
Abstract
Ferroptosis, a regulated form of cellular death characterized by the iron-mediated accumulation of lipid peroxides, provides a novel avenue for delving into the intersection of cellular metabolism, oxidative stress, and disease pathology. We have witnessed a mounting fascination with ferroptosis, attributed to its pivotal roles across diverse physiological and pathological conditions including developmental processes, metabolic dynamics, oncogenic pathways, neurodegenerative cascades, and traumatic tissue injuries. By unraveling the intricate underpinnings of the molecular machinery, pivotal contributors, intricate signaling conduits, and regulatory networks governing ferroptosis, researchers aim to bridge the gap between the intricacies of this unique mode of cellular death and its multifaceted implications for health and disease. In light of the rapidly advancing landscape of ferroptosis research, we present a comprehensive review aiming at the extensive implications of ferroptosis in the origins and progress of human diseases. This review concludes with a careful analysis of potential treatment approaches carefully designed to either inhibit or promote ferroptosis. Additionally, we have succinctly summarized the potential therapeutic targets and compounds that hold promise in targeting ferroptosis within various diseases. This pivotal facet underscores the burgeoning possibilities for manipulating ferroptosis as a therapeutic strategy. In summary, this review enriched the insights of both investigators and practitioners, while fostering an elevated comprehension of ferroptosis and its latent translational utilities. By revealing the basic processes and investigating treatment possibilities, this review provides a crucial resource for scientists and medical practitioners, aiding in a deep understanding of ferroptosis and its effects in various disease situations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Programmed cell death (PCD) plays a critical role in various cellular processes, including embryogenesis, cell growth, cellular immunity, and other biological processes [1,2,3]. Over the past half-century, several pathways of cell death, such as apoptosis, pyroptosis, and autophagy, have been discovered [4]. In 2003, Erastin and RAS-selective lethal 3 (RSL3) were found to effectively inhibit NRAS-mutant HT-1080, but without the release of mitochondrial cytochrome c, activation of caspase, or fragmentation of chromatin in human foreskin fibroblasts [5]. It was not until 2012 when Dr. Brent R. Stockwell discovered that iron chelation weakens the effect of Erastin in NRAS-mutant HT-1080, leading to the identification of "ferroptosis", an iron-dependent cell death form [6, 7] (Fig. 1).

Timeline diagram depicting essential discoveries in the field of ferroptosis research. The exploration of ferroptosis originated from the identification of system xc-, which was initially reported in 1980. Nevertheless, the specific term "ferroptosis" was officially coined and introduced in the scientific community in 2012
With the progress of research, ferroptosis was identified as an iron-dependent programmed cell death. Distinct from apoptosis, necrosis, and autophagy, the morphological feature of cells in ferroptosis include mitochondria shrinkage and membrane density increased [7, 8]. The unique process of ferroptosis is the dysregulation of iron metabolism and the accumulation of reactive oxygen species (ROS) [9, 10]. The sufficient concentration oxidation of polyunsaturated fatty acids (PUFAs) and phospholipids, the dysregulation of iron metabolism, and the loss of antioxidant defense system execute the ferroptosis [11] and the mechanism of ferroptosis involves a complicated interplay between multiple cellular pathways, including iron metabolism, lipid metabolism, and antioxidant defense mechanisms [12].
Due to involving various and complicated signaling, ferroptosis plays an important role in the occurrence and development of major chronic diseases and different roles in different disease contexts. A growing body of evidence suggests that the imbalance of ferroptosis affects, development and aging [13, 14], and is closely related to the tumor [8, 15, 16], ischemic diseases [17,18,19,20,21,22], neurodegenerative disease [23, 24], organ transplantation [25, 26], cardiovascular disease [27,28,29], autoimmune functions [15, 30], infection [31, 32], iron-overload disease [33], and so on (Fig. 2). Of note, inducing ferroptosis can significantly enhance the sensitivity of chemotherapy drugs to suppress tumor [34, 35], on the other hand, the occurrence of ferroptosis can aggravate the severity of the disease [20, 36]. Although many compounds targeting the key ferroptosis regulators, like glutathione peroxidase 4 (GPX4) and solute carrier family 7 member 11 (SLC7A11, Cystine transporter, also commonly known as xCT), no compounds targeting ferroptosis can be applied to any diseases clinically. Recently, the structure of erastin-bound xCT-4F2hc (4F2 cell-surface antigen heavy chain, SLC3A2, also called CD98) complex had been solved [37], which provides a molecular basis for drugs development targeting on SLC3A2.

The involvement of ferroptosis in various human diseases. Ferroptosis has played important roles in multiple system diseases, such as lung diseases, nervous system diseases, heart diseases, breast diseases, gastric diseases, liver diseases, pancreatic diseases, kidney diseases, intestines diseases, reproductive diseases, skin diseases, musculoskeletal system diseases and so on
In the subsequent sections, our attention converges on the explication of ferroptosis mechanisms, coupled with the accentuation of its pertinent disease-associated targets and bioactive compounds. This assumes pivotal importance, given its potential to create innovative avenues for therapeutic interventions within disorders wherein ferroptosis assumes a key position. This review uncovered the hidden insights about ferroptosis, with the main goal of highlighting its important status as a newly recognized therapeutic target and its deep relevance to various disease states, and aiding researchers in achieving a clearer comprehension of the initiation, progression, and involvement of ferroptosis in various diseases.
Mechanisms of ferroptosis
Distinct from conventional cell death forms like apoptosis and necrosis, ferroptosis uniquely hinges on dysregulated iron metabolism and ROS generation [9, 10], featuring an intricate interplay across multiple cellular pathways encompassing iron and lipid metabolism, alongside antioxidant defenses [12]. Dysregulated iron metabolism, characterized by the accumulation of labile iron ions in the cytoplasm, plays a central role in ferroptosis by catalyzing the Fenton reaction, which leads to the production of highly reactive hydroxyl radicals (•OH) from hydrogen peroxide (H2O2) [38, 39]. These •OH entities interact with cellular membrane PUFAs, kindling lipid peroxidation and ensuing oxidative impairment [40]. Central to ferroptosis, lipid peroxidation arises from PUFAs accruement in cellular membranes [41, 42], predominantly in phospholipids like phosphatidylethanolamine (PE), phosphatidylcholine (PC), and cardiolipin (CL) [43,44,45]. This accrual engenders lipid peroxidation process, thus destabilizing cellular membranes, leading to cellular damage and ultimately cell death [46]. The cellular antioxidant defense system [47, 48], including enzymes such as superoxide dismutase (SOD) [10, 49], GPX4 [42, 50], catalase [51], alongside non-enzymatic antioxidants such as glutathione (GSH) [50] and vitamin E [52], orchestrates ferroptosis regulation. These constituents synergistically counteract ROS and lipid hydroperoxides, forestalling lipid peroxidation and consequent ferroptosis (Fig. 3). Beyond iron and lipid metabolism, and antioxidant defense mechanisms, several other pathways contribute to ferroptosis modulation. These encompass cellular metabolism [53], the activity of lipid metabolism enzymes [54], and the modulation of cellular redox status [55]. Furthermore, the interplay between ferroptosis and other types of cell death is an active area of research that continues to expand our understanding of the mechanism of ferroptosis [49, 56, 57]. A deeper understanding of the molecular and cellular mechanisms underlying ferroptosis increase the potential to uncover novel therapeutic targets and strategies for the treatment of various diseases associated with dysregulated iron metabolism and oxidative stress.

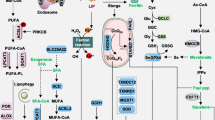
Several intrinsic or cell-autonomous mechanisms profoundly impact cellular susceptibility to ferroptosis. This non-exhaustive compilation encompasses metabolic pathways that intricately regulate iron levels, polyunsaturated fatty acids (PUFA), glutathione peroxidase 4 (GPX4), and ferroptosis suppressor protein 1 (FSP1). Abbreviations: TF: transferrin; TFR1: transferrin receptor 1; NRF2: nuclear factor erythroid 2–related factor 2; IREB2: Iron Responsive Element Binding Protein 2; HSPB1: heat shock protein beta 1; PKC: protein kinase C; Actin cytockeleton: a collection of actin filaments with their accessory and regulatory proteins; Ferritin: a protein that stores iron; SFXN1: siderofexin 1; MUFA: Monounsaturated fatty acids; Acetyl-CoA: acetyl coenzyme; HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme; IPP: isopentenyl pyrophosphate; FPP: Fertilization promoting peptide; GGPP: geranylgeranyl pyrophosphate; CoQ: coenzyme-Q; CoQH2: reduced coenzyme Q; ROS: Reactive oxygen species; GSH: glutathione; GSSG: glutathione disulfide; NADPH: nicotinamide adenine dinucleotide phosphate; NADP + : Nicotinamide Adenine Dinucleotide Phosphate; MESH1: metazoan SpoT homolog-1
The roles of iron metabolism
Dietary iron, predominantly in oxidized ferric (Fe3+) form, is assimilated by duodenal and proximal jejunal enterocytes through the divalent metal transporter 1 (DMT1) [58,59,60]. To be physiologically absorbed, Fe3+ must be converted to a ferrous (Fe2+) form or bind to co-factors, such as heme [60]. Upon entry into cells, Fe2+ associates with transferrin (Tf), which facilitates the translocation of iron into circulation via the iron exporter ferroportin (FPN). Inside the cells, iron is internalized in endosomes via transferrin receptor 1 (TfR1) and then translocated to the cytosol by DMT1, constituting the labile iron pool (LIP)—a crucial source of Fe2+ and a key regulator of iron metabolism [61,62,63,64,65,66]. Mitochondrial iron comes from endosomes through the DMT1 and mitoferrin interaction, or from the LIP, facilitated by DMT1, mitoferrin, and siderofexin (SFXN1) [67,68,69]. Superfluous iron from the LIP is sequestered in ferritin, of which the lysosomal degradation can replenish the LIP. Cellular iron efflux is mediated by FPN, with hepatocytes and spleen macrophages acting as pivotal iron storage sites [70]. Among the multitude of processes and signaling pathways regulating systemic iron metabolism, the hepcidin-mediated ferroportin internalization and degradation, or the hepcidin-FPN axis, is the paramount mechanism, governing dietary iron absorption and senescent red blood cell recycling [71].
Integral to the basic physiological processes such as oxygen transport, energy synthesis, immune response, DNA replication, and the tricarboxylic acid cycle (TCA), iron's centrality is indisputable [72, 73]. Intriguingly, this iron-sulfur cluster (ISC) -dependent electron transport concurrently augments endogenous ROS generation within mitochondria [72]. While ROS plays an essential role in preserving cellular equilibrium and signaling, the overload of ROS initiates oxidative damage and deleterious outcomes [74, 75]. Concomitantly, iron can also catalyze reactions to induce excessive ROS production via the Fenton reaction, underscoring the delicate balancing of iron metabolism [74, 75]. Therefore, any disturbance in the dynamics of iron import, sequestration, or export can destabilize cellular iron homeostasis, impacting the propensity toward ferroptosis. Substantial evidence suggests that amplified iron import, ferritin degradation (a key iron storage protein), and iron derivative accumulation contribute to ROS production together, thereby igniting the ferroptosis cascade [76, 77].
The orchestration of ROS production via the iron-catalyzed Fenton reaction serves is critical to ferroptosis. Notably, iron-bearing proteins such as Cytochrome P450 enzymes, Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), and subunits of the mitochondrial electron transport chain generate superoxide radicals (O2•−). Following this, SOD facilitates the conversion of O2•− to H2O2. As a result, heme and containing proteins are oxidized by O2•− and H2O2, leading to the release of reactive Fe2+ and the expansion of LIP. This catalysis prompts the Fenton reaction, which, in turn, yields •OH. These •OH then interact with polyunsaturated lipids, causing lipid radicals (L•), lipid peroxidation, and final ferroptosis [78]. Thereafter, L• reacts with additional polyunsaturated lipids, generating lipid hydroperoxide (LOOH) and more L•. Upon interaction with Fe2+ and Fe3+, LOOH converts into LO• and lipid peroxy radical (LOO•) [79, 80]. Arachidonate-15-lipoxygenase and other iron-containing lipoxygenases (LOXs) catalyze the reaction between O2 and polyunsaturated lipids, forming LOOH, with iron integral to the catalytic subunit of LOX. Ferroptosis is typically triggered by iron-dependent LOXs and expanded by the iron-fueled Fenton reaction. Nonetheless, the concentration of iron to initiate ferroptosis remains unclear, necessitating further investigation.
