
Abstract
Vascular calcification (VC) is highly correlated with cardiovascular disease morbidity and mortality, but anti-VC treatment remains an area to be tackled due to the ill-defined molecular mechanisms. Regardless of the type of VC, it does not depend on a single cell but involves multi-cells/organs to form a complex cellular communication network through the vascular microenvironment to participate in the occurrence and development of VC. Therefore, focusing only on the direct effect of pathological factors on vascular smooth muscle cells (VSMCs) tends to overlook the combined effect of other cells and VSMCs, including VSMCs-VSMCs, ECs-VMSCs, Macrophages-VSMCs, etc. Extracellular vesicles (EVs) are a collective term for tiny vesicles with a membrane structure that are actively secreted by cells, and almost all cells secrete EVs. EVs docked on the surface of receptor cells can directly mediate signal transduction or transfer their contents into the cell to elicit a functional response from the receptor cells. They have been proven to participate in the VC process and have also shown attractive therapeutic prospects. Based on the advantages of EVs and the ability to be detected in body fluids, they may become a novel therapeutic agent, drug delivery vehicle, diagnostic and prognostic biomarker, and potential therapeutic target in the future. This review focuses on the new insight into VC molecular mechanisms from the perspective of crosstalk, summarizes how multi-cells/organs interactions communicate via EVs to regulate VC and the emerging potential of EVs as therapeutic methods in VC. We also summarize preclinical experiments on crosstalk-based and the current state of clinical studies on VC-related measures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With the acceleration of the global aging process and the increasing prevalence of metabolic diseases, the prevalence of vascular calcification (VC) is increasing dramatically [1, 2]. VC is common in patients with atherosclerosis, diabetes, chronic kidney disease (CKD), and other advanced diseases [3]. VC can reduce the elasticity and compliance of the vascular wall, increase the instability of atherosclerotic plaque, aggravate cardiac afterload, and finally lead to intravascular thrombosis, vascular occlusion, left ventricular hypertrophy, heart failure, and acute myocardial infarction [4]. It is an independent predictor of cardiovascular morbidity and mortality and a harbinger of future coronary heart disease, stroke, lower limb amputations, and other diseases [4, 5].
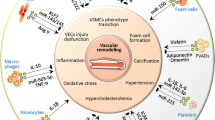
Initially, VC was thought to be a passive process due to the pathological mineral deposition in the vascular system. VC is now widely recognized as an active, dynamic, and complex pathological process regulated by numerous factors, with VSMCs playing a key driving role [6]. In addition to VSMCs, endothelial cells, pericytes, macrophages, bone marrow-derived mesenchymal stem cells, and so on, can also be involved in VC [7,8,9,10]. Current understanding of the pathogenesis of VC includes calcium and phosphorus imbalance [11,12,13], VSMCs transdifferentiation [14, 15], extracellular matrix [16], EVs [17], bone homeostasis imbalance [18,19,20,21,22], inflammation [23], epigenetics (DNA methylation and demethylation, histone modification, non-coding RNA) [24,25,26,27,28,29], autophagy [30], oxidative stress [31], mitochondrial dysfunction [24], iron death [32] and pyroptosis [33], etc. Despite some progress in very recent years, VC remains difficult to operate and has poor outcomes and prognoses because the exact mechanisms of VC remain unclear [34].
Crosstalk refers to information communication between cells, the interaction between information substances, and the interaction between information transduction pathways or channels. Therefore, crosstalk is a multi-level and time–space characterization of the network involving information communication, coordination and interaction. It has been demonstrated that the occurrence of any type of VC does not occur in a single cell/organ, but involves multi-cells/organs and systems [35]. Crosstalk enables cell-to-cell/organ-to-organ interactions in VC through a variety of biomolecules and signaling pathways. For example, endothelial cells can interact with VSMCs through myoendothelial gap junction (MEGJ), release various biomolecules and EVs under pathological stimulation, and then affect the occurrence and development of VC [36].
In recent years, EVs have been found to play a crucial role in cross-border crosstalk. There is compelling evidence that EVs play an important regulatory role not only in diseases such as bone diseases, tumours and metabolic diseases [37,38,39], but also in cardiovascular diseases (CVD). EVs are secreted from cells into circulation in a regulated manner and are captured by distant cells to exert their biological effects [39, 40]. In fact, EVs carry biologically active substances, such as nucleic acids, proteins, lipids, and so on [41,42,43,44,45], which can affect target cells through the following mechanisms:(1) EVs activate target cell downstream signaling pathways through specific binding of surface ligands to target cell receptors;(2) EVs directly transfer the receptor in the activated state to the target cells, thereby activating downstream signaling pathways; and (3) EVs transfer their contents into target cells through direct fusion with the plasma membrane or endocytosis, and mediate target cell biological effects by regulating downstream signaling pathways in target cells [46, 47]. Under different physiopathological conditions, the amount of EVs secreted by maternal cells and their contents are altered and the results of EVs targeting cells such as VSMCs or ECs are different. In this review, we summarize a new perspective of EVs-mediated intercellular/interorgan material communication to elucidate the molecular mechanism of VC and "cross-talk"-based therapeutic strategies.
Overview of extracellular vesicles
Discovery and nomenclature of EVs
Extracellular vesicles(EVs) are nanoparticles with lipid bilayer structure and no nucleus; and are a heterogeneous population of membrane-bound vesicles (heterogeneity in size, content, function, and origin) [40]. Exosomes were originally discovered by Dr. Rose Johnstone in order to understand the biological process of reticulocyte transformation into mature red blood cells and thought that they might be one of the ways of cell excretion of waste products, but recent studies have shown that they have multiple functions, contrary to the original view that exosomes are waste excretion [48].
The classification of extracellular vesicles (EVs) is constantly evolving, and in the past different names have been used in the litetature to denote these EVs: according to their size, they are called microparticles, microvesicles, nanovesicles and nanopartices; according to their possible functions, they are called matrix vesicles, argosomes, tolerosomes; as they are secreted outside the cell, they are called etosomes, exosomes, exovesicles, exosome-like vesicles, and so on [42, 49, 50]. In fact, there are no universal markers to distinguish between different types of EVs, and each has overlapping characteristics [51]. Therefore, the biological classification and nomenclature of EVs is complex and controversial [50], and isolating specific EV subtypes is challenging. The International Society for Extracellular Vesicles (ISEV) recommends the use of characteristics such as the size (< 200 nm for small vesicles and > 200 nm for medium/large vesicles) or density and biochemical composition (EVs expressing CD63/CD81 or Annexin A5), among other physicochemical properties, to characterize EVs [51].
Biogenesis, release and internalization of EVs
According to their mechanisms of occurrence, EVs can be divided into three types: Exosomes (30–150 nm, endocytic pathway of EVs origin), Microvesicles (100–1000 nm EVs sprouting from the plasma membrane), Apoptotic bodies (Abs, 1–5 μm EVs from apoptotic cells) [52,53,54]. Exosomes originate from endocytosis. Endosomal membranes bud inward to form intraluminal vesicles (ILVs) in the lumen of multivesicular bodies (MVBs) [55]. ILV is released as exosomes by plasma membrane fusion, involving ESCRT (endosomal sorting complex required for transport)-dependent mechanisms, ESCRT-independent mechanisms and other nonclassical pathways [56,57,58]. The ESCRT-independent exosome synthesis pathway requires ceramide [59,60,61]. Release of cellular exosomes was significantly reduced after inhibition of neurtral sphingomyelinase (n-SMase) [62], a key enzyme in ceramide production [59,60,61]. GW4869 is a non-competitive neutral sphingomyelinase inhibitor that prevents ILVs formation capable of blocking exosome production [63,64,65,66]. Several studies have shown that calcification can be significantly ameliorated after the use of GW4869 at the cellular level or in mice models [67,68,69], or that GW4869 partially reverses substances that act as inhibitors of calcification [70, 71]. These two reviews provide a detailed assessment of the major drugs currently interfering with the biosynthesis or release of EVs [65, 72]. By using confocal microscopy it was observed that some MVBs can also be degraded by interacting with lysosomes [73]. Unlike the exosomes, microvesicles are formed by direct germination and division of the plasma membrane [74]. During microvesicle biogenesis, the interaction between the cytoskeleton and the plasma membrane weakens and several proteins such as calpain or lipid transferases are activated [58]. This remodeling of the cytoskeleton is associated with an increase in intracellular calcium concentration, which in turn induces phosphatidylserine externalization, bud formation, and microvesicle secretion [75]. Unlike exosomes and microvesicles, which are secreted during normal cellular processes, apoptotic bodies are only released upon apoptosis [58]. Apoptotic bodies are formed due to cytoskeleton destruction of apoptotic cells, swelling of plasma membrane and separation of plasma membrane [55, 76].
CD29 is a marker for both microvesicles and exosomes, while CD29+EVs can be used to distinguish between microvesicles and exosomes when combined with other protein markers; for example, Tetraspanins, including CD63, are thought to be mostly present in exosomes but mostly absent in microvesicles [77]. Furthermore, proteins of the tetraspanin family such as CD9 and CD81 are also abundant in exosomes and are also widely considered as markers [78]. The membrane lipid bilayer of EVs also contains integrins, cell adhesion molecules, etc. [78, 79] The first two types of extracellular vesicles are currently being studied, and some researchers refer to these collectively as EVs. Differences in their surface molecules and size may affect the ability to be internalized and studies have shown the mechanisms by which EVs are internalized: ligand-receptor interaction, micropinocytosis, phagocytosis, and endocytosis mediated by target cells via clathrin or caveolin [79,80,81]; they can also fuse with the cell membrane to release their contents into the cytosol of the target cell [82] (Fig. 1).

Biogenesis, release and internalization of EVs. EVs can be divided into three types according to their mechanisms: Exosomes (30–150 nm), Microvesicles (100–1000 nm), Apoptotic bodies (Abs, 1–5 μm). Exosomes originate from endocytosis. Endosomal membranes bud inward to form intraluminal vesicles (ILVs) in the lumen of multivesicular bodies (MVBs). ILV is released as exosomes by plasma membrane fusion, involving ESCRT (endosomal sorting complex required for transport)-dependent mechanisms, ESCRT-independent mechanisms and other nonclassical pathways. Some MVBs can also be degraded by interacting with lysosomes. Proteins of the tetraspanin family such as CD9, CD63 and CD81, are abundant in exosomes and are widely recognized as markers of exosomes. Microvesicles are formed by direct germination and division of the plasma membrane. Abs are formed due to the cytoskeleton destruction of apoptotic cells, swelling of the plasma membrane and separation of the plasma membrane. Mechanisms of EVs internalization by target cells: Endocytosis mediated by clathrin or caveolin; fusion with plasma membrane; micropinocytosis and phagocytosis; ligand-receptor interaction. After being taken up by target cells, the contents (such as proteins, mRNAs, non-coding RNAs, and lipids) of EVs are released to regulate the translation, metabolism, growth and development of target cells
EVs come from a wide range of sources as many types of cells can secrete EVs, for example, macrophage, BMSC, VSMC, ECs, VICs, blood cells, etc., and are relatively stable present in a variety of body fluids, including blood, urine, saliva, bile and breast milk, etc. [57]. Numerous studies have shown that EVs can be released in both physiological and pathological settings and act as intercellular communication vehicles to transfer a wide range of biomolecules such as proteins, DNA, mRNAs, non-coding RNAs (ncRNAs: miRNAs, lncRNAs, circRNAs) and lipids from their parent cells to proximal or distal target cells/organs, regulating their activities including translation, metabolism, growth and development, and dynamically reflecting the state of the disease [39, 40]. Therefore, EVs as a medium play an important role in information exchange, which differs from the traditional way of information exchange.
Overview of the current knowledge of vascular calcification molecular mechanism
Classification and associated risk factors of VC
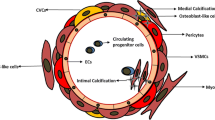
The occurrence of vascular calcification is a dynamic process in many aspects, which depends on the change in the microenvironment and the occurrence site. The pathological manifestation is to induce abnormal deposition of calcium, phosphorus, and other minerals in the form of hydroxyapatite on the vascular wall [3]. Abnormal mineralization can occur in all vascular. There are many classification methods for VC according to different causes. VC can be divided into five categories according to its site of occurrence and pathophysiological mechanisms: intimal, media, adventitia, valvular calcification (ValvC), and calciphylaxis [83] (Fig. 2).

Classification of vascular calcification (VC) and associated risk factors. VC can be divided into five categories according to its site of occurrence and pathophysiological mechanisms: intimal, media, adventitia, valvular calcification (ValvC), and calciphylaxis
Intimal calcification occurs frequently in atherosclerotic lesions and is associated with hyperlipidemia, macrophage infiltration and vascular inflammation [84]. Medial calcification mainly occurs in aging, diabetes and CKD diseases development, and VSMCs play a crucial role in medial calcification [3, 85]. Adventitial calcification is mainly thought to be related to pericytes, but the mechanism of adventitial calcification is still unclear and less studied [35]. Valvular calcification mainly occurs in aortic and mitral valves, and risk factors are often related to hypertension, aging, diabetes, and CKD [86, 87]. Calciphylaxis is a rare vascular calcification syndrome with severe pain, ulcer or necrotic skin injury in multiple parts of the body due to subcutaneous adipose tissue and microvascular occlusion of the dermis [88, 89]. Once calciphylaxis occurs, the disease progresses rapidly and the survival rate is low [88]. This disease has not been accurately recognized at present and usually occurs in patients on long-term dialysis for end-stage renal disease [88].
Calcification also can be divided into physiological calcification and pathological calcification [90]. Physiological calcification occurs in the normal physiological process of bone and tooth [90]. During the initial stages of mineralization of hard tissues such as bone, cartilage and dentin, hard tissue forming cells secrete matrix vesicles to function [91, 92]. For example, bone matrix vesicles are released during bone formation and development by microvilli sprouting at the tip of osteoblasts and participate in the initiation of mineralization by adjusting the ratio of pyrophosphate to phosphate, providing sites for hydroxyapatite crystal nucleation and interacting with extracellular matrix to promote the formation of hydroxyapatite in its cavity [92,93,94]. This series of processes is known as matrix vesicle-mediated mineralization. Pathological calcification is closely related to diabetes, chronic kidney disease (CKD), aging, advanced atherosclerosis, fatty liver, etc. [3] The mechanisms that promote the initiation and progression of VC are similar to those of physiological bone formation and include osteogenic/chondrogenic transdifferentiation, reduced availability of calcification inhibitors, the release of EVs, and remodeling of the extracellular matrix [95].
VSMCs-derived MVs mediated mineral deposition
VSMCs are the predominant cell type in the vessel wall, are plastic, and can switch between a contractile and a synthetic phenotype [96]. VSMCs exhibit a contractile phenotype to maintain an anti-VC microenvironment under physiological conditions. Upon abnormal stimulation, VSMCs will switch from a contractile phenotype to a synthetic phenotype (osteogenic/cartilaginous phenotype), which is a necessary condition for VSMC calcification [96, 97]. The phenotypic switch is driven by enhanced expression of osteogenic markers such as alkaline phosphatase (ALP), runt-related transcriptionfactor2 (Runx2), osteocalcin (OCN), bone morphogenetic protein (BMP), osteopontin (OPN), collagen type 1 (COL1) and bone sialoproteins, etc. [40, 98] In addition, there is also suppression of VSMC lineage markers such as sm22α and α-smooth muscle actin (α-SMA), etc. [3, 14, 99] Like Bone cells, VSMCs can also produce matrix EVs-like structures (Matrix vesicles, MVs). Matrix vesicles (30-300 nm) are a specialized type of extracellular vesicles [100]. The analysis of size, shape, lipid and protein content showed that the matrix vesicles were anchored exosomes secreted by cells and that they were homologous structures [101, 102]. Currently, there are no comprehensive definitions and no studies have been done to distinguish MVs from exosomes and microvesicles [101], so they should not be confused. MVs may represent a mixture of exosomes and microvesicles that contain specific components needed to direct ECM mineralization [103, 104].
VSMCs spontaneously release MVs under physiological conditions, and MVs are loaded with mineralization inhibitors (such as endogenous inhibitor MGP, cycle inhibitor Fetuin-A) to prevent mineral nucleation, which is an adaptive response aimed at preventing intracellular calcium overload [105, 106]. Matrix Gla protein (MGP), a vitamin K-dependent protein, is considered a natural calcification inhibitor and is highly expressed in VSMCs and chondrocytes [107]. MGP carboxylated by Vit K2 can not only prevent crystal formation, but also block BMP signal transduction to inhibit VC [107]. Therefore, reducing VC requires maintaining Vit K2 levels and avoiding the use of Vit K antagonists such as warfarin [108].
In contrast, under pathological stimulation (such as high extracellular calcium), VSMCs can secrete mineralized MVs in which MGP expression is absent and the activity of calcification-promoting factor matrix metalloproteinases 2 (MMP2) is increased [109]. When the tunica media calcifies, the degradation of elastin leads to the decrease of elastic fiber cross-linking to promote the deposition of hydroxyapatite crystals, and the elastic degradation products can further promote the transformation of VSMC osteogenic phenotype and aggravate VC [110]. In addition to the loss of calcium-induced inhibitors, VSMC-derived MVs are also involved in key events in the calcification process. Chen et al. [111] suggested that mineralization in VSMC requires both active MVs and the interaction of MVs with COL 1(ECM) to promote the release and aggregation of hydroxyapatite crystals, and the activity of annexin plays an important role in both steps. Previously, Annexin was shown to be a pro-calcification marker for calcifying EVs [109, 112]. AnxA6 and phosphatidylserine forms a protein-lipid complex that nucleates hydroxyapatite, leading to mineral deposition initiating micromineralization [109, 113]. Under the condition of continuous calcium overload, AnxA6 can shuttle to the plasma membrane to regulate calcium homeostasis and vesicle release to maintain intracellular calcium homeostasis and limit cell damage caused by calcium overload [109]. It has been shown that the mineralization of VSMC-MVs is a pathological reaction of intracellular calcium homeostasis disorder. Disturbance of calcium and phosphorus homeostasis is the premise of bone mineralization and vascular calcification development [11, 114]. MVs regulate ion channels that affect the concentration of internal and external calcium and phosphorus to regulate matrix mineralization. In addition, pathological stimulation such as Ca2+ and Pi can induce VSMC dedifferentiation and apoptosis, and then VSMC secretes MVs and releases apoptotic bodies into the extracellular matrix, which promotes calcium and phosphorus deposition, thus forming the initial focus of VC and promoting calcification [11, 27].
