
Abstract
New drugs may in some cases need to be tested in paediatric and pregnant patients. However, it is difficult to recruit such patients and there are many ethical issues around their inclusion in clinical trials. Modelling and simulation can help to plan well-designed clinical trials with a reduced number of participants and to bridge gaps where recruitment is difficult. Physiologically based pharmacokinetic (PBPK) models for small molecule drugs have been used to aid study design and dose adjustments in paediatrics and pregnancy, with several publications in the literature. However, published PBPK models for monoclonal antibodies (mAb) in these populations are scarce. Here, the current status of mAb PBPK models in paediatrics and pregnancy is discussed. Seven mAb PBPK models published for paediatrics were found, which report good prediction accuracy across a wide age range. No mAb PBPK models for pregnant women have been published to date. Current challenges to the development of such PBPK models are discussed, including gaps in our knowledge of relevant physiological processes and availability of clinical data to verify models. As the availability of such data increases, it will help to improve our confidence in the PBPK model predictive ability. Advantages for using PBPK models to predict mAb PK in paediatrics and pregnancy are discussed. For example, the ability to incorporate ontogeny and gestational changes in physiology, prediction of maternal, placental and foetal exposure and the ability to make predictions from in vitro and preclinical data prior to clinical data being available.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Pharmaceutical companies are legally required and incentivised by regulators to consider studying new drugs in paediatric patients. However, approval of monoclonal antibodies (mAbs) in paediatrics has lagged behind adults and currently many mAbs are only licenced for older children or adolescents [1], which can contribute to off label use in young children. Children generally have higher mAb clearance and volume of distribution (Vss) than adults, once body weight has been accounted for, and hence require higher weight-based doses to maintain the same exposure as in adults [2]. Age has also been identified as a covariate for mAb PK in some population PK analyses, after body weight has been accounted for [2], indicating that selection of a paediatric dose level based on body weight alone may not be adequate, particularly in the very young where physiology is changing rapidly.
Most mAbs are contraindicated for use in pregnancy due to the lack of clinical safety data in pregnant women and should be stopped prior to conception [3, 4]. Due to the long half-life (t1/2) of mAbs, this could be < 1 year before conception. However, there are several situations where mAb use during pregnancy is warranted. For example, inflammatory bowel disease (IBD) and asthma, where taking the patient off the medication can be associated with negative pregnancy outcomes and reduced fertility [5, 6]. Hence, the benefit of continuing treatment to maintain disease remission may outweigh the risk to the foetus. Additionally, maternal autoantibodies in diseases such as lupus, antiphospholipid syndrome and Hashimoto disease can cross the placenta and cause severe complications such as congenital lupus, thyroid dysfunction, foetal cardiac block and death [7]. Anti-neonatal Fc receptor (FcRn) mAbs are one type of treatment aimed at reducing the transfer of autoantibodies and improving pregnancy outcomes in these types of disease [8, 9]. Reports of the effect of pregnancy on mAb PK are very limited and will be discussed in detail later. Inconsistent changes in mAb PK and amount of mAb transferred across the placenta to the foetus during pregnancy have been reported [10,11,12,13,14,15,16,17,18,19,20,21,22], indicating that careful consideration is required to determine suitable doses in pregnant women.
Clinical trials are usually required to determine suitable dosing regimens in paediatric and pregnant patients. However, it is difficult to recruit these patients due to a raft of ethical issues. In pregnancy, the balance between the benefit of treatment to the mother has to be weighed against potential risk to the foetus. Starting doses should be high enough to be effective but not so high as to cause safety concerns. In addition, studies need to be adequately powered to achieve the objectives but not unnecessarily expose too many patients. Hence, efficient study design is essential. Modelling and simulation techniques including physiologically based pharmacokinetic (PBPK) modelling can be used to bridge gaps where subject recruitment is difficult and to help optimise clinical trial design and are supported by regulatory authorities [23, 24].
PBPK models are mathematical models comprising ordinary differential equations which describe compartments representing specific tissues linked by blood flow [25]. They use drug-specific data with physiological data to predict drug exposure in blood and tissues. PBPK models can also be linked to pharmacodynamic (PD) models to predict drug effects. As the physiological and anatomical data are separate from the drug-specific parameters, ontogeny and changes in pregnancy can be incorporated and their impact on drug exposure and dose requirement assessed at different ages/stages of gestation. PBPK models are being increasingly used in paediatric study design for small molecule drugs [26, 27] and there are several publications of PBPK models for small molecules in pregnancy [28]. However, reports of such models for mAbs are very limited. This review will focus on the status of PBPK models for mAbs in paediatrics and pregnancy as well as the opportunities and challenges with using these models. The issues discussed herein apply to mAbs and their derivatives (Fc fusion proteins, mAb fragments, bispecifics and antibody drug conjugates) but mAbs is used as a catchall term herein.
OVERVIEW OF MAB PBPK MODELS
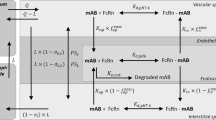
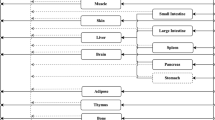
The whole-body PBPK model is made up of a number of compartments corresponding to the different tissues in the body linked by blood and lymph flow (Fig. 1). The tissues can also be lumped into one or a few compartments [29]. Compared to small molecule models, there are several additional physiological processes that need to be considered for mAbs. Many mAb PBPK models and articles discussing important physiological processes governing mAb PK have been published (see [30,31,32,33,34] as examples) and hence only a high-level overview will be given here.

Typical full PBPK model structure for mAbs (A); each tissue compartment is split into sub-compartments representing the interstitial, endosomal and vascular spaces (B). L, lymph flow; Q, blood flow; σv, vascular reflection coefficient; σi, lymphatic reflection coefficient
MAb distribution is slow and permeability limited; hence, PBPK models for mAbs use a multi-compartment structure for each tissue with sub-compartments representing vascular, endosomal, interstitial and intracellular spaces. MAbs and endogenous immunoglobulin G (IgG) are too large and polar to diffuse through the cell membranes; instead, their distribution is governed mainly by convection through pores between endothelial cells to gain access to the interstitial fluid [35]. In addition, mAbs can be taken up into endothelial cells by pinocytosis or endocytosis and transferred across the cells by transcytosis to be released into the interstitial space. One of the most important aspects governing mAb disposition is binding to FcRn within the endosomes of endothelial, epithelial and immune cells [36]. Binding occurs at low pH in the endosome and prevents the mAb being degraded within the lysosome; instead, it is returned to the cell surface and is released into the blood or interstitial fluid [36]. Similarly, endogenous IgG binds to FcRn, which gives its classic long t1/2 and mAbs compete with endogenous IgG for binding to FcRn. Hence, levels of endogenous IgG in patients can have an impact on mAb PK.
MAbs are designed to tightly bind to their target receptors/soluble targets and may be eliminated through this binding [37]. This target-mediated drug disposition (TMDD) leads to higher clearance at dose levels where receptor binding is not saturated [37]. Production of anti-drug antibodies (ADA) and binding to Fc-γ receptors on immune cells lead to increased mAb clearance [38, 39], although these complex immune responses are not usually modelled mechanistically in PBPK models. Renal filtration could also be important for smaller mAb fragments [40].
Understanding the ontogeny of these physiological processes and changes throughout pregnancy and foetal development are important for modelling the PK of mAbs in paediatric and pregnant patients. One of the main challenges to PBPK modelling of mAbs in paediatrics and pregnancy is that the data available for many of the key physiological processes are limited. Changes in tissue volumes, blood flows, body weight, etc. are the same as those already accounted for in small molecule PBPK models [41,42,43,44,45]. The availability of physiological data specific to mAbs will be discussed below and is presented in Table I.
-
1.