Iron intricately interweaves with the foundational metabolism of glucose, lipids, and amino acids, all of which exhibit pertinent links to ferroptosis [81]. Iron insufficiency is recognized to influence glucose metabolism by affecting glucose utilization, amplifying glucose absorption and transportation via glucose transporter protein type 1(GLUT1). In contrast, iron surplus induces a decrease in insulin sensitivity and the emergence of insulin resistance, culminating in diminished glucose uptake and transport in vitro, but a contrasting impact in vivo [82,83,84,85,86,87]. Although the explicit role of iron in glucose metabolism remains elusive, these insights imply that glucose is the major metabolic regulator during iron perturbations. Concurrently, iron deficiency impinges on lipid metabolism, which attenuates the rate-limiting enzyme in fatty acid oxidation—Carnitine palmitoyl transferase 1 (CPT-1)—in fetal liver [88]. Moreover, iron surplus initiates the inhibition of hepatic expression of peroxisome proliferator-activated receptor α, while hydroxyl radicals and nitrate anions implicated in the oxidation of PUFAs are also products of the Fenton reaction [89]. Thus, iron deficiency undermines fatty acid oxidation and desaturation while fostering lipogenesis [88,89,90,91]. Iron also engages in amino acid transport and synthesis, e.g., 4-hydroxyproline is derived from proline through the iron-dependent dioxygenase prolyl-4-hydroxylase, and cysteine dioxygenase, a key player in cysteine catabolism, is iron enzyme [92, 93]. Though iron plays a critical role in amino acid metabolism, the regulatory details await further exploration [79, 92,93,94,95].
Numerous iron-associated metabolic pathways have been pinpointed to either promote or inhibit ferroptosis. Following iron uptake and the subsequent conversion of Fe3+ to Fe2+, facilitated by the Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3), free Fe2+ concentrations escalate, which triggers ferroptosis by propelling the Fenton reaction and lipid peroxidation [74]. Ferritinophagy, the process of ferritin degradation, also yields free Fe2+ capable of inducing ferroptosis [96]. Additionally, increased cytoplasmic Fe2+ level, caused by ferritinophagy, have been discovered to enhance the expression of SFXN1 on the mitochondrial membrane [96]. SFXN1, reciprocally, expedites the transfer of Fe2+ from the cytoplasm to the mitochondria, precipitating mitochondrial ROS production and ferroptosis [97]. Apelin-13, a peptide hormone, is reported to increase the expression of SFXN1 and nuclear receptor coactivator 4 (NCOA4), inducing ferroptosis via ferritinophagy and the shuttling of Fe2+ into mitochondria [98, 99] (Fig. 4).

Iron metabolism in ferroptosis. Abbreviations: STEAP3: Six-Transmembrane Epithelial Antigen of Prostate 3; TRPML1: transient receptor potential mucolipin 1; DMT-1: divalent metal transporter 1; NCOA4: Nuclear receptor coactivator 4; FPN: ferroportin
While progress has been made in exploring the mechanisms of iron homeostasis, the functions of iron are not fully understood yet. The roles of iron-mediated ROS production and iron-containing enzymes in this process are still uncertain. The roles of iron homeostasis and proteins following lipid peroxidation in ferroptosis are still elusive, of which, however, the involvement in various diseases like cancer, neurodegenerative diseases, and ischemia–reperfusion injury-related diseases has been noted. Hence, treatments to suppress ferroptosis signals could potentially benefit iron overload diseases. Iron chelating agents are being studied as potential therapies for ferroptosis diseases, though more in vivo studies are needed to clarify the mechanisms and the effect. Future challenges include developing an effective and safe iron chelator. Further studies into the mechanisms of iron-dependent lipid peroxidation are required to identify more treatment targets for diseases associated with ferroptosis, as well as whether iron overload alone can cause ferroptosis in different cells or tissues.
Lipid peroxidation
Lipid peroxidation, a critical mechanism in ferroptosis, is a procedure in which oxidizing agents, like free radicals, target lipids that possess carbon–carbon double bonds, particularly in PUFAs [100,101,102,103]. Lipid peroxidation includes three sequential phases: inception, perpetuation, and cessation [104,105,106]. Initiating with the inception phase, prooxidants, such as hydroxyl radicals, pluck an electron from allylic hydrogen, yielding a carbon-centric L•. Transiting to the perpetuation phase, this lipid radical swiftly amalgamates with oxygen, thus generating a LOO•. Subsequently, the LOO• detaches a hydrogen atom from a distinct lipid molecule, producing a nascent lipid radical and LOOH, which perpetuates the chain reaction. Ending in the cessation phase, antioxidants, like vitamin E, donate a hydrogen atom to the LOO•, thus producing a corresponding vitamin E radical. This nascent radical then interacts with another LOO•, resulting in the synthesis of non-radical derivatives. It is noteworthy that, once catalyzed, lipid peroxidation induces a cascade of chain reactions until cessation derivatives are generated [104, 107, 108].
The link between lipid peroxidation and ferroptosis arises from the fact that the accumulation of lipid peroxides to lethal levels during the ferroptosis process [43, 50]. Specifically, the oxidation of PUFAs is crucial for the execution of ferroptosis [40, 45, 109]. The process is facilitated by lipoxygenases and iron [44]. Importantly, lipid peroxidation in ferroptosis is delicately regulated by several systems, including the glutathione/GPX4 system and the ferroptosis suppressor protein 1 (FSP1)/CoQ10 system, which neutralize peroxidized lipids and thus inhibit ferroptosis [9]. One of the obvious results of lipid dysregulation is ferroptosis, therefore, investigating lipid peroxidation holds significance in regulating ferroptosis.
However, ferroptosis and lipid peroxidation are intertwined yet distinct biological processes. Ferroptosis constitutes a specialized form of regulated cell death marked by the iron-dependent accumulation of lipid peroxides, eventually results in cell membrane deterioration and cell death [9]. In contrast, lipid peroxidation encompasses a broader biochemical phenomenon involving the oxidative breakdown of lipids within cell membranes, often instigated by various oxidative stresses, such as toxins, ultraviolet etc. [107]. While ferroptosis is a specific outcome resulting from disrupted cellular redox balance, lipid peroxidation is a multifaceted process that can occur under diverse conditions, not always leading to cell death. Ferroptosis is thus a subset of the broader lipid peroxidation landscape, characterized by intricate molecular mechanisms and distinctive cellular consequences (Fig. 5).

Lipid peroxidation in ferroptosis. Abbreviations: ACSL-4: acyl-CoA synthetase long chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LysoPL: lysophospholipase
Fatty acids
Fatty acids command a cardinal role in ferroptosis. As indispensable nutrients, they play critical functions in cellular and physiological processes, encompassing energy metabolism and signaling pathways [110]. Four major categories of fatty acids exist: saturated, monounsaturated, polyunsaturated, and trans fats, with PUFAs peroxides reported to exhibit a close association with ferroptosis [111]. PUFAs, containing multiple double bonds (C = C), are predominantly derived from the diet and are pivotal components of cell membranes [112, 113]. They participate in diverse processes, such as inflammation, vascular function, platelet aggregation, synaptic plasticity, cellular growth, immune response, and cellular proliferation [114, 115]. The abundance of double bonds in PUFAs enhances their vulnerability to oxidation, given the susceptibility of the C-H bond in PUFAs to such an oxidative attack [116]. Investigations have underscored that membrane PUFAs are the principal targets of oxidative stress caused by ROS. PUFAs have been found to produce free radicals during their interaction with these ROS, subsequently triggering a cascade that amplifies the extent of damage [78, 104, 117].
Noteworthily, the exogenous introduction of monounsaturated fatty acids (MUFAs), which directly contest with PUFAs, has exhibited an impressive capacity to attenuate erastin-induced ferroptosis [115, 118]. This implies that PUFAs, in contrast to MUFAs, assume a critical role in lipid peroxidation processes and ferroptosis. It has been reported that MUFAs can maintain a state of ferroptosis resistance by curtailing lipid peroxidation in a long-chain acyl-coenzyme A synthases (ACSLs)-dependent manner [9]. Further investigations validated that ACSL3, specifically, is in charge of producing ferroptosis resistance and attenuating saturated fatty acid lipotoxicity [119]. These findings suggest that exogenous MUFAs may change the constitution of the cell membrane by replacing membrane PUFAs and that the replaced PUFAs may be sequestered in cytoplasmic lipid droplets, restraining their pro-ferroptosis activity [120]. Additionally, it has been demonstrated that PUFAs can provoke cancer cell death through escalated ROS production and formation of lipid peroxides [121, 122]. Consequently, the scrupulous regulation of PUFAs and the development of targeted delivery methodologies for PUFAs, as well as techniques to amplify or inhibit ROS and lipid peroxidation production in specific contexts, could provide promising trajectories for therapeutic interventions in various ailments.
Ether phospholipids
Ether phospholipids (ePLs), by their unique properties and susceptibility to peroxidation, have been implicated within the matrix of ferroptosis. As a divergent class of phospholipids from the ester phospholipids, ePLs harbor an ether bond at the sn-1 position of the glycerol backbone which is the major difference with an ester bond [123]. Investigations provide a deep understanding of the role of ePLs, particularly plasmalogens, in regulating lipid peroxidation and ferroptosis [123, 124].
ePLs are notably vulnerable to peroxidation by lipoxygenases, potentially catalyzing the accumulation of lipid peroxides and contributing to ferroptosis. This susceptibility hinges on the presence of an ether bond at the sn-1 position of the glycerol backbone of ether phospholipids, which is more vulnerable to ROS assault than the ester bond found in other typical phospholipids [125, 126]. The metabolic reduction of oxidized ether phospholipids, the elimination of lipid peroxides from the membrane, and the suppression of the ether lipid peroxidase have been shown to guard against ferroptosis [127]. The selective vulnerability of certain cells or tissues to ferroptosis is also tied to the levels of ether phospholipids within their membranes.
The proteins related to ePLs are also investigated intensively. Cui et al. reported that sensitization to ferroptosis across various cancer cell lines following TMEM189 deletion. This suggests an unanticipated anti-ferroptosis role for TMEM189, distinguishing it from other ePL biosynthesis genes like glyceronephosphate O-acyltransferase (GNPAT), fatty Acyl-CoA Reductase 1 (FAR1), alkylglycerone phosphate synthase (AGPS), and 1-acylglycerol-3-phosphate o-acyltransferase 3 (AGPAT3) [123, 128]. Cui et al. postulated a mechanistic link where plasmalogens produced by TMEM189 downgrade FAR1 via negative feedback regulation, resulting in the suppression of ferroptosis [123, 128]. However, Zou et al. claimed that TMEM189 deficiency showed no significant link to ferroptosis [124]. The root of this discrepancy seems to lie in the cell lines utilized in the two studies. Further lipidomic analyses in these TMEM189-depleted cell lines will offer clarity on its precise role in the modulation of ferroptosis [129]. Recently, Liang et al. constructed a comprehensive whole-genome CRISPR activation screen and subsequent mechanistic investigation, identified phospholipid-modifying enzymes MBOAT1 and MBOAT2 as potent suppressors of ferroptosis [130]. These enzymes inhibit ferroptosis by reshaping the cellular phospholipid composition, independently of GPX4 or FSP1. Their transcriptional upregulation is governed by sex hormone receptors, estrogen receptor (ER) and androgen receptor (AR). Employing ER or AR antagonists in tandem with ferroptosis induction effectively impedes the growth of ER + breast cancer and AR + prostate cancer, even in cases of resistance to individual hormonal therapies. In summary, the interplay between ether phospholipids and ferroptosis is intricate, involving a delicate balance between susceptibility to lipid peroxidation and protective mechanisms against it. To fully understand the role of ether phospholipids in ferroptosis and their potential as therapeutic targets for diseases characterized by dysregulation of this process, further research is necessary.
ACSL4 and LPCAT3
Enzymes catalyzing the incorporation of PUFAs into phospholipids, such as ACSL4 and lysophosphatidylcholine acyltransferase 3 (LPCAT3), are paramount in the orchestration of ferroptosis [45, 131, 132]. ACSL4 plays an fundamental role in the metabolic process of membrane PUFAs, notably arachidonic acid (AA) and adrenic acid (ADA) [133]. This enzyme is critical in the conversion of these fatty acids into their respective CoA thioesters, which subsequently integrate into phospholipids under the guidance of LPCAT3. Both in vivo and in vitro evidence demonstrates that disruption of these enzymatic functions results in heightened resistance to ferroptosis stimuli [45]. Importantly, in the context of hepatocellular carcinoma, ACSL4-dependent mechanisms may have both tumor-promoting and tumor-inhibitory effects [134]. Additionally, evidence derived from both in vivo and in vitro studies corroborate that the ablation of LPCAT3 render a resilience against RSL3-mediated ferroptosis [43, 45, 132]. Therefore, the roles of these enzymes in cellular susceptibility to ferroptosis are pivotal, with implications for cancer progression and therapeutic interventions [135, 136].
LOXs and PEBP1
In general, two pathways could regulate lipid peroxidation, non-enzymatic autoxidation and enzyme-mediated reactions [44, 104, 137, 138]. In the presence of free Fe2+ and H2O2, Fe3+ is generated and hydroxyl radicals initiate the lipid peroxidation process by abstracting hydrogen from the bis-allylic position of PUFAs [107, 139, 140]. LOXs are non-heme iron-containing dioxygenases that catalyze the stereospecific addition of oxygen onto PUFAs, such as AA and linoleic acids, resulting in lipid peroxidation [141]. Structurally, LOX possesses a unique U-shaped fatty acid binding channel that allows easy access to PUFA substrates [142, 143]. Although several studies have shown that LOX inhibitors/knockout effectively inhibit ferroptosis in various disease models [137, 144], study have also reported that LOX inhibitors/knockout failed to inhibit RSL3-induced ferroptosis in renal carcinoma cells [44]. Further research is still needed to elucidate whether LOXs also participate in GPX4 inhibition during ferroptosis.