Gene mutations of proteins in MVs can produce correspond matrix mineralization abnormalities, such as enzyme activity imbalance and ion concentration imbalance [115]. Activation of tissue non-specific alkaline phosphatase (TNAP) in VSMCs precedes vascular calcification in atherosclerosis. TNAP may shift the Pi/PPi balance toward calcification by hydrolyzing the calcification inhibitor inorganic pyrophosphate (PPi) and generating inorganic phosphate (Pi) required for mineralization, which is critical for MVs-mediated hydroxyapatite formation [116]. Anderson et al. [117] demonstrated inadequate bone mineralization in TNAP-/-knockout mice and proposed two major hypothesized mechanisms: the first stage is primarily from an inability of initial mineral crystals within MVs to self-nucleate and to proliferate beyond the protective confines of the MVs membrane; the failure of the second stage of mineral formation may be due to the excess of the mineral inhibitor pyrophosphate in the extracellular-fluid around MVs. The activity of TNAP is necessary for both physiological bone mineralization and the induction of pathological calcification. MVs contain another phosphatase, PHOSPHO1, which acts as an alternative supplier of Pi, and plays a role in the initiation of matrix mineralization and may also be involved in MVs-mediated calcification [118,119,120]. The pathological induction of vascular wall by TNAP and PHOSPHO1 may be an important cause of atherosclerotic calcification [116].
In summary, VSMCs-derived MVs are involved in the initiation of VC. Apoptosis [121, 122], pyroptosis [33], oxidative stress [31, 123], inflammation [23, 124], autophagy [30, 125], microRNAs [27, 126], aging [14, 127], ferroptosis [32, 128], Metabolic reprogramming [129, 130], and epigenetic changes caused by reduced ATP production in mitochondrial insufficiency [24, 25, 131, 132] have also been reported to play an important role in the regulation of VC. The signaling pathways associated with VC such as Msx2, mTOR, MAPK, Wnt, and TGF-β pathways have been extensively elucidated [14, 133].
Current status and clinical treatment for VC
The current treatment of calcified lesions is mainly mechanical treatment with "coronary artery spinning" and "Intravascular lithotripsy (IVL)" [34, 134,135,136]. In addition to the treatment of calcification, the risk factors associated with it, such as diabetes, chronic kidney disease, hyperlipidemia and smoking, should be treated and controlled [137,138,139,140]. Sodium thiosulphate [141,142,143,144], bisphosphonates [145,146,147], SNF472 [148,149,150], phosphate binders [151, 152], calcimimetics [153, 154], Denosumab [155] and TNAP inhibitors [156,157,158] can potentially intervene in VC, but their clinical applications need to be further explored in depth. Prevention and treatment of VC focuses on early monitoring and prevention, correcting disturbances in calcium and phosphorus metabolism, avoiding hypercalcemia, and preventing secondary hyperparathyroidism or hypoparathyroidism. There is a lack of effective therapeutic measures to prevent and treat VC. It has been recognized that the VC process is a complex network, involving multiple cells involved in regulation, such as VSMC to osteogenic phenotype transformation, endothelial dysfunction, EndMT, stem/progenitor cell to osteoblast differentiation, etc. [14, 159, 160]. However, studies only focus on the direct effects of various pathological factors on vascular wall cells (e.g., VSMCs, ECs and macrophages) will tend to ignore the comprehensive effects of multiple cells. Once formed, VC is difficult to reverse, and although there are many studies on how to overcome VC, the complexity and diversity of VC pathophysiology hinders the discovery of optimal drug targets and drug development. Thus, it remains a major clinical challenge, and further insights into the mechanisms of vascular calcification are necessary to potentially trigger the development of multiple types of therapeutic agents. In the past, a large number of studies have focus on the role of EVs in mediating mineral deposition, whereas in recent years it has been found that EVs can mediate cell-to-cell/organ-to-organ crosstalk to propagate calcification. Using the biological characteristics of EVs, the molecular mechanism of VC is elucidated from a new perspective, which may provide a new direction for the treatment and prevention of VC in the future. Improving the understanding of crosstalk between VSMCs and cells/organs will not only help to regulate the risk factors related to the occurrence and development of VC, but also may provide insight into the development of VC treatment.
The crosstalk in vascular calcification
Extracellular vesicles mediated crosstalk
In addition to the aggregation of calcified EVs to produce microcalcifications in the ECM as elaborated above, EVs also play an important mediator role in multi-organ/cellular regulation of VC. The vascular wall can be divided into three layers: tunica intima, tunica media and tunica adventitia, and has a variety of functionally active cells [161]. The development of vascular calcification is not dictated by cells in one of the layers of the vessel wall, but rather it tends to function as a whole with cellular crosstalk. Under physiological conditions, vascular cell-derived EVs regulate the homeostasis of the vascular wall, but when subjected to pathological stimuli, such as risk factors associated with cardiovascular diseases: aging, diabetes, CKD, smoking, lipoprotein(a) and inflammation, etc., the biological properties of EVs can be altered to acquire pro-calcification potential and increase their release [17, 46, 162,163,164,165], and mediate communication between vascular wall cells through the EVs thereby affecting environmental homeostasis and VSMC phenotype to promote calcification [166].
Among the many biomolecules in EVs, ncRNAs are key regulators of their ability to mediate intercellular communication. MicroRNAs are regulatory molecules that inhibit protein expression, and they have been confirmed to be involved in the regulation of phenotypic transformation, senescence and calcification of VSMCs [27]. Several studies have shown that under pathological stimulation, different types of cell-derived EVs can regulate the phenotypic switch of VSMCs to promote VC by delivering miRNAs to recipient cells to upregulate the expression of Runx2 and activate various signaling pathways such as Wnt/β-catenin [167, 168]. And that transported miRNAs can further accelerate the development of VC through autophagy, oxidative stress, inflammation, immune response and other possible mechanisms [168]. A recent study has shown that functional molecule miR-23a-3p in EVs from atherosclerotic plaques can induce endothelial cell inflammation and propagate AS distantly through the circulation [169]. This study indicates that EVs are also regulated in the closed loop and affect neighboring cells, exacerbating disease progression. Similarly, there can be closed loop regulation between VMSCs through EVs.
In addition to the autocrine stimulation described above, EVs also play a key role in paracrine, such as the exchange of information between endothelial cells (ECs) and VSMCs [170,171,172,173]. And EVs mediate crosstalk between organs such as the bone-vessel axis [10] and so on. Furthermore, EVs derived from different cells also have potential improvement effects on cardiovascular diseases, such as progenitor cells(stem cells or mesenchymal cells)-EVs [174,175,176], BMSC-EVs [177], Cardiosphere-derived cells (CDCs)-EVs [178, 179] and so on. Therefore, we next focused on summarizing how EVs mediate communication between vascular wall cells (VSMCs-VSMCs, ECs-VSMCs, and Macrophages-VSMCs), bone-vascular, liver-vascular, and adipose-vascular under microenvironmental changes to regulate VC.
Bone-vascular crosstalk
In recent years, studies have found that the skeleton is not only an "inert organ" that accepts nerve and humoral regulation but also an "endocrine organ" that participates in the regulation of the whole body. The bone acts on itself by autocrine and paracrine means, and also acts on extraskeletal organs by remote secretion via the circulatory system. Osteoporosis and vascular calcification are two pathological phenomena that seriously threaten the health of middle-aged and elderly people. Osteoporosis in the elderly is often accompanied by VC. With the aging of the body, the loss of calcium in bones increases, bone mass decreases, and osteoporosis gradually forms, while the calcium deposition in vascular wall tissues increases, resulting in increased rigidity and reduced compliance of blood vessels. This contradictory phenomenon is called the "calcification paradox" between bone and blood vessels [180]. Both VC and bone mineralization are actively regulated processes, and these two reviews discuss in detail the many similarities in pathophysiology and progression between osteoporosis and VC, and summarize clinical data supporting their interaction [95, 181]. However, whether osteoporosis drives VC or vice versa has not been established, and the specific mechanisms of their co-occurrence need to be further clarified. Previous studies have found that VC is accompanied by the expression of many bone metabolic proteins (osteopontin, adiponectin, bone morphogenetic protein 2[BMP2], osteoprotegerin, etc.) [18,19,20,21], suggesting the existence of a bone-vessel axis. It is believed that the bone-vessel axis may be regulated directly or indirectly by various hormones and physiological processes, but the specific mechanisms have not been fully elucidated.
Role of EVs in the regulation of the bone-vascular axis
With the development of aging and oxidative stress, the increase of miR-183 family (miR-96, miR-182, and miR-183–5 p) in bone matrix EVs (B-EVs) can inhibit the proliferation and osteogenic differentiation of Bone marrow mesenchymal stem cells (BMSCs) and induce stem cell aging [182,183,184,185]. In addition to the effects of B-EVs secretion on themselves, B-EVs also carry out long-distance cell-to-cell/organ-to-organ crosstalk. Surprisingly, a recent study by Wang et al. [10] suggests that EVs may help resolve this "calcification paradox". Osteoclastic bone resorption activity increases relative or absolute with bone aging and menopause, and B-EVs are found to be released from bone matrix into bone marrow during bone resorption and transported through blood circulation to vascular walls. It was found that EVs from aged bone matrix (AB-EVs) not only directly stimulated VSMC mineralization, but also found that the calcium content of AB-EVs was higher than that of YB-EVs(young bone-EVs), and increased serum calcium and inorganic phosphorus in both acute and chronic vascular calcification models, suggesting that AB-EVs can directly transport a large amount of calcium to circulation and increase phosphate in blood may be an important mechanism for AB-EVs to promote VC [10].
Another important mechanism is to reveal that AB-EVs mediates VC by transferring miR-483-5p and miR-2861, which are messengers of the "calcification paradox": miR-483-5p enrichment promotes adipogenesis (increased PPARγ expression) but not osteogenesis in BMSCs, and exacerbates calcification of VSMCs; it also stimulates RUNX2 expression and osteogenic transdifferentiation of VSMCs by transferring miR-2861 into the circulation and depositing it in blood vessels [10]. OCYB-CM (bone-resorption conditioned media from osteoclasts with young bone slices) and its EVs had no effect on VSMC calcification [10]. Intravenous or intramedullary injection of AB-EVs promoted bone fat imbalance and aggravated VC induced by vitamin D3 in young or old mice; it has also been found that the use of the bone resorption inhibitor alendronate (ALE) down-regulates the release of AB-EVs and reduces aging and ovariectomy-induced bone lipid imbalance and VC [10].
In a word, a new mechanism behind the "calcification paradox" related to age and menopause, namely the new mechanism of AB-EVs mediated "calcification paradox," provides a new idea for the prevention and treatment of osteoporosis and VC in the elderly, and further enriches the understanding of bone-vascular axis.
Kidney-gut-bone-vascular axis
In addition, intestinal microbiota plays a key regulator role in bone and cardiovascular homeostasis, and intestinal microbiota imbalance may induce osteoporosis and vascular calcification, which is envisioned as one of the pathogenesis involved in the bone-vascular axis. Chronic kidney disease is associated with a dysbacteriosis of the gut, which may contribute to bone and vascular disease in patients with CKD (the "kidney-gut-bone-vascular axis"), and potential mechanisms include: Decreased carbohydrate fermentation and increased protein fermentation result in gut-derived inflammation, deficiency of vit K and short-chain fatty acids (SCFAs) [186].
BMSCs-VSMCs crosstalk
Bone marrow mesenchymal stem cells (BMSCs) are pluripotent stem cells that can differentiate into a variety of cell types in vivo, such as: osteoblasts, chondroblasts, adipocytes, VSMCs, ECs and cardiomyocytes; and BMSCs and derived EVs have attracted great interest because they show great therapeutic potential as "miracle drugs." In previous studies, we have found that BMSC derived EVs may have the effect of relieving VC induced by high phosphorus (Pi), but the mechanism is not completely understood [187]. Guo et al. [187] found that BMSC-derived EVs attenuated VC induced by high Pi by changing the miRNA profile involving mTOR, MAPK and Wnt signaling pathways related to VC, suggesting that miRNAs may be key regulatory points for BMSC-EVs to inhibit VC. BMSC-EVs inhibit the osteogenic transdifferentiation and calcification of HA-VSMC by transferring miR-15a/15b/16 and inhibiting its common target gene nuclear factors of activated T cells 3 (NFATc3), thereby down-regulating the expression of osteocalcin (OCN) [188]. Liu et al. [189] proposed a novel mechanism that BMSC-EVs can also directly down-regulate NFAT5 by delivering miR-381-3p, improving apoptosis and VC in CKD-VC. Recent studies suggest that BMSC-EVs can regulate the NONHSAT 084969.2/NF-κB axis to inhibit Pi-induced VSMC transdifferentiation and calcification [190]. It also inhibits high phosphate-induced aortic calcification and improves renal function through the SIRT6-HMGB1 deacetylation pathway [191]. These studies suggest that BMSC-EVs may be a potential strategy in the treatment of CKD-VC.
Advanced glycation end products (AGEs) are proteins or lipids that become glycated after exposure to diabetes, which is the main cause of diabetic vascular complications, including diabetic vascular calcification [192]. Advanced Glycation End Product-Bovine Serum Albumin (AGEs-BSA) down-regulated and up-regulated the expression of miR-146a and thiotoxin-interacting protein(TXNIP) in VSMC, respectively, thereby increasing the production of ROS and promoting VSMC to differentiate into osteogenic phenotype [193]. However, miR-146a expression was increased in BMSC-EVs treated with AGEs-BSA, and EVs migrated into VSMC to inhibit AGEs-BSA-mediated VC by binding to its target, TXNIP [193]. Moreover, BMSC-EVs have an improved effect on cardiomyocyte function [177, 194], Intervertebral disc degeneration(IDD) and bone defect/damage [195].
In conclusion, it is further confirmed that EVs carrying multiple signaling molecules are important carriers for skeletal cells to regulate body functions, indicating the existence of bone-vascular axis regulation. The signaling pathway and gene expression of VSMCs are affected after uptake of EVs, which regulates calcification. And BMSCs-EVs showed great therapeutic prospects in VC.
Liver-vascular crosstalk
The liver is the main organ of lipid metabolism and patients with metabolic-related fatty liver disease have varying degrees of abnormal lipid metabolism, leading to dyslipidemia, phenotypically elevated LDL cholesterol and triglycerides, which in turn accelerates the development of cardiovascular disease (CVD) [196]. Non-alcoholic fatty liver disease (NAFLD) is a disease prevalent in obesity and diabetes, caused by a combination of genetic and lifestyle factors, and shares common risk factors with CVD, including insulin resistance, central obesity, T2DM, dyslipidemia, and hypertension, among others. In recent years, the term "metabolic dysfunction associated fatty liver disease (MAFLD)" has been coined and is replacing the name "NAFLD" as it better reflects the underlying pathogenesis and cardiometabolic significance of NAFLD [197, 198].
In a large study of a young and middle-aged population, it was shown that AFLD and NAFLD are both metabolic liver diseases and that the early liver disease they cause, whether obese or non-obese, are strongly associated with and accelerate coronary artery calcification (CAC) [199]. The results of a recent large Korean cohort study showed a significant positive association between both NAFLD and MAFLD and the prevalence and incidence of CAC, with MAFLD being more strongly associated [200]. Several clinical studies have shown that NAFLD is strongly associated with CVD, and NAFLD patients are more likely to die from CVD than from liver-related disease [201,202,203]. NAFLD is not only limited to the progression of liver function deterioration but is becoming an independent risk factor for CVD [204].
In recent years, liver-derived sEVs have been found to play an important role in intercellular/interorgan communication [205]. The amount of hepatocyte-derived EVs release and the expression of miRNAs in their contents changed in different liver disease states [206]. For example, steatotic hepatocytes under NAFLD released more EVs, which mediates the development of inflammation, fibrosis and angiogenesis [207], and significantly alters the expression profile of miRNAs [208]. Steatotic hepatocyte-derived EVs promote endothelial cells inflammation and AS formation by delivering miRNA-1 to inhibits KLF4 and activates NF-κB, while anti-miR-1 attenuates this effect [208]. Furthermore, steatosis hepatocytes secreted sEVs abundant in novel-miR-7 in the circulation, which promotes hyperpermeability of coronary microvascular endothelial cells by directly regulating the lysosomal-associated membrane protein 1 (LAMP1)/Cathepsin B/NLRP3 inflammasome axis [209]. This may also be one of the mechanisms leading to the microvascular complications of NAFLD. Previously, the NLRP3 inflammasome has been shown to be involved in the regulation of VC [210,211,212].
These studies suggest that hepatocyte-derived EVs play an important role in the cross-talk between the liver and cardiovascular, providing new ideas about the mechanism of the link between NAFLD and CVD. It also suggests a possible link between hepatocyte-derived EVs and VC, and a liver-vascular axis may exist. However, basic studies of hepatocyte-derived EVs mediating cardiovascular calcification are scarce, and there is a need for more in-depth mechanistic exploration. Future studies devoted to EVs may not only contribute to the development of NAFLD but also largely reduce the occurrence of CVD and thus reduce the number of NAFLD patients who die from CVD.
Adipose-vascular crosstalk
Adipose tissue (AT) is composed of a heterogeneous population of cells that regulate energy metabolism and immune responses. AT can divided into visceral adipose tissue and subcutaneous adipose tissue, among which abnormal deposits of visceral adipose tissue causing a higher incidence of cardiovascular disease [213]. The AT can act as an endocrine organ, secreting a variety of adipokines such as adiponectin, leptin, visfatin, omentin, etc. [214]. The AT-derived adipokines can mediate crosstalk between adipose tissue and cardiovascular via autocrine, paracrine and endocrine mechanisms [215]. For example, adiponectin inhibits beta-glycerophosphate (β-GP)-induced VC via the JAK2/STAT3 signaling pathway [216] and omentin-1 inhibits VC by activating AMPK and Akt signaling pathways [217]. In addition to the traditional soluble mediators described above, adipose tissue-derived EVs have also been shown to be an insoluble mediator that can regulate adjacent or distant target organs/cells.
Adipocyte derived EVs act as effective messengers between ADSCs and macrophages to maintain the metabolic and immune homeostasis of the body while driving a vicious cycle between M1 macrophages and hypertrophic adipocytes to cause immunometabolic imbalance [218]. Gan et al. [219] showed for the first time that miR-130b-3p was enriched in dysfunctional adipocyte-derived sEVs and inhibited multiple anti-apoptotic/cardioprotective molecules in cardiomyocytes, suggesting a novel mechanism for exacerbating MI/R injury in the diabetic heart. In addition, Human adipose tissue-derived mesenchymal stem cell-derived EVs (ASCs-EVs) could be taken up by endothelial cells, transfer miR-125a to endothelial cells and promoted angiogenesis by inhibiting DLL4 [220], and platelet-derived growth factor (PDGF) also can stimulate the secretion of ASCs-EVs and alter its protein composition to enhance the pro-angiogenic potential [221]. Perivascular adipose tissue (PVAT) is the adipose tissue that surrounds the aorta and releases various factors that regulate vascular function in a paracrine or autocrine manner [222]. PVAT causes proinflammation, the proliferation of VSMCs and vasoconstriction through the release of proinflammatory adipokines or cytokines that are associated with cardiovascular risk, can induce the development of AS, and are thought to be associated with VC [223]. Li et al. [224] found that PVAT-derived EVs and their abundantly loaded miR-221-3p could be taken up by neighboring VSMCs, leading to arterial remodeling and promoting the transformation of VSMCs from a contractile to a synthetic phenotype. Previously, miR-221 has been shown to be involved in the regulation of VC [225].
So far, the effect of adipose tissue-derived EVs on VC has not been fully explored, but the above studies show that AT-derived EVs may be involved in the regulation of VC as a new mediator, and we need to invest more research in the future.