Ontogeny of key physiological processes in paediatrics
The current understanding and gaps in our knowledge of the ontogeny of key physiological processes for mAb disposition in paediatrics have been reviewed recently [46, 47] and hence only a brief overview is given here.
Extracellular fluid volume decreases rapidly following birth whereas plasma volume gradually increases [45, 48]. Taken together, these processes lead to a higher proportion of total body available for mAb distribution and a higher Vss/kg in young children compared to adults [2].
The rate of extravasation is ~ 3-fold higher in young children compared to adults, leading to an increased rate of mAb distribution [46]. The glomerular capillary endothelium has fewer pores compared to mature capillaries in adult dogs [49]; if this also holds true for endothelium in other tissues and across species, it would lead to reduced mAb permeability in young children. However, children have higher capillary densities in the skin, muscle and adipose and a larger skin surface area/volume than adults [46]. In addition, the proportion of leaky tissues in the body, which have larger gaps through the endothelial walls, is greater in young children [46]. Hence, although capillary permeability is reduced, the extent of distribution of mAbs is greater in young children compared to adults [2]. In addition, the rate of distribution is faster in children, as observed for IgG, where the transcapillary escape rate is ~ 3-fold higher in neonates [46].
Measured lymph flow data in children are lacking; however, animal data shows a ~ 2-fold higher lymph flow in neonatal compared to adult animals [50, 51]. This contributes to faster distribution and absorption following subcutaneous (SC) and intramuscular (IM) administration [52].
Endogenous IgG levels at birth in full-term neonates are similar to adults [53, 54]. Following birth, the total IgG levels fall as the maternal IgG is eliminated and the neonatal synthesis takes over. A nadir in IgG concentrations occurs at about 3 months and then concentrations increase to adult levels by about 10 years [55].
One of the most important determinants of mAb clearance is binding to FcRn and hence understanding its ontogeny is crucial. Unfortunately, absolute abundance of FcRn in children is currently unavailable. Rodent mRNA and western blot data show increases in FcRn abundance in some or all tissues following birth [56, 57]. However, these data are highly variable and the mouse data suggest age-related changes in FcRn abundance in the liver, lungs and kidney do not affect mAb disposition [57]. A lower FcRn abundance in neonates would contribute to increased mAb clearance compared to adults, particularly in the first couple of months after birth when endogenous IgG levels are high and there is increased competition for binding to FcRn.
Unfortunately, there is a lack of knowledge about extracellular proteolysis of IgG and mAbs in children. However, the majority of IgG and mAb elimination occurs intracellularly and is dependent on endocytosis, phagocytosis and pinocytosis. Phagocytosis, dependent mainly on binding to Fcγ receptors, is only a minor contribution to elimination of most mAbs and hence age-dependent expression and activity differences will be of less importance to overall clearance of most mAbs [46, 47]. Currently, there is no information on how pinocytosis and endocytosis rates vary in children versus adults.
Clearance via TMDD will be dependent on target binding affinity and the concentration and turnover of target receptors and receptor-mAb complexes. Data for target levels are often limited in paediatric patients; for instance, no data are available for erythropoietin receptor in neonates [55]. In contrast, data from paediatric patients showed no trends between TNF-α level and age either in healthy subjects or those with inflammatory diseases [55]. Target levels are variable between individuals with different disease types and severity and will need to be assessed for each target and disease individually.
Understanding the ontogeny of each specific process that affects mAb PK is clearly of great importance; however, defining the extent of their direct effect is difficult with many compensating processes occurring. Observed differences in mAb PK between children and adults will be a result of the net effect of all ontogeny processes and could be lower than the impact of any process on its own. Hence, understanding the interplay of all the physiological processes is key.
-
2.
Development of key physiological processes throughout gestation
A brief discussion of the available data required for PBPK models for mAbs in pregnancy is provided below. Similar to paediatrics, alterations in mAb PK during pregnancy will be due to the net effect of changes in multiple physiological and anatomical processes which may compensate for each other.
During pregnancy, the levels of IgG in the mother decrease, particularly in the third trimester (Fig. 2). This is due primarily to the increase in plasma volume and also to transport of IgG across the placenta to the foetus [58]. In the foetus, IgG comes almost exclusively from the placental transfer of maternal IgG, with foetal IgG representing ~ 0.2% of total IgG in cord blood [59]. Transfer is very low in the first trimester and then increases rapidly in trimesters 2 and 3 to reach adult levels at term (Fig. 2B). Foetal IgG levels reach 50% of maternal levels by 28–32 weeks, with the majority being transferred in the final few weeks prior to birth [58]. As the IgG levels decline in the mother but increase in the foetus, the foetal:maternal IgG ratio increases throughout gestation, with foetal levels exceeding those in the mother after 35 weeks of gestation [60] and being approximately 1.5-fold higher in the average infant at term [54] (Fig. 2C). Maternal IgG levels increase back to non-pregnant levels by 4–6 months after birth [61].

A IgG plasma concentrations in pregnant women throughout gestation, black solid and grey dashed lines represent the mean and SD for IgG plasma concentrations in non-pregnant women [53]; B IgG plasma concentration in the foetus throughout gestation, black solid and grey dashed lines represent the mean and SD for IgG plasma concentrations in adults [53]; C foetal:maternal IgG plasma concentration ratio throughout gestation, black line represents the line of unity. Data are from mainly Caucasian women and foetuses, those of other ethnicities have been screened out where possible. Data are plotted against the mid-point if a range of gestation weeks was reported. Closed circles and error bars represent values reported as means or medians and SD. Open circles represent values reported for individual subjects. References for data are given in the Supplementary Material along with the key for the colours of mean study data
Foetal placental villi have continuous endothelium similar to muscle and heart capillaries, with pores of ~ 6 nm in diameter, which restricts movement of molecules > 65 kDa [62, 63]. As the endothelial pores are smaller than IgG, very little convective movement of IgG and mAbs across placental endothelium is expected. Indeed, other types of antibodies (IgA, IgE, IgD and IgM) with large molecular size are unable to cross the placenta [54, 64]. The placental transfer of endogenous IgG and mAbs is instead due to endocytosis and transcytosis, dependent on binding to FcRn. The rate of endocytosis in human placental chorionic villi has been measured in vitro [65]; unfortunately, such data in foetal endothelial cells are lacking. In vitro data show transfer of infliximab, adalimumab and etanercept across ex vivo perfused placentas but no transfer of certolizumab pegol and abciximab, which do not bind to FcRn [9, 13, 66, 67]. Nipocalimab, an anti-FcRn mAb which binds to FcRn with high affinity at both pH 6 and pH 7.4, was not released into the foetal side of the perfused placenta and inhibited the transfer of adalimumab [9]. Fc-γRIIb is also proposed to contribute to foetal IgG absorption, although this is still under debate [68,69,70].
Absolute abundance of FcRn in the pregnant woman, foetus and placenta is currently lacking. In pregnant women, the IgG t1/2 is similar to non-pregnant women (~ 21 days) [71]. Although body weight increases in pregnancy, there is only a small increase in mAb Vss due to their restricted distribution [10, 11, 72]. This suggests IgG catabolism and FcRn abundance are similar in pregnant and non-pregnant women. In addition, dose changes for adalimumab and infliximab are not required in pregnancy. In contrast, increased clearance of mAbs has been reported in the pregnant rat (< 15-fold) in conjunction with increased excretion of radioactivity in the urine, suggesting increased IgG catabolism during pregnancy [73, 74]. Unfortunately, there are currently no human data on protease activity and expression in pregnancy [10, 72] and hence, it is difficult to discern if the changes in physiology have minor effects on mAb PK or if there are compensating mechanisms acting in opposite directions resulting a small net effect.