The well-known tumor suppressor protein p53 has been implicated in the intricate regulation of ferroptosis. p53 functions include amplifying ferroptosis by impeding the transcription of SLC7A11-an integral constituent of system xc− or by upregulating both spermidine/spermine N1-acetyltransferase 1 (SAT1) and glutaminase 2 [8, 144,145,146]. Conversely, p53 is also capable of curtailing ferroptosis via the suppression of dipeptidyl-peptidase 4 (DPP4) activity or through the elicitation of Cyclin-dependent kinase inhibitor 1A/p21 (CDKN1A/p21) transcription [147, 148], e.g., p53 can upregulate 15-LOX and thereby increase the sensitivity of cells to induced ferroptosis [144]. p53-mediated ferroptosis in response to TBH is independent of ACSL4, and the specific phospholipids accountable for p53-linked ferroptosis remain unidentified [149].
While LOXs predominantly target free PUFAs for oxidation, phospholipids embedded within the cellular membrane housing PUFAs transpire as the main targets during ferroptosis [44]. Notwithstanding this knowledge, the precise mechanistic pathway employed by LOXs to manipulate membrane phospholipids remains elusive. Preliminary data suggest a robust interaction between 15-LOX and phosphatidylethanolamine-binding protein 1 (PEBP1), a protein proposed to modulate the Raf-1-facilitated mitogen-activated protein kinase (MAPK) signaling cascade [150, 151]. Subsequent investigations hypothesize a stable complex formed between 15-LOX and PEBP1 that can modulate PUFAs, thus invoking ferroptosis [137]. Locostatin, a compound known to escalate oxidized PE concentrations and promote ferroptosis upon RSL3 treatment, is postulated to bolster the formation of the 15-LOX/PEBP1 complex [137]. Various disease models also revealed the accumulation of 15-LOX/PEBP1 complex resulted in elevated oxidized PEs and ferroptosis [137]. Further validation of PEBP1’s integral role in orchestrating ferroptosis arises from the observation that selective ferroptosis inhibitors-ferrostatin-1 (Fer-1), liproxstatin-1, and α-tocopherol-also engage with the 15-LOX2/PEBP1 complex [7, 152]. Whereas corroborating evidence emphasizes PEBP1's fundamental role in producing oxidized PEs, no discernible effects on free ETE (eicosatetraenoic acid) have been reported. Intriguingly, Fer-1 selectively hinders the formation of 15-hydroperoxy (Hp)-arachidonoyl-phosphatidylethanolamine (15-HpETE-PE) but not 15-HpETE, implying that Fer-1 specifically targets the 15-LOX2/PEBP1 complex, leaving free 15-LOX2 unimpeded [153]. These investigations corroborate that the collaboration between LOXs and PEBP1 is crucial in governing lipid peroxidation and the progression of ferroptosis.
Other oxygenases
Other oxygenases, such as NOXs and cytochrome p450 oxidoreductase (POR), are also involved in ferroptosis. While NOXs induce superoxide radicals, the extent of their requirement for ferroptosis remains contested [147, 154,155,156,157]. POR, identified as a ferroptosis contributor, facilitates electron transfer from NADPH to cytochrome p450, possibly promoting lipid peroxidation. Notably, POR's ubiquitous presence in various cancer cell lines suggests its potential significance in lipid peroxidation and ferroptosis [158, 159]. Further, an ER-resident oxidoreductase, NADH-cytochrome b5 reductase 1 (CYB5R1), and POR have been implicated in lipid peroxidation through H2O2 production and iron-dependent Fenton reaction [160]. Despite the common belief that LOXs primarily induce lipid peroxidation, their expression is limited in certain cancer cell lines. Intriguingly, POR is expressed in most cancer cells, suggesting an underestimation of POR's role in ferroptosis [159]. A comprehensive understanding of each enzyme's contribution to ferroptosis could pave the way for developing targeted therapeutic agents for related diseases.
Role of GPX4
Glutathione is a small molecule found in most cells. It is made up of three amino acids: glutamate, cysteine, and glycine. Glutathione is one of the most important antioxidants in cells, as it is responsible for neutralizing a variety of harmful substances [161, 162]. Glutathione exists in reduced GSH and oxidized (GSSG) states [163]. In the reduced state, glutathione can donate a reducing equivalent to unstable molecules like ROS. Once the electron is donated, glutathione becomes oxidized and is turned into GSSG. The ratio of GSH to GSSG within cells is usually used as a measure of cellular oxidative stress [164]. Glutathione serves as a cofactor for the enzyme GPX4, which helps to reduce lipid peroxides and prevent lipid peroxidation [50]. When glutathione is depleted, GPX4 cannot function effectively, leading to an accumulation of lipid peroxides and increased susceptibility to ferroptosis.
System xc−/GSH/GPX4 axis is the main mechanism responsible for the catalyzation of phospholipid hydroperopxides [7, 165, 166]. The key component of the xc−/GSH/GPX4 axis is system xc−, which is a highly selective uptake system for cystine (oxidized cysteine) and cystathionine [167,168,169]. System xc− exchanges cysteine and glutamate in and out of the cell at a 1:1 ratio [7]. The xCT light chain, which is the substrate-related subunit of system xc−, is subject to complicated transcriptional control. Under oxidative stress and cysteine deprivation conditions, xCT is upregulated by apoptosis-inducing factor-4 (ATF4) [170]. It has also been reported that p53 can inhibit xCT expression and increase sensitivity to ferroptosis [8, 171].
Once cystine was taken up by the cell, it is converted to cysteine by GSH and/or thioredoxin reductase 1, which is then used for GSH synthesis [172]. Besides, other mechanisms, such as the transsulfuration pathway and the neutral amino acid transporter, also contribute to cysteine production [173, 174]. Cysteine plays a significant role by contributing the essential redox-active thiol group central to its multifaceted functions. Within cells where GSH is produced, intracellular cysteine concentrations are relatively modest, thereby typically governing GSH synthesis due to the confined availability of cysteine. During instances of heightened demand for GSH synthesis, there is an intensified cellular uptake of cysteine from the more abundant extracellular environment. Interestingly, the predominant extracellular form of cysteine is cystine, characterized by its oxidized state. Subsequent to cellular entry, cystine can undergo reduction to cysteine by cystine reductase, thereafter being channeled towards GSH or protein synthesis. The distinctive recognition of these compounds by specific transporters plays a pivotal role, as the relative concentrations of cysteine and cystine in the plasma modulate the ability of cells to import either substance, contingent upon the unique profiles of transporter expression [175, 176].
GPX4 takes part in several physiological processes and is considered as the main inhibitory gene of ferroptosis [177]. GPX4 catalyzes lipid peroxides and is crucial for preventing the accumulation of lipid peroxides and subsequent ferroptosis [178]. The GPX4 pathway regulates ferroptosis in several ways: 1) Reduction of lipid peroxides: GPX4 converts lipid peroxides into their corresponding alcohols, which are less toxic and less likely to cause ferroptosis. Inhibition of GPX4 activity leads to the accumulation of lipid peroxides, which triggers ferroptosis. 2) Maintenance of membrane integrity: The cell membrane is particularly susceptible to lipid peroxidation, which can lead to membrane damage and subsequent ferroptosis. GPX4 helps to maintain membrane integrity by reducing lipid peroxides in the cell membrane. 3) Regulation of iron metabolism: Iron is a key mediator of ferroptosis, as it catalyzes lipid peroxidation through the Fenton reaction [178]. GPX4 can also regulate iron metabolism by binding to iron ions and preventing their participation in the Fenton reaction. Overall, the GPX4 pathway plays a crucial role in regulating ferroptosis by reducing lipid peroxides, maintaining membrane integrity, and regulating iron metabolism (Fig. 6).

The role of GPX4 in ferroptosis. Abbreviations: Glu: glutamic acid; Gln: Glutamine; Cys: cysteine; Gly: Glycine; P53: a tumor suppressor protein; KEAP1: Kelch-like ECH-associated protein; 12-LOX: 12-lipoxygenase; GLS2: glutaminase 2; γ-GC: γ-glutamylcysteine; GSS: glutathione synthetase; GSR: glutathione reductase
Role of FSP1
Studies have indicated that the sensitivity of different cell lines to inhibitors of GPX4 varies significantly, suggesting the existence of unexplored downregulatory mechanisms of ferroptosis beyond GPX4 [179]. Using synthetic lethal CRISPR-Cas9 screening, researchers have identified FSP1 as another key factor in ferroptosis resistance [180, 181]. Initially referred to as AIF-like mitochondrion-associated inducer of death (AMID) or Apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as FSP1), FSP1 was the first gene named for ferroptosis [182]. However, unlike AIF, FSP1 is predominantly found in the cytosol, with a potential affinity towards the mitochondrial outer membrane, although it lacks a long N-terminal mitochondrial targeting sequence as seen in AIF [183].
Subsequent studies have confirmed that FSP1 expression confers resistance to ferroptosis but not apoptosis [184]. Further research has revealed that myristoylation of FSP1 accelerates its accumulation on the plasma membrane, where it acts as an oxidoreductase and lipophilic radical-trapping antioxidant, reducing CoQ10 to ubiquinol, thus preventing the peroxidation of PUFAs in the lipid bilayer, and suppressing ferroptosis [181]. Doll’s group has demonstrated that the FSP1-CoQ10-NAD(P)H pathway operates independently with the GPX4 pathway, functioning to either directly scavenge lipid radicals by reducing ubiquinone to ubiquinol, or indirectly regenerate oxidized-tocopheryl radical, thereby suppressing ferroptosis [181]. Such observation elucidates the protective role of extra-mitochondrial ubiquinone in tissues and cells, which has been a long-standing puzzle due to the canonical function of ubiquinone in the mitochondrial electron transport chain [185]. However, the regulation of FSP1 oxidoreductase activity or how its subcellular localization impacts its involvement in various physiological and pathological processes, remains to be further elucidated [180, 181, 183, 186]. Recently, FSP1 was reported that it can convert Vitamin K into the reduced form, hydroquinone (VKH2) [187, 188]. Nevertheless, the versatility of FSP1 in oxidizing and reducing substrates, including NADH, NADPH, ubiquinone, and α-tocopherol, implies the sophisticated control of FSP1 activity (Fig. 7).

The role of FSP1 in ferroptosis. Abbreviations: VK: Vitamin K
The prospect of exploiting FSP1 as a therapeutic node to bolster the effectiveness of ferroptosis-based interventions and radiotherapy, notably in the milieu of Kelch-like ECH-associated protein 1 (KEAP1) and Kirsten rat sarcoma virus (KRAS) mutant lung malignancies, has elicited substantial scientific interest [189, 190]. A seminal exploration subjected 30,000 pharmacologically pertinent compounds to rigorous screening, seeking agents capable of precipitating cellular death in cells singularly dependent on FSP1, consequently spotlighting iFSP1 as a robust inhibitor [181]. Another investigation suggested that ferroptosis sensitizer 1 (FSEN1) proficiently inhibits FSP in vitro while also thwarting ferroptosis within the confines of cultured cancer cells [191]. Nonetheless, the necessity for additional investigation is underscored to validate whether FSEN1 can inhibit FSP1 in vivo. It is noteworthy that the applicability of FSEN1 is constricted to human FSP1 [191], thereby decreasing the utility in the scrutiny of mouse FSP1 or neoplastic growth within Genetically Engineered Mouse Models. Anticipated investigative endeavors must strive to establish whether other FSP1 inhibitors unearthed in this study can inhibit mFSP1 and their repercussions on preclinical tumor progression paradigms [191]. Conversely, amplifying FSP1 activity within models of traumatic pathologies, such as ischemia–reperfusion injury, carries immense therapeutic promise. Yet, this field remains relatively unexplored, emphasizing the urgency for concentrated research endeavors to bridge this knowledge gap.
Other pathways regulating ferroptosis
While the central mechanism governing ferroptosis centers around iron metabolism, lipid peroxidation, GPX4, and FSP1 pathway, it is increasingly apparent that a multitude of ancillary pathways also significantly contribute to the modulation of this distinctive form of cellular death. Recent investigations have unveiled the role of the Hippo-Yes-associated protein (YAP) pathway, AMP-activated protein kinase (AMPK) signaling, and hypoxia pathway in ferroptosis. Fascinatingly, cells cultured at heightened densities demonstrate escalated resistance to ferroptosis triggered by cysteine deprivation and GPX4 inhibition [192,193,194]. The Hippo-YAP pathway, illustrious for its orchestration of cell proliferation, stress recognition, and organ size moderation, has been scrutinized for its correlation with ferroptosis [195, 196]. Findings delineate that E-cadherin-mediated cell–cell contacts kindle the Hippo signaling pathway via the neurofibromatosis 2 (NF2) tumor suppressor protein, thus curbing nuclear translocation and activity of the transcriptional co-regulator YAP in epithelial cells [193]. YAP, along with its akin homolog TAZ, targets numerous regulators of ferroptosis, encompassing ACSL4 and transferrin receptor TfR1, postulating that the dynamism of the Hippo pathway may modulate cellular responsiveness to ferroptosis, thereby escalating susceptibility upon Hippo suppression and YAP activation [156, 193].
