem Eng Asp. 2016;504:350–7.
Liu X, Gao C, Shen J, Möhwald H. Multilayer microcapsules as anti-cancer drug delivery vehicle: deposition, sustained release, and in vitro bioactivity. Macromol Biosci. 2005;5:1209–19.
Gao C, Donath E, Möhwald H, Shen J. Spontaneous deposition of water-soluble substances into microcapsules: phenomenon, mechanism, and application. Angew Chemie Int Ed. 2002;41:3789–93.
Yang K-N, Zhang C-Q, Wang W, Wang PC, Zhou J-P, Liang X-J. pH-responsive mesoporous silica nanoparticles employed in controlled drug delivery systems for cancer treatment. Cancer Biol Med Chinese Anti-Cancer Assoc. 2014;11:34–43.
Mioni R, Mioni G. A mathematical model of pH, based on the total stoichiometric concentration of acids, bases and ampholytes dissolved in water. Scand J Clin Lab Invest. 2015;75:452–69.
Ide M, El-Roz M, De Canck E, Vicente A, Planckaert T, Bogaerts T, et al. Quantification of silanol sites for the most common mesoporous ordered silicas and organosilicas: total versus accessible silanols. Phys Chem Chem Phys. 2013;15:642–50.
Acknowledgments
The research is a Polish-Irish collaborative project supported by the Polish National Centre for Science (project OPUS 15 no. 2018/29/B/NZ7/00533).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Prokopowicz, M., Żeglinski, J., Szewczyk, A. et al. Surface-Activated Fibre-Like SBA-15 as Drug Carriers for Bone Diseases. AAPS PharmSciTech 20, 17 (2019). https://doi.org/10.1208/s12249-018-1243-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-018-1243-5