FcRn is located in the syncytiotrophoblasts, placental and foetal endothelial cells, foetal epithelial cells and placental macrophages [70, 75,76,77,78,79], although some data are conflicting. The FcRn expression level in these cells and how it changes throughout pregnancy are required for PBPK models. Rat western blot and mRNA data show placental expression of FcRn increases throughout pregnancy, consistent with the increase in foetal plasma IgG levels [80]. Human data also shows increasing placental FcRn staining throughout Trimester 3, which correlates with newborn plasma IgG levels [81]. As IgG is only transported across the placenta via binding to FcRn and maternal IgG is found in the foetus from as early as 13 weeks of gestation [82], this indicates that FcRn must be present in the placenta by the end of trimester 1. The rapid increase in foetal IgG in trimester 3 correlates with the increased staining for placental FcRn throughout trimester 3. Currently, there are no quantitative measurements of FcRn in foetal endothelial cells and its location there is still under debate. Little is known about the IgG homeostasis in the foetus. The role FcRn plays in the extent of catabolism in foetal endosomes requires additional research.
The placenta has no lymphatic vessels [83]. Lymph flow data in pregnant women and the foetus are limited. Maternal lymph flow increases to the uterus in trimester 3 (sheep data) and the mammary glands during lactation (rat data) [84]. Edema due to increased extravasation of fluid is common in pregnancy [85], with interstitial volume increasing by ~ 2.5 L during pregnancy [86, 87]. During pregnancy, there is a mild and sustained inflammatory state where cytokines and vascular endothelial growth factor (VEGF) levels are increased [88]. Cytokines and VEGF increase capillary permeability, interstitial fluid volume, decreased colloid osmotic pressure (COP) and consequently increase extravasation of albumin and IgG [88, 89]. These changes in pregnancy may result in increased rate and extent of mAb distribution and absorption following SC and IM administration. Data for interstitial COP and interstitial hydrostatic pressure (IHP) are very limited [89]. Pregnant sheep have increased IHP with decreased interstitial COP; however, in pregnant guinea pigs, IHP remained relatively constant and there was a decrease in COP throughout gestation [90].
Foetal total body water increases throughout gestation although as a proportion of body weight it decreases with increasing age [42]. In addition, there is a gradual reduction in extracellular water as a proportion of total body water [42], which reduces the volume a mAb can distribute into following transfer to the foetus. The foetal lymphatic system starts developing by gestation week 6 [91]; however, lymph flow has never been measured in the human foetus. Animal data suggest foetal lymph flow is fivefold faster than in adult animals [92]. Fluid loss from capillaries is high in the foetus, matching the high lymph flow [93]. This high fluid loss is partly driven by high capillary permeability, where immature foetal blood vessels are more leaky than in adults [93]. Plasma protein levels are low in the foetus leading to reduced COP and increased fluid movement out of capillaries [93, 94]. Indeed, reduced plasma COP and raised interstitial COP have been reported in foetal guinea pig and sheep SC tissue [90]. Lower plasma COP has also been reported in the human foetus, increasing linearly throughout gestation from 30% of maternal COP at 26 weeks gestation to ~ 77% at term [95,96,97]. Together these effects would lead to increased rate and extent of distribution of mAbs transported to the foetus.
TMDD may change during pregnancy depending on the target receptor. For instance, levels of TNF-α are decreased in pregnancy [98], leading to reduced disease activity and even remission [99, 100]. Infliximab exposure increases during pregnancy after accounting for increased body weight [10], which could be due to a reduction in TMDD clearance. However, adalimumab exposure remains similar to non-pregnant women [10]. TMDD in the placenta needs to be considered for certain targets, e.g. TNF-α is highly expressed in term placenta [101]. In addition, some mAb targets may be present within the developing foetus; hence, their ontogeny would need to be considered within PBPK models if the extent of mAb transferred across the placenta is insufficient to saturate target binding in the foetus.
PUBLISHED PAEDIATRIC MAB PBPK MODELS
Seven PBPK models for mAbs in paediatrics have been published to date [55, 102,103,104,105,106,107] (search criteria are detailed in the Supplementary Material). All models were able to predict mAb PK with reasonable accuracy, with PK parameters and/or plasma concentrations generally predicted within 2-fold of observed values (Table II). Several of the published models include verification using only data from older children (> 4 years [105, 107]) or infants > 1 year [103, 104], when the effects of ontogeny will be less noticeable. However, good prediction accuracy has also been reported for models that were verified with data from neonates [55, 102]. Malik and Edginton [106] developed PBPK models for pagibaximab, palivizumab, MEDI8897 and IVIG in premature infants, with 90% of the predicted concentrations within 1.5-fold of the observed values [106]. Hanke et al., [103] prospectively used a PBPK model for the fusion protein asunercept to make dose recommendations in paediatrics.
The published paediatric mAb PBPK models incorporate ontogeny of organ volumes, blood flows, lymph flows, interstitial volumes and haematocrit. Ontogeny of lymph flow differs between the models, one used a uniform scalar in young children (0–1 year) [106] and others allometrically scale adult lymph flows [55, 104], both resulting in increased lymph flow in paediatrics compared to adults, in line with animal data [50, 51]. Other PBPK models determined lymph flow as a percentage of blood flow [102, 105, 107]; consequently, ontogeny of lymph flow follows that of blood flow. Ontogeny of physiology governing extravasation (e.g. capillary surface area) of mAbs has been incorporated into a couple of PBPK models [104, 106]; however, further data are required to determine organ-specific ontogenies. Allometry was used to describe age-related differences in clearance of mAbs in lysosomes and non-specific clearance routes in one model [55], whereas degradation rate in lysosomes was fitted using paediatric infliximab data in another model [107] and no ontogeny of catabolism was included in other models. Most of the PBPK models incorporate competitive binding of IgG and mAbs to FcRn [55, 102,103,104,105,106]; however, ontogeny of paediatric IgG concentrations is only included in two of the models [55, 104]. Malik and Edginton [106] incorporated FcRn recycling within hematopoietic cells and their ontogeny. The ontogeny of FcRn is a key area of debate and currently only one of the published PBPK models incorporated ontogeny of FcRn (by fitting of clinical paediatric data for bevacizumab [104]). The fitted ontogeny showed an inverse correlation between FcRn concentration/kg and age [104], which is consistent with physiological data described earlier. The authors consistently agree that more information is required to define the ontogeny of FcRn abundance along with other physiological processes important for mAb PK, particularly for accurate predictions in the very young.
Physiology and anatomy of children changes rapidly in the first months of life and because mAb exposure is usually monitored over many days/weeks, continuous maturation of physiology may need to be considered within PBPK models aimed at predicting mAb exposure in very young children. Continual maturation of physiology has been incorporated in a few of the paediatric mAb PBPK models [102, 106, 107].
Population covariates and variability in physiological parameters can be incorporated into PBPK models, allowing prediction of mAb exposure in representative populations rather than just for an ‘average’ person. To date, three PBPK models have predicted population variability in paediatric mAb exposure with reasonable success [55, 103, 107].
PUBLISHED PREGNANCY MAB PBPK MODELS AND AVAILABILITY OF CLINICAL DATA FOR PBPK MODEL VERIFICATION
There are several published PBPK models predicting small molecule exposure in pregnancy [28]; however, there are currently no models for mAbs in pregnant women. Clinical data are required to verify PBPK model predictions; however, suitable data for mAb exposure in pregnancy are scarce, often from individual case studies. The available data are mainly for anti-TNF-α mAbs.
Maternal exposure data throughout gestation are available for 4 mAbs, but the effect of pregnancy on exposure appears mAb specific [10,11,12]. Infliximab exposure significantly increased (median values 3-fold higher in trimester 3 compared to pre-conception [10]), vedolizumab significantly decreased (median values 2-fold lower in trimester 3 compared to trimester 1 [11]) and adalimumab and etanercept remained stable during pregnancy [10,11,12]. Increases in infliximab exposure could be driven by the normal reduction in inflammatory response in pregnancy, resulting in reduced TMDD clearance [11]. Seow et al. report that most women change SC administration site and also fat distribution changes during pregnancy, which could alter SC absorption and possibly contributes to the stable exposure of adalimumab [10]. In addition, changes in ADA formation during pregnancy could affect mAb exposure; however, very little information on ADA levels in pregnancy has been reported.