Energy and metabolic stress under normal physiological conditions are crucial for maintaining homeostasis [197]. Disturbances in energy production can result in excessive ROS and cell death [198, 199]. However, interventions mimicking energy stress have been shown to prevent ferroptosis and lipid peroxidation, an effect credited to AMPK, an energy-sensing kinase [112, 200]. The activation of AMPK during glucose deprivation initiates a protective mechanism against ferroptosis, mainly inhibiting PUFA biosynthesis [44, 45]. These findings suggest that such an energy stress program can protect against renal ischemia–reperfusion damage and potentially guard against organ damage related to energy failure.
Initial investigations, suggesting minimal alterations to erastin-induced ferroptosis sensitivity in a 1% oxygen environment, challenged the presumption that hypoxia induces ferroptosis [201]. Hypoxia escalates ROS production via mitochondrial complex III and augments cellular H2O2 levels, enabling the Fenton reaction [202]. Concurrently, in renal clear cell carcinoma, activation of hypoxia-inducible factors (HIFs) amplifies ferroptosis sensitivity due to GPX4 inhibition, particularly via the HIF2α isoform. Hypoxia initiates HIF2-mediated expression of the hypoxia-inducible lipid droplet-associated protein (HILPDA), resulting in polyunsaturated lipid enrichment [179]. This HIF2-HILPDA-driven heightened sensitivity to ferroptosis suggests an evolutionary mechanism to eradicate hypoxic tumors in the early stages.
Along with the progress, the role of ferroptosis in a proliferating array of disease processes becomes increasingly evident, thereby illuminating novel therapeutic approaches. Operating in concert with other strategies, ferroptosis enriches current treatment paradigms, providing potential solutions to drug resistance challenges. Notwithstanding, our understanding of ferroptosis remains embryonic, with numerous unresolved enigmas left. While it is acknowledged that ferroptosis is initiated by the peroxidation of PUFAs in the cellular membrane and organellar membranes such as the endoplasmic reticulum, the precise mechanisms through which these processes lead to cell death remain uncertain. Furthermore, a thorough investigation into the underlying initiatory and regulatory mechanisms of ferroptosis, the participants involved, and most critically, the complicated interplay between various cell types, persists as an active research domain. Complicating the traditional understanding of ferroptosis, the potential regulation of this process by other metallic ions, such as copper, challenges the dominant position of iron [203]. Thus, deciphering the exact molecular mechanisms and elucidating the role of upstream iron metabolism genes in ferroptosis becomes essential. Furthermore, the identification of distinctive ferroptosis markers is of profound significance to future investigations. In conclusion, the advent of ferroptosis research has inaugurated a promising landscape in disease research, offering considerable potential in devising highly targeted therapies. Nonetheless, much remains to be discovered about the mechanisms of ferroptosis and its role in various diseases, which are important future research directions.
Physiological functions of ferroptosis
To investigate the biological processes in which ferroptosis is involved, several markers have been developed, including those that detect lipid peroxidation, mitochondrial morphologies, specific gene expression, and TfR1 expression and location [204, 205]. Through the combination of these approaches, ferroptosis has been shown to be critical in tumor suppression, immune surveillance, development, and aging.
Ferroptosis in tumor suppression and immune functions
The first evidence linking ferroptosis and tumors was the discovery that p53, a well-known tumor suppressor, sensitizes tumor cells to ferroptosis by inhibiting the expression of SLC7A11, a key component of the cystine/glutamate antiporter that mediates cystine transport and represses ROS-induced ferroptosis [8, 206,207,208]. In human tumors, high expression of SLC7A11 can dampen ferroptosis and diminish the inhibition of tumor growth in xenograft models by acetylation-defective mutant p53 (K117R; K161R; K162R encoding the so-called p53 3KR) [8]. Further investigations revealed that mammalian lipoxygenase family member arachidonate 12-Lipoxygenase (ALOX12) is crucial for p53-dependent ferroptosis. Inactivation or missense mutations of ALOX12, even haploinsufficiency, can ablate p53-mediated tumor growth suppression [149, 209, 210]. Mechanistically, ALOX12 has been identified as a bona fide binding partner of SLC7A11, and its lipoxygenase activity is inhibited in a dosage-dependent manner by SLC7A11 level, which is downregulated by p53 [211]. A nonsynonymous single-nucleotide polymorphism at codon 47 (S47) in tumor protein p53 (TP53 or p53), which is restricted to individuals of African descent, has been found to impair ferroptosis and, therefore, p53-dependent tumor suppression [171]. In cells with S47 mutation, the level of glutamine synthase 2 (GLS2), a glutaminase that converts glutamine into glutamate to induce ferroptosis, is markedly decreased, and the negative regulation of p53 to SLC7A11 is compromised compared to wild-type cells [146, 171]. Moreover, in cells and mice with S47 mutation, the cellular abundance of antioxidants GSH and CoA is elevated, leading to decreased ferroptosis sensitivity [212]. Additionally, the S47 variant of TP53, which has been shown to ablate ferroptosis in cells and mice, also results in iron accumulation in macrophages, altering macrophage cytokine profiles and causing increased susceptibility to bacterial infection and limitation of malarial infection. A recent study found that ALOX12 activation induced by a photosensitizer in cancer cells significantly increases lipid reactive oxygen species and promotes ferroptosis, independent of ACSL4 [213].
MLL4 is an epigenetic regulator and one of the most frequently mutated genes in cancer biology. Depletion of MLL4 in mice promotes features of human precancerous neoplasms. On one hand, MLL4 deficiency suppresses the expression of key lipoxygenases, such as ALOX12, ALOX12B, and ALOXE3, which are involved in driving ferroptosis. On the other hand, lower expression of MLL4 is significantly associated with decreased expression levels of anti-ferroptosis regulators, such as GPX4, SCD1, and GCH1 [214].
The tumor suppressor BRCA1-associated protein 1 (BAP1) is a nuclear de-ubiquitinating enzyme that is responsible for histone 2A modification and gene transcription regulation. BAP1 can regulate ferroptosis primarily through SLC7A11 [215, 216]. Specifically, BAP1 reduces ubiquitinated H2A occupancy on the promoter of SLC7A11, resulting in the repression of SLC7A11 expression. This abrogates cystine uptake and induces ferroptosis [215, 217].
Cysteine desulfurase (NFS1) is an iron-sulfur cluster biosynthetic enzyme that is essential for cancer cell survival when exposed to oxygen [218]. Suppression of NFS1 limits iron-sulfur cluster availability, promoting the iron-starvation response [219] increasing ferroptosis susceptibility [184, 218, 219].
Similar to previous studies that have found excessive accumulation of oxidized PUFA-containing lipids can induce ferroptosis, acidic cancer cells exposed to PUFAs also undergo ferroptosis [220]. PUFAs elevate susceptibility to ferroptosis in the presence of ferroptosis inducers erastin and RSL3, which may be due to diminished upregulation of GPX4 and SLC7A11, as well as apparent downregulation of dihydrofolate reductase (DHFR) and FSP1 [221]. However, unlike acidic cancer cells, uptake of PUFAs from the tumor microenvironment impairs the antitumor ability of CD8+ T cells in a mouse melanoma model B16 [222]. PUFAs promote the expression of CD36 on CD8+ T cells from human and murine cells, which then activates lipid peroxidation and ferroptosis, reducing cytotoxic cytokine production and antitumor function of CD8+ T cells.
Of note, in melanoma and ovarian mouse models, CD8+ T cells, when activated by anti-PD-L1 antibody, have been found to drive tumor cell lipid peroxidation and ferroptosis, and this enhanced ferroptosis can promote the anti-tumor function of immunotherapy in turn [223]. In this process, interferon-γ (IFNγ) derived from activated CD8+ T cells has been shown to defer the expression of SLC3A2 and SLC7A11, inhibiting tumor cell cystine import and sensitizing tumor cells to ferroptosis. Furthermore, in a melanoma mouse model, IFNγ and AA, one of the PUFAs, have been identified as an anti-tumor combination [15]. IFNγ released from T cells is an activator of the ferroptosis regulator ACSL4 and can accelerate the incorporation of AA into phospholipids, subsequently inducing immunogenic tumor ferroptosis. This suggests that AA found in the tumor microenvironment could potentially be used together with IFNγ as a physiological inducer of ferroptosis.
While ferroptosis is known to serve as a guard in tumor suppression in most research, it appears to play an opposite role in immune functions. Apart from its impact on cytokine production in immune cells such as macrophages and CD8+ T cells, ferroptosis also regulates the homeostasis of follicular helper T (TFH) cells [224]. Upregulation of GPX4 by selenium addition has been shown to result in a higher number of TFH cells and elevate humoral immune response in immunized mice and young adults following influenza vaccination. Although evidence suggests that ferroptosis is involved in immunity, further investigation is needed to uncover more links between ferroptosis and immune functions.
Ferroptosis in development and aging
Due to the delayed development of ferroptosis detection methodologies, the physiological function of ferroptosis remains to be fully understood. Recently, a mouse monoclonal antibody called HNEJ-1 has been designed to specifically identify the most sensitive lipid peroxidation marker, 4-hydroxy-2-nonenal (HNE). This antibody has been used to monitor ferroptosis in different developmental stages of animal models [225]. In Fisher-344 rats, ranging from E9.5 to 2.5 years of age, a significant age-dependent increase in ferroptosis and iron accumulation has been observed in various organs [225]. This increase is also enhanced in a naturally accelerated aging animal model, the Senescence Accelerated Mouse-Prone 8 (SAMP8) mice [225]. Ferroptosis has also been found to occur during rat embryonic erythropoiesis, with its level decreasing as erythrocytes enucleate during the process of maturation. This maturation process is reduced in the presence of ferroptosis inhibitors, Lipro-1 and Fer-1. Inhibition of ferroptosis by melatonin, through neutralizing lipid peroxidation toxicity, has been shown to delay age-related cataract formation [226].
In addition to rats, ferroptosis also affects aging and development in other organisms such as C. elegans and Magnaporthe oryzae. In C. elegans, a reduction in GSH and an increase in ferrous iron typically occur in late life, and suppression of ferroptosis using lipid peroxidation inhibitor liproxstatin or iron chelator salicylaldehyde isonicotinoyl hydrazone has been shown to protect against GSH depletion toxicity, dramatically restrain age-related cell death, and improve the lifespan and healthspan of C. elegans [227]. Regarding to M. oryzae, ferroptosis is crucial for the developmental cell death of conidia during appressorium maturation in rice blast [228]. Inhibition of ferroptosis has been found to dampen the ability of M. oryzae to invade the host.
Ferroptosis in pathologies
Since the discovery of ferroptosis, evidence has implicated it in a broad array of pathological states including various types of cancer, ischemia–reperfusion (I/R) injury, neurodegenerative disorders, etc. As such, the elucidation of ferroptosis regulatory mechanisms and their relation to human disease has drawn substantial scientific attention. Consequently, therapeutic strategies to modulate ferroptosis, either as inducers to eradicate cancer cells or as inhibitors to protect neurons or ischemic tissues, have unfolded as a promising avenue of translational research.
Ferroptosis and tumor
Neoplasms encompass an array of genetically divergent subclones. In recent years, burgeoning evidence has underscored the cardinal role of ferroptosis in curbing neoplastic proliferation. A plethora of tumor-suppressive and oncogenic signaling pathways have been identified, which respectively promote or inhibit ferroptosis, offering potential perspectives in cancer therapeutics (Tables 1 & 2).
Tumor progression
Cancer is a disease characterized by the uncontrolled proliferation of abnormal cells, exhibiting features of unregulated cell growth, invasive expansion, and metastatic potential [290]. Recent years have witnessed remarkable strides in cancer diagnosis and holistic therapeutic approaches such as surgery, chemotherapy, radiation therapy, targeted therapy, and immunotherapy, consequently mitigating cancer mortality rates [291]. Nevertheless, these therapeutic modalities continue to grapple with impediments such as drug resistance, adverse side-effects, and inability to conclusively extirpate metastatic lesions, and the recurrence and metastasis rates of certain tumors persist at elevated levels [10]. For example, the yearly recurrence rate of hepatocellular carcinoma (HCC) post-surgical resection equals or exceeds 10% and escalates to between 70 and 80% after five years [292]. The five-year survival rate for pancreatic ductal adenocarcinoma (PDAC) stands at 10% [293]. Therefore, the exploration of novel therapeutic strategies remains a pressing necessity.