Crosstalk among vascular wall cells
VSMCs-VSMCs crosstalk
It is well known that VSMCs are the main cells in the tunica media of vascular wall and play a key role in the occurrence and development of VC [226]. It was found that under calcification-promoting conditions, VSMCs can be induced to produce calcification-promoting EVs, which act on adjacent normal VSMCs leading to a conversion to an osteogenic phenotype that promotes VC [17, 227], suggesting the existence of a closed-loop regulation.
Regulation of the release of EVs
Phingomyelin phosphodiesterase 3 (SMPD3, also known as Neutral sphingomyelinase 2,nSM2) has been shown to regulate the release of exosomes [228]. Increased SMPD3 expression under high extracellular calcium promotes the secretion of calcifying exosomes [17], and inhibition of SMPD3 expression by Barley-ß-glucans prevents VSMC calcification [229]. Lysosomal sphingolipids such as ceramide (CER) may promote mTOR activation on lysosomes, and lysosomal CER-mTOR signaling may prevent lysosomal fusion with MVBs within VSMCs and promote exosome secretion to induce arterial medial calcification (AMC) [230]. Increased CER production via the SMPD3 pathway may induce EVs biosynthesis to promote AMC [17]. And increased expression of the SMPD1 gene in the lysosomes of VSMCs [231]; deletion of the specific lysosomal ASAH1 gene leading to reduced TRPML1 channel-mediated Ca2+ release [232]; and absence of mucolipin-1 product of the Mcoln1 gene leading to abnormal localization of lysosomes [233], all of which can lead to reduced lysosomal-MVB interactions and increased MVBs fusion with the plasma membrane and EVs release, thereby promoting AMC. This may be a new molecular mechanism involved in the development of AMC. In a recent study, nano-sized hydroxyapatite (nHAp) was first demonstrated to promote the release of calcifying-exosomes through autophagy-lysosomal damage and accelerate VC [234]. Targeting this novel autophagy-lysosome-exosome pathway may help regulate the development of VC.
In addition, Kapustin et al. [235] found that circulating vitamin K–dependent coagulation proteins, PT in particular, a novel inhibitor of circulating vascular calcification and low circulating levels of PT in calcified patients, can bind to the surface of VSMC exosomes via PS and can also be loaded into exosomes by cellular internalization and recycling via the late endosome/multivesicular body (LE/MVB) compartment. The gradual loading of PT and PT activation products into exosomes inhibits procoagulant activity and nucleation site formation on the exosomes surface through interaction with Gla/PS, thus preventing exosome-mediated VC [235]. Thus, anticoagulation therapy with warfarin may enhance VC through impaired carboxylation of vitamin K–dependent coagulation factors delivered to VSMC-derived exosomes, which play a dual role in calcification and coagulation [235].
In this study, a novel mechanism for influencing intracellular calcium levels through calcium uptake by EVs as extracellular calcium was proposed: Extracellular Ca2+ entry into VSMCs is mediated by EVs through clathrin- and caveolin-mediated uptake, resulting in an increase in cytosolic Ca2+ [236]. Taken together, the above indicates that there are two mechanisms for calcium uptake through calcium channels: EVs-dependent and EVs-independent. Elevated cytosolic Ca2+ increases oxidative stress by promoting the expression of Nox5, and ROS can promote the release of EVs [236]. Contractile VSMCs are induced by PDGF and high Ca2+, and are dedifferentiated into synthetic VSMCs mediated by Nox5, which undergo a series of changes including apoptosis and osteogenic differentiation, secretion of more extracellular matrix (ECM) and EVs, all of which lead to increased calcification [236]. Studies have demonstrated for the first time that Nox5 is the molecular link between Ca2+, oxidative stress, and EVs release, and have confirmed Nox5 as a key regulator of VSMC phenotypic switching and calcification [236]. The risk of cardiovascular events is significantly increased in smokers, which is mainly caused by nicotine in cigarette components, which promotes VSMC calcification through α7 nAchR increasing intracellular Ca2+ and increasing Ca2+-dependent Nox5-induced ROS, thus increasing EVs release [164]. It was also found that the expression level of Nox5 in carotid artery of smokers was high and correlated with VC level [164]. In addition, pretreatment with vitamin K inhibited nicotine-mediated ROS production in VSMCs and decreased EVs release [164]. It was also found that the expression level of Nox5 in carotid artery of smokers was high and correlated with VC level. PCSK9 may contribute to the osteogenic phenotype of SMCs by mediating the release of more Ca2+-and ALP-rich EVs during VC associated with CKD [237]. High concentrations of Nε-Carboxymethyl-lysine (CML, a key active component of AGEs in serum) can significantly promote the release of SMCs-derived EVs and promote the recruitment of Sortilin to EVs, thereby exacerbating diabetic VC, while anti-sortilin treatment can significantly reduce VC caused by EVs [69]. Sortilin is a type I membrane glycoprotein encoded by SORT1 gene and belongs to vesicle sorting receptor. It participates in the loading and transportation of extracellular vesicles, which can transport the calcification-promoting protein TNAP into EVs [238], and can also promote the transportation of sortilin into EVs to form EVs with high mineralization capacity by forming homodimers containing intermolecular disulfide bonds [239], thus promoting VC. Sortilin plays an important role not only in the pathogenesis of cardiovascular diseases but also in metabolic diseases and cancers [240]. Furmanik et al. [241] demonstrated that endoplasmic reticulum (ER) stress mediated increased release of Grp 78-loaded EVs to promote VSMC calcification, but not through apoptosis; Warfarin can induce endoplasmic reticulum stress through PERK-ATF4 pathway, increase the release of EVs, and promote calcification [241].
Regulation of bioactive molecules in EVs
In addition, EVs can also transfer functional bioactive molecules (such as miRNA, proteins, etc.) to regulate the development of VC. Regulation of VC by changing protein content in EVs, For example, the GFOGER peptide (a specific six-amino-acid repeat in COL1 sequence) reduces VC by modulating the content of proteins associated with the osteogenic phenotype in VSMC-derived EVs (e.g. ANK-binding kinase 1 and casein kinase II) and inhibiting osteogenic transdifferentiation of VSMCs [242]. Previous studies have shown that GFOGER peptide partially inhibits VC by preventing EVs-COL1 interaction [243].
Moreover, more attention has been paid to miRNAs, which can be regulated by changing the amount of miRNAs in EVs. For example, the miRNA profile in EVs derived from calcified VSMCs was markedly altered, with 987 and 92 miRNAs significantly upregulated and downregulated [244]. It has been reported that Curcumin (CUR, a natural polyphenol compound with lipid-lowering, anti-inflammatory and antioxidant effects on cardiovascular system) may inhibit VC by increasing the content of miR-92b-3p loaded in VSMC-derived EVs and reducing the expression of its target KLF4, thus affecting the expression of RUNX2 [245]. In a recent study, Melatonin attenuated osteogenic differentiation and senescence of VSMC or calcified vascular smooth muscle (CVSMCs) by upregulating miR-204/miR-211 in EVs secreted by VSMCs or CVSMCs [71].
In conclusion, microenvironment changes (high calcium, ROS, endoplasmic reticulum stress, lysosome activity and autophagy-lysosomal damage, etc.) can promote VC by increasing the release of VSMC-EVs, and carry more abundant pro-calcification substances. They can mediate mineral deposition to initiate calcification, as well as propagate calcification through the transfer of bioactive molecules (such as miRNAs and proteins) or regulation of signal transduction (Fig. 3). Altering the nature and biogenesis of VSMC-derived EVs may be an effective strategy to limit vascular calcification. These studies provide strong evidence that EVs regulate VC through information exchange between VSMCs-VSMCs. And in the future treatment and prevention of VC has a vital role.

Potential mechanism of VSMCs-derived EVs regulating osteogenic differentiation and calcification of VSMCs. VSMCs spontaneously releases EVs under physiological conditions, which are loaded with the calcification inhibitors MGP and Fetuin-A. Pathological stimuli can alter the biological characteristics of EVs, promoting the formation and release of EVs with calcification potential. Calcified EVs contain less MGP and Fetuin-A and more MMP-2, annexins, TNAP, Ca2+ and Pi. When calcified EVs are released into the extracellular matrix (ECM) and interact with it to promote the release and aggregation of hydroxyapatite crystals and form microcalcifications. TNAP may shift the Pi/PPi balance toward calcification by hydrolyzing the calcification inhibitor inorganic pyrophosphate (PPi) and generating inorganic phosphate (Pi) required for mineralization, which is critical for EVs mediated hydroxyapatite formation. Sortlin regulates TNAP entry into EVs, increasing the calcification potential of EVs. EVs contain another phosphatase, PHOSPHO1, which acts as an alternative supplier of Pi, and plays a role in the initiation of matrix mineralization. Calcified EVs formed under pathological stimuli regulate VC through the following mechanisms:(1) promoting extracellular mineral deposition;(2) promoting osteogenic phenotype transformation of VSMC;(3) transferring microRNAs between cells; and (4) regulating signaling pathways
ECs-VSMCs crosstalk
Vascular endothelial cells (ECs) are also the main cells involved in vascular system diseases [246] and are located in the intima, which means that they act as "frontrunners" that can be directly stimulated by pathological triggers in the circulating blood (such as hyperglycemia, hyperphosphatemia, uremic toxins) [247,248,249]. It has been proved that VC often occurs in the tunica media in patients with diabetic mellitus or CKD [250]. However, how does high glucose and high phosphorus in circulation transfer from the intima to the media and thus affect calcification/aging of VSMC that are not contact with blood directly? Therefore, most of the studies on arterial media calcification focus on ECs in recent years. ECs can synthesize and secrete several Gas signals (e.g., H2S, NO, and CO) and small molecule peptides (e.g., CNP, ADM, PTH, and ET), and so on, which directly affect VSMCs to promote VC formation and development [251]. Increasing evidence suggests that ECs are involved in VC through the following mechanisms: endothelial dysfunction [252, 253], endothelial-mesenchymal transition (EndMT) [23], autocrine/paracrine pathways [254], angiogenesis [255], and mechanotransduction [256]. Importantly, a series of studies have proved that ECs can also communicate with VSMCs through EVs. For example, CXCR6 promote calcification by downregulating miR-29b in aortic endothelial cell (AEC)-derived exosomes [257]. ECs-derived EVs also can regulate signal transduction pathways, such as triggering a pro-inflammatory, hypertrophic and senescent phenotypes in VSMCs through a mechanism involving high-mobility group box proteins (HMGB) [170].
Inflammatory stimuli and CKD and/or senescence-induced endothelial damage lead to increased BMP2 expression in HUVEC and increase the release of their microvesicles loaded with abundant Ca2+ and BMP2 to promote osteogenic transformation and calcification of VSMCs [258]. The study further suggests that the number of circulating microvesicles in plasma increases with age and that these microvesicles may be produced by aging ECs to induce VC [162]. Several studies have demonstrated that the pro-calcific potential of aged HUVEC-derived microvesicles is related to the substances carried: carries more calcium; can carry extra calcium-binding proteins (such as annexin A2 and A6); more bone related protein (BMP2) [18, 109, 162]. These studies demonstrate that aging is inextricably linked to ECs-EVs. Next, we have summarized how ECs, when stimulated by high glucose and high phosphorus in circulation, regulate VSMCs through EVs.
Crosstalk in CKD-related VC
Some studies have shown that ECs-EVs are closely related to CKD-VC. Guava FACS analysis of EV from dialysis patients at T0 showed that only endothelium-derived EVs were up-regulated among most circulating EVs derived from monocytes/macrophages, platelets and endothelial cells in CKD patients compared with healthy recipients, indicating that endothelial cells are the main participants in circulating EVs in CKD patients [259]. A recent study suggests that by transcriptomic analysis of circulating sEVs from CKD mice models as well as from CKD patients, possibly from endothelial cells, the calcification-protecting miRNAs that target VEGFA signaling in CKD-driven vascular calcification are lacking: miR-16-5p, miR-17-5p, miR-20a-5p, and miR-106b-5p [67]. And the sEVs biogenesis system inhibitor GW4869 [63,64,65], was used to ameliorate aortic VC in CKD model mice [67]. The areas under the ROC curves for miR-16-5p, miR-17-5p, miR-20a-5p and miR-106b-5p, which further predicted aortic vascular calcification in CKD patients, were 0.7630, 0.7704, 0.7407 and 0.7704, respectively, suggesting that the lower expression levels of these four miRNAs could predict abdominal aortic calcification [67]. Alique et al. [260] found in vitro that uremic toxin such as indoxyl sulfate (IS) induces stress, premature senescence in endothelial cells and increases the release of microvesicles to cause osteogenic transformation of VSMCs and an inflammatory response that promotes the development of VC. In addition, uremic toxins-treated senescent endothelial cell-derived EVs, and uremic rat plasma-derived EVs (hereinafter referred to as uraemic EV, EVUR) promote calcium-and phosphorus-induced osteogenic differentiation and calcification of VSMC [172]. The mechanism has been demonstrated that EVUR accelerates and increases pro-calcifying-mediated (CM) expression of runx2, osterix, and osteocalcin, and decreases gene expression of sm22α in VC; also increased protein expression of the phosphate transporter PIT-1 in VSMCs and induced phosphorylation of AKT and ERK [172]. Reducing the expression of PiT-1 and inhibiting AKT and ERK signaling pathways alone can block the calcification-promoting effect of EVUR [172].
In addition, EVUR significantly down-regulated miR-143 and miR-145, whereas transfection of the mimetics of miR-143/145 alone into EVUR blocked the pro-calcific effects of EVUR [172]. And the combined use of miR-143 and miR-145 mimics was similar to the combined use of miR-221 and miR-222 inhibitors, which could more significantly block the aggravating effects of EVUR on CM-induced calcification [172]. For Hp-EC-EVs (hyperphosphatemia-induced-endothelial extracellular vesicles) miRNA-seq analysis showed that miRNAs expression were changed: 12 down-regulated miRs (has-miR193b-5p, hsa-miR-941, hsa-miR-99b-5p, hsa-miR-365a-5p, hsa-miR-30c-2-3p, hsa-miR-30a-3p, hsa-miR-30a-5p, hsa-miR-486-5p, hsa-miR-7706, hsa-miR-10a-5p, hsa-miR-10b-5p and including hsa-miR-143-3p as confirmed above) and an up-regulated hsa-miR-3182, suggesting that these miRs may be potential markers for CDK-VC; KEGG analysis of Top-20 signaling pathways showed that calcium signaling pathway, cAMP signaling pathway, and ABC transporters may be closely related to VC [261]. A recent study also shown that miR-670-3p released from ECs-EVs plays an important role in regulating VC and may be a potential target for arterial calcification in patients with CKD [68]. These studies show that ECs and their derived EVs play a key role in CKD and aging, and help to elucidate the mechanism of CKD-VC from the perspective of ECs-EVs.
Crosstalk in diabetes-related VC
AGEs activate ECs by binding to its receptor RAGE, and then induce the production of reactive oxygen species (ROS), which can damage ECs and ultimately lead to apoptosis [262, 263]. The interaction between ECs and VSMCs mediated by EVs have also received considerable attention in the HG environment. One study showed that EVs derived from high glucose-induced HUVEC(HG-HUVEC-EVs) delivered Notch3(a key regulator of VSMC proliferation and phenotypic transformation) to VSMC through mTOR signaling pathway, which promoted VSMC calcification/aging [173]. In another study, HG-HUVEC-EVs contain functional VCAN, which may promote VSMC calcification/senescence by regulating mitochondrial function [264]. In addition, Li et al. [264] revealed that HG-HUVEC-EVs also increased the levels of MDA(which is produced by lipid peroxidation of polyunsaturated fatty acids and MDA levels increases under oxidative stress) and LDH(intracellular enzyme existing in cytoplasm, the leakage of which from cells is considered to be an important indicator of cell damage) and significantly reduced the activity of SOD (an important antioxidant enzyme involved in antioxidant defense) in VSMCs. Surprisingly, unlike previous studies showing that AGEs are harmful to vascular cells, the latest study by Guo et al. [70] shows that AGEs can inhibit diabetic media calcification by stimulating HUVEC to secrete miR-126-5p-rich sEVs, targeting BMPR1 and thus blocking smad1/5/9 signaling pathway. Although EVs and their cargo may be potential therapeutic targets for diabetic calcification, this contradiction means that further in vivo studies and clinical applications are needed to assess the therapeutic value of EVs in patients with VC.
ECs-other cells crosstalk
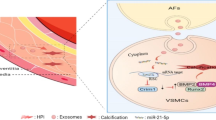
Some studies have shown that the EVs-mediated crosstalk between other cells and ECs/other cells also mediates cardiovascular calcification. For example, valvular interstitial cells (VICs) are the most abundant cells in the aortic valve and have the ability to convert to an osteogenic phenotype and calcify [265, 266]. Although the mechanism of this process has not been fully elucidated, VICs can inhibit VEC (valvular endothelial cell) EndMT and osteogenesis, suggesting that valve homeostasis is maintained by appropriate VIC-VEC interactions [267]. In calcific aortic disease, VICs secrete pro-calcific EVs to interact with ECs and thereby remodel the ECM [268]. Annexins II, V, and VI are up-regulated in pro-calcific VICs-derived EVs, and these annexins mediate calcium inward flow into the EVs [109]. Elevated circulating calcium and phosphorus levels in ESRD patients may increase the levels of annexin VI and calcium loaded in VICs-EVs to promote aortic valve calcification [269]. Telocytes (TCs) are novel mesenchymal cells that are widely distributed in the extracellular matrix of any tissue and can participate in cellular communication through direct homo- and heterocellular junctions or release EVs [270, 271]. Interestingly, Yang et al. [272] showed for the first time that TCs-derived EVs co-cultured with calcified VICs could improve aortic valve calcification through the miR-30b/Runx2/Wnt/β-catenin axis. These studies suggest that calcified EVs secreted by VICs may be mediators of heart valve calcification, and may also be mediators of VICs-VECs interaction mediating valve calcification. Aortic valve calcification remains an unresolved clinical problem and requires more attention to the mechanistic role of VICs-EVs in the development of aortic valve calcification.
In conclusion, EVs perform critical functions in intercellular communication by transferring their bioactive substances to receptor cells or activating signaling pathways in target cells. New perspective of ECs-derived EVs reveals the relationship between endothelial cells and vascular calcification, and can better understand the complex mechanism of how some substances induce VC development through ECs-VSMCs crosstalk without direct contact with VSMCs.