The extent of mAb transfer across the placenta reported at birth (newborn:maternal plasma mAb concentration ratio (N:M ratio)) for various mAbs is shown in Fig. 3. The ratio ranges from generally higher concentrations in the newborn compared to the mother (adalimumab, infliximab, natalizumab, rituximab and ustekinumab [13,14,15,16,17,18,19]), similar to observations for endogenous IgG, to much lower levels in the newborn (certolizumab pegol, etanercept and tocilizumab [12,13,14, 20,21,22]). For certolizumab pegol, this is expected due to the lack of binding to FcRn and hence no transfer across the placenta [20]. The lower N:M ratio for etanercept compared to mAbs like infliximab and adalimumab may be due to the lower binding affinity of etanercept to FcRn [13, 66]. Vedolizumab N:M ratio is often < 1, which is surprising given that it is an IgG1 mAb and hence should be transported across the placenta similarly to IgG. The lower vedolizumab N:M ratio may be due to high binding to integrin receptors in the placenta [11, 15]. A strong positive correlation between vedolizumab, adalimumab and infliximab maternal and newborn mAb levels at birth was reported [11, 16, 108]. In addition, there is a negative correlation between newborn and maternal mAb concentrations at birth and the duration between last dose and birth [11, 16, 108].

Newborn:maternal plasma concentration ratio at birth for a variety of mAbs. Newborn data include both cord and neonatal plasma concentrations. Blue line represents the line of unity. Grey bars represent the range of newborn:maternal plasma concentration ratios reported in clinical studies for individual mother-baby pairs. References for data are given in the Supplementary Material. Data from one mother-baby pair excluded for infliximab as ratio = 35 [16]
It is important to consider how long transferred mAbs remain in the newborn following birth. This could have important implications on immune development and when live vaccines can be administered [4, 6, 7, 109]. Indeed, one infant who was exposed to infliximab in utero died from disseminated Bacillus Calmette-Guérin (BCG) following routine BCG vaccination at 3 months of age [110]. Hence, live vaccines are not usually administered until > 6 months of age in infants exposed to mAbs in utero [4, 6, 7, 109]. The available data range from > 2 years in one case study for adalimumab [111] to < 4 weeks for certolizumab pegol and tocilizumab [20, 22] (Table III). The duration that mAbs remain in the newborn post birth may be affected by breastfeeding. Tocilizumab and etanercept concentrations have been measured in breast milk; however, serum concentrations in breastfed infants who were exposed in utero were undetectable by 4–6 weeks of age [12, 22, 112]. In contrast, certolizumab pegol was not measurable in breast milk [14]; hence, breastfeeding during mAb treatment likely provides little mAb transfer to the infant.
ADVANTAGES OF MAB PBPK MODELS FOR PAEDIATRICS AND PREGNANCY
PBPK models and allometry are both reported to work reasonably well for predicting mAb exposure in older children and adolescents [55, 105, 113, 114], although the value of allometric exponents is still debated [113, 114]. However, in young children when physiology is changing rapidly, modelling techniques such as PBPK have the advantage of allowing ontogeny of key physiological processes to be incorporated. Simple modelling techniques are not available to predict foetal exposure. PBPK models allow prediction of exposure in the mother as well as transfer of mAbs, endogenous IgG and/or autoantibodies across the placenta and consequently exposure in the foetus. In addition, physiology can be allowed to mature during the simulated time scale, which is important for mAbs that have a long t1/2.
TMDD cannot be reliably scaled by simple allometry [114]. However, TMDD can be included in PBPK models, allowing changes in target levels during paediatric development and pregnancy to be accounted for. Indeed, TMDD has been incorporated into several paediatric mAb PBPK models [55, 103, 105]. In addition, physiological/anatomical changes and target level variations in different disease conditions can be considered [55, 105]. As demonstrated in paediatric PBPK models for infliximab and etanercept [55], where altered endogenous IgG levels in Kawasaki disease patients compared to healthy children were incorporated. PD models can be linked to PBPK models, where response is driven by the plasma exposure or the concentrations in specific tissues at the site of action, and hence, the impact of ontogeny on drug effect is also predicted. PBPK modelling offers a clear advantage in cases where disease pathology varies between adults and children or during gestation and for mAbs where target burden is high and consequently PK non-linear.
Early in drug development when clinical data is scarce, PBPK models can be used to make initial predictions by harnessing the available in vitro and preclinical data [115], whereas population PK approaches require clinical data for model development. Sensitivity analysis can be performed with PBPK models to evaluate the most important drug characteristics/physiological processes determining mAb exposure and help focus future research.
CHALLENGES FOR MAB PBPK MODELS IN PAEDIATRICS AND PREGNANCY
Although there are clear opportunities for using PBPK models to predict mAb exposure in paediatrics and pregnancy, there are also challenges to their development. To develop PBPK models, ontogeny or changes with gestation of the key physiological processes need to be incorporated. However, data for many processes are still emerging. Even where ontogeny is known, it can be difficult to quantify the effect of specific processes on mAb PK and translate this into PBPK models; hence, the published PBPK models for mAbs in paediatrics often consider the ontogeny of specific processes differently. One of the most important determinants of mAb PK is binding to FcRn. Indeed, Malik and Edginton [106] reported that only a 10% decrease in FcRn abundance in children compared to adults would have a moderate effect on pagibaximab clearance using their PBPK model. Sensitivity of mAb PK to FcRn concentration will be PBPK model and mAb specific, depending on the assumptions in the model and the binding affinity of the mAb to FcRn. Unfortunately, the absolute abundance of FcRn in children, the foetus and placenta have not been reported to date. However, in its absence data from other measurement techniques and preclinical species can be used to inform models. Reports of FcRn absolute abundance in adults and preclinical animals are increasing and hopefully data for children, the placenta and the foetus will follow. As FcRn abundance data become available, they can easily be incorporated into existing PBPK model platforms to refine predictions. A fuller understanding of the ontogeny of physiological processes will help to improve confidence in model predictions in the future.
Levels of target receptors can change with disease severity and may change with age [116]. In addition, there could be functional differences in the paediatric form of target receptors leading to differences in the binding and pharmacological activity and exposure of mAbs in children when compared to adults. Differences in target expression and/or binding need to be considered in PBPK models for mAbs. This is an additional complication compared to small molecule PBPK models, where determining dose levels that can provide equivalent paediatric and adult exposure is often adequate.
The availability of published clinical data which can be used to verify PBPK models for mAbs in paediatrics, particularly in the very young, and pregnancy are limited. However, this issue is not exclusive to PBPK modelling and applies to all modelling techniques. More data are continually emerging in the literature, which will allow further verification and increased confidence in PBPK modelling in the future.
To allow truly bottom-up modelling for mAbs, good in vitro-in vivo extrapolation (IVIVE) techniques are required, allowing the prediction of PK from physiochemical and in vitro data, rather than relying on clinical data to inform the models [115]. IVIVE techniques for mAbs are not as established as those for small molecule drugs. However, there is much ongoing work to develop such IVIVE techniques for mAbs and incorporate them into PBPK models [117,118,119]. For example, non-specific charge-based interactions, self-association, FcRn binding affinity and FcRn-dependent transcytosis of mAbs all correlate with in vivo clearance [120, 121]. The ex vivo placental perfusion assay has been used to measure the transfer of mAbs and IgG across the placenta [9, 13, 66]. Ex vivo placental transfer data correlates with in vivo cord:maternal ratio for small molecule drugs [122]. However, transfer of mAbs is slow in this system with very little occurring in the first 6 h. Hence, there is currently no IVIVE method to utilise these data to predict the extent of placental transfer of mAbs in vivo [13]. As the IVIVE methods are refined, we will have a better ability to harness the data from in vitro assays to predict what will happen in vivo.