In recent years, emerging research has highlighted the connection between tumor development and ferroptosis [294]. Various oncogenic signaling cascades have been found to conduct the symphony of ferroptosis in malignant cells, and ferroptosis intersects with the functionalities of numerous tumor suppressors, such as the retinoblastoma protein (RB1) and the breast cancer 1 (BRCA1)-associated protein 1 (BAP1) [215, 257]. Compared to their non-malignant counterparts, the proliferation of cancer cells (particularly cancer stem cells) demonstrates a heightened dependency on iron due to its indispensable role in rapid cell multiplication and metabolic activity [295]. By destabilizing iron metabolism within tumorous cells and regulating iron-dependent signaling pathways, it is plausible to provoke ferroptosis in these cells, thereby suppressing tumor expansion and metastasis, and augmenting the efficacy of traditional oncologic treatments [296].
In a recent study, Wang et al. and other researchers discovered that castration-resistant prostate cancer cells are particularly sensitive to ferroptosis, highlighted that the RB/E2F/ACSL4 molecular pathway is a critical regulator of this process [257, 297,298,299]. Inactivation of the RB1 tumor suppressor gene is common in metastatic castration-resistant prostate cancer, RB1 loss/E2F activation upregulated expression of ACSL4 and enriched ACSL4-dependent AA-containing phospholipids [257].
Numerous other key regulators in neoplastic development have been linked to ferroptosis. The role of Serum/glucocorticoid regulated kinase 2 (SGK2) in promoting prostate cancer metastasis via ferroptosis inhibition was identified by Cheng et al. in 2023 [258, 300]. SGK2 overexpression phosphorylates the Thr-24 and Ser-319 sites of forkhead box O1 (FOXO1) and relieves the inhibitory effect of FOXO1 on GPX4. Moreover, CCAAT/enhancer-binding protein gamma (CEBPG) was established as a novel transcriptional modulator of ferroptosis in ovarian cancer, regulating ferroptosis via transcriptional control of SLC7A11 [254].
Certain neoplasms appear highly reliant on ferroptosis defensive mechanisms for survival under metabolic and oxidative stress. Therefore, disruption of those defenses would be deadly to such cancer cells while sparing normal cells. In 2023, Wang et al. identified heat shock protein family A member 8 (HSPA8) as a crucial host factor that modulates hepatitis B virus (HBV) replication and ferroptosis in liver cancer [238]. HSPA8 suppressed ferroptosis in liver cancer cells by upregulating the expression of SLC7A11/GPX4, decreasing erastin-mediated reactive oxygen species, and accumulating Fe2+ in cells in vitro and in vivo [238]. Su et al. identified BTB domain and CNC homology 1 (BACH1) as a cellular factor that strongly interacts with P53R175H [252], and p53R175H acts as a repressor for ferroptosis by abrogating BACH1-mediated downregulation of SLC7A11 to enhance tumor growth [252]. In addition, Chang et al. revealed that STC2 could interact with protein methyltransferase 5 (PRMT5) and activate PRMT5 to participate in SLC7A11 mediated ferroptosis [259]. Ovarian cancer (OC) is the seventh most common malignant tumor and ranks eighth among the causes of cancer death in females [301]. Anandhan et al. also showed that nuclear factor erythroid 2–related factor 2 (NRF2) maintains iron homeostasis by controlling HERC2 (E3 ubiquitin ligase for NCOA4 and F-Box and Leucine-Rich Repeat Protein 5 FBXL5) and vesicle associated membrane protein 8 (VAMP8) (mediates autophagosome-lysosome fusion) [255]. Taken together, the modulation of the iron metabolism pathway serves as a therapeutic means to trigger cancer cell ferroptosis.
Therapeutic potential of targeting ferroptosis in cancer
Despite remarkable strides in oncological therapeutics, resistance remains a formidable challenge [302]. A multitude of preclinical and clinical studies are centered on circumventing drug resistance [303]. Intriguingly, ferroptosis has been linked to cancer therapy resistance, and induction of ferroptosis can potentially reverse this resistance. In recent years, certain drugs and compounds have been found to have the ability to induce ferroptosis and demonstrate anti-tumor activity [294].
Wen et al. discovered in 2023 that baicalin affects NRF2 stability through ubiquitin degradation, thereby suppressing NRF2 downstream targets GPX4 and xCT, thereby eliciting ferroptosis [250]. Wogonin is a flavonoid with anticancer activity against various cancers, including pancreatic cancer [304]. In 2023, Liu et al. showed that wogonin upregulates the levels of Fe, lipid peroxidation, and superoxide, and decreases the protein expression levels of ferroptosis suppressor genes, and downregulates level of glutathione in pancreatic cancer cells [267]. Ponicidin could suppress pancreatic cancer cell proliferation via inducing ferroptosis by inhibiting the gamma-glutamyl cycle and regulating the polyunsaturated fatty acid metabolism in SW1990 cells [269]. For several decades, lung cancer has been one of the most common cancers. Many studies have found some antitumor reagents can play an important role in the treatment of lung cancer through ferroptosis [305]. For example, GPX4 inhibitor-Bufotalin (BT), through facilitating the ubiquitination and degradation of GPX4, induces ferroptosis of non-small cell lung cancer (NSCLC) cells [280]. Timosaponin AIII (Tim-AIII), A steroid saponin, can bind to the heat shock protein 90 (HSP90), which further promotes the ubiquitination of GPX4 and thereby degrades GPX4 [279].
Sorafenib, a tyrosine kinase inhibitor, shows an obvious antitumor effect as a ferroptosis inducer in multiple cancers [306]. In 2023, Xu et al. found that activating transcription factor 2 (ATF2) was significantly upregulated by Sorafenib [271]. In this study, heat shock protein family H (Hsp110) member 1 (HSPH1) was identified as a target of ATF2, which can interact with SLC7A11 (cystine/glutamate transporter) and increase its protein stability [271]. In addition, Kang et al. also found salinomycin-induced ferroptosis in renal cell carcinomas (RCCs) [277]. The Disulfide Isomerase Family A Member 4 (PDIA4), as a mediator of salinomycin, suppressed PDIA4 by increasing its autophagic degradation, increasing the sensitivity of RCCs to ferroptosis [277].
As discussed, several drugs (including wogonin, ponicidin, sorafenib and salinomycin) have proferroptotic activity in preclinical models [229, 267, 269, 277]. In the future, targeting ferroptosis with specific drugs is anticipated to play a crucial role in cancer treatment [307]. With advancing understanding of the molecular mechanisms underlying ferroptosis and ongoing research efforts, the potential impact of targeting ferroptosis in cancer therapy can be envisaged in the following aspects: Firstly, targeting ferroptosis holds promise as a strategy to overcome drug resistance, a major obstacle in cancer treatment. By modulating iron metabolism and the signaling pathways related to iron dependency, drugs designed to induce ferroptosis may bypass the resistance mechanisms associated with conventional therapies, exerting pronounced cytotoxic effects on resistant tumor cells [308,309,310]. Secondly, targeting ferroptosis may enhance treatment efficacy and improve patient outcomes [278, 311]. Given the significant role of ferroptosis in tumor growth, invasion, and metastasis, interventions that interfere with tumor cell iron metabolism and induce ferroptosis have the potential to effectively suppress tumor progression and dissemination, thereby improving treatment responses and prognoses, ultimately leading to better survival rates and quality of life for patients [312, 313]. Furthermore, targeting ferroptosis could offer new avenues for personalized cancer therapy [314]. The heterogeneity of tumors and individual variability often render conventional treatment modalities suboptimal for all patients. By targeting iron metabolism and signaling pathways, drugs designed to induce ferroptosis can enable tailored treatment approaches based on individual patient characteristics, providing more precise and effective therapeutic strategies [315,316,317]. Lastly, targeting ferroptosis may emerge as a critical component of combination therapies. Combinatorial approaches have become a major trend in cancer treatment, as they can enhance therapeutic efficacy while reducing side effects. By integrating drugs targeting ferroptosis with other treatment modalities such as chemotherapy, immunotherapy, or targeted therapies, synergistic effects can be achieved, further augmenting treatment responses [318,319,320]. In summary, targeting ferroptosis with specific drugs holds tremendous potential in future cancer treatment. This approach offers the prospects of overcoming drug resistance, improving treatment efficacy, enabling personalized therapy, and integrating with other treatment modalities, thereby paving the way for enhanced outcomes and advancements in cancer care.
Ferroptosis and ischemic/reperfusion related diseases
I/R injury is a complex physiological event that occurs when blood supply to a tissue or organ is disrupted and then subsequently restored [321, 322]. This process, while seemingly paradoxical, can lead to significant tissue damage and cell death, often exceeding the initial injury caused by ischemia alone[323, 324]. The initial ischemic phase can be induced by a variety of causes, such as a blockage in the blood vessels due to a clot or plaque, or a systemic reduction in blood flow due to shock, cardiac arrest or organ surgeries [321]. The lack of blood flow deprives the tissue of oxygen and nutrients, leading to a state of hypoxia and nutrient deprivation. This can result in cellular dysfunction and, if prolonged, irreversible cell damage and death [325]. The subsequent reperfusion stage is necessary to deliver oxygen and nutrients to the ischemic tissue, however, it paradoxically leads to further tissue damage. This process is due to the sudden influx of oxygen and nutrients, which can result in the overproduction of ROS and the initiation of inflammatory responses [326, 327]. The ROS can cause oxidative damage to cellular components, while the inflammatory responses can lead to further cell death and tissue damage [328, 329].
The type of cells and tissues affected by I/R injury can vary widely, and include the heart (as in myocardial infarction), brain (as in stroke), kidneys (as in acute kidney injury), liver (as in hepatic I/R injury), and intestines (as in mesenteric ischemia) [322, 330,331,332,333,334]. At the cellular level, I/R injury can lead to various forms of cell death, including necrosis, apoptosis, and autophagy [335, 336]. Recently, ferroptosis has been implicated in I/R injury [337,338,339]. It has been proposed that the oxidative stress and inflammation caused by I/R injury may trigger ferroptosis, thereby exacerbating tissue damage [48]. This has led to the hypothesis that targeting ferroptosis could be a novel therapeutic strategy for mitigating I/R injury. We have summarized the potential therapeutic targets on I/R injury in Table 3.
Myocardial I/R injury
Acute myocardial infarction (MI), a paramount life-threatening coronary event, afflicts in millions of individuals annually, and these numbers continue to rise worldwide [390,391,392]. Despite the mitigating mortality and morbidity rates concomitant with the rapid evolution of medical technologies, the heart failure precipitated by MI continues to remain alarmingly high, imposing a substantial financial and societal burden on individuals and communities [393, 394]. I/R injury is an important pathological process during MI treatment [395]. MI-induced myocardial ischemia results in inadequate oxygen supply to the myocardial cells, while oxidative stress during reperfusion exacerbates cellular damage [396]. Studies have found that insufficient oxygen supply and oxidative stress caused by ischemia lead to the excessive accumulation of intracellular iron ions, increasing the likelihood of ferroptotic cell death [396]. Iron contribute to myocardial cell injury through oxidative stress reactions and lipid peroxidation mechanisms [397]. Subsequently, MI is commonly remedied with prompt and efficacious myocardial reperfusion, typically through thrombolytic therapy or primary percutaneous coronary intervention (PPCI) [398]. Reperfusion therapy exacerbate damage to the myocardial tissue, through oxidative stress, inflammatory reaction, disorder of energy metabolism, causing cell death, myocardial stunning, arrhythmia, myocardial vertigo [399, 400]. Xiao-Hui Ma and colleagues have elucidated the role of ischemia in inducing a specific oxidative-reductive reaction involving PUFAs-containing phospholipids within myocardial cells [401]. This reaction serves as a pivotal initiating signal for the robust initiation of oxidative damage during reperfusion [401]. They have proposed ALOX15 as the primary mediator responsible for the ischemia-induced peroxidation of phospholipids [401]. Additionally, another study has provided evidence demonstrating that 15-hydroperoxyeicosatetraenoic acid (15-HpETE), an intermediate metabolite derived from AA through the action of ALOX15, acts as a critical trigger for ferroptosis in cardiac myocytes [339]. Other targeted therapeutic strategies associated with various genes associated with ferroptosis have also been studied in myocardial I/R injury models. Research has revealed that inhibition of MALT1 can reduce I/R-induced myocardial iron efflux by enhancing the NRF2/SLC7A11 pathway [340]. Inhibiting the expression of key ferroptotic genes NOX4, P53, and LOX can reduce ferroptosis in myocardial cells and improve cardiac function [341]. By modulating the PUM2/PRDX6 axis, it is possible to suppress myocardial iron deposition, thereby alleviating I/R-induced cardiac injury and improving cardiac function [342]. Tang et al. identified a novel pathway involving USP7/P53/TfR1 in the hearts of rats subjected to I/R treatment, where upregulation of USP7 promoted iron deposition through activation of the P53/TfR1 pathway [343]. Small molecule drugs targeting ferroptosis have shown promising potential in myocardial I/R injury. The ALOX15-specific inhibitor ML351 has been shown to elevate the protein level of Pgc1α, suppress cardiomyocyte death, protect damaged myocardium, and promote cardiac function recovery [339]. Xanthohumol (XN), an isoenoic flavonoid derived from hops, exhibits cardioprotective effects by mitigating ferroptosis through lipid peroxidation and ROS generation, chelating iron ions, modulating NRF2 protein levels, and regulating GPX4 protein expression [402]. Another study demonstrated that dapagliflozin, the sodium glucose co-transporter 2 (SGLT2) inhibitor, exerts inhibitory effects on ACSL4, which suppresses ferrosome formation, by upregulating the SLC7A11/GPX4 axis and ferritin heavy chain (FTH) expression [403]. Further research and understanding of the mechanism of ferroptosis, especially identifying effective compounds targeting ferroptosis, in myocardial I/R injury will help reveal the pathogenesis of myocardial I/R injury and provide for the development of more effective treatment strategies.