Macrophages (Immune Cells)-VSMCs crosstalk
Macrophages are a type of innate immune cell that reside in the intima and adventitia of blood vessels [273]. Their primary function is to clear cellular debris, lipids, and other foreign substances from within the blood vessels, thereby maintaining vascular health [274]. And remarkable plasticity and functional heterogeneity are important features of macrophages [275]. Macrophages are involved in almost all the stages of vascular calcification, and different subtypes of macrophages play different roles. M1 macrophages (pro-inflammatory) produce pro-inflammatory factors such as IL-1β, IL-6 and TNF-α to promote osteogenic transdifferentiation of VSMCs [276,277,278]. Similarly, products of oxidative stress such as ROS can induce a switch in VSMC phenotype toward pro-calcification [279]. M1 macrophage can directly release oncostatin M (OSM) to promote the differentiation of VSMCs into osteoblastic phenotypes through the JAK3-STAT3 pathway [280]. In contrast to M1 macrophages, M2 macrophages are anti-inflammatory and are thought to be protective against VC. M2 macrophages express low or no pro-inflammatory mediators (e.g., TNFα) and highly express anti-inflammatory mediators (e.g., IL-10 and TGF-β) [281]. In addition, M2 macrophages can increase the concentration of the calcification inhibitor pyrophosphate/ATP to inhibit hydroxyapatite crystal nucleation [282]. In plaque progression, M1 macrophages may promote microcalcification formation by consistently expressing pro-inflammatory cytokines such as TNF-α, IL-6 and MMPs, whereas these macrophages transform into M2 macrophages in plaque regression and secrete of anti-inflammatory cytokines such as IL-10 and TGF-β may promote macrocalcification [283, 284]. Oncostatin M and statins may facilitate plaque regression by enhancing M2 polarization of macrophages, leading to plaque macrocalcification and stabilization [283]. Collectively, M1 and M2 have nearly opposite effects in regulating the development of VC, suggesting that using the plasticity of macrophages to impel a polar shift in different subsets could offer a new breakthrough for calcification regression. Macrophages can promote VC through various mechanisms such as release of reactive oxygen species, pro-inflammatory cytokines, polarization drift of macrophages, phenotypic transformation of macrophages and matrix vesicles. The potential mechanisms of macrophage involvement in calcification are evaluated in these two reviews [8, 285].
Molecular imaging shows coexistence of macrophages with microcalcifications [286]. In inflammation-mediated calcification, inflammation precedes microcalcification and is mediated by the release of EVs from macrophages, and macrophage-derived EVs may be a bridge to mineral deposition in these tissues. Furthermore, it was found that macrophage-derived EVs also play a role in crosstalk. Therefore, we next summarize the role of macrophage-derived EVs in mediating mineral deposition and acting as mediators of communication.
Macrophage-derived EVs mediate mineral deposition to form microcalcifications
S100A9-positive macrophages were found in early atherosclerotic plaques and it was confirmed that S100A9 is associated with vascular inflammation and calcified EVs [287]. To further characterize macrophage-derived EVs as having a potent role in calcification, it is proposed that pro-inflammatory S100A9, a calcium-binding protein identified as a biomarker of acute cardiovascular events [288], has a key role in macrophage EVs-mediated microcalcification. S100A9 was found to interact with annexin V to form the PS-Annexin V-S100A9 membrane complex as a nucleation center for hydroxyapatite [288]. Calcium/Phosphorus stimulation promoted macrophages to release MVs enriched in calcium, ALP, S100A9 and annexin V, which directly promoted the formation of microcalcifications [289].
Macrophage-derived EVs-mediated microcalcification has been shown to be associated with the development of diabetic vascular calcification [290]. Macrophage S100A9 also plays a key regulatory role in diabetic vascular calcification. Increased secretion of S100A9 and increased expression of receptor for advanced glycosylation end-products (RAGE) protein in human primary macrophages under high glucose (25 mmol/L) conditions; the S100A9-RAGE signaling pathway promotes macrophage expression of proinflammatory and osteogenic factors and induces the release of calcified EVs from proinflammatory macrophages; and revealed a novel mechanism by which the S100A9-RAGE axis may regulate proinflammatory and pro-osteogenic macrophage activation through crosstalk between NRF-2 (nuclear factor 2 erythroid related factor 2) and NF-κB (nuclear factor-κB) pathways [290]. These findings suggest that macrophage S100A9 is a key regulator of diabetic-mediated vascular calcification. Modulation of S100A9 and Annexin V may inhibit the formation of hydroxyapatite nucleation and unstable microcalcifications in macrophage-derived MVs [289].
EVs transfer bioactive molecules or trigger signaling pathways
Pro-inflammatory macrophage-derived EVs deliver miR-241, which is associated with inflammatory response, to VIC and downregulate TWIST1 expression to promote aortic valve calcification (AVC) [291]. Recent studies have confirmed that miR-32 is an important molecule for macrophage-derived EVs to inhibit VSMC autophagy to promote T2D VC [292]. In addition, upregulation of galectin-3 (a marker of cardiovascular disease) in macrophages promotes migration of VSMC-derived EVs to the intima and induces the formation of diabetic vascular intimal calcification [293]. Yaker et al. [294] recently showed that lipopolysaccharide-treated macrophage-derived EVs induced inflammation and oxidative stress in VSMCs, thereby promoting the development of VC.
To date, studies of immune cell-derived EVs in inflammation-mediated cardiovascular calcification have focused on macrophages in the innate immune response. However, other innate immune cells (such as Dendritic cells, Mast cells, Neutrophils and Natural Killer cells) and adaptive immune-mediated inflammation related vascular calcification studies will also be an exciting area for future and provide a deeper understanding of inflammation-mediated calcification. Although their role in cardiovascular calcification remains poorly understood, and crosstalk between macrophages and other immune cells has rarely been described [295]. Therefore, an in-depth study of the molecular mechanisms of immune cell-derived EVs in response to VC is warranted.
In summary, the relationship between EVs-mediated intercellular communication was briefly summarized in Fig. 4. And the summary of bioactive cargo in EVs derived from different cells and their regulatory pathway in Table 1.

Extracellular vesicle-mediated intercellular crosstalk in vascular calcification. Different cell-derived EVs mediate communication between cells, such as by delivering miRNAs and osteogenic-associated proteins, or by regulating signaling pathways and so on, thereby regulating VC
Circulating EVs as potential predictors in cardiovascular events
Circulating EVs include EVs from platelets, endothelial cells, neutrophils, monocytes, lymphocytes, erythrocytes, and their precursor cells. With the occurrence of CVD, circulating EVs of platelets and endothelial cells, among other sources, are significantly higher than in normal persons [297, 298]. Oggero et al. [299] during 3.5 years of follow-up (median) showed that in a population at risk for major adverse cardiovascular event or death (MACE) and receiving atorvastatin therapy (compared with controls and placebo, respectively), the biology of plasma-derived EVs was significantly different in size, level, and surface characteristics. MACE patients had preceding higher levels of CD14+ and CD14+/CD41+EVs, and higher CD14+ extracellular vesicles were associated with a 3.7-fold increased risk of MACE on matched analysis [299]. Patients treated with atorvastatin had both reduced size of extracellular vesicles and the proportion of CD146+ extracellular vesicles [299]. Kanhai et al. [300] conducted a large prospective cohort study including 1060 patients with CVD and found that elevated levels of Cystatin C, Serpin F2 and CD14 in plasma-derived EVs were associated with an elevated risk of future vascular events and death in patients with clinically manifest vascular disease. And CD144+ in endothelial cells-derived EVs as an independent predictor of cardiovascular event occurrence may contribute to risk stratification of coronary heart disease in populations with coronary risk factors [301]. In addition, several studies have found that circulating miRNAs can also predict the risk of cardiovascular events. Patients with low expression of miR-126 and miR-199 have a high incidence of cardiovascular events, and ECs-derived EVs are the major source of circulating miR-126 and miR-199, while platelet-derived EVs are another pathway of miR-126 origin [302, 303]. Platelet-derived EVs are potent regulators of immune cells and have shown strong immunomodulatory capacity on VSMCs, leading to phenotypic changes, migration and proliferation, which may accelerate the progression of vascular diseases [304, 305]. It has been shown that platelet-derived EVs promote VSMC dedifferentiation may via Src/Lamtor1/mTORC1 signaling pathway [296]. A recent study, Koide et al. [67] found that low expression levels of miR-16-5p, miR-17-5p, miR-20a-5p, and miR-106b-5p in circulating sEVs, possibly derived from endothelial cells, in patients with CKD predicted calcification of abdominal aorta. Compared with histopathology, blood and body fluid-based EVs tests are highly acceptable to patients and facilitate monitoring and reflecting the overall condition of the disease. Compared with traditional markers, EVs have the advantages of targeting, packaging greater information and easy storage, etc. Therefore, EVs-related biomolecules are circulating markers with considerable clinical diagnostic value.
Treatment strategy of vascular calcification based on "crosstalk"
EVs also have a "disease-curing" effect. EVs can improve VC by mechanisms such as acting as direct targets or transferring bioactive substances between cells or modulating signaling pathways.
Chen et al. [306] showed that the calcium channel blocker verapamil inhibited the production and activity of calcifying EVs and also reduced the calcifying capacity of COL 1 to prevent ECM mineralization. BGP-15 is a new anti-diabetic drug candidate that increased MGP content and decreased Annexin A2 content in EVs and prevented calcium deposition in the ECM [307]. Alendronate (ALE), a bone resorption inhibitor, down-regulated the release of aged bone-EVs in VD3-treated aged mice to reduce the ovariectomy-induced VC [10]. Bisphosphonates (BiPs) are commonly used to treat bone loss and are also a strategy intended to prevent pathological calcification. Retrospective clinical data examining the effects of BiP therapy on cardiovascular calcification have revealed conflicting findings and intriguing paradoxes [308,309,310]. Ruiz et al. [311] established an AS model in APOE-/-mice and a 3D collagen hydrogel incubated with calcifying EVs to mimic an in vitro fibrous cap calcification model to determine whether BiP treatment altered microcalcification formation. In both models, it was shown that one of the mechanisms by which BiP modulates cardiovascular mineralization is to alter the kinetics of EVs-mediated microcalcification formation in a time-dependent manner, depending on whether BiP treatment was initiated before or after the expected onset of microcalcification formation [311]. PT and PT activated products can inhibit exosome-mediated VC by preventing nucleation site formation on the exosomal surface [235]. In addition to treating calcified EVs as a direct target for drugs [55], EVs may also be a promising strategy to treat VC by transferring bioactive substances or modulating signaling pathway. For example, BMSC-EVs are enriched in anti-calcification miRNAs such as miR-15a/15b/16 [188], miR-381-3p [189] and miR-146a [193], which can be transferred to VSMCs to improve VC. BMSC-EVs also improved VC by modulating the NONHSAT 084969.2/NF-κB axis [190] and the SIRT6-HMGB1 deacetylation pathway [191]. Curcumin may inhibit VC by increasing miR-92b-3p loading in VSMC-derived EVs and decreasing expression of its target KLF4 [245]. Melatonin attenuates osteogenic differentiation and senescence of VSMCs or calcified VSMCs by upregulating miR-204/miR-211 in EVs secreted by VSMCs [71]. AGEs inhibit diabetic medial calcification by stimulating HUVEC to secrete miR-126-5p-rich sEVs [70]. TC-EVs ameliorates aortic valve calcification via the miR-30b/Runx2/Wnt/β-catenin axis [272]. Furthermore, GFOGER peptide inhibits osteogenic transformation of VSMCs by modulating the content of proteins associated with osteogenic phenotype in VSMC-EVs, and also partially inhibits VC by preventing EVs-COL 1 interaction [242, 243].
There are other potential therapies for VC associated with EVs. The above sortilin protein can transport calcifying protein to EVs, and can also form homodimers containing intermolecular disulfide bonds to promote sortilin transport to EVs, thus forming EVs with high mineralization capacity. Anti-sortilin treatment significantly reduced VC due to EVs [69, 238, 239]. Modification of electrospun poly (ε-caprolactone) (PCL) vascular grafts with heparin to enhance their antithrombotic properties, whereas heparinization can caused severe graft calcification [312]. Human placenta-derived MSCs (hP-MSCs) derived sEVs were loaded onto heparin-functionalized vascular grafts (PCL-Hep) to form PCL-Hep/sEVs [312]. HP-MSC-sEVs significantly inhibited calcification and improved patency of vascular grafts by immunoregulation in hyperlipidemia rats, and more surprisingly, hP-MSCs-derived EVs have potent immunomodulatory effects by inducing a switch from a pro-inflammatory and AS-causing phenotype to an anti-inflammatory and anti-osteogenic phenotype in macrophages [312]. The preparation of immunomodulatory vascular grafts improves vascular performance and function by modulating EVs, which provides a therapeutic measure.
Elevated levels of fibroblast growth factor 23 (FGF23) and phosphate have been recognised as cardiotoxins causing left ventricular hypertrophy and cardiac fibrosis, among others, and their elevated levels are strongly associated with cardiovascular disease in CDK patients [313,314,315]. FGF23 can promote MSC of vascular progenitor cells towards osteoblastic differentiation and calcification, leading to excessive deposition of calcified ECM [316]. By testing uremic plasma from 19 end-stage renal disease dialysis patients, it was found that using the novel dialysis technology HCO membrane compared to conventional high-flux haemodialysis (HFL), HCO membrane can effectively remove pro-inflammatory factors including FGF23 to alter plasma composition and thereby protect vascular progenitor cells from calcification [316]. Another study showed that in vitro, conversion of hemodialysis patients from bicarbonate hemodialysis (BHD) to mixed online hemodiafiltration (mOL-HDF) treatment significantly reduced the expression of miR-223 in plasma-derived EVs, which was associated with osteogenic differentiation of VSMCs [259]. This technique may parallel the altered extracellular vesicle production in the anti-calcification mechanism, but further research is needed to determine whether the novel dialysis technique can actually reduce cardiovascular loss and contribute to angiogenesis and improve the prognosis of dialysis patients [316]. We summarize the potential benefits of EVs as therapeutic agents for calcification in Table 2.
The above demonstrates that EVs are an attractive cell-free therapeutic product that shows positive effects on VC. It also illustrates the excellent clinical potential of EVs, particularly in terms of different cell sources and their application in next-generation diagnostic and therapeutic platforms. EVs not only play a role in the treatment of diseases by themselves, but also show a broad application prospect as drug carriers based on their advantages. EVs have the following objective advantages: Firstly, safety: they are naturally occurring secretory membrane vesicles with lower toxicity and lower immunogenicity, which can avoid immune rejection and greatly improve safety [317, 318]; Secondly, EVs can cross biological barriers (e.g. blood–brain barrier) [319]; Thirdly, stability: EVs can protect their contents from degradation and thus prevent degradation and failure of the active ingredient [40]; Finally, controllable: altering the cellular microenvironment can regulate the function of EVs [40]. The drug loading strategies for loading drugs into EVs can be divided into 1. exogenous loading systems is to load drugs directly into EVs: physical methods (such as adsorption, electroporation, liposome fusion, etc.), chemical methods (such as chemical coupling, etc.); 2. endogenous loading systems is to load drugs into EVs-secreting cells: cell co-culture, direct modification of parental cells, genetic engineering, etc. [320]. By these methods drugs are directly transferred into EVs or genes encoding proteins of interest are transferred into cells secreting EVs for better treatment of calcification. And the in vitro construction of engineered vesicles with specific expressed active substances to mimic the effects of EVs may also be future therapeutic tools to improve VC.
In addition, we list crosstalk-based preclinical studies on VC in Table 3, and these reflect the fact that the development of VC has been shown to involve multi-cells and multi-organs co-regulation in animal models of VC and that the novelty of EVs will drive the research of VC from single cells to multi-cells, multi-organs and even multi-systems. We list the current state of clinical trials of drugs and therapeutic and preventive approaches (e.g., IVL, parathyroidectomy, dialysis, etc.) related to VC in Table 4. Few clinical trials related to VC of EVs as therapeutic delivery systems have been carried out, which also reflects that EVs have great room for application in VC. Due to the use of allogeneic EVs, translating basic therapies into clinical practice remains challenging [321]. There is growing evidence that EVs do play a role in disease progression, and we still require strong research efforts to make clinical practice possible in the future.
Conclusions and perspectives
Current understanding of the pathogenesis of VC includes calcium and phosphorus imbalance, VSMCs transdifferentiation, extracellular matrix, EVs, bone homeostasis imbalance, inflammation, epigenetics (DNA methylation and demethylation, histone modification, non-coding RNA), autophagy, oxidative stress, mitochondrial dysfunction, iron death and pyroptosis, etc. The main therapeutic agents for VC are statins, sodium thiosulfate, bisphosphonates, SNF472, phosphate binders, calcimimetics, Denosumab, and TNAP inhibitors, etc. And the main therapeutic means are "coronary artery spinning" and "Intravascular lithotripsy (IVL)". To date, there is no definitive and feasible treatment for VC due to its irreversible process and complex pathology, and these measures mentioned above may have an interventional effect on VC, but their clinical application needs to be further explored. We have recognised that VC involves multicellular and multiorgan co-regulation and that EVs are a crucial mediator in crosstalk. In recent years, EVs have received widespread attention as their unique functions and extensive cargos have gradually been discovered. EVs are secreted in a controlled manner, so that changes in the microenvironment of the original cells cause changes in the production and contents of EVs. In VC, EVs have a dual role as both "disease-causing" and "disease-curing" agents. In this review, we address the mechanism of multi-organ/cellular regulation of VC from a new perspective of EVs, and a deeper understanding of the mechanism of VC can help in the discovery of optimal drug targets and drug development. In addition, EVs have potential therapeutic roles for calcification due to their properties as carrier systems, however, the use of EVs as a nanodrug delivery system for the treatment of calcification is still in the preclinical research phase or lacks extensive clinical studies. It is because researchers still face many issues and challenges, limitations in the preparation, engineering, and analytical techniques of EVs pose technical barriers to clinical translation. No extracellular vesicle products have received regulatory approval and remain challenging, and their associated risk–benefit ratios remain controversial, necessitating robust research efforts and preclinical and clinical approaches to assess their safety and efficacy.
In conclusion, there is an imbalance between calcification inhibitors and promoters in calcifying EVs: some inhibitors such as fetuin-A, MGP and some miRNAs were decreased, while some promoters such as annexin, Ca2+, Pi, TNAP and some miRNAs were increased. The mechanisms for regulating EVs under microenvironmental changes are as follows: (1) Regulation of Calcified EVs Release (2) Regulation of the interaction between EVs with ECM (3) EVs load more Ca2+ (4) Regulation of the content of bioactive molecules in EVs (such as, proteins and miRNAs, etc.) More importantly, EVs play a crucial role in cell-to-cell /organ-to-organ crosstalk, which may amplify and accelerate the progression of vascular calcification. We conclude that the EVs, in turn, can adjust VC through the following mechanisms: (1) Promote extracellular mineral deposition; (2) Promote osteogenic differentiation and senescence of VSMC; (3) Transfer bioactive molecules between cells; (4) Regulate autophagy, oxidative stress, endoplasmic reticulum stress mitochondrial dysfunction, inflammation and MAPK/mTOR/Wnt signaling pathways, etc. However, what we have discovered is only the tip of the iceberg in this complex network and is far from sufficient. Through EVs as mediators, the crosstalk between other cells (such as ECs)-VSMCs in VC is also reflected in the effect of VSMCs on ECs, which can also amplify the effect of ECs on VSMCs through feedback loops. Not only that, EVs of different cellular origin regulate VSMC calcification in turn calcifying VSMCs can also feedback to regulate other neighbouring cells. Understanding that EVs of different cellular origins regulate cardiovascular calcification can lead to a better understanding of the complex mechanisms underlying VC development. RNA cargo in EVs is more extensively studied as proteomics technologies lag behind sequencing technologies. Proteins are similarly functionally important and more extensive analysis will be necessary at a later date. In vitro organ culture, co-culture, and 3d culture of multiple cell types are the most promising tools to overcome the limitations of traditional in vitro models, and their widespread use in the coming years may help us to identify potential targets, ameliorate and prevent calcification in EVs-mediated VC.