CONCLUSION
With the limitations of recruiting paediatric and pregnant patients into clinical trials and the requirement for treatment options for these sensitive groups, there is clearly a need for suitable modelling and simulation techniques to bridge the gaps and help optimise clinical trial design. PBPK models predicting mAb exposure in children and adolescents have shown reasonable prediction accuracy in children as young as newborns. However, there are currently no published PBPK models for mAbs in pregnant women.
As discussed, there are many opportunities and advantages to using PBPK models for mAbs in paediatrics and pregnancy. Data for key physiological processes affecting mAb PK in paediatrics and pregnant women are still emerging. In addition, IVIVE techniques are being developed so that in vitro data can be harnessed in PBPK models. As more clinical data for mAbs in paediatrics and pregnant women become available, they can be used to validate PBPK models and build confidence in the predictions.
In conclusion, although PBPK modelling for mAbs in paediatrics is currently in its infancy, there is wide scope for using such models to aid in clinical study design/dose recommendations. In addition, it is clear that although there are still gaps in our knowledge, development of PBPK models for mAbs in pregnancy would be helpful to determine suitable dose regimens in these sensitive patients.
References
Liu XI, Dallmann A, Wang YM, Green DJ, Burnham JM, Chiang B, et al. Monoclonal antibodies and Fc-fusion proteins for pediatric use: dosing, immunogenicity, and modeling and simulation in data submitted to the US Food and Drug Administration. J Clin Pharmacol. 2019;59(8):1130–43. https://doi.org/10.1002/jcph.1406.
Malik PRV, Temrikar ZH, Chelle P, Edginton AN, Meibohm B. Pediatric dose selection for therapeutic proteins. J Clin Pharmacol. 2021;61(Suppl 1):S193-s206. https://doi.org/10.1002/jcph.1829.
Ghalandari N, Dolhain R, Hazes JMW, Siezen CLE, van der Laan JW, Crijns H, et al. The pre- and post-authorisation data published by the European medicines agency on the use of biologics during pregnancy and lactation. Br J Clin Pharmacol. 2020;86(3):580–90. https://doi.org/10.1111/bcp.14145.
Hyrich KL, Verstappen SM. Biologic therapies and pregnancy: the story so far. Rheumatology (Oxford). 2014;53(8):1377–85. https://doi.org/10.1093/rheumatology/ket409.
Pfaller B, José Yepes-Nuñez J, Agache I, Akdis CA, Alsalamah M, Bavbek S, et al. Biologicals in atopic disease in pregnancy: an EAACI position paper. Allergy. 2021;76(1):71–89. https://doi.org/10.1111/all.14282.
Roseira J, Ramos J. A narrative review on anti-tumor necrosis factor α therapies in inflammatory bowel disease during pregnancy: immunoglobulin placental translocation and its impact. Acta Med Port. 2019;32(4):305–12. https://doi.org/10.20344/amp.11482.
Ellinger I, Fuchs R. HFcRn-mediated transplacental immunoglobulin G transport: protection of and threat to the human fetus and newborn. Wien Med Wochenschr. 2012;162(9–10):207–13. https://doi.org/10.1007/s10354-012-0085-0.
Bussel JB, Vander Haar EL, Berkowitz RL. New developments in fetal and neonatal alloimmune thrombocytopenia. Am J Obstet Gynecol. 2021;225(2):120–7. https://doi.org/10.1016/j.ajog.2021.04.211.
Roy S, Nanovskaya T, Patrikeeva S, Cochran E, Parge V, Guess J et al. M281, an anti-FcRn antibody, inhibits IgG transfer in a human ex vivo placental perfusion model. Am J Obstet Gynecol. 2019;220(5):498.e1-.e9. https://doi.org/10.1016/j.ajog.2019.02.058.
Seow CH, Leung Y, Vande Casteele N, Ehteshami Afshar E, Tanyingoh D, Bindra G, et al. The effects of pregnancy on the pharmacokinetics of infliximab and adalimumab in inflammatory bowel disease. Aliment Pharmacol Ther. 2017;45(10):1329–38. https://doi.org/10.1111/apt.14040.
Flanagan E, Gibson PR, Wright EK, Moore GT, Sparrow MP, Connell W, et al. Infliximab, adalimumab and vedolizumab concentrations across pregnancy and vedolizumab concentrations in infants following intrauterine exposure. Aliment Pharmacol Ther. 2020;52(10):1551–62. https://doi.org/10.1111/apt.16102.
Murashima A, Watanabe N, Ozawa N, Saito H, Yamaguchi K. Etanercept during pregnancy and lactation in a patient with rheumatoid arthritis: drug levels in maternal serum, cord blood, breast milk and the infant’s serum. Ann Rheum Dis. 2009;68(11):1793–4. https://doi.org/10.1136/ard.2008.105924.
Eliesen GAM, van Drongelen J, van Hove H, Kooijman NI, van den Broek P, de Vries A, et al. Assessment of placental disposition of infliximab and etanercept in women with autoimmune diseases and in the ex vivo perfused placenta. Clin Pharmacol Ther. 2020;108(1):99–106. https://doi.org/10.1002/cpt.1827.
Mahadevan U, Wolf DC, Dubinsky M, Cortot A, Lee SD, Siegel CA et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11(3):286–92; quiz e24. https://doi.org/10.1016/j.cgh.2012.11.011.
Mitrova K, Pipek B, Bortlik M, Bouchner L, Brezina J, Douda T et al. Differences in the placental pharmacokinetics of vedolizumab and ustekinumab during pregnancy in women with inflammatory bowel disease: a prospective multicentre study. Therap Adv Gastroenterol. 2021;14:17562848211032790. https://doi.org/10.1177/17562848211032790.
Julsgaard M, Christensen LA, Gibson PR, Gearry RB, Fallingborg J, Hvas CL, et al. Concentrations of adalimumab and infliximab in mothers and newborns, and effects on infection. Gastroenterology. 2016;151(1):110–9. https://doi.org/10.1053/j.gastro.2016.04.002.
Zelinkova Z, de Haar C, de Ridder L, Pierik MJ, Kuipers EJ, Peppelenbosch MP, et al. High intra-uterine exposure to infliximab following maternal anti-TNF treatment during pregnancy. Aliment Pharmacol Ther. 2011;33(9):1053–8. https://doi.org/10.1111/j.1365-2036.2011.04617.x.
Friedrichs B, Tiemann M, Salwender H, Verpoort K, Wenger MK, Schmitz N. The effects of rituximab treatment during pregnancy on a neonate. Haematologica. 2006;91(10):1426–7.
Haghikia A, Langer-Gould A, Rellensmann G, Schneider H, Tenenbaum T, Elias-Hamp B, et al. Natalizumab use during the third trimester of pregnancy. JAMA Neurol. 2014;71(7):891–5. https://doi.org/10.1001/jamaneurol.2014.209.
Mariette X, Förger F, Abraham B, Flynn AD, Moltó A, Flipo RM, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77(2):228–33. https://doi.org/10.1136/annrheumdis-2017-212196.
Moriyama M, Wada Y, Minamoto T, Kondo M, Honda M, Murakawa Y. Unexpectedly lower proportion of placental transferred tocilizumab relative to whole immunoglobulin G: a case report. Scand J Rheumatol. 2020;49(2):165–6. https://doi.org/10.1080/03009742.2019.1639821.
Saito J, Yakuwa N, Kaneko K, Takai C, Goto M, Nakajima K, et al. Tocilizumab during pregnancy and lactation: drug levels in maternal serum, cord blood, breast milk and infant serum. Rheumatology (Oxford). 2019;58(8):1505–7. https://doi.org/10.1093/rheumatology/kez100.
Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5. https://doi.org/10.1016/j.xphs.2018.10.033.
Cole S, Hay JL, Luzon E, Nordmark A, Rusten IS. European regulatory perspective on pediatric physiologically based pharmacokinetic models. Int J Pharmacokinet. 2017;2(2):113–24. https://doi.org/10.4155/ipk-2016-0025.
Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2(8): e63. https://doi.org/10.1038/psp.2013.41.
Templeton IE, Jones NS, Musib L. Pediatric dose selection and utility of PBPK in determining dose. AAPS J. 2018;20(2):31. https://doi.org/10.1208/s12248-018-0187-8.
Yellepeddi V, Rower J, Liu X, Kumar S, Rashid J, Sherwin CMT. State-of-the-art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin Pharmacokinet. 2019;58(1):1–13. https://doi.org/10.1007/s40262-018-0677-y.
Chaphekar N, Dodeja P, Shaik IH, Caritis S, Venkataramanan R. Maternal-fetal pharmacology of drugs: a review of current status of the application of physiologically based pharmacokinetic models. Front Pediatr. 2021;9: 733823. https://doi.org/10.3389/fped.2021.733823.
Cao Y, Jusko WJ. Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2012;39(6):711–23. https://doi.org/10.1007/s10928-012-9280-2.
Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52(2):83–124. https://doi.org/10.1007/s40262-012-0027-4.
Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67–86. https://doi.org/10.1007/s10928-011-9232-2.
Li L, Gardner I, Dostalek M, Jamei M. Simulation of monoclonal antibody pharmacokinetics in humans using a minimal physiologically based model. AAPS J. 2014;16(5):1097–109. https://doi.org/10.1208/s12248-014-9640-5.
Glassman PM, Balthasar JP. Physiologically-based modeling of monoclonal antibody pharmacokinetics in drug discovery and development. Drug Metab Pharmacokinet. 2019;34(1):3–13. https://doi.org/10.1016/j.dmpk.2018.11.002.
Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709. https://doi.org/10.1007/s10928-007-9065-1.
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14. https://doi.org/10.1186/2040-2384-2-14.
Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I- related receptor FcRn. Annu Rev Immunol. 2000;18:739–66. https://doi.org/10.1146/annurev.immunol.18.1.739.
Levy G. Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248–52. https://doi.org/10.1038/clpt.1994.134.
Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. https://doi.org/10.1208/s12248-012-9340-y.
Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. https://doi.org/10.1038/nri2206.
Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol. 2012;52(1 Suppl):54s–62s. https://doi.org/10.1177/0091270011413894.
Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6):365–96. https://doi.org/10.2165/11597440-000000000-00000.
Abduljalil K, Johnson TN, Rostami-Hodjegan A. Fetal physiologically-based pharmacokinetic models: systems information on fetal biometry and gross composition. Clin Pharmacokinet. 2018;57(9):1149–71. https://doi.org/10.1007/s40262-017-0618-1.
Dallmann A, Ince I, Solodenko J, Meyer M, Willmann S, Eissing T, et al. Physiologically based pharmacokinetic modeling of renally cleared drugs in pregnant women. Clin Pharmacokinet. 2017;56(12):1525–41. https://doi.org/10.1007/s40262-017-0538-0.
Xia B, Heimbach T, Gollen R, Nanavati C, He H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. 2013;15(4):1012–24. https://doi.org/10.1208/s12248-013-9505-3.
Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45(10):1013–34. https://doi.org/10.2165/00003088-200645100-00005.
Malik P, Edginton A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin Drug Metab Toxicol. 2018;14(6):585–99. https://doi.org/10.1080/17425255.2018.1482278.
Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Paediatr Drugs. 2020;22(2):199–216. https://doi.org/10.1007/s40272-020-00382-7.
Friis-Hansen B. Body water compartments in children: changes during growth and related changes in body composition. Pediatrics. 1961;28:169–81.
Hay DA, Evan AP. Maturation of the glomerular visceral epithelium and capillary endothelium in the puppy kidney. Anat Rec. 1979;193(1):1–21. https://doi.org/10.1002/ar.1091930102.
Johnson SA, Vander Straten MC, Parellada JA, Schnakenberg W, Gest AL. Thoracic duct function in fetal, newborn, and adult sheep. Lymphology. 1996;29(2):50–6.
Taylor PM, Boonyaprakob U, Waterman V, Watson D, Lopata E. Clearances of plasma proteins from pulmonary vascular beds of adult dogs and pups. Am J Physiol. 1967;213(2):441–9. https://doi.org/10.1152/ajplegacy.1967.213.2.441.
Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti-respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56(9):4927–36. https://doi.org/10.1128/aac.06446-11.
Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog. Allergy. 1969;13:1–110. https://doi.org/10.1159/000385919.
Allansmith M, McClellan BH, Butterworth M, Maloney JR. The development of immunoglobulin levels in man. J Pediatr. 1968;72(2):276–90. https://doi.org/10.1016/s0022-3476(68)80324-5.
Pan X, Stader F, Abduljalil K, Gill KL, Johnson TN, Gardner I, et al. Development and application of a physiologically-based pharmacokinetic model to predict the pharmacokinetics of therapeutic proteins from full-term neonates to adolescents. AAPS J. 2020;22(4):76. https://doi.org/10.1208/s12248-020-00460-1.
Tian Z, Sutton BJ, Zhang X. Distribution of rat neonatal Fc receptor in the principal organs of neonatal and pubertal rats. J Recept Signal Transduct Res. 2014;34(2):137–42. https://doi.org/10.3109/10799893.2013.865745.
Limothai W, Meibohm B, editors. Effect of developmental growth and FcRn expresion on the pharmacokinetics of monocolnal antibodies in mice. World Conference on Pharmacometrics; 2016; Brisbane.
Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 1996;36(5):248–55. https://doi.org/10.1111/j.1600-0897.1996.tb00172.x.
Sarvas H, Seppälä I, Kurikka S, Siegberg R, Mäkelä O. Half-life of the maternal IgG1 allotype in infants. J Clin Immunol. 1993;13(2):145–51. https://doi.org/10.1007/bf00919271.
Palfi M, Selbing A. Placental transport of maternal immunoglobulin G. Am J Reprod Immunol. 1998;39(1):24–6. https://doi.org/10.1111/j.1600-0897.1998.tb00329.x.
Ailus KT. A follow-up study of immunoglobulin levels and autoantibodies in an unselected pregnant population. Am J Reprod Immunol. 1994;31(4):189–96. https://doi.org/10.1111/j.1600-0897.1994.tb00866.x.
Leach L, Firth JA. Structure and permeability of human placental microvasculature. Microsc Res Tech. 1997;38(1–2):137–44. https://doi.org/10.1002/(sici)1097-0029(19970701/15)38:1/2%3c137::Aid-jemt14%3e3.0.Co;2-q.
Pang V, Bates DO, Leach L. Regulation of human feto-placental endothelial barrier integrity by vascular endothelial growth factors: competitive interplay between VEGF-A(165)a, VEGF-A(165)b. PIGF and VE-cadherin Clin Sci (Lond). 2017;131(23):2763–75. https://doi.org/10.1042/cs20171252.
Avrech OM, Samra Z, Lazarovich Z, Caspi E, Jacobovich A, Sompolinsky D. Efficacy of the placental barrier for immunoglobulins: correlations between maternal, paternal and fetal immunoglobulin levels. Int Arch Allergy Immunol. 1994;103(2):160–5. https://doi.org/10.1159/000236622.
Ockleford CD, Clint JM. The uptake of IgG by human placental chorionic villi: a correlated autoradiographic and wide aperture counting study. Placenta. 1980;1(2):91–111. https://doi.org/10.1016/s0143-4004(80)80018-x.