Cerebral I/R injury
Ischemic stroke, also known as cerebral infarction, is a neurological disorder caused by localized cerebral, spinal cord, or retinal infarction [404]. It is a major public health issue with a high incidence, resulting in disability and death, with millions of new cases reported annually [405]. Survivors often experience long-term physical, cognitive, and emotional impairments [405]. Additionally, it also imposes a significant economic burden on healthcare systems and societies. Ischemic stroke and subsequent reperfusion injury elicit oxidative stress, which results in aberrant intracellular iron ion accumulation, consequently triggering ferroptosis [406]. Guo et al. also found that rats with cerebral I/R injury had severe brain damage and neurological deficits, accompanied by typical molecular features of ferroptosis, including GSH disturbances, abnormal accumulation of iron, and increased lipid peroxides. These observations underscore the significance of comprehending and intervening in the mechanisms underlying ferroptosis, offering potential avenues to enhance therapeutic efficacy in the context of stroke management. Hu et al. showed that upregulation of CDGSH iron-sulfur domain 2 alleviates cerebral I/R injury through activation of the NRF2/HO-1 pathway, which is a key factor in maintaining cellular redox homeostasis and lipid and iron metabolism [356]. Another study found that in cells exposed to I/R injury, the knockdown of Retinoid X receptor γ (RXRγ) resulted in the downregulation of GPX4 expression and the upregulation of COX-2 and ROS levels [407]. Researchers therefore suggest that the transcriptional activation of GPX4, mediated by RXRγ, may contribute to the inhibition of ferroptosis in the context of cerebral I/R injury [407]. Furthermore, the absence of NCOA4 significantly abrogated ferritinophagy induced by I/R injury, thereby suppressing ferroptosis [362]. Numerous inhibitors of ferroptosis have shown promising effects in ameliorating stroke. Srs11-92 (AA9), a Fer-1 analog, reduced oxidative stress and neuroinflammation in neurons subjected to OGD/R by activating the NRF2 pathway [408]. Researchers believe that AA9 has potential as a therapeutic candidate for protecting against neuronal damage in stroke and other neurological diseases, by targeting NRF2-mediated oxidative stress and neuroinflammation [408]. Dl-3-n-butylphthalide, a compound derived from celery seed, regulates ferroptosis through SLC7A11/GSH/GPX4 pathway to achieve neuroprotective effect on I/R injury [409]. The administration of proanthocyanidins (PC), a class of organic antioxidants, upregulates the expression of GPX4 and SLC7A11 while downregulating the expression of TFR1, thereby exerting an inhibitory effect on ferroptosis. Proanthocyanidins (PC), as organic antioxidants, upregulate the expression of GPX4 and SLC7A11 while downregulating the expression of TFR1, thereby inhibiting ferroptosis and ameliorating cerebral I/R injury [410]. The continued investigation and development of ferroptosis inhibitors hold great promise for improving the treatment and management of stroke, providing new avenues for reducing the devastating consequences of this cerebrovascular disorder. In summary, ferroptosis plays a crucial role in cerebral I/R injury, and understanding its mechanisms can aid in the development of new therapeutic strategies to protect brain cells from oxidative stress and cell death. However, further research is still needed to explore the specific mechanisms and potential therapeutic targets of ferroptosis in cerebral I/R injury.
Hepatic I/R injury
Hepatic I/R injury is mainly caused by liver surgery, such as partial hepatic resection and liver transplantation, where severe hepatic I/R injury after liver transplantation leads to acute or chronic rejection and even transplant failure by inducing inflammation and oxidative stress [411, 412]. Hepatic I/R injury is frequently associated with inflammation and oxidative stress, which can precipitate systemic inflammatory response syndrome (SIRS) or multiple organ dysfunction syndrome (MODS), exacerbating the patient's condition and leading to organ damage and functional impairment) [413]. Several studies have explored the involvement of ferroptosis in hepatic I/R injury and its potential as a therapeutic target [414]. The study by Ye et al. provides confirmation that MCTR1 attenuates hepatic ischemia–reperfusion injury caused by ferroptosis through the promotion of NRF2 expression [415]. Guo et. Declared that transmembrane member 16A (TMEM16A), a component of hepatocyte Ca2+-activated chloride channel, exacerbates hepatic I/R injury through the promotion of GPX4-dependent ferroptosis, and interrupting the TMEM16A-GPX4 interaction or inhibiting TMEM16A in liver cells may represent promising therapeutic strategies for the treatment of hepatic I/R injury [416]. The HECT domain-containing ubiquitin E3 ligase HUWE1 (also known as MULE) has emerged as a promising protective factor in mitigating acute liver injury by counteracting abnormal iron accumulation and inhibiting ferroptosis [373]. There is currently limited development and application of small molecule drugs targeting ferroptosis in the treatment of hepatic ischemia–reperfusion injury. Neutrophil membrane-coated taurine nanoparticles increased the expression of SLC7A11 and GPX4, and decreased the expression of Ptgs2, suggesting that nano-taurine has a targeted therapeutic effect on hepatic I/R injury by inhibiting inflammation, oxidative stress and ferroptosis [417]. Dimethyl fumarate (DMF), a therapeutic agent utilized in the treatment of relapsing–remitting multiple sclerosis, demonstrates inhibitory effects on ferroptosis through activation of the NRF2/SLC7A11/HO-1 axis, thereby conferring protection against hepatic I/R injury [418]. Although the relationship between hepatic I/R injury and ferroptosis has been confirmed, the development and application of therapies and drugs targeting ferroptosis are still limited. Further research is needed to explore the regulation of iron metabolism, oxidative stress, and other molecular targets associated with ferroptosis, aiming to discover more effective treatment strategies and opportunities for the management of hepatic I/R injury.
Renal I/R injury
Renal I/R injury can be triggered by multiple factors including renal artery obstruction, hypotension, shock, and surgical interventions [419, 420]. This injury culminates in renal tissue ischemia, hypoxia, disruption of tubular and vascular architecture, provoking inflammatory response and cellular death, ultimately culminating in renal dysfunction [420]. Renal I/R injury can cause electrolyte imbalances and discomfort, while requiring patients to undergo multiple treatments like hemodialysis or kidney transplantation. This significantly affects their quality of life and imposes a financial burden [421]. The molecular mechanisms regulating iron metabolism and ferroptosis have been found to play a crucial role in the development and treatment of renal I/R injury [422]. A recent study suggests that miR-20a-5p has potential therapeutic applications in kidney transplantation by inhibiting ACSL4-dependent ferroptosis [374]. TRIM21 exhibits elevated expression in kidney tissues undergoing renal I/R injury. Downregulation of TRIM21 mitigated renal I/R injury and protected renal function [375]. The involvement of cold-inducible RNA-binding protein (CIRBP) in acute kidney injury has been suggested in another research, which proposed that ferritinophagy-mediated ferroptosis may be responsible for the enhanced ischemic kidney injury observed in the presence of CIRBP [379]. In ischemic kidney injury, iron chelators such as deferoxamine, deferiprone, and lipophilic antioxidants have been shown to inhibit lipid peroxidation and protect against cell damage [423]. These agents can target lipoprotein-1 (Lip-1), ferristatin-1, as well as vitamins and flavonoids, which are involved in antioxidant defense [423, 424]. These studies also suggest that molecular mechanisms regulating iron metabolism and ferroptosis may play an important role in the treatment of acute ischemic kidney injury. Some pharmacological agents targeting ferroptosis have also been investigated. Cyanidin-3-glucoside (C3G), a typical flavonoid, can activate AMPK pathway to inhibit ferroptosis in renal tubular cells after I/R injury [425]. Qi et al. found that the regulatory effect of MGZ on the Mitoneet-mediated iron apoptosis pathway, highlighting its potential role in renal protection [426]. Methods such as interfering in iron metabolism, modulating antioxidant defenses, and inhibiting iron-related pathways have shown promising results in preclinical studies aimed at alleviating renal I/R injury and preserving renal function (Table 4). However, further research is needed to fully understand the underlying mechanisms driving renal I/R-induced ferroptosis and to develop effective treatment strategies.
In conclusion, emerging evidence strongly supports the pivotal role of ferroptosis in the pathogenesis of I/R injury, highlighting its potential as a promising therapeutic target. However, the regulatory mechanisms underlying ferroptosis in the context of I/R injury remain incompletely elucidated. Further research is warranted to unravel the new pharmacological mechanisms, toxicity profiles, side effects, and optimal dosages of ferroptosis inhibitors through rigorous preclinical and clinical investigations. Therefore, it is imperative to comprehensively understand the regulatory mechanisms governing ferroptosis in I/R injury and identify safe and effective targeting strategies for modulating ferroptosis regulators to mitigate I/R injury.
Ferroptosis and neurodegenerative diseases
Neurodegenerative afflictions, encompassing Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Amyotrophic Lateral Sclerosis (ALS), constitute a cohort of incapacitating disorders marked by the progressive neuronal attrition and the attendant regression in cognitive and motor functionalities. Despite prodigious research, the integral mechanisms instigating and fostering these diseases remain elusive. Recently, ferroptosis has been unveiled as a plausible mechanism bearing implications for the pathogenesis of neurodegenerative diseases [7, 462]. The incorporation of ferroptosis in the context of neurodegenerative diseases has garnered increasing recognition due to the burgeoning evidence associating dysregulated iron metabolism, compromised antioxidant defenses [463], and amplified lipid peroxidation [464] with the pathogenesis of these disorders. Prior studies have manifested alterations in iron distribution and accrual in specific cerebral regions affected by neurodegeneration [465,466,467,468]. Furthermore, heightened levels of lipid peroxidation markers and diminished antioxidant capacity have been discerned in the brains of individuals afflicted with neurodegenerative diseases [469, 470], suggesting a potential role of ferroptosis in the selective neuronal loss (Tables 5 & 6).
Alzheimer’s disease
AD manifests as a catastrophic neurodegenerative disorder typified by the incremental loss of cognitive faculties, memory deterioration, and behavioral metamorphoses. It represents the predominant form of dementia, impacting millions globally [514]. Despite exhaustive research efforts, the precise mechanisms piloting AD pathogenesis remain enigmatic. Recently, ferroptosis has surfaced as a prospective mechanism with implications for the genesis and advancement of AD. Recent evidence posits that cerebral iron correlates with hastened cognitive decline in individuals exhibiting Alzheimer's pathology [515]. Scott and colleagues have delineated variations in cerebrospinal fluid ferritin levels [470]. Furthermore, the aggregation of amyloid-beta (Aβ) plaques, which constitute the signature pathologies of AD, have been associated with ferroptosis-linked mechanisms [516]. Aβ accumulation may incite oxidative stress and lipid peroxidation, heightening neuronal susceptibility to ferroptosis. Deteriorated antioxidant defenses and diminished activity of crucial enzymes engaged in lipid peroxide detoxification have been witnessed in AD, further corroborating the participation of ferroptosis in neuronal expiration [502, 517]. Deciphering the role of ferroptosis in AD might yield novel insights into the disease trajectory and prospective therapeutic interventions. Interfering with ferroptosis pathways might represent a propitious strategy for attenuating neurodegeneration and cognitive degradation in AD. Diverse pharmacological methodologies, including iron chelators, antioxidants, and ferroptosis inhibitors, have demonstrated promise in preclinical explorations by diminishing neurotoxicity and enhancing cognitive function in AD animal prototypes [476, 499, 500, 503, 518]. However, numerous challenges and unresolved queries persist. Augmented research is imperative to illuminate the precise molecular mechanisms underpinning ferroptosis in AD and its contribution to the progressive neurodegeneration witnessed in afflicted individuals. Additionally, fine-tuning therapeutic interventions targeting ferroptosis, including the development of selective and efficacious drugs, determination of an appropriate treatment window, and managing potential off-target ramifications, is crucial for successful clinical translation.
Parkinson’s disease
PD represents a chronic, relentlessly progressive neurodegenerative disorder distinguished by the degradation of dopaminergic neurons within the substantia nigra territory of the brain. This neuronal death culminates in the characteristic motor symptoms of PD, encompassing tremors, rigidity, and bradykinesia. Evolving evidence proposes that ferroptosis may constitute a critical determinant in the pathogenesis of Parkinson's disease. In postmortem cerebral evaluations from individuals afflicted with PD, an elevation of iron regulatory protein 1 (IRP1) activity was discerned within the substantia nigra (SN). This amplified activity could provoke a diminution in ferritin concentrations and an intensification in neuronal iron assimilation, culminating in escalated TfR1 expression. Consequently, the melanized neurons within the SN become increasingly vulnerable to oxidative damage affiliated with iron [519]. Augmented DMT1 concentrations, in conjunction with diminished Cp ferroxidase activity, have been documented in both PD patients and animal representations of PD. These manifestations are posited to contribute to the noticeable amplification in iron levels [520]. Alpha-synuclein (α-Syn), abundantly expressed within the brain and implicated in numerous pivotal synaptic processes of neurons, can bind to Fe2+ or Fe3+ to fabricate the α-Syn-iron complex. The up-regulation of DMT1 ensuing from α-Syn overexpression also exerts a profound influence on the enhancement of iron uptake and the dysfunction of iron metabolism evidenced in PD [521]. Furthermore, the distorted expression and functionality of proteins involved in iron homeostasis have been detected in PD, further substantiating the association between iron dysregulation and the disease [504, 509, 510, 522, 523].