Availability of data and materials
Not applicable.
References
Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005;4:4. https://doi.org/10.1186/1475-2840-4-4.
Liu W, Zhang Y, Yu CM, Ji QW, Cai M, Zhao YX, et al. Current understanding of coronary artery calcification. J Geriatr Cardiol. 2015;12(6):668–75.
Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25(4):267–74. https://doi.org/10.1016/j.tcm.2014.10.021.
Henaut L, Mentaverri R, Liabeuf S, Bargnoux AS, Delanaye P, Cavalier E, et al. Pathophysiological mechanisms of vascular calcification. Ann Biol Clin (Paris). 2015;73(3):271–87. https://doi.org/10.1684/abc.2015.1044.
Wang X, Chen X, Chen Z, Zhang M. Arterial calcification and its association with stroke: implication of risk, prognosis, treatment response, and prevention. Front Cell Neurosci. 2022;16: 845215. https://doi.org/10.3389/fncel.2022.845215.
Schinke T, Karsenty G. Vascular calcification–a passive process in need of inhibitors. Nephrol Dial Transplant. 2000;15(9):1272–4. https://doi.org/10.1093/ndt/15.9.1272.
Collett GD, Canfield AE. Angiogenesis and pericytes in the initiation of ectopic calcification. Circ Res. 2005;96(9):930–8. https://doi.org/10.1161/01.RES.0000163634.51301.0d.
Waring OJ, Skenteris NT, Biessen EAL, Donners M. Two-faced Janus: the dual role of macrophages in atherosclerotic calcification. Cardiovasc Res. 2022;118(13):2768–77. https://doi.org/10.1093/cvr/cvab301.
Shi J, Yang Y, Cheng A, Xu G, He F. Metabolism of vascular smooth muscle cells in vascular diseases. Am J Physiol Heart Circ Physiol. 2020;319(3):H613–31. https://doi.org/10.1152/ajpheart.00220.2020.
Wang Z-X, Luo Z-W, Li F-X-Z, Cao J, Rao S-S, Liu Y-W, et al. Aged bone matrix-derived extracellular vesicles as a messenger for calcification paradox. Nat Commun. 2022;13(1):1453.
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109(6):697–711. https://doi.org/10.1161/CIRCRESAHA.110.234914.
Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011;6(4):913–21. https://doi.org/10.2215/CJN.06040710.
Carrillo-Lopez N, Panizo S, Alonso-Montes C, Martinez-Arias L, Avello N, Sosa P, et al. High-serum phosphate and parathyroid hormone distinctly regulate bone loss and vascular calcification in experimental chronic kidney disease. Nephrol Dial Transplant. 2019;34(6):934–41. https://doi.org/10.1093/ndt/gfy287.
Lee SJ, Lee I-K, Jeon J-H. Vascular calcification—new insights into its mechanism. Int J Mol Sci. 2020;21(8):2685. https://doi.org/10.3390/ijms21082685.
Ouyang L, Yu C, Xie Z, Su X, Xu Z, Song P, et al. Indoleamine 2,3-dioxygenase 1 deletion-mediated kynurenine insufficiency in vascular smooth muscle cells exacerbates arterial calcification. Circulation. 2022;145(24):1784–98. https://doi.org/10.1161/circulationaha.121.057868.
Chen Y, Mao C, Gu R, Zhao R, Li W, Ma Z, et al. Nidogen-2 is a novel endogenous ligand of LGR4 to Inhibit vascular calcification. Circ Res. 2022;131(12):1037–54. https://doi.org/10.1161/circresaha.122.321614.
Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116(8):1312–23. https://doi.org/10.1161/CIRCRESAHA.116.305012.
Yang P, Troncone L, Augur ZM, Kim SSJ, McNeil ME, Yu PB. The role of bone morphogenetic protein signaling in vascular calcification. Bone. 2020;141: 115542. https://doi.org/10.1016/j.bone.2020.115542.
Sun WL, Wang N, Xu Y. Impact of miR-302b on calcium-phosphorus metabolism and vascular calcification of rats with chronic renal failure by regulating BMP-2/Runx2/osterix signaling pathway. Arch Med Res. 2018;49(3):164–71. https://doi.org/10.1016/j.arcmed.2018.08.002.
Rashdan NA, Sim AM, Cui L, Phadwal K, Roberts FL, Carter R, et al. Osteocalcin regulates arterial calcification via altered wnt signaling and glucose metabolism. J Bone Miner Res. 2020;35(2):357–67. https://doi.org/10.1002/jbmr.3888.
Rochette L, Meloux A, Rigal E, Zeller M, Malka G, Cottin Y, et al. The role of osteoprotegerin in vascular calcification and bone metabolism: the basis for developing new therapeutics. Calcif Tissue Int. 2019;105(3):239–51. https://doi.org/10.1007/s00223-019-00573-6.
Di Bartolo BA, Schoppet M, Mattar MZ, Rachner TD, Shanahan CM, Kavurma MM. Calcium and osteoprotegerin regulate IGF1R expression to inhibit vascular calcification. Cardiovasc Res. 2011;91(3):537–45. https://doi.org/10.1093/cvr/cvr084.
Sanchez-Duffhues G, Garcia de Vinuesa A, van de Pol V, Geerts ME, de Vries MR, Janson SG, et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. 2019;247(3):333–46. https://doi.org/10.1002/path.5193.
Ouyang L, Su X, Li W, Tang L, Zhang M, Zhu Y, et al. ALKBH1-demethylated DNA N6-methyladenine modification triggers vascular calcification via osteogenic reprogramming in chronic kidney disease. J Clin Invest. 2021;131(14):e146985.
Maleszewska M, Gjaltema RA, Krenning G, Harmsen MC. Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22alpha expression in endothelial cells in response to interleukin-1beta and transforming growth factor-beta2. Cell Signal. 2015;27(8):1589–96. https://doi.org/10.1016/j.cellsig.2015.04.008.
Gu J, Lu Y, Deng M, Qiu M, Tian Y, Ji Y, et al. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp Mol Med. 2019;51(7):1–14. https://doi.org/10.1038/s12276-019-0272-9.
Wang SS, Wang C, Chen H. MicroRNAs are critical in regulating smooth muscle cell mineralization and apoptosis during vascular calcification. J Cell Mol Med. 2020;24(23):13564–72. https://doi.org/10.1111/jcmm.16005.
Yu C, Li L, Xie F, Guo S, Liu F, Dong N, et al. LncRNA TUG1 sponges miR-204-5p to promote osteoblast differentiation through upregulating Runx2 in aortic valve calcification. Cardiovasc Res. 2018;114(1):168–79. https://doi.org/10.1093/cvr/cvx180.
Ma C, Gu R, Wang X, He S, Bai J, Zhang L, et al. circRNA CDR1as promotes pulmonary artery smooth muscle cell calcification by upregulating CAMK2D and CNN3 via sponging miR-7-5p. Mol Ther Nucleic Acids. 2020;22:530–41. https://doi.org/10.1016/j.omtn.2020.09.018.
Zhou X, Xu SN, Yuan ST, Lei X, Sun X, Xing L, et al. Multiple functions of autophagy in vascular calcification. Cell Biosci. 2021;11(1):159. https://doi.org/10.1186/s13578-021-00639-9.
Hu CT, Shao YD, Liu YZ, Xiao X, Cheng ZB, Qu SL, et al. Oxidative stress in vascular calcification. Clin Chim Acta. 2021;519:101–10. https://doi.org/10.1016/j.cca.2021.04.012.
Ye Y, Chen A, Li L, Liang Q, Wang S, Dong Q, et al. Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney Int. 2022;102(6):1259–75. https://doi.org/10.1016/j.kint.2022.07.034.
Pang Q, Wang P, Pan Y, Dong X, Zhou T, Song X, et al. Irisin protects against vascular calcification by activating autophagy and inhibiting NLRP3-mediated vascular smooth muscle cell pyroptosis in chronic kidney disease. Cell Death Dis. 2022;13(3):283. https://doi.org/10.1038/s41419-022-04735-7.
Hill JM, Kereiakes DJ, Shlofmitz RA, Klein AJ, Riley RF, Price MJ, et al. Intravascular lithotripsy for treatment of severely calcified coronary artery disease. J Am Coll Cardiol. 2020;76(22):2635–46. https://doi.org/10.1016/j.jacc.2020.09.603.
Bardeesi ASA, Gao J, Zhang K, Yu S, Wei M, Liu P, et al. A novel role of cellular interactions in vascular calcification. J Transl Med. 2017;15(1):95. https://doi.org/10.1186/s12967-017-1190-z.
Zhang YX, Tang RN, Wang LT, Liu BC. Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Prolif. 2021;54(3): e12980. https://doi.org/10.1111/cpr.12980.
Marar C, Starich B, Wirtz D. Extracellular vesicles in immunomodulation and tumor progression. Nat Immunol. 2021;22(5):560–70. https://doi.org/10.1038/s41590-021-00899-0.
Xie H, Wang Z, Zhang L, Lei Q, Zhao A, Wang H, et al. Extracellular vesicle-functionalized decalcified bone matrix scaffolds with enhanced pro-angiogenic and pro-bone regeneration activities. Sci Rep. 2017;7:45622. https://doi.org/10.1038/srep45622.
Isaac R, Reis FCG, Ying W, Olefsky JM. Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab. 2021;33(9):1744–62. https://doi.org/10.1016/j.cmet.2021.08.006.
Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. https://doi.org/10.3402/jev.v4.27066.
Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A. 2017;114(43):E9066–75. https://doi.org/10.1073/pnas.1704862114.
Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–89. https://doi.org/10.1146/annurev-cellbio-101512-122326.
Li S, Li Y, Chen B, Zhao J, Yu S, Tang Y, et al. exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018;46(D1):D106–12. https://doi.org/10.1093/nar/gkx891.
Record M, Silvente-Poirot S, Poirot M, Wakelam MJO. Extracellular vesicles: lipids as key components of their biogenesis and functions. J Lipid Res. 2018;59(8):1316–24. https://doi.org/10.1194/jlr.E086173.
Mathieu M, Martin-Jaular L, Lavieu G, Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9–17. https://doi.org/10.1038/s41556-018-0250-9.
Yaker L, Kamel S, Ausseil J, Boullier A. Effects of chronic kidney disease and uremic toxins on extracellular vesicle biology. Toxins (Basel). 2020;12(12):811. https://doi.org/10.3390/toxins12120811.
Yellon DM, Davidson SM. Exosomes: nanoparticles involved in cardioprotection? Circ Res. 2014;114(2):325–32. https://doi.org/10.1161/CIRCRESAHA.113.300636.
Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 1987;262(19):9412–20.
Andaloussi EL, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–57. https://doi.org/10.1038/nrd3978.
Pocsfalvi G, Stanly C, Vilasi A, Fiume I, Capasso G, Turiak L, et al. Mass spectrometry of extracellular vesicles. Mass Spectrom Rev. 2016;35(1):3–21. https://doi.org/10.1002/mas.21457.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. https://doi.org/10.1080/20013078.2018.1535750.
Carnino JM, Ni K, Jin Y. Post-translational modification regulates formation and cargo-loading of extracellular vesicles. Front Immunol. 2020;11:948. https://doi.org/10.3389/fimmu.2020.00948.
Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019;8(7):727. https://doi.org/10.3390/cells8070727.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977. https://doi.org/10.1126/science.aau6977.
Blaser MC, Aikawa E. Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Front Cardiovasc Med. 2018;5:187. https://doi.org/10.3389/fcvm.2018.00187.
Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. https://doi.org/10.1007/s00018-017-2595-9.
Xie S, Zhang Q, Jiang L. Current knowledge on exosome biogenesis, cargo-sorting mechanism and therapeutic implications. Membranes (Basel). 2022;12(5):498. https://doi.org/10.3390/membranes12050498.
Kalra H, Drummen G, Mathivanan S. Focus on extracellular vesicles: introducing the next small big thing. Int J Mol Sci. 2016;17(2):170. https://doi.org/10.3390/ijms17020170.
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244–7. https://doi.org/10.1126/science.1153124.
Crivelli SM, Giovagnoni C, Zhu Z, Tripathi P, Elsherbini A, Quadri Z, et al. Function of ceramide transfer protein for biogenesis and sphingolipid composition of extracellular vesicles. J Extracell Vesicles. 2022;11(6): e12233. https://doi.org/10.1002/jev2.12233.
Horbay R, Hamraghani A, Ermini L, Holcik S, Beug ST, Yeganeh B. Role of ceramides and lysosomes in extracellular vesicle biogenesis, cargo sorting and release. Int J Mol Sci. 2022;23(23):15317. https://doi.org/10.3390/ijms232315317.
Shamseddine AA, Airola MV, Hannun YA. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul. 2015;57:24–41. https://doi.org/10.1016/j.jbior.2014.10.002.
Airola MV, Shanbhogue P, Shamseddine AA, Guja KE, Senkal CE, Maini R, et al. Structure of human nSMase2 reveals an interdomain allosteric activation mechanism for ceramide generation. Proc Natl Acad Sci U S A. 2017;114(28):E5549–58. https://doi.org/10.1073/pnas.1705134114.
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–93. https://doi.org/10.1038/nn.4132.
Catalano M, O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles. 2020;9(1):1703244. https://doi.org/10.1080/20013078.2019.1703244.
Tallon C, Hollinger KR, Pal A, Bell BJ, Rais R, Tsukamoto T, et al. Nipping disease in the bud: nSMase2 inhibitors as therapeutics in extracellular vesicle-mediated diseases. Drug Discov Today. 2021;26(7):1656–68. https://doi.org/10.1016/j.drudis.2021.03.025.
Koide T, Mandai S, Kitaoka R, Matsuki H, Chiga M, Yamamoto K, et al. Circulating extracellular vesicle-propagated microRNA signature as a vascular calcification factor in chronic kidney disease. Circ Res. 2023;132(4):415–31. https://doi.org/10.1161/CIRCRESAHA.122.321939.
Lin X, Shan SK, Xu F, Zhong JY, Wu F, Duan JY, et al. The crosstalk between endothelial cells and vascular smooth muscle cells aggravates high phosphorus-induced arterial calcification. Cell Death Dis. 2022;13(7):650. https://doi.org/10.1038/s41419-022-05064-5.
Jing L, Li L, Ren X, Sun Z, Bao Z, Yuan G, et al. Role of sortilin and matrix vesicles in nepsilon-carboxymethyl-lysine-induced diabetic atherosclerotic calcification. Diabetes Metab Syndr Obes. 2020;13:4141–51. https://doi.org/10.2147/DMSO.S273029.
Guo B, Shan SK, Xu F, Lin X, Li FX, Wang Y, et al. Protective role of small extracellular vesicles derived from HUVECs treated with AGEs in diabetic vascular calcification. J Nanobiotechnology. 2022;20(1):334. https://doi.org/10.1186/s12951-022-01529-z.
Xu F, Zhong JY, Lin X, Shan SK, Guo B, Zheng MH, et al. Melatonin alleviates vascular calcification and ageing through exosomal miR-204/miR-211 cluster in a paracrine manner. J Pineal Res. 2020;68(3): e12631. https://doi.org/10.1111/jpi.12631.
Hao Y, Song H, Zhou Z, Chen X, Li H, Zhang Y, et al. Promotion or inhibition of extracellular vesicle release: Emerging therapeutic opportunities. J Control Release. 2021;340:136–48. https://doi.org/10.1016/j.jconrel.2021.10.019.
Fei X, Li Z, Yang D, Kong X, Lu X, Shen Y, et al. Neddylation of Coro1a determines the fate of multivesicular bodies and biogenesis of extracellular vesicles. J Extracell Vesicles. 2021;10(12): e12153. https://doi.org/10.1002/jev2.12153.
Tricarico C, Clancy J, D’Souza-Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases. 2017;8(4):220–32. https://doi.org/10.1080/21541248.2016.1215283.
Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda). 2005;20:22–7. https://doi.org/10.1152/physiol.00029.2004.
Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73(10):1907–20. https://doi.org/10.1016/j.jprot.2010.06.006.
Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, et al. Reassessment of exosome composition. Cell. 2019;177(2):428–45 e18. https://doi.org/10.1016/j.cell.2019.02.029.
Salunkhe S, Dheeraj, Basak M, Chitkara D, Mittal A. Surface functionalization of exosomes for target-specific delivery and in vivo imaging & tracking: Strategies and significance. J Control Release. 2020;326:599–614. https://doi.org/10.1016/j.jconrel.2020.07.042.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–28. https://doi.org/10.1038/nrm.2017.125.
Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3. https://doi.org/10.3402/jev.v3.24641.
Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–87. https://doi.org/10.1111/j.1600-0854.2010.01041.x.
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106(5):1604–11. https://doi.org/10.1182/blood-2004-03-1095.
Zeng ZL, Yuan Q, Zu X, Liu J. Insights into the role of mitochondria in vascular calcification. Front Cardiovasc Med. 2022;9: 879752. https://doi.org/10.3389/fcvm.2022.879752.
Mehta A, Vasquez N, Ayers CR, Patel J, Hooda A, Khera A, et al. Independent association of lipoprotein(a) and coronary artery calcification with atherosclerotic cardiovascular risk. J Am Coll Cardiol. 2022;79(8):757–68. https://doi.org/10.1016/j.jacc.2021.11.058.
Lanzer P, Hannan FM, Lanzer JD, Janzen J, Raggi P, Furniss D, et al. Medial arterial calcification: JACC State-of-the-art review. J Am Coll Cardiol. 2021;78(11):1145–65. https://doi.org/10.1016/j.jacc.2021.06.049.
Clemente A, Traghella I, Mazzone A, Sbrana S, Vassalle C. Vascular and valvular calcification biomarkers. Adv Clin Chem. 2020;95:73–103. https://doi.org/10.1016/bs.acc.2019.08.002.
Urena-Torres P, D’Marco L, Raggi P, Garcia-Moll X, Brandenburg V, Mazzaferro S, et al. Valvular heart disease and calcification in CKD: more common than appreciated. Nephrol Dial Transplant. 2020;35(12):2046–53. https://doi.org/10.1093/ndt/gfz133.
Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378(18):1704–14. https://doi.org/10.1056/NEJMra1505292.
Garcia-Lozano JA, Ocampo-Candiani J, Martinez-Cabriales SA, Garza-Rodriguez V. An update on calciphylaxis. Am J Clin Dermatol. 2018;19(4):599–608. https://doi.org/10.1007/s40257-018-0361-x.
Golub EE. Biomineralization and matrix vesicles in biology and pathology. Semin Immunopathol. 2011;33(5):409–17. https://doi.org/10.1007/s00281-010-0230-z.