Porter C, Armstrong-Fisher S, Kopotsha T, Smith B, Baker T, Kevorkian L, et al. Certolizumab pegol does not bind the neonatal Fc receptor (FcRn): consequences for FcRn-mediated in vitro transcytosis and ex vivo human placental transfer. J Reprod Immunol. 2016;116:7–12. https://doi.org/10.1016/j.jri.2016.04.284.
Miller RK, Mace K, Polliotti B, DeRita R, Hall W, Treacy G. Marginal transfer of ReoPro (Abciximab) compared with immunoglobulin G (F105), inulin and water in the perfused human placenta in vitro. Placenta. 2003;24(7):727–38. https://doi.org/10.1016/s0143-4004(03)00101-2.
Pentsuk N, van der Laan JW. An interspecies comparison of placental antibody transfer: new insights into developmental toxicity testing of monoclonal antibodies. Birth Defects Res B Dev Reprod Toxicol. 2009;86(4):328–44. https://doi.org/10.1002/bdrb.20201.
Ishikawa T, Takizawa T, Iwaki J, Mishima T, Ui-Tei K, Takeshita T, et al. Fc gamma receptor IIb participates in maternal IgG trafficking of human placental endothelial cells. Int J Mol Med. 2015;35(5):1273–89. https://doi.org/10.3892/ijmm.2015.2141.
Latvala S, Jacobsen B, Otteneder MB, Herrmann A, Kronenberg S. Distribution of FcRn across species and tissues. J Histochem Cytochem. 2017;65(6):321–33. https://doi.org/10.1369/0022155417705095.
Gitlin D, Kumate J, Urrusti J, Morales C. The selectivity of the human placenta in the transfer of plasma proteins from mother to fetus. J Clin Invest. 1964;43(10):1938–51. https://doi.org/10.1172/jci105068.
Stone RH, Hong J, Jeong H. Pharmacokinetics of monoclonal antibodies used for inflammatory bowel diseases in pregnant women. J Clin Toxicol. 2014;4(4). https://doi.org/10.4172/2161-0495.1000209.
Hubbard JJ, Laurenzana EM, Williams DK, Gentry WB, Owens SM. The fate and function of therapeutic antiaddiction monoclonal antibodies across the reproductive cycle of rats. J Pharmacol Exp Ther. 2011;336(2):414–22. https://doi.org/10.1124/jpet.110.175083.
Ishii S, Arizono H, Nagao T, Kudo S, Kondo S, Kiyoki M. Pharmacokinetics of a new human monoclonal antibody against cytomegalovirus. Second communication: distribution and elimination of the new monoclonal antibody, regavirumab after repeated administration in rats, and placental transfer and milk-passage study after single administration to pregnant and lactating rats. Arzneimittelforschung. 1994;44(7):899-908.
Simister NE, Story CM, Chen HL, Hunt JS. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. 1996;26(7):1527–31. https://doi.org/10.1002/eji.1830260718.
Leach JL, Sedmak DD, Osborne JM, Rahill B, Lairmore MD, Anderson CL. Isolation from human placenta of the IgG transporter, FcRn, and localization to the syncytiotrophoblast: implications for maternal-fetal antibody transport. J Immunol. 1996;157(8):3317–22.
Kristoffersen EK, Matre R. Co-localization of the neonatal Fc gamma receptor and IgG in human placental term syncytiotrophoblasts. Eur J Immunol. 1996;26(7):1668–71. https://doi.org/10.1002/eji.1830260741.
Kiskova T, Mytsko Y, Schepelmann M, Helmer H, Fuchs R, Miedl H, et al. Expression of the neonatal Fc-receptor in placental-fetal endothelium and in cells of the placental immune system. Placenta. 2019;78:36–43. https://doi.org/10.1016/j.placenta.2019.02.012.
Antohe F, Rădulescu L, Gafencu A, Gheţie V, Simionescu M. Expression of functionally active FcRn and the differentiated bidirectional transport of IgG in human placental endothelial cells. Hum Immunol. 2001;62(2):93–105. https://doi.org/10.1016/s0198-8859(00)00244-5.
Wang Y, Jiang X, He J, Diraviyam T, Zhang X. Quantitative investigation on correlation between IgG and FcRn during gestation and lactating periods in rat. Am J Reprod Immunol. 2016;75(2):81–5. https://doi.org/10.1111/aji.12465.
Lozano NA, Lozano A, Marini V, Saranz RJ, Blumberg RS, Baker K, et al. Expression of FcRn receptor in placental tissue and its relationship with IgG levels in term and preterm newborns. Am J Reprod Immunol. 2018;80(3): e12972. https://doi.org/10.1111/aji.12972.
Berg T, Nilsson BA. The foetal development of serum levels of IgG and IgM. Acta Paediatr Scand. 1969;58(6):577–83. https://doi.org/10.1111/j.1651-2227.1969.tb04765.x.
Mikhael M, Khan YS. Anatomy, abdomen and pelvis, lymphatic drainage. StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.; 2022.
Breslin JW, Yang Y, Scallan JP, Sweat RS, Adderley SP, Murfee WL. Lymphatic vessel network structure and physiology. Compr Physiol. 2018;9(1):207–99. https://doi.org/10.1002/cphy.c180015.
Cataldo Oportus S, de Paiva Rodrigues L, Pereira de Godoy JM, Guerreiro Godoy Mde F. Lymph drainage in pregnant women. Nurs Res Pract. 2013;2013:364582. https://doi.org/10.1155/2013/364582.
Hytten FE, Robertson EG. Maternal water metabolism in pregnancy. Proc R Soc Med. 1971;64(10):1072.
Forsum E, Forsberg AM, Nilsson E, Bergström J, Hultman E. Electrolytes, water, RNA, total creatine and calculated resting membrane potential in muscle tissue from pregnant women. Ann Nutr Metab. 2000;44(4):144–9. https://doi.org/10.1159/000012837.
Soeters PB, Wolfe RR, Shenkin A. Hypoalbuminemia: pathogenesis and clinical significance. JPEN J Parenter Enteral Nutr. 2019;43(2):181–93. https://doi.org/10.1002/jpen.1451.
Bungum L, Tollan A, Oian P. Antepartum to postpartum changes in transcapillary fluid balance. Br J Obstet Gynaecol. 1990;97(9):838–42. https://doi.org/10.1111/j.1471-0528.1990.tb02580.x.
Brace RA, Christian JL. Transcapillary Starling pressures in the fetus, newborn, adult, and pregnant adult. Am J Physiol. 1981;240(6):H843–7. https://doi.org/10.1152/ajpheart.1981.240.6.H843.
Bailey RP, Weiss L. Ontogeny of human fetal lymph nodes. Am J Anat. 1975;142(1):15–27. https://doi.org/10.1002/aja.1001420103.
Bellini C, Boccardo F, Bonioli E, Campisi C. Lymphodynamics in the fetus and newborn. Lymphology. 2006;39(3):110–7.
Eiby YA, Lingwood BE, Wright IMR. Plasma leak from the circulation contributes to poor outcomes for preterm infants: a working hypothesis. Front Neurol. 2021;12:636740-. https://doi.org/10.3389/fneur.2021.636740.
Sola A, Gregory GA. Colloid osmotic pressure of normal newborns and premature infants. Crit Care Med. 1981;9(8):568–72. https://doi.org/10.1097/00003246-198108000-00002.
Delivoria-Papadopoulos M, Battaglia FC, Meschia G. A comparison of fetal versus maternal plasma colloidal osmotic pressure im man. Proc Soc Exp Biol Med. 1969;131(1):84–7. https://doi.org/10.3181/00379727-131-33809.
Wu PY, Rockwell G, Chan L, Wang SM, Udani V. Colloid osmotic pressure in newborn infants: variations with birth weight, gestational age, total serum solids, and mean arterial pressure. Pediatrics. 1981;68(6):814–9.