Amyotrophic lateral sclerosis
ALS epitomizes a relentlessly progressive neurodegenerative disorder typified by the selective compromise of cortical and spinal motor neurons, instigating paralysis and ultimately, mortality [524]. Although comprehension of the underlying pathophysiological mechanisms of ALS remains incomplete, the accretion and amassment of ubiquitinated proteinaceous inclusions within motor neurons are broadly recognized as the quintessential neuropathological characteristic of this disease [525]. The majority of ALS instances, roughly 90%, materialize sporadically and fail to exhibit a clear correlation with familial lineage. The residual 10% of cases are tethered to familial inheritance patterns and are typically associated with autosomal dominant mutations. The most prevalently observed mutations transpire within genes such as superoxide dismutase 1 gen (SOD1), TAR DNA-binding protein 43 (TDP-43), FUS, and C9orf72. In a murine model of GPX4 neuronal inducible knockout, the specific depletion of GPX4 within neurons precipitated rapid paralysis, severe muscular atrophy, and ultimately, mortality, thereby evincing symptoms evocative of ALS [526]. A recent study unveiled the depletion of GPX4 in postmortem spinal cord samples from both sporadic and familial ALS patients, revealing a potential involvement of GPX4 in the pathogenesis of ALS [489]. Moreau et al. demonstrated that the administration of deferiprone to ALS patients engendered a significant reduction in iron concentration within the cervical spinal cord [527]. However, the potential influence of ferroptosis inhibition on enhancing the quality of life and survival rate among ALS patients remains undetermined and demands further inquiry.
Huntington’s disease
HD manifests as an inheritable neurodegenerative disorder typified by the gradual degeneration of specific neuronal populations within the brain. It is initiated by a mutation in the huntingtin gene (HTT), culminating in the synthesis of an aberrant form of the huntingtin protein [528]. HD is characterized by an extensive array of motor, cognitive, and psychiatric symptoms that progressively intensify over time. Song et al. unveiled that ALOX5-mediated ferroptosis serves as a distinct cell death trajectory in response to oxidative stress in Huntington's disease [491]. Klepac et al. identified a significant diminution (28%) in plasma GSH concentrations among individuals afflicted with HD compared to age and sex-congruent controls [529]. Magnetic resonance imaging revealed an accumulation of iron within the cerebral regions of HD patients [530]. Nevertheless, the pathway inciting ferroptosis within the brain remains largely ambiguous. The potential to procure similar outcomes through alternative strategies for ferroptosis inhibition, such as the modulation of GPX4, lipid peroxidation, and iron-storage proteins, has yet to be explored. Moreover, the question of whether ferroptosis inhibition can effectively decelerate the progression of HD remains unaddressed and necessitates further exploration in prospective investigations.
Ferroptosis and cardiovascular diseases
Cardiovascular diseases (CVDs) encompass a wide range of conditions affecting the heart and blood vessels, contributing to acute illnesses that result in numerous fatalities worldwide [531]. The death of fully differentiated cardiomyocytes plays a crucial role in the development of various cardiovascular conditions. In this study, we provide a comprehensive perspective on the molecular mechanisms underlying ferroptosis in the pathogenesis of several cardiovascular diseases, including hypertension, atherosclerosis, myocardial infarction (MI), pulmonary hypertension (PH), cardiomyopathy, and heart failure (HF) [39, 532].
When examining cardiomyopathy, our investigation focused on several subtypes, namely Diabetic cardiomyopathy (DCM), Hypertrophic cardiomyopathy, post-transplant cardiomyopathy, Septic cardiomyopathy, Doxorubicin-induced cardiomyopathy (DIC), and radioactive cardiomyopathy [533,534,535,536,537]. The primary objective was to explore the association between the diverse spectrum of CVDs and ferroptosis, while also identifying potential novel compounds that target iron metabolism and ferroptosis within the context of CVDs (Tables 7 & 8).
Myocardial infarction
MI culminates in cardiac detriment precipitated by cellular death and inadequate self-regeneration of cardiomyocytes [596]. Previous investigation has elucidated that ferroptosis participates in MI, which involves lipoprotein receptor-related protein 6 (LRP6) and circRNA1615 [597]. LPR6 and circRNA1615 function as a modulator of ferroptosis via autophagy regulation [597]. Ferroptosis, in concert with hypoxia, assumes a pivotal role in acute myocardial infarction (AMI), prompting Liu et al. to delineate key genes associated with AMI, ferroptosis, and hypoxia that might serve as novel biomarkers or prospective therapeutic targets for AMI [598]. Gao et al. unveiled that lncRNA Gm47283 orchestrates its effect by targeting miR-706 and Ptgs2, thus modulating Ptgs2 expression and downstream ferroptosis, thereby establishing itself as a primary risk factor for MI [541].
Understanding the intricate and complex interplay between MI and ferroptosis is crucial in identifying potential therapeutic strategies. Targeting the molecular mechanisms involved in ferroptosis, such as iron metabolism, peroxidation, and antioxidant systems, may offer new approaches to mitigate the damage caused by myocardial infarction and improve patient outcomes. As shown in Table 7, Malic enzyme 2 (ME2), Adaptor protein HIP-55, and fibronectin type III domain containing 5 (FNDC5)/irisin have also been suggested as potential targets mediating ferroptosis in MI.
Atherosclerosis
Atherosclerosis typifies a chronic inflammatory disease hallmarked by dysregulated lipid metabolism and endothelial malfunction [599, 600]. Vinchi et al. have explicated the interplay between ferroptosis and the pathogenesis of Atherosclerosis [601]. They found that GPX4 mitigates the evolution of atherosclerosis via curtailing lipid peroxidation and diminishing the sensitivity of vascular cells to oxidized lipids [602]. Qing-Xin-Jie-Yu Granule (QXJYG), a traditional Chinese medicinal compound constituted of quintuple Chinese medicinal constituents, could inhibit ferroptosis through the regulation of the GPX4/xCT pathway for atherosclerosis [594]. Currently, investigations into the therapeutic efficacies of Chinese medicine on cardiovascular diseases mediated by ferroptosis are sparse [568, 593]. Thus, elucidating the role and mechanism of Chinese medicine in impeding ferroptosis might shed light on the treatment of cardiovascular diseases.
Pulmonary hypertension
PH is a condition characterized by elevated arterial blood pressure in the pulmonary circulation, placing increased strain on the heart and ultimately leading to heart failure [603]. Patients with PH commonly experience progressive shortness of breath, which is the predominant symptom observed. Unfortunately, the prognosis for individuals with pulmonary hypertension is generally poor, as treatment options are limited and the disease significantly impacts their quality of life [604]. The pathogenesis of PH involves multiple complex cellular processes and pathological changes. The pathogenesis of PH involves complex cellular processes and pathological changes. Notably, various types of PH are associated with diverse inflammatory responses. In animal models, several immunomodulatory interventions have demonstrated the ability to modulate the progression and advancement of the disease [605]. These findings highlight the importance of understanding the intricate cellular mechanisms involved in the development of PH and suggest potential avenues for therapeutic interventions.
Disruption of signaling pathways involving ROS and nitric oxide (NO) can contribute to the proliferation of pulmonary arterial endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), leading to DNA damage, metabolic dysregulation, and vascular remodeling [606]. Growing evidence supports the role of ferroptosis in the development and progression of PH, highlighting the potential of antioxidant therapy as a significant area of investigation for PH treatment. miRNAs have been found to modulate the process of ferroptosis and regulate the expression of target genes involved in iron metabolism in PH patients. Specifically, six differentially regulated miRNAs (miR-483-5p, miR-27a-3p, miR-27b-3p, miR-26b-5p, miR-199a-5p, and miR-23b-3p) have been implicated in PH, indicating their role in the regulation of iron-related pathways [607]. In a study by Xie et al., it was observed that ferroptosis is upregulated in PAECs from rats with monocrotaline (MCT)-induced PH. The authors proposed that pulmonary endothelial ferroptosis triggers an inflammatory response through the HMGB1/TLR4/NLRP3 inflammasome signaling pathway in rats. Pharmacological inhibition of ferroptosis using Ferrostatin 1 (Fer-1) was found to mitigate the progression of MCT-induced pulmonary vascular remodeling and protect the right ventricle from the effects of PH [608].
Considering these findings, the utilization of ferroptosis inhibitors in PH treatment and the exploration of innovative therapies based on the regulation of iron-dependent cell death hold promise for the management of PH.
Cardiomyopathy
Diabetic cardiomyopathy
DCM is a common complication of diabetes mellitus (DM) and is associated with an increased risk of heart failure and mortality among diabetic individuals [609]. The disease is characterized by left ventricular hypertrophy and diastolic dysfunction in the early stages, progressing to dominant heart failure with reduced systolic function in advanced stages. The pathogenesis of DCM is multifactorial, primarily involving insulin resistance and hyperglycemia [610].
Insulin resistance, an emblematic feature of type 2 diabetes, instigates compromised glucose uptake and utilization by cardiomyocytes, culminating in energy depletion and perturbed cardiac metabolism [611]. Conversely, hyperglycemia contributes to the genesis of advanced glycation end products (AGEs), oxidative stress, and inflammation, thereby exacerbating cardiac dysfunction and provoking structural remodeling [612]. Despite hyperglycemia governing numerous pathways within DCM, the amplification of ROS is perceived as the central mechanism underlying adverse remodeling [613]. The induction of ferroptosis precipitates an elevation in intracellular levels of lipid ROS, consequently inciting cellular death [614]. Recent evidence increasingly implicates ferroptosis as a significant player in the progression of DCM [545]. Intriguingly, sulforaphane-activated NRF2 can repress ferroptosis in cardiomyocytes via the modulation of SLC7A11 levels, indicating a novel therapeutic strategy for DCM [545].
As our comprehension of the pathophysiological mechanisms underlying DCM and ferroptosis continues to advance, innovative therapeutic approaches targeting ferroptosis pathways may emerge. By directing interventions toward pivotal regulators of ferroptosis, such as iron metabolism and lipid peroxidation, it may be plausible to ameliorate the deleterious effects of ferroptosis and augment cardiac function in individuals afflicted with DCM.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy delineates a gradually evolving compensatory mechanism for cardiac functionality, predominantly arising in the context of chronic stress overload [536]. It is denoted by a surge in total myocardium and heightened contractility, thereby maintaining regular blood circulation [615]. Evidence points to ferritin's pivotal role in guarding against cardiac ferroptosis, mediated by SLC7A11. Under a high-iron diet, ferritin-deficient mice demonstrated severe heart damage and hypertrophic cardiomyopathy with a distinctive iron death molecular signature, while SLC7A11 overexpression in these mice forestalled cardiac iron death and remodeling [563]. Wang et al. conducted bioinformatics analysis into the pathogenesis of hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) by focusing on the mechanisms of ferroptosis [616]. Their findings revealed that three hub genes, namely POSTN, IGFBP5, and FMOD, have the potential to serve as valuable biomarkers or therapeutic targets in the field of cardiomyopathies. Nevertheless, the exact characteristics of these gene mechanisms associated with ferroptosis remain largely uncertain, especially when considering their implications in myocardial diseases. There might still be underlying mechanisms awaiting clarification to provide an explanation for this phenomenon. In contrast to DCM, the role of ferroptosis in HCM seems to be more intricate, and the precise impact of ferroptosis on HCM remains undisclosed.
Doxorubicin-induced cardiomyopathy
Doxorubicin (DOX), a widely used chemotherapeutic agent for various malignancies, possesses significant cardiac toxicity as its most notable side effect, often leading to cardiomyopathy [617,618,619]. Consequently, there is considerable potential for the development of therapeutic approaches aimed at addressing or mitigating the cardiac damage caused by this drug. Doxorubicin-induced cardiomyopathy (DIC) arises from a complex interplay of various mechanisms, including DNA damage, oxidative stress, intracellular signaling, transcription factors, epigenetic regulatory factors, autophagy, and metabolic inflammation [620, 621].