Hasegawa T, Hongo H, Yamamoto T, Abe M, Yoshino H, Haraguchi-Kitakamae M, et al. Matrix vesicle-mediated mineralization and osteocytic regulation of bone mineralization. Int J Mol Sci. 2022;23(17):9941. https://doi.org/10.3390/ijms23179941.
Iwayama T, Bhongsatiern P, Takedachi M, Murakami S. Matrix vesicle-mediated mineralization and potential applications. J Dent Res. 2022;101(13):1554–62. https://doi.org/10.1177/00220345221103145.
Thouverey C, Malinowska A, Balcerzak M, Strzelecka-Kiliszek A, Buchet R, Dadlez M, et al. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics. 2011;74(7):1123–34. https://doi.org/10.1016/j.jprot.2011.04.005.
Ansari S, de Wildt BWM, Vis MAM, de Korte CE, Ito K, Hofmann S, et al. Matrix Vesicles: Role in Bone Mineralization and Potential Use as Therapeutics. Pharmaceuticals (Basel). 2021;14(4):289. https://doi.org/10.3390/ph14040289.
Cannata-Andia JB, Carrillo-Lopez N, Messina OD, Hamdy NAT, Panizo S, Ferrari SL, et al. Pathophysiology of Vascular calcification and bone loss: linked disorders of ageing? Nutrients. 2021;13(11):3835. https://doi.org/10.3390/nu13113835.
Cao G, Xuan X, Hu J, Zhang R, Jin H, Dong H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal. 2022;20(1):180. https://doi.org/10.1186/s12964-022-00993-2.
Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114(4):590–600. https://doi.org/10.1093/cvr/cvy010.
da Silva RA, da SFG, da CFCJ, Zambuzzi WF. Osteogenic gene markers are epigenetically reprogrammed during contractile-to-calcifying vascular smooth muscle cell phenotype transition. Cell Signal. 2020;66:109458. https://doi.org/10.1016/j.cellsig.2019.109458.
Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118(17):1748–57. https://doi.org/10.1161/circulationaha.108.783738.
Hasegawa T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem Cell Biol. 2018;149(4):289–304. https://doi.org/10.1007/s00418-018-1646-0.
Shapiro IM, Landis WJ, Risbud MV. Matrix vesicles: Are they anchored exosomes? Bone. 2015;79:29–36. https://doi.org/10.1016/j.bone.2015.05.013.
Li T, Yu H, Zhang D, Feng T, Miao M, Li J, et al. Matrix vesicles as a therapeutic target for vascular calcification. Front Cell Dev Biol. 2022;10: 825622. https://doi.org/10.3389/fcell.2022.825622.
Azoidis I, Cox SC, Davies OG. The role of extracellular vesicles in biomineralisation: current perspective and application in regenerative medicine. J Tissue Eng. 2018;9:2041731418810130. https://doi.org/10.1177/2041731418810130.
Zazzeroni L, Faggioli G, Pasquinelli G. Mechanisms of arterial calcification: the role of matrix vesicles. Eur J Vasc Endovasc Surg. 2018;55(3):425–32. https://doi.org/10.1016/j.ejvs.2017.12.009.
Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15(11):2857–67. https://doi.org/10.1097/01.ASN.0000141960.01035.28.
Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol. 2005;16(10):2920–30. https://doi.org/10.1681/ASN.2004100895.
Bjorklund G, Svanberg E, Dadar M, Card DJ, Chirumbolo S, Harrington DJ, et al. The Role of Matrix Gla Protein (MGP) in vascular calcification. Curr Med Chem. 2020;27(10):1647–60. https://doi.org/10.2174/0929867325666180716104159.
Poterucha TJ, Goldhaber SZ. Warfarin and Vascular Calcification. Am J Med. 2016;129(6):635 e1-4. https://doi.org/10.1016/j.amjmed.2015.11.032.
Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109(1):e1–12. https://doi.org/10.1161/CIRCRESAHA.110.238808.
Hosaka N, Mizobuchi M, Ogata H, Kumata C, Kondo F, Koiwa F, et al. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif Tissue Int. 2009;85(6):523–9. https://doi.org/10.1007/s00223-009-9297-8.
Chen NX, O’Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. 2008;23(11):1798–805. https://doi.org/10.1359/jbmr.080604.
Rogers MA, Buffolo F, Schlotter F, Atkins SK, Lee LH, Halu A, et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv. 2020;6(38):eabb1244. https://doi.org/10.1126/sciadv.abb1244.
Veschi EA, Bolean M, Strzelecka-Kiliszek A, Bandorowicz-Pikula J, Pikula S, Granjon T, et al. Localization of annexin a6 in matrix vesicles during physiological mineralization. Int J Mol Sci. 2020;21(4):1367. https://doi.org/10.3390/ijms21041367.
Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. 2020;40(5):1078–93. https://doi.org/10.1161/ATVBAHA.120.313131.
Jing L, Li L, Sun Z, Bao Z, Shao C, Yan J, et al. Role of matrix vesicles in bone-vascular cross-talk. J Cardiovasc Pharmacol. 2019;74(5):372–8. https://doi.org/10.1097/FJC.0000000000000720.
Bobryshev YV, Orekhov AN, Sobenin I, Chistiakov DA. Role of bone-type tissue-nonspecific alkaline phosphatase and PHOSPO1 in vascular calcification. Curr Pharm Des. 2014;20(37):5821–8. https://doi.org/10.2174/1381612820666140212193011.
Anderson HC, Sipe JB, Hessle L, Dhamyamraju R, Atti E, Camacho NP, et al. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol. 2004;164(3):841–7. https://doi.org/10.1016/s0002-9440(10)63172-0.
Yadav MC, Bottini M, Cory E, Bhattacharya K, Kuss P, Narisawa S, et al. Skeletal Mineralization deficits and impaired biogenesis and function of chondrocyte-derived matrix vesicles in Phospho1(-/-) and Phospho1/Pi t1 double-knockout mice. J Bone Miner Res. 2016;31(6):1275–86. https://doi.org/10.1002/jbmr.2790.
Roberts S, Narisawa S, Harmey D, Millan JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22(4):617–27. https://doi.org/10.1359/jbmr.070108.
Mebarek S, Abousalham A, Magne D, le Do D, Bandorowicz-Pikula J, Pikula S, et al. Phospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizations. Int J Mol Sci. 2013;14(3):5036–129. https://doi.org/10.3390/ijms14035036.
Cui L, Zhou Q, Zheng X, Sun B, Zhao S. Mitoquinone attenuates vascular calcification by suppressing oxidative stress and reducing apoptosis of vascular smooth muscle cells via the Keap1/Nrf2 pathway. Free Radic Biol Med. 2020;161:23–31. https://doi.org/10.1016/j.freeradbiomed.2020.09.028.
Boraldi F, Lofaro FD, Quaglino D. Apoptosis in the extraosseous calcification process. Cells. 2021;10(1):131. https://doi.org/10.3390/cells10010131.
Nguyen NT, Nguyen TT, Park KS. Oxidative stress related to plasmalemmal and mitochondrial phosphate transporters in vascular calcification. Antioxidants (Basel). 2022;11(3):494. https://doi.org/10.3390/antiox11030494.
Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34(4):715–23. https://doi.org/10.1161/ATVBAHA.113.302070.
Phadwal K, Feng D, Zhu D, MacRae VE. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol Ther. 2020;206: 107430. https://doi.org/10.1016/j.pharmthera.2019.107430.
Panizo S, Naves-Diaz M, Carrillo-Lopez N, Martinez-Arias L, Fernandez-Martin JL, Ruiz-Torres MP, et al. MicroRNAs 29b, 133b, and 211 regulate vascular smooth muscle calcification mediated by high phosphorus. J Am Soc Nephrol. 2016;27(3):824–34. https://doi.org/10.1681/ASN.2014050520.
Sutton NR, Malhotra R, St Hilaire C, Aikawa E, Blumenthal RS, Gackenbach G, et al. Molecular mechanisms of vascular health: insights from vascular aging and calcification. Arterioscler Thromb Vasc Biol. 2023;43(1):15–29. https://doi.org/10.1161/ATVBAHA.122.317332.
Drueke TB, Massy ZA. Vascular calcification in chronic kidney disease: contribution of ferroptosis? Kidney Int. 2022;102(6):1209–11. https://doi.org/10.1016/j.kint.2022.08.031.
Wang P-W, Pang Q, Zhou T, Song X-Y, Pan Y-J, Jia L-P, et al. Irisin alleviates vascular calcification by inhibiting VSMC osteoblastic transformation and mitochondria dysfunction via AMPK/Drp1 signaling pathway in chronic kidney disease. Atherosclerosis. 2022;346:36–45. https://doi.org/10.1016/j.atherosclerosis.2022.02.007.
Ma WQ, Sun XJ, Zhu Y, Liu NF. PDK4 promotes vascular calcification by interfering with autophagic activity and metabolic reprogramming. Cell Death Dis. 2020;11(11):991. https://doi.org/10.1038/s41419-020-03162-w.
Hou YC, Lu CL, Yuan TH, Liao MT, Chao CT, Lu KC. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int J Mol Sci. 2020;21(3):980. https://doi.org/10.3390/ijms21030980.
Ding Q, Shao C, Rose P, Zhu YZ. Epigenetics and vascular senescence-potential new therapeutic targets? Front Pharmacol. 2020;11: 535395. https://doi.org/10.3389/fphar.2020.535395.
Liu Y, Shanahan CM. Signalling pathways and vascular calcification. Front Biosci (Landmark Ed). 2011;16(4):1302–14. https://doi.org/10.2741/3790.
Jazar DA, Thakker R, Salehin S, Hasan SM, Jabri A, Albaeni A, et al. Use of coronary intravascular lithotripsy: a comprehensive review of literature. Curr Probl Cardiol. 2022;47(11): 101076. https://doi.org/10.1016/j.cpcardiol.2021.101076.
Gonzalvez-Garcia A, Jimenez-Valero S, Galeote G, Moreno R, Lopez de Sa E, Jurado-Roman A. “RotaTripsy”: combination of rotational atherectomy and intravascular lithotripsy in heavily calcified coronary lesions: a case series. Cardiovasc Revasc Med. 2022;35:179–84. https://doi.org/10.1016/j.carrev.2021.04.011.
Rozenbaum Z, Takahashi T, Kobayashi Y, Bliagos D, Menegus M, Colombo A, et al. Contemporary technologies to modify calcified plaque in coronary artery disease. Prog Cardiovasc Dis. 2021;69:18–26. https://doi.org/10.1016/j.pcad.2021.07.003.
Ahmed HM, Blaha MJ, Nasir K, Jones SR, Rivera JJ, Agatston A, et al. Low-risk lifestyle, coronary calcium, cardiovascular events, and mortality: results from MESA. Am J Epidemiol. 2013;178(1):12–21. https://doi.org/10.1093/aje/kws453.
Lehmann N, Mohlenkamp S, Mahabadi AA, Schmermund A, Roggenbuck U, Seibel R, et al. Effect of smoking and other traditional risk factors on the onset of coronary artery calcification: results of the Heinz Nixdorf recall study. Atherosclerosis. 2014;232(2):339–45. https://doi.org/10.1016/j.atherosclerosis.2013.11.045.
Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7(9):528–36. https://doi.org/10.1038/nrcardio.2010.115.
Chen NX, Moe SM. Pathophysiology of vascular calcification. Curr Osteoporos Rep. 2015;13(6):372–80. https://doi.org/10.1007/s11914-015-0293-9.
Djuric P, Dimkovic N, Schlieper G, Djuric Z, Pantelic M, Mitrovic M, et al. Sodium thiosulphate and progression of vascular calcification in end-stage renal disease patients: a double-blind, randomized, placebo-controlled study. Nephrol Dial Transplant. 2020;35(1):162–9. https://doi.org/10.1093/ndt/gfz204.
Adirekkiat S, Sumethkul V, Ingsathit A, Domrongkitchaiporn S, Phakdeekitcharoen B, Kantachuvesiri S, et al. Sodium thiosulfate delays the progression of coronary artery calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25(6):1923–9. https://doi.org/10.1093/ndt/gfp755.
Mathews SJ, de Las FL, Podaralla P, Cabellon A, Zheng S, Bierhals A, et al. Effects of sodium thiosulfate on vascular calcification in end-stage renal disease: a pilot study of feasibility, safety and efficacy. Am J Nephrol. 2011;33(2):131–8. https://doi.org/10.1159/000323550.
Saengpanit D, Chattranukulchai P, Tumkosit M, Siribumrungwong M, Katavetin P, Sitprija V, et al. Effect of sodium thiosulfate on arterial stiffness in end-stage renal disease patients undergoing chronic hemodialysis (Sodium Thiosulfate-Hemodialysis Study): a randomized controlled trial. Nephron. 2018;139(3):219–27. https://doi.org/10.1159/000488009.
Lomashvili KA, Monier-Faugere MC, Wang X, Malluche HH, O’Neill WC. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 2009;75(6):617–25. https://doi.org/10.1038/ki.2008.646.
Uitto J, Li Q. Vascular mineralization in pseudoxanthoma elasticum: etidronate to the rescue? J Am Coll Cardiol. 2018;71(10):1127–9. https://doi.org/10.1016/j.jacc.2018.01.018.
O’Neill WC, Lomashvili KA. Recent progress in the treatment of vascular calcification. Kidney Int. 2010;78(12):1232–9. https://doi.org/10.1038/ki.2010.334.
Raggi P, Bellasi A, Bushinsky D, Bover J, Rodriguez M, Ketteler M, et al. Slowing progression of cardiovascular calcification with SNF472 in patients on hemodialysis: results of a randomized phase 2b study. Circulation. 2020;141(9):728–39. https://doi.org/10.1161/CIRCULATIONAHA.119.044195.
Raggi P, Bellasi A, Sinha S, Bover J, Rodriguez M, Ketteler M, et al. Effects of SNF472, a novel inhibitor of hydroxyapatite crystallization in patients receiving hemodialysis - subgroup analyses of the CALIPSO trial. Kidney Int Rep. 2020;5(12):2178–82. https://doi.org/10.1016/j.ekir.2020.09.032.
Zabirnyk A, Ferrer MD, Bogdanova M, Perez MM, Salcedo C, Kaljusto ML, et al. SNF472, a novel anti-crystallization agent, inhibits induced calcification in an in vitro model of human aortic valve calcification. Vascul Pharmacol. 2019;122–123: 106583. https://doi.org/10.1016/j.vph.2019.106583.
Penne EL, van der Weerd NC, Grooteman MP, Mazairac AH, van den Dorpel MA, Nube MJ, et al. Role of residual renal function in phosphate control and anemia management in chronic hemodialysis patients. Clin J Am Soc Nephrol. 2011;6(2):281–9. https://doi.org/10.2215/CJN.04480510.
Isaka Y, Hamano T, Fujii H, Tsujimoto Y, Koiwa F, Sakaguchi Y, et al. Optimal phosphate control related to coronary artery calcification in dialysis patients. J Am Soc Nephrol. 2021;32(3):723–35. https://doi.org/10.1681/ASN.2020050598.
Joki N, Nikolov IG, Caudrillier A, Mentaverri R, Massy ZA, Drueke TB. Effects of calcimimetic on vascular calcification and atherosclerosis in uremic mice. Bone. 2009;45(Suppl 1):S30–4. https://doi.org/10.1016/j.bone.2009.03.653.
Singh A, Tandon S, Tandon C. An update on vascular calcification and potential therapeutics. Mol Biol Rep. 2021;48(1):887–96. https://doi.org/10.1007/s11033-020-06086-y.
Suzuki S, Suzuki M, Hanafusa N, Tsuchiya K, Nitta K. Denosumab recovers aortic arch calcification during long-term hemodialysis. Kidney Int Rep. 2021;6(3):605–12. https://doi.org/10.1016/j.ekir.2020.12.002.
Andleeb H, Hussain M, Abida Ejaz S, Sevigny J, Farman M, Yasinzai M, et al. Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h-TNAP): a therapeutic target against vascular calcification. Bioorg Chem. 2020;101: 103999. https://doi.org/10.1016/j.bioorg.2020.103999.
Opdebeeck B, Neven E, Maudsley S, Leysen H, Walter D, Geryl H, et al. A Proteomic screen to unravel the molecular pathways associated with warfarin-induced or TNAP-inhibited arterial calcification in rats. Int J Mol Sci. 2023;24(4):3657. https://doi.org/10.3390/ijms24043657.
Opdebeeck B, Neven E, Millan JL, Pinkerton AB, D’Haese PC, Verhulst A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone. 2020;137: 115392. https://doi.org/10.1016/j.bone.2020.115392.
Zhang L, Yao J, Yao Y, Bostrom KI. Contributions of the endothelium to vascular calcification. Front Cell Dev Biol. 2021;9: 620882. https://doi.org/10.3389/fcell.2021.620882.
Leszczynska A, Murphy JM. Vascular calcification: is it rather a stem/progenitor cells driven phenomenon? Front Bioeng Biotechnol. 2018;6:10. https://doi.org/10.3389/fbioe.2018.00010.
Mazurek R, Dave JM, Chandran RR, Misra A, Sheikh AQ, Greif DM. Vascular cells in blood vessel wall development and disease. Adv Pharmacol. 2017;78:323–50.
Alique M, Ruíz-Torres MP, Bodega G, Noci MV, Troyano N, Bohórquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (Albany NY). 2017;9(3):778–89.
Carmona A, Guerrero F, Buendia P, Obrero T, Aljama P, Carracedo J. Microvesicles derived from indoxyl sulfate treated endothelial cells induce endothelial progenitor cells dysfunction. Front Physiol. 2017;8:666. https://doi.org/10.3389/fphys.2017.00666.
Petsophonsakul P, Burgmaier M, Willems B, Heeneman S, Stadler N, Gremse F, et al. Nicotine promotes vascular calcification via intracellular Ca2+-mediated, Nox5-induced oxidative stress, and extracellular vesicle release in vascular smooth muscle cells. Cardiovasc Res. 2022;118(9):2196–210. https://doi.org/10.1093/cvr/cvab244.
Rogers MA, Atkins SK, Zheng KH, Singh SA, Chelvanambi S, Pham TH, et al. Lipoprotein(a) induces vesicular cardiovascular calcification revealed with single-extracellular vesicle analysis. Front Cardiovasc Med. 2022;9: 778919. https://doi.org/10.3389/fcvm.2022.778919.
Aikawa E. Extracellular vesicles in cardiovascular disease: focus on vascular calcification. J Physiol. 2016;594(11):2877–80. https://doi.org/10.1113/JP272112.
Krohn JB, Hutcheson JD, Martinez-Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. 2016;594(11):2895–903. https://doi.org/10.1113/JP271338.
Zhang C, Zhang K, Huang F, Feng W, Chen J, Zhang H, et al. Exosomes, the message transporters in vascular calcification. J Cell Mol Med. 2018;22(9):4024–33. https://doi.org/10.1111/jcmm.13692.
Peng M, Sun R, Hong Y, Wang J, Xie Y, Zhang X, et al. Extracellular vesicles carrying proinflammatory factors may spread atherosclerosis to remote locations. Cell Mol Life Sci. 2022;79(8):430. https://doi.org/10.1007/s00018-022-04464-2.