Baum JD, Eisenberg C, Franklin FA, Jr., Meschia G, Battaglia FC. Studies on colloid osmotic pressure in the fetus and newborn infant. With observations on the effects of membranes of various pore sizes. Biol Neonate. 1971;18(3):311–20. https://doi.org/10.1159/000240371.
van der Giessen J, Binyamin D, Belogolovski A, Frishman S, Tenenbaum-Gavish K, Hadar E, et al. Modulation of cytokine patterns and microbiome during pregnancy in IBD. Gut. 2020;69(3):473–86. https://doi.org/10.1136/gutjnl-2019-318263.
Hazes JM, Coulie PG, Geenen V, Vermeire S, Carbonnel F, Louis E, et al. Rheumatoid arthritis and pregnancy: evolution of disease activity and pathophysiological considerations for drug use. Rheumatology (Oxford). 2011;50(11):1955–68. https://doi.org/10.1093/rheumatology/ker302.
Rook GA, Steele J, Brealey R, Whyte A, Isenberg D, Sumar N, et al. Changes in IgG glycoform levels are associated with remission of arthritis during pregnancy. J Autoimmun. 1991;4(5):779–94. https://doi.org/10.1016/0896-8411(91)90173-a.
Haider S, Knöfler M. Human tumour necrosis factor: physiological and pathological roles in placenta and endometrium. Placenta. 2009;30(2):111–23. https://doi.org/10.1016/j.placenta.2008.10.012.
Basu S, Lien YTK, Vozmediano V, Schlender JF, Eissing T, Schmidt S, et al. Physiologically based pharmacokinetic modeling of monoclonal antibodies in pediatric populations using PK-Sim. Front Pharmacol. 2020;11:868. https://doi.org/10.3389/fphar.2020.00868.
Hanke N, Kunz C, Thiemann M, Fricke H, Lehr T. Translational PBPK modeling of the protein therapeutic and CD95L inhibitor asunercept to develop dose recommendations for its first use in pediatric glioblastoma patients. Pharmaceutics. 2019;11(4). https://doi.org/10.3390/pharmaceutics11040152.
Hardiansyah D, Ng CM. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically-based pharmacokinetic modelling. MAbs. 2018;10(7):1144–56. https://doi.org/10.1080/19420862.2018.1494479.
Malik PRV, Edginton AN. Physiologically-based pharmacokinetic modeling vs. allometric scaling for the prediction of infliximab pharmacokinetics in pediatric patients. CPT Pharmacometrics Syst Pharmacol. 2019;8(11):835–44. https://doi.org/10.1002/psp4.12456.
Malik PRV, Edginton AN. Integration of ontogeny into a physiologically based pharmacokinetic model for monoclonal antibodies in premature infants. J Clin Pharmacol. 2020;60(4):466–76. https://doi.org/10.1002/jcph.1540.
Chang HP, Shakhnovich V, Frymoyer A, Funk RS, Becker ML, Park KT et al. A population physiologically-based pharmacokinetic model to characterize antibody disposition in pediatrics and evaluation of the model using infliximab. Br J Clin Pharmacol. 2021. https://doi.org/10.1111/bcp.14963.
Julsgaard M, Baumgart DC, Baunwall SMD, Hansen MM, Grosen A, Bibby BM, et al. Vedolizumab clearance in neonates, susceptibility to infections and developmental milestones: a prospective multicentre population-based cohort study. Aliment Pharmacol Ther. 2021;54(10):1320–9. https://doi.org/10.1111/apt.16593.
Ling J, Koren G. Challenges in vaccinating infants born to mothers taking immunoglobulin biologicals during pregnancy. Expert Rev Vaccines. 2016;15(2):239–56. https://doi.org/10.1586/14760584.2016.1115351.
Cheent K, Nolan J, Shariq S, Kiho L, Pal A, Arnold J. Case Report: Fatal case of disseminated BCG infection in an infant born to a mother taking infliximab for Crohn’s disease. J Crohns Colitis. 2010;4(5):603–5. https://doi.org/10.1016/j.crohns.2010.05.001.
Labetoulle R, Roblin X, Paul S. Prolonged persistence of adalimumab transferred from mother to infant during pregnancy. Ann Intern Med. 2018;169(1):60–1. https://doi.org/10.7326/l17-0629.
Berthelsen BG, Fjeldsøe-Nielsen H, Nielsen CT, Hellmuth E. Etanercept concentrations in maternal serum, umbilical cord serum, breast milk and child serum during breastfeeding. Rheumatology (Oxford). 2010;49(11):2225–7. https://doi.org/10.1093/rheumatology/keq185.
Xu Y, Langevin BA, Zhou H, Xu Z. Model-aided adults-to-children pharmacokinetic extrapolation and empirical body size-based dosing exploration for therapeutic monoclonal antibodies-is allometry a reasonable choice? J Clin Pharmacol. 2020;60(12):1573–84. https://doi.org/10.1002/jcph.1677.
Germovsek E, Cheng M, Giragossian C. Allometric scaling of therapeutic monoclonal antibodies in preclinical and clinical settings. MAbs. 2021;13(1):1964935. https://doi.org/10.1080/19420862.2021.1964935.
Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–23. https://doi.org/10.1517/17425250802691074.
Gill KL, Machavaram KK, Rose RH, Chetty M. Potential sources of inter-subject variability in monoclonal antibody pharmacokinetics. Clin Pharmacokinet. 2016;55(7):789–805. https://doi.org/10.1007/s40262-015-0361-4.
Jones HM, Zhang Z, Jasper P, Luo H, Avery LB, King LE, et al. A physiologically-based pharmacokinetic model for the prediction of monoclonal antibody pharmacokinetics from in vitro data. CPT Pharmacometrics Syst Pharmacol. 2019;8(10):738–47. https://doi.org/10.1002/psp4.12461.
Glassman PM, Balthasar JP. Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J Pharmacokinet Pharmacodyn. 2016;43(4):427–46. https://doi.org/10.1007/s10928-016-9482-0.
Jones HM, Tolsma J, Zhang Z, Jasper P, Luo H, Weber GL, et al. A physiologically-based pharmacokinetic model for the prediction of “half-life extension” and “catch and release” monoclonal antibody pharmacokinetics. CPT Pharmacometrics Syst Pharmacol. 2020;9(9):534–41. https://doi.org/10.1002/psp4.12547.
Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10(2):244–55. https://doi.org/10.1080/19420862.2017.1417718.
Chung S, Nguyen V, Lin YL, Lafrance-Vanasse J, Scales SJ, Lin K, et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs. 2019;11(5):942–55. https://doi.org/10.1080/19420862.2019.1605270.
Hutson JR, Garcia-Bournissen F, Davis A, Koren G. The human placental perfusion model: a systematic review and development of a model to predict in vivo transfer of therapeutic drugs. Clin Pharmacol Ther. 2011;90(1):67–76. https://doi.org/10.1038/clpt.2011.66.
Steenholdt C, Al-Khalaf M, Ainsworth MA, Brynskov J. Therapeutic infliximab drug level in a child born to a woman with ulcerative colitis treated until gestation week 31. J Crohns Colitis. 2012;6(3):358–61. https://doi.org/10.1016/j.crohns.2011.10.002.
Acknowledgements
We thank Eleanor Savill for her assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
KL Gill performed the research, and KL Gill and HM Jones both wrote the paper.
Corresponding author
Ethics declarations
Conflict of Interest
Katherine L Gill and Hannah M Jones are employees of Certara UK Limited.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gill, K.L., Jones, H.M. Opportunities and Challenges for PBPK Model of mAbs in Paediatrics and Pregnancy. AAPS J 24, 72 (2022). https://doi.org/10.1208/s12248-022-00722-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-022-00722-0