Emerging evidence increasingly implicates ferroptosis as a pivotal process in the progression of DIC. Wang et al. demonstrated that miR-21-5p effectively inhibits apoptosis and oxidative stress in primary cardiomyocytes and mouse heart tissue exposed to DOX, offering potential leads for novel treatments in cardiovascular diseases [622]. Notably, Na Ta et al. discovered that the mitochondrial outer membrane protein FUNDC2 governs the occurrence of iron-mediated cell death, shielding cells from this fate in Fundc2-knockout mice and MEF cells. Further investigations revealed that FUNDC2 modulates the stability of mito-GSH, GPX4, and SLC25A11, all of which are crucial in DOX-induced ferroptosis and subsequent cardiomyopathy [549]. Wang et al. unveiled that PRMT4 exerts inhibitory effects on the NRF2/GPX4 signaling pathway, accelerating ferroptosis in DIC. This compelling evidence suggests that targeting PRMT4 could potentially serve as a preventive strategy to DIC [548].
Additionally, various models of cardiomyopathy induced under different conditions were examined, alongside an exploration of the potential influence of pharmacological interventions on ferroptosis in these disease models, as outlined in Table 7. The development of ferroptosis inhibitors, coupled with a deeper understanding of the iron-dependent cell death process, holds the promise of breakthroughs in the treatment strategies for cardiomyopathy. Therefore, researchers can delve into the regulatory mechanisms and signaling pathways associated with ferroptosis to better understand its role and impact in cardiovascular diseases. This line of inquiry will shed light on the relationship between ferroptosis and the development of cardiovascular conditions, offering fresh insights and strategies for early disease diagnosis, prevention, and treatment.
Ferroptosis and autoimmune diseases
Autoimmune diseases impact approximately 8–9% of the global population, yet the underlying mechanisms remain inadequately explored [623]. However, the study of ferroptosis offers a fresh vantage point for investigating these conditions, introducing a novel perspective into the realm of autoimmune disease research. One of the contributing factors to the development of autoimmunity is the aberrant initiation of cell death and inadequate clearance of deceased cells, leading to the exposure or release of intracellular contents that activate the immune system [624]. Ferroptosis plays a substantial role in influencing both the quantity and functionality of immune cells625. Numerous autoimmune diseases, such as Systemic Lupus Erythematosus (SLE), Rheumatoid Arthritis (RA), Inflammatory Bowel Disease (IBD), and Multiple Sclerosis (MS), are intricately associated with ferroptosis [625]. Although different autoimmune diseases may exhibit shared clinical manifestations, each possesses distinct characteristics. For example, RA patients primarily experience polyarthritis affecting the joints of the hands, while major extra-articular organs, such as the kidneys, are rarely involved. Conversely, individuals with SLE may suffer from organ damage caused by excessive production of multiple autoantibodies and subsequent deposition of immune complexes composed of antibodies and antigens in various organs, including the kidneys [625]. Therefore, investigating the potential mechanisms could offer valuable insights for therapeutic approaches targeting autoimmune diseases[626, 627] (Tables 9 & 10).
Systemic lupus erythematosus
SLE is a severe, debilitating autoimmune disease that affects multiple organs and body systems. The prevalence of SLE worldwide is estimated to be as high as 150 per 100,000 individuals [665]. The disease is characterized by autoantibodies against nuclear antigens (ANA), which are caused by a dysregulation of the immune system [666]. Recent investigations have revealed that neutrophils derived from lupus-prone mice or individuals with SLE undergo cell death through the process of ferroptosis [667]. Notably, the presence of autoantibodies and interferon α in the serum acts as a stimulant for neutrophil ferroptosis. This stimulation leads to an increased binding of the transcriptional suppressor CREMα to the GPX4 promoter, resulting in the suppression of GPX4 expression. Consequently, this cascade of events promotes the accumulation of lipid-ROS [165, 166]. In mice, the presence of neutrophil-specific GPX4 haploinsufficiency leads to the development of a phenotype resembling SLE. Additionally, inhibiting ferroptosis in vivo slows down the progression of the disease in lupus-susceptible MRL/lpr mice. These findings shed light on the involvement of neutrophil ferroptosis in the underlying causes of SLE [667]. Additionally, the effective suppression of lipid ROS levels in neutrophils and the significant inhibition of lupus development in a murine model have been observed through the use of the ferroptosis inhibitor liproxstatin-1 [667]. Furthermore, the proliferation of pathogenic T cells, specifically T follicular helper (Tfh) cells, plays a crucial role in the pathogenesis of SLE [668]. Iron overload promotes the expansion of Tfh cells, secretion of pro-inflammatory cytokines, and antibody production in mice prone to lupus. Mice subjected to a high-iron diet exhibited an increased proportion of Tfh cells and antigen-specific germinal center responses [669]. At the molecular level, overexpression of miR-21 inhibits 3-hydroxybutyrate dehydrogenase-2 (BDH2), leading to iron accumulation and enhanced activity of Fe2+-dependent TET enzymes. This, in turn, results in hydroxymethylation of the BCL6 gene and differentiation of Tfh cells. In summary, maintaining iron homeostasis is crucial for controlling the proliferation of pathogenic T cells, might provide novel therapeutic potential in treating SLE [669].
Rheumatoid arthritis
The primary pathogenesis of RA involves immune dysfunction and inflammation, leading to notable pathological changes such as synovitis, progressive cartilage degradation, and subchondral bone destruction [670]. While the exact mechanism of RA remains unknown, immune cells and fibroblast-like synoviocytes (FLS) are believed to play significant roles in disease progression [671, 672]. For instance, in FLS associated with RA, glycine has been shown to enhance s-adenosylmethionine (SAM) levels, leading to SAM-mediated GPX4 promoter methylation and decreased FTH1 expression. These actions regulate the ferroptosis process [632]. Furthermore, the inhibition of system xc- by Erastin has been demonstrated to induce damage to cartilage tissue by upregulating the expression of matrix metalloproteinase 13 (MMP-13) in chondrocytes and suppressing type II collagen expression, thereby exacerbating RA [655]. umor Necrosis Factor (TNF), a pivotal pro-inflammatory cytokine in the pathogenesis of RA, has been found to inhibit ferroptosis by upregulating SLC7A11, glutamate-cysteine ligase catalytic subunit (GCLM), and glutamate-cysteine ligase regulatory subunit (GCLC). This, in turn, promotes cystine uptake and cellular GSH biosynthesis [633]. In a Collagen-Induced Arthritis (CIA) mouse model, low doses of an undisclosed compound (IKE) along with the TNF antagonist etanercept induced ferroptosis in fibroblasts and attenuated the progression of arthritis [633]. These findings elucidate the mechanisms by which TNF modulates resistance to ferroptosis and suggest the therapeutic potential of ferroptosis-focused therapies targeting dysregulated fibroblasts across a broader range of diseases [633].
Inflammatory bowel disease
IBD is a progressive and recurrent condition with a rising global incidence, encompassing both Crohn's Disease (CD) and Ulcerative Colitis (UC) [673]. These diseases are characterized by extensive cell death in the gut and colon due to chronic inflammation [673, 674]. In an experimental colitis model induced by Dextran Sodium Sulfate (DSS), upregulation of HO-1 within the inflamed colon has been observed, leading to anti-inflammatory and antioxidative effects[675]. NF-κB is involved in the production of cytokines and chemokines in inflammatory cells, as well as the regulation of Endoplasmic Reticulum (ER) stress signaling and ferroptosis processes [676,677,678]. One study suggests that phosphorylated NF-κB-p65 protects intestinal epithelial cells from ferroptosis by alleviating endoplasmic reticulum stress, potentially indicating therapeutic targets for UC treatment involving ferroptosis and NF-κB-p65 phosphorylation [679]. Curculigoside (CUR), the main active constituent of Rhizoma Curculiginis, exhibits diverse biological activities and has shown protective effects on intestinal epithelial cell death, GSH levels, Malondialdehyde (MDA) content, and Lactate Dehydrogenase (LDH) activity. These effects are significantly diminished upon knockdown of GPX4 [680]. CUR prevents ferroptosis in UC by inducing GPX4, highlighting the potential of GPX4 as a therapeutic target for UC [680, 681]. Studies indicate that ferroptosis inhibitors such as Liproxstatin-1 (Lip1), Fer-1, and Deferoxamine (DFO) alleviate disease symptoms and prevent colon length reduction in DSS-induced colitis in mice, emphasizing the beneficial impact of ferroptosis inhibition on IBD [679, 681, 682]. Overall, targeting ferroptosis inhibition may offer a new avenue for the treatment of IBD.
Multiple sclerosis
MS is characterized by chronic inflammation in the central nervous system, marked by neuroinflammation, demyelination, oligodendrocyte depletion, and neurodegeneration [683]. Microglia, known for their ability to alter transcriptional profiles and exhibit diverse inflammatory phenotypes, play a crucial role in the development of MS [684]. The ferroptosis inducer RSL3 has been found to reduce inflammation in microglia and peritoneal macrophages (PM) in response to lipopolysaccharide (LPS) stimulation, while conditioned medium from cells undergoing ferroptosis significantly amplifies inflammation in these cells [645]. Interestingly, despite their resistance to ferroptosis, BV2 cells and PMs exhibit reduced inflammation by increasing the abundance of NRF2 protein. Treatment with RSL3 and Fer-1 concurrently decreases systemic inflammation in vivo [645]. However, the precise mechanism of ferroptosis in MS remains to be fully elucidated.
Overall, cytokines, such as Tumor Necrosis Factor-alpha (TNF-α) and Interferon-alpha (IFN-α), modulate ferroptosis in different ways, contributing uniquely to the pathogenesis of autoimmune diseases [628, 633]. Therefore, it is crucial to understand the intricate interactions between different cell death pathways and the significance of these interactions in the context of autoimmune diseases.
Ferroptosis and infection
Infection embodies a dynamic interaction entailing the complicatedly interplay and conflict between invading pathogens and the host organism [685]. The infection process commences once these pathogens breach the host's defenses via diverse avenues, often culminating in substantial detriment to host cells [686]. Recent research accentuates the cardinal role of ferroptosis in the context of pathogenic infections, as expounded extensively in several recent studies [685, 687, 688]. Consequently, this discourse aims to encapsulate our current understanding of the nexus between ferroptosis and pathogenic infections, emphasizing the underpinning molecular mechanisms, principal regulators, and prospective therapeutic approaches.
A range of pathogens—encompassing bacteria, viruses, fungi, and parasites—typically cause diseases via three mechanisms: direct cellular damage, toxin activity, and immune response [689]. Emerging evidence underscores a robust association between pathogenic infections and ferroptosis [690] (Table 11). On one side of this balance, the host organism can curtail infection by inciting ferroptosis; for instance, oxidative degradation of cellular lipids can suppress hepatitis C virus (HCV) replication [691]. Conversely, certain pathogens may bolster their proliferation and survival by inducing ferroptosis; mycobacterium tuberculosis (Mtb), for example, initiates ferroptosis to augment its pathogenicity and dissemination [692]. These pathogens orchestrate ferroptosis by impeding lipid peroxidation [693]. Comprehending the potential signaling mechanisms of ferroptosis in the context of pathogenic infections will undoubtedly pave the way for the development of novel therapeutic agents (Table 12).
Bacterial infection
Bacteria can provoke host tissue deterioration and organ impairment through the activation of ferroptosis [42]. For instance, Pseudomonas aeruginosa (P. aeruginosa), an important opportunistic pathogen, is the main cause of ventilator-associated pneumonia, urinary tract, blood flow and chronic infection [734,735,736]. However, P. aeruginosa show natural resistance to many classes of antibiotics [737]. Not only that, the effectiveness of antimicrobials in treating P. aeruginosa infection has gradually declined in recent years [738, 739]. Hopefully, regulate ferroptosis to intervene in the development of various resistance mechanisms in P. aeruginosa has emerged as a promising treatment option [740]. P. aeruginosa possesses the capability to express lipoxygenase (pLoxA), which catalyzes the oxidation of host AA-phosphatidylethanolamine (AA-PE) to 15-hydroperoxy-AA-PE (15-Ho-AA-PE), thereby instigating ferroptosis in human bronchial epithelial cells [741]. Concurrently, it undermines the host’s GPX4 defenses by mobilizing lysosomal chaperon-mediated autophagy (CMA)14. In response, the host organism activates the inducible nitric oxide synthase/nitric oxide (iNOS/NO•) driven anti-ferroptosis mechanism to halt lipid peroxidation [705]. Consequently, pLoxA and iNOS/NO• may serve as potential therapeutic targets for P. aeruginosa-associated afflictions, such as cystic fibrosis. Another investigation demonstrated that ferric chloride could relieve P. aeruginosa-mediated cell death [728]. Baicalein, a mammalian lipoxygenases inhibitor, markedly relieves animal mortality, PAO1 colonization, intestinal epithelial cell death, and generation of ferroptotic oxidized phosphatidylethanolamine (PEox) signals [704]. These studies underscore that P. aeruginosa propagation occurs through ferroptosis, thereby motivating us to explore strategies to curb P. aeruginosa infection by focusing on ferroptosis-induced necrosis.
Ferroptosis is also critical to the pathogenic mechanism of Mtb, which is the main pathogenic factor of tuberculosis [693]. Tuberculosis is one of the world's deadliest infections and, along with malaria and HIV/AIDS, has the most significant socio-economic impact on humanity [742]. It is reported that ferroptosis plays a major role in cell death and tissue necrosis induced by Mtb [55]. Protein tyrosine