Boyer MJ, Kimura Y, Akiyama T, Baggett AY, Preston KJ, Scalia R, et al. Endothelial cell-derived extracellular vesicles alter vascular smooth muscle cell phenotype through high-mobility group box proteins. J Extracell Vesicles. 2020;9(1):1781427. https://doi.org/10.1080/20013078.2020.1781427.
Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14(3):249–56. https://doi.org/10.1038/ncb2441.
Freise C, Querfeld U, Ludwig A, Hamm B, Schnorr J, Taupitz M. Uraemic extracellular vesicles augment osteogenic transdifferentiation of vascular smooth muscle cells via enhanced AKT signalling and PiT-1 expression. J Cell Mol Med. 2021;25(12):5602–14. https://doi.org/10.1111/jcmm.16572.
Lin X, Li S, Wang YJ, Wang Y, Zhong JY, He JY, et al. Exosomal Notch3 from high glucose-stimulated endothelial cells regulates vascular smooth muscle cells calcification/aging. Life Sci. 2019;232: 116582. https://doi.org/10.1016/j.lfs.2019.116582.
Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4(3):214–22. https://doi.org/10.1016/j.scr.2009.12.003.
Chen L, Wang Y, Pan Y, Zhang L, Shen C, Qin G, et al. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem Biophys Res Commun. 2013;431(3):566–71. https://doi.org/10.1016/j.bbrc.2013.01.015.
Kervadec A, Bellamy V, El Harane N, Arakelian L, Vanneaux V, Cacciapuoti I, et al. Cardiovascular progenitor-derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J Heart Lung Transplant. 2016;35(6):795–807. https://doi.org/10.1016/j.healun.2016.01.013.
Chen Q, Liu Y, Ding X, Li Q, Qiu F, Wang M, et al. Bone marrow mesenchymal stem cell-secreted exosomes carrying microRNA-125b protect against myocardial ischemia reperfusion injury via targeting SIRT7. Mol Cell Biochem. 2020;465(1–2):103–14. https://doi.org/10.1007/s11010-019-03671-z.
Ibrahim AG, Cheng K, Marban E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports. 2014;2(5):606–19. https://doi.org/10.1016/j.stemcr.2014.04.006.
Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J. 2017;38(3):201–11. https://doi.org/10.1093/eurheartj/ehw240.
Persy V, D’Haese P. Vascular calcification and bone disease: the calcification paradox. Trends Mol Med. 2009;15(9):405–16. https://doi.org/10.1016/j.molmed.2009.07.001.
Lampropoulos CE, Papaioannou I, D’Cruz DP. Osteoporosis–a risk factor for cardiovascular disease? Nat Rev Rheumatol. 2012;8(10):587–98. https://doi.org/10.1038/nrrheum.2012.120.
Li G, Luna C, Qiu J, Epstein DL, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. 2009;130(11–12):731–41. https://doi.org/10.1016/j.mad.2009.09.002.
Kim KM, Park SJ, Jung SH, Kim EJ, Jogeswar G, Ajita J, et al. miR-182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J Bone Miner Res. 2012;27(8):1669–79. https://doi.org/10.1002/jbmr.1604.
Ke K, Sul OJ, Rajasekaran M, Choi HS. MicroRNA-183 increases osteoclastogenesis by repressing heme oxygenase-1. Bone. 2015;81:237–46. https://doi.org/10.1016/j.bone.2015.07.006.
Davis C, Dukes A, Drewry M, Helwa I, Johnson MH, Isales CM, et al. MicroRNA-183-5p increases with age in bone-derived extracellular vesicles, suppresses bone marrow stromal (Stem) cell proliferation, and induces stem cell senescence. Tissue Eng Part A. 2017;23(21–22):1231–40. https://doi.org/10.1089/ten.TEA.2016.0525.
Evenepoel P, Dejongh S, Verbeke K, Meijers B. The role of gut dysbiosis in the bone-vascular axis in chronic kidney disease. Toxins (Basel). 2020;12(5):285. https://doi.org/10.3390/toxins12050285.
Guo Y, Bao S, Guo W, Diao Z, Wang L, Han X, et al. Bone marrow mesenchymal stem cell-derived exosomes alleviate high phosphorus-induced vascular smooth muscle cells calcification by modifying microRNA profiles. Funct Integr Genomics. 2019;19(4):633–43. https://doi.org/10.1007/s10142-019-00669-0.
Luo F, Guo W, Liu W. Exosomes derived from bone marrow mesenchymal stem cells inhibit human aortic vascular smooth muscle cells calcification via the miR-15a/15b/16/NFATc3/OCN axis. Biochem Biophys Res Commun. 2022;635:65–76. https://doi.org/10.1016/j.bbrc.2022.09.076.
Liu Y, Guo Y, Bao S, Huang H, Liu W, Guo W. Bone marrow mesenchymal stem cell-derived exosomal microRNA-381-3p alleviates vascular calcification in chronic kidney disease by targeting NFAT5. Cell Death Dis. 2022;13(3):278. https://doi.org/10.1038/s41419-022-04703-1.
Liu Y, Bao S, Guo W, Liu W. Bone mesenchymal stem cell derived exosomes alleviate high phosphorus-induced calcification of vascular smooth muscle cells through the NONHSAT 084969.2/NF-κB axis. Aging (Albany NY). 2021;13(12):16749–62.
Wei W, Guo X, Gu L, Jia J, Yang M, Yuan W, et al. Bone marrow mesenchymal stem cell exosomes suppress phosphate-induced aortic calcification via SIRT6-HMGB1 deacetylation. Stem Cell Res Ther. 2021;12(1):235. https://doi.org/10.1186/s13287-021-02307-8.
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114(6):597–605. https://doi.org/10.1161/CIRCULATIONAHA.106.621854.
Wang Y, Ma WQ, Zhu Y, Han XQ, Liu N. Exosomes derived from mesenchymal stromal cells pretreated with advanced glycation end product-bovine serum albumin inhibit calcification of vascular smooth muscle cells. Front Endocrinol (Lausanne). 2018;9:524. https://doi.org/10.3389/fendo.2018.00524.
Ning W, Li S, Yang W, Yang B, Xin C, Ping X, et al. Blocking exosomal miRNA-153-3p derived from bone marrow mesenchymal stem cells ameliorates hypoxia-induced myocardial and microvascular damage by targeting the ANGPT1-mediated VEGF/PI3k/Akt/eNOS pathway. Cell Signal. 2021;77: 109812. https://doi.org/10.1016/j.cellsig.2020.109812.
Liang W, Han B, Hai Y, Sun D, Yin P. Mechanism of action of mesenchymal stem cell-derived exosomes in the intervertebral disc degeneration treatment and bone repair and regeneration. Front Cell Dev Biol. 2021;9: 833840. https://doi.org/10.3389/fcell.2021.833840.
Duell PB, Welty FK, Miller M, Chait A, Hammond G, Ahmad Z, et al. Nonalcoholic fatty liver disease and cardiovascular risk: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(6):e168–85. https://doi.org/10.1161/ATV.0000000000000153.
Zhou XD, Cai J, Targher G, Byrne CD, Shapiro MD, Sung KC, et al. Metabolic dysfunction-associated fatty liver disease and implications for cardiovascular risk and disease prevention. Cardiovasc Diabetol. 2022;21(1):270. https://doi.org/10.1186/s12933-022-01697-0.
C G, Y U, M-H Z, J G. MAFLD: What is Different from NAFLD? Clin Mol Hepatol. 2022. https://doi.org/10.3350/cmh.2022.0367.
Chang Y, Ryu S, Sung KC, Cho YK, Sung E, Kim HN, et al. Alcoholic and non-alcoholic fatty liver disease and associations with coronary artery calcification: evidence from the Kangbuk Samsung Health Study. Gut. 2019;68(9):1667–75. https://doi.org/10.1136/gutjnl-2018-317666.
Sung KC, Yoo TK, Lee MY, Byrne CD, Zheng MH, Targher G. Comparative associations of nonalcoholic fatty liver disease and metabolic dysfunction-associated fatty liver disease with coronary artery calcification: a cross-sectional and longitudinal cohort study. Arterioscler Thromb Vasc Biol. 2023;43(3):482–91. https://doi.org/10.1161/ATVBAHA.122.318661.
Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313(22):2263–73. https://doi.org/10.1001/jama.2015.5370.
Mantovani A, Scorletti E, Mosca A, Alisi A, Byrne CD, Targher G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism. 2020;111S: 154170. https://doi.org/10.1016/j.metabol.2020.154170.
Adams LA, Anstee QM, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66(6):1138–53. https://doi.org/10.1136/gutjnl-2017-313884.
Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69(9):1691–705. https://doi.org/10.1136/gutjnl-2020-320622.
Zhang X, Ji X, Wang Q, Li JZ. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell. 2017;9(2):164–77. https://doi.org/10.1007/s13238-017-0436-0.
Babuta M, Szabo G. Extracellular vesicles in inflammation: focus on the microRNA cargo of EVs in modulation of liver diseases. J Leukoc Biol. 2022;111(1):75–92. https://doi.org/10.1002/JLB.3MIR0321-156R.
Hernandez A, Arab JP, Reyes D, Lapitz A, Moshage H, Banales JM, et al. Extracellular vesicles in NAFLD/ALD: from pathobiology to therapy. Cells. 2020;9(4):817. https://doi.org/10.3390/cells9040817.
Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72(1):156–66. https://doi.org/10.1016/j.jhep.2019.09.014.
Zuo R, Ye LF, Huang Y, Song ZQ, Wang L, Zhi H, et al. Hepatic small extracellular vesicles promote microvascular endothelial hyperpermeability during NAFLD via novel-miRNA-7. J Nanobiotechnology. 2021;19(1):396. https://doi.org/10.1186/s12951-021-01137-3.
Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, et al. Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (Nucleotide-Binding Domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-kappaB (Nuclear Factor kappaB) signals. Arterioscler Thromb Vasc Biol. 2020;40(3):751–65. https://doi.org/10.1161/ATVBAHA.119.313414.
Wen C, Yang X, Yan Z, Zhao M, Yue X, Cheng X, et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168(3):2242–7. https://doi.org/10.1016/j.ijcard.2013.01.211.
The E, de Graaf DM, Zhai Y, Yao Q, Ao L, Fullerton DA, et al. Interleukin 38 alleviates aortic valve calcification by inhibition of NLRP3. Proc Natl Acad Sci U S A. 2022;119(36): e2202577119. https://doi.org/10.1073/pnas.2202577119.
Cosson E, Nguyen MT, Rezgani I, Tatulashvili S, Sal M, Berkane N, et al. Epicardial adipose tissue volume and coronary calcification among people living with diabetes: a cross-sectional study. Cardiovasc Diabetol. 2021;20(1):35. https://doi.org/10.1186/s12933-021-01225-6.
Zhao S, Kusminski CM, Scherer PE. Adiponectin, leptin and cardiovascular disorders. Circ Res. 2021;128(1):136–49. https://doi.org/10.1161/CIRCRESAHA.120.314458.
Xiao X, Liu YZ, Cheng ZB, Sun JX, Shao YD, Qu SL, et al. Adipokines in vascular calcification. Clin Chim Acta. 2021;516:15–26. https://doi.org/10.1016/j.cca.2021.01.009.
Lu Y, Ma Y, Wang R, Sun J, Guo B, Wei R, et al. Adiponectin inhibits vascular smooth muscle cell calcification induced by beta-glycerophosphate through JAK2/STAT3 signaling pathway. J Biosci. 2019;44(4):86. https://doi.org/10.1007/s12038-019-9895-1.
Xu F, Li F-X-Z, Lin X, Zhong J-Y, Wu F, Shan S-K, et al. Adipose tissue-derived Omentin-1 attenuates arterial calcification. adipose tissue-derived Omentin-1 attenuates arterial calcification via AMPK/Akt signaling pathway. J Biosci. 2019;11(20):8760–76.
Zhou Z, Tao Y, Zhao H, Wang Q. Adipose extracellular vesicles: messengers from and to macrophages in regulating immunometabolic homeostasis or disorders. Front Immunol. 2021;12: 666344. https://doi.org/10.3389/fimmu.2021.666344.
Gan L, Xie D, Liu J, Lau WB, Christopher TA, Lopez B, et al. Small extracellular microvesicles mediated pathological communications between dysfunctional adipocytes and cardiomyocytes as a novel mechanism exacerbating ischemia/reperfusion injury in diabetic mice. Circulation. 2020;141(12):968–83. https://doi.org/10.1161/CIRCULATIONAHA.119.042640.
Liang X, Zhang L, Wang S, Han Q, Zhao RC. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci. 2016;129(11):2182–9. https://doi.org/10.1242/jcs.170373.
Lopatina T, Bruno S, Tetta C, Kalinina N, Porta M, Camussi G. Platelet-derived growth factor regulates the secretion of extracellular vesicles by adipose mesenchymal stem cells and enhances their angiogenic potential. Cell Commun Signal. 2014;12:26. https://doi.org/10.1186/1478-811X-12-26.
Costa RM, Neves KB, Tostes RC, Lobato NS. Perivascular adipose tissue as a relevant fat depot for cardiovascular risk in obesity. Front Physiol. 2018;9:253. https://doi.org/10.3389/fphys.2018.00253.
Villacorta L, Chang L. The role of perivascular adipose tissue in vasoconstriction, arterial stiffness, and aneurysm. Horm Mol Biol Clin Investig. 2015;21(2):137–47. https://doi.org/10.1515/hmbci-2014-0048.
Li X, Ballantyne LL, Yu Y, Funk CD. Perivascular adipose tissue-derived extracellular vesicle miR-221-3p mediates vascular remodeling. FASEB J. 2019;33(11):12704–22. https://doi.org/10.1096/fj.201901548R.
Mackenzie NC, Staines KA, Zhu D, Genever P, Macrae VE. miRNA-221 and miRNA-222 synergistically function to promote vascular calcification. Cell Biochem Funct. 2014;32(2):209–16. https://doi.org/10.1002/cbf.3005.
Voelkl J, Lang F, Eckardt KU, Amann K, Kuro OM, Pasch A, et al. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell Mol Life Sci. 2019;76(11):2077–91. https://doi.org/10.1007/s00018-019-03054-z.
Chen NX, O’Neill KD, Moe SM. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018;93(2):343–54. https://doi.org/10.1016/j.kint.2017.07.019.
Pavlic A, Bahram Sangani N, Kerins J, Nicolaes G, Schurgers L, Reutelingsperger C. Vascular smooth muscle cell neutral sphingomyelinase 2 in the release of exosomes and vascular calcification. Int J Mol Sci. 2022;23(16):9178. https://doi.org/10.3390/ijms23169178.
Arcidiacono MV, Carrillo-Lopez N, Panizo S, Castro-Grattoni AL, Valcheva P, Ulloa C, et al. Barley-ss-glucans reduce systemic inflammation, renal injury and aortic calcification through ADAM17 and neutral-sphingomyelinase2 inhibition. Sci Rep. 2019;9(1):17810. https://doi.org/10.1038/s41598-019-54306-8.
Bhat OM, Yuan X, Kukreja RC, Li PL. Regulatory role of mammalian target of rapamycin signaling in exosome secretion and osteogenic changes in smooth muscle cells lacking acid ceramidase gene. FASEB J. 2021;35(7): e21732. https://doi.org/10.1096/fj.202100385R.
Bhat OM, Yuan X, Cain C, Salloum FN, Li PL. Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: a ceramide-mediated vasculopathy. J Cell Mol Med. 2020;24(1):539–53. https://doi.org/10.1111/jcmm.14761.
Bhat OM, Li G, Yuan X, Huang D, Gulbins E, Kukreja RC, et al. Arterial medial calcification through enhanced small extracellular vesicle release in smooth muscle-specific asah1 gene knockout mice. Sci Rep. 2020;10(1):1645. https://doi.org/10.1038/s41598-020-58568-5.
Bhat OM, Yuan X, Camus S, Salloum FN, Li PL. Abnormal lysosomal positioning and small extracellular vesicle secretion in arterial stiffening and calcification of mice lacking mucolipin 1 gene. Int J Mol Sci. 2020;21(5):1713. https://doi.org/10.3390/ijms21051713.
Liu Q, Luo Y, Zhao Y, Xiang P, Zhu J, Jing W, et al. Nano-hydroxyapatite accelerates vascular calcification via lysosome impairment and autophagy dysfunction in smooth muscle cells. Bioact Mater. 2022;8:478–93. https://doi.org/10.1016/j.bioactmat.2021.06.004.
Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, et al. Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arterioscler Thromb Vasc Biol. 2017;37(3):e22–32. https://doi.org/10.1161/ATVBAHA.116.308886.
Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, et al. Reactive oxygen-forming nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res. 2020;127(7):911–27. https://doi.org/10.1161/circresaha.119.316159.
Lupo MG, Bressan A, Donato M, Canzano P, Camera M, Poggio P, et al. PCSK9 promotes arterial medial calcification. Atherosclerosis. 2022;346:86–97. https://doi.org/10.1016/j.atherosclerosis.2022.01.015.
Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126(4):1323–36. https://doi.org/10.1172/JCI80851.
Itoh S, Mizuno K, Aikawa M, Aikawa E. Dimerization of sortilin regulates its trafficking to extracellular vesicles. J Biol Chem. 2018;293(12):4532–44. https://doi.org/10.1074/jbc.RA117.000732.
Goettsch C, Kjolby M, Aikawa E. Sortilin and its multiple roles in cardiovascular and metabolic diseases. Arterioscler Thromb Vasc Biol. 2018;38(1):19–25. https://doi.org/10.1161/ATVBAHA.117.310292.
Furmanik M, van Gorp R, Whitehead M, Ahmad S, Bordoloi J, Kapustin A, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (Glucose-Regulated Protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol. 2021;41(2):898–914. https://doi.org/10.1161/atvbaha.120.315506.
Mansour A, Darwiche W, Yaker L, Da Nascimento S, Gomila C, Rossi C et al. GFOGER peptide modifies the protein content of extracellular vesicles and inhibits vascular calcification. Front Cell Dev Biol. 2020;8. https://doi.org/10.3389/fcell.2020.589761.
Hodroge A, Trecherel E, Cornu M, Darwiche W, Mansour A, Ait-Mohand K, et al. Oligogalacturonic acid inhibits vascular calcification by two mechanisms: inhibition of vascular smooth muscle cell osteogenic conversion and interaction with collagen. Arterioscler Thromb Vasc Biol. 2017;37(7):1391–401. https://doi.org/10.1161/ATVBAHA.117.309513.
Pan W, Liang J, Tang H, Fang X, Wang F, Ding Y, et al. Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int J Biochem Cell Biol. 2020;118: 105645. https://doi.org/10.1016/j.biocel.2019.105645.
Chen C, Li Y, Lu H, Liu K, Jiang W, Zhang Z et al. Curcumin attenuates vascular calcification via the exosomal miR-92b-3p/KLF4 axis. Exp Biol Med (Maywood). 2022:15353702221095456. https://doi.org/10.1177/15353702221095456.
Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol. 2017;37(9):e108–14. https://doi.org/10.1161/ATVBAHA.117.309813.
Williams SB, Goldfine AB, Timimi FK, Ting HH, Roddy MA, Simonson DC, et al. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation. 1998;97(17):1695–701. https://doi.org/10.1161/01.cir.97.17.1695.
Asenjo-Bueno A, Alcalde-Estevez E, El Assar M, Olmos G, Plaza P, Sosa P, et al. Hyperphosphatemia-induced oxidant/antioxidant imbalance impairs vascular relaxation and induces inflammation and fibrosis in old mice. Antioxidants (Basel). 2021;10(8):1308. https://doi.org/10.3390/antiox10081308.
Cunha RSD, Santos AF, Barreto FC, Stinghen AEM. How do uremic toxins affect the endothelium? Toxins (Basel). 2020;12(6):412. https://doi.org/10.3390/toxins12060412.
Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35(23):1515–25. https://doi.org/10.1093/eurheartj/ehu163.
Yuan C, Ni L, Zhang C, Hu X, Wu X. Vascular calcification: New insights into endothelial cells. Microvasc Res. 2021;134:104105.
Tesauro M, Mauriello A, Rovella V, Annicchiarico-Petruzzelli M, Cardillo C, Melino G, et al. Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med. 2017;281(5):471–82. https://doi.org/10.1111/joim.12605.
Van den Bergh G, Van den Branden A, Opdebeeck B, Fransen P, Neven E, De Meyer GRY, et al. Endothelial dysfunction aggravates arterial media calcification in warfarin administered rats. FASEB J. 2022;36(5): e22315. https://doi.org/10.1096/fj.202101919R.
Zhou Y-B, Zhou H, Li L, Kang Y, Cao X, Wu Z-Y, et al. Hydrogen Sulfide Prevents Elastin Loss and Attenuates Calcification Induced by High Glucose in Smooth Muscle Cells through Suppression of Stat3/Cathepsin S Signaling Pathway. Int J Mol Sci. 2019;20(17):4202. https://doi.org/10.3390/ijms20174202.
Hackett L, Millar NL, Lam P, Murrell GAC. Are the symptoms of calcific tendinitis due to neoinnervation and/or neovascularization? J Bone Joint Surg. 2016;98(3):186–92. https://doi.org/10.2106/jbjs.O.00417.
Zhou J, Li Y-S, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. 2014;34(10):2191–8. https://doi.org/10.1161/atvbaha.114.303422.
Wang S, Wu J, Li X, Tan R, Chen L, Yang L, et al. CXCR6 mediates pressure overload-induced aortic stiffness by increasing macrophage recruitment and reducing exosome-miRNA29b. J Cardiovasc Transl Res. 2022. https://doi.org/10.1007/s12265-022-10304-2.
Buendia P, Montes de Oca A, Madueno JA, Merino A, Martin-Malo A, Aljama P, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. 2015;29(1):173–81. https://doi.org/10.1096/fj.14-249706.
Cavallari C, Dellepiane S, Fonsato V, Medica D, Marengo M, Migliori M, et al. Online hemodiafiltration inhibits inflammation-related endothelial dysfunction and vascular calcification of uremic patients modulating miR-223 expression in plasma extracellular vesicles. J Immunol. 2019;202(8):2372–83. https://doi.org/10.4049/jimmunol.1800747.
Alique M, Bodega G, Corchete E, Garcia-Menendez E, de Sequera P, Luque R, et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput Struct Biotechnol J. 2020;18:953–66. https://doi.org/10.1016/j.csbj.2020.04.006.
Peng Z, Duan Y, Zhong S, Chen J, Li J, He Z. RNA-seq analysis of extracellular vesicles from hyperphosphatemia-stimulated endothelial cells provides insight into the mechanism underlying vascular calcification. BMC Nephrol. 2022;23(1):192. https://doi.org/10.1186/s12882-022-02823-6.
Jing C, Zhang G, Liu Z, Xu Q, Li C, Cheng G, et al. Peroxidasin promotes diabetic vascular endothelial dysfunction induced by advanced glycation end products via NOX2/HOCl/Akt/eNOS pathway. Redox Biol. 2021;45: 102031. https://doi.org/10.1016/j.redox.2021.102031.
Wautier JL, Schmidt AM. Protein glycation: a firm link to endothelial cell dysfunction. Circ Res. 2004;95(3):233–8. https://doi.org/10.1161/01.RES.0000137876.28454.64.
Li S, Zhan JK, Wang YJ, Lin X, Zhong JY, Wang Y, et al. Exosomes from hyperglycemia-stimulated vascular endothelial cells contain versican that regulate calcification/senescence in vascular smooth muscle cells. Cell Biosci. 2019;9:1. https://doi.org/10.1186/s13578-018-0263-x.
Ruiz JL, Hutcheson JD, Aikawa E. Cardiovascular calcification: current controversies and novel concepts. Cardiovasc Pathol. 2015;24(4):207–12. https://doi.org/10.1016/j.carpath.2015.03.002.
Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114(1 Suppl):I547–52. https://doi.org/10.1161/CIRCULATIONAHA.105.001115.
Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. 2015;242(1):251–60. https://doi.org/10.1016/j.atherosclerosis.2015.07.008.
Bakhshian Nik A, Hutcheson JD, Aikawa E. Extracellular vesicles as mediators of cardiovascular calcification. Front Cardiovasc Med. 2017;4:78. https://doi.org/10.3389/fcvm.2017.00078.
Cui L, Rashdan NA, Zhu D, Milne EM, Ajuh P, Milne G, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J Cell Physiol. 2017;232(11):2985–95. https://doi.org/10.1002/jcp.25935.
Kondo A, Kaestner KH. Emerging diverse roles of telocytes. Development. 2019;146(14):dev175018.
Cretoiu D, Xu J, Xiao J, Cretoiu SM. Telocytes and their extracellular vesicles-evidence and hypotheses. Int J Mol Sci. 2016;17(8):1322. https://doi.org/10.3390/ijms17081322.
Yang R, Tang Y, Chen X, Yang Y. Telocytes-derived extracellular vesicles alleviate aortic valve calcification by carrying miR-30b. ESC Heart Fail. 2021;8(5):3935–46. https://doi.org/10.1002/ehf2.13460.
Hernandez GE, Iruela-Arispe ML. The many flavors of monocyte/macrophage–endothelial cell interactions. Curr Opin Hematol. 2020;27(3):181–9. https://doi.org/10.1097/moh.0000000000000573.
Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118(4):653–67. https://doi.org/10.1161/circresaha.115.306256.
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–40. https://doi.org/10.1002/jcp.26429.
Kurozumi A, Nakano K, Yamagata K, Okada Y, Nakayamada S, Tanaka Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone. 2019;124:53–61. https://doi.org/10.1016/j.bone.2019.04.006.
Deuell KA, Callegari A, Giachelli CM, Rosenfeld ME, Scatena M. RANKL Enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: dependence on IL-6 and TNF-α. J Vasc Res. 2012;49(6):510–21. https://doi.org/10.1159/000341216.
Ceneri N, Zhao L, Young BD, Healy A, Coskun S, Vasavada H, et al. Rac2 modulates atherosclerotic calcification by regulating macrophage Interleukin-1β production. Arterioscler Thromb Vasc Biol. 2017;37(2):328–40. https://doi.org/10.1161/atvbaha.116.308507.
Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283(22):15319–27. https://doi.org/10.1074/jbc.M800021200.
Zhang X, Li J, Qin J-J, Cheng W-L, Zhu X, Gong F-H, et al. Oncostatin M receptor β deficiency attenuates atherogenesis by inhibiting JAK2/STAT3 signaling in macrophages. J Lipid Res. 2017;58(5):895–906. https://doi.org/10.1194/jlr.M074112.
Borland SJ, Morris TG, Borland SC, Morgan MR, Francis SE, Merry CLR, et al. Regulation of vascular smooth muscle cell calcification by syndecan-4/FGF-2/PKCα signalling and cross-talk with TGFβ. Cardiovasc Res. 2017;113(13):1639–52. https://doi.org/10.1093/cvr/cvx178.
Villa-Bellosta R, Hamczyk MR, Andrés V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am J Physiol Cell Physiol. 2016;310(10):C788–99. https://doi.org/10.1152/ajpcell.00370.2015.
Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. J Atheroscler Thromb. 2018;25(4):294–303. https://doi.org/10.5551/jat.RV17020.
Lopez-Mejias R, Gonzalez-Gay MA. IL-6: linking chronic inflammation and vascular calcification. Nat Rev Rheumatol. 2019;15(8):457–9. https://doi.org/10.1038/s41584-019-0259-x.
Li Y, Sun Z, Zhang L, Yan J, Shao C, Jing L, et al. Role of macrophages in the progression and regression of vascular calcification. Front Pharmacol. 2020;11:661. https://doi.org/10.3389/fphar.2020.00661.
New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108(11):1381–91. https://doi.org/10.1161/CIRCRESAHA.110.234146.
Xiao X, Yang C, Qu SL, Shao YD, Zhou CY, Chao R, et al. S100 proteins in atherosclerosis. Clin Chim Acta. 2020;502:293–304. https://doi.org/10.1016/j.cca.2019.11.019.
McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, et al. S100A8 and S100A9 in human arterial wall. Implications for atherogenesis J Biol Chem. 2005;280(50):41521–9. https://doi.org/10.1074/jbc.M509442200.
New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, et al. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113(1):72–7. https://doi.org/10.1161/CIRCRESAHA.113.301036.
Kawakami R, Katsuki S, Travers R, Romero DC, Becker-Greene D, Passos LSA, et al. S100A9-RAGE axis accelerates formation of macrophage-mediated extracellular vesicle microcalcification in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2020;40(8):1838–53. https://doi.org/10.1161/ATVBAHA.118.314087.
Li X-F, Wang Y, Zheng D-D, Xu H-X, Wang T, Pan M, et al. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am J Transl Res. 2016;8(12):5773–83.
Cao J, Chen C, Chen Q, Gao Y, Zhao Z, Yuan Q, et al. Extracellular vesicle miR-32 derived from macrophage promotes arterial calcification in mice with type 2 diabetes via inhibiting VSMC autophagy. J Transl Med. 2022;20(1):307. https://doi.org/10.1186/s12967-022-03502-8.
Sun Z, Li L, Zhang L, Yan J, Shao C, Bao Z, et al. Macrophage galectin-3 enhances intimal translocation of vascular calcification in diabetes mellitus. Am J Physiol Heart Circ Physiol. 2020;318(5):H1068–79. https://doi.org/10.1152/ajpheart.00690.2019.
Yaker L, Tebani A, Lesueur C, Dias C, Jung V, Bekri S, et al. extracellular vesicles from lps-treated macrophages aggravate smooth muscle cell calcification by propagating inflammation and oxidative stress. Front Cell Dev Biol. 2022;10: 823450. https://doi.org/10.3389/fcell.2022.823450.
Passos LSA, Lupieri A, Becker-Greene D, Aikawa E. Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis. 2020;306:59–67. https://doi.org/10.1016/j.atherosclerosis.2020.02.016.
Liu JT, Bao H, Fan YJ, Li ZT, Yao QP, Han Y, et al. Platelet-derived microvesicles promote VSMC dedifferentiation after intimal injury via Src/Lamtor1/mTORC1 signaling. Front Cell Dev Biol. 2021;9: 744320. https://doi.org/10.3389/fcell.2021.744320.
Chiva-Blanch G, Padró T, Alonso R, Crespo J, Perez de Isla L, Mata P, et al. Liquid Biopsy of extracellular microvesicles maps coronary calcification and atherosclerotic plaque in asymptomatic patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2019;39(5):945–55. https://doi.org/10.1161/atvbaha.118.312414.
Jayachandran M, Litwiller RD, Owen WG, Heit JA, Behrenbeck T, Mulvagh SL, et al. Characterization of blood borne microparticles as markers of premature coronary calcification in newly menopausal women. Am J Physiol Heart Circ Physiol. 2008;295(3):H931–8. https://doi.org/10.1152/ajpheart.00193.2008.
Oggero S, Godec T, van Gorp R, Pinto AL, Schurgers LJ, Reutelingsperger C, et al. Role of plasma extracellular vesicles in prediction of cardiovascular risk and alterations in response to statin therapy in hypertensive patients. J Hypertens. 2022;40(8):1522–9. https://doi.org/10.1097/HJH.0000000000003178.
Kanhai DA, Visseren FL, van der Graaf Y, Schoneveld AH, Catanzariti LM, Timmers L, et al. Microvesicle protein levels are associated with increased risk for future vascular events and mortality in patients with clinically manifest vascular disease. Int J Cardiol. 2013;168(3):2358–63. https://doi.org/10.1016/j.ijcard.2013.01.231.
Nozaki T, Sugiyama S, Koga H, Sugamura K, Ohba K, Matsuzawa Y, et al. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J Am Coll Cardiol. 2009;54(7):601–8. https://doi.org/10.1016/j.jacc.2009.05.022.
Jansen F, Yang X, Proebsting S, Hoelscher M, Przybilla D, Baumann K, et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J Am Heart Assoc. 2014;3(6): e001249. https://doi.org/10.1161/JAHA.114.001249.
de Boer HC, van Solingen C, Prins J, Duijs JM, Huisman MV, Rabelink TJ, et al. Aspirin treatment hampers the use of plasma microRNA-126 as a biomarker for the progression of vascular disease. Eur Heart J. 2013;34(44):3451–7. https://doi.org/10.1093/eurheartj/eht007.
Vajen T, Benedikter BJ, Heinzmann ACA, Vasina EM, Henskens Y, Parsons M, et al. Platelet extracellular vesicles induce a pro-inflammatory smooth muscle cell phenotype. J Extracell Vesicles. 2017;6(1):1322454. https://doi.org/10.1080/20013078.2017.1322454.
Schurgers LJ, Akbulut AC, Kaczor DM, Halder M, Koenen RR, Kramann R. Initiation and propagation of vascular calcification is regulated by a concert of platelet- and smooth muscle cell-derived extracellular vesicles. Front Cardiovasc Med. 2018;5:36. https://doi.org/10.3389/fcvm.2018.00036.
Chen NX, Kircelli F, O’Neill KD, Chen X, Moe SM. Verapamil inhibits calcification and matrix vesicle activity of bovine vascular smooth muscle cells. Kidney Int. 2010;77(5):436–42. https://doi.org/10.1038/ki.2009.481.
Nagy A, Petho D, Gesztelyi R, Juhasz B, Balla G, Szilvassy Z, et al. BGP-15 Inhibits hyperglycemia-aggravated VSMC calcification induced by high phosphate. Int J Mol Sci. 2021;22(17):9263. https://doi.org/10.3390/ijms22179263.
Perkins RM, Kirchner HL, Matsushita K, Bucaloiu ID, Norfolk E, Hartle JE. Bisphosphonates and mortality in women with CKD and the presence or absence of cardiovascular disease. Clin J Am Soc Nephrol. 2014;9(5):874–80. https://doi.org/10.2215/cjn.07790713.
Hartle JE, Tang X, Kirchner HL, Bucaloiu ID, Sartorius JA, Pogrebnaya ZV, et al. Bisphosphonate therapy, death, and cardiovascular events among female patients with CKD: a retrospective cohort study. Am J Kidney Dis. 2012;59(5):636–44. https://doi.org/10.1053/j.ajkd.2011.11.037.
Elmariah S, Delaney JAC, O’Brien KD, Budoff MJ, Vogel-Claussen J, Fuster V, et al. Bisphosphonate use and prevalence of valvular and vascular calcification in women. J Am Coll Cardiol. 2010;56(21):1752–9. https://doi.org/10.1016/j.jacc.2010.05.050.
Ruiz JL, Hutcheson JD, Cardoso L, Bakhshian Nik A, Condado de Abreu A, Pham T et al. Nanoanalytical analysis of bisphosphonate-driven alterations of microcalcifications using a 3D hydrogel system and in vivo mouse model. Proc Nat Acad Sci. 2021;118(14). https://doi.org/10.1073/pnas.1811725118.
Wei Y, Wu Y, Zhao R, Zhang K, Midgley AC, Kong D, et al. MSC-derived sEVs enhance patency and inhibit calcification of synthetic vascular grafts by immunomodulation in a rat model of hyperlipidemia. Biomaterials. 2019;204:13–24. https://doi.org/10.1016/j.biomaterials.2019.01.049.
Vogt I, Haffner D, Leifheit-Nestler M. FGF23 and Phosphate-Cardiovascular Toxins in CKD. Toxins (Basel). 2019;11(11):647. https://doi.org/10.3390/toxins11110647.
Nasrallah MM, El-Shehaby AR, Salem MM, Osman NA, El Sheikh E, Sharaf El Din UA. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol Dial Transplant. 2010;25(8):2679–85. https://doi.org/10.1093/ndt/gfq089.
Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83(6):1159–68. https://doi.org/10.1038/ki.2013.3.
Schaub T, Janke D, Zickler D, Lange C, Girndt M, Schindler R, et al. High cut-off dialysis mitigates pro-calcific effects of plasma on vascular progenitor cells. Sci Rep. 2021;11(1):1144. https://doi.org/10.1038/s41598-020-80016-7.
Saleh AF, Lázaro-Ibáñez E, Forsgard MAM, Shatnyeva O, Osteikoetxea X, Karlsson F, et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale. 2019;11(14):6990–7001. https://doi.org/10.1039/c8nr08720b.
Zhu X, Badawi M, Pomeroy S, Sutaria DS, Xie Z, Baek A, et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6(1):1324730. https://doi.org/10.1080/20013078.2017.1324730.
Saint-Pol J, Gosselet F, Duban-Deweer S, Pottiez G, Karamanos Y. Targeting and crossing the blood-brain barrier with extracellular vesicles. Cells. 2020;9(4):851. https://doi.org/10.3390/cells9040851.
Chen Y, Yu L. Extracellular vesicles: from bench to bedside. Curr Med. 2022;1(1). https://doi.org/10.1007/s44194-022-00001-2.
van Niel G, Carter DRF, Clayton A, Lambert DW, Raposo G, Vader P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat Rev Mol Cell Biol. 2022;23(5):369–82. https://doi.org/10.1038/s41580-022-00460-3.
Yan J, Pan Y, Shao W, Wang C, Wang R, He Y, et al. Beneficial effect of the short-chain fatty acid propionate on vascular calcification through intestinal microbiota remodelling. Microbiome. 2022;10(1):195. https://doi.org/10.1186/s40168-022-01390-0.
Acknowledgements
Thanks to all authors for their contributions to the manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (No. 82270939), Natural Science Foundation of Hunan Province (2022JJ70036), Postgraduate Scientific Research Innovation Project of Hunan Province (CX20210917), and the Clinical Rsearch 4310 Program of the First Affiliated Hospotal of the University of South China (20214310NHYCG02).
Author information
Authors and Affiliations
Contributions
ZL, WZ and SQ conceptualized the study; YQ, YSQ, and CQ searched and analyzed the literature. YSQ wrote the original manuscript. ZL revised the manuscript. ZL and YSQ generated the figures. JH and XH supervised this work and edited the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., Zeng, Z., Yuan, Q. et al. Vascular calcification: from the perspective of crosstalk. Mol Biomed 4, 35 (2023). https://doi.org/10.1186/s43556-023-00146-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-023-00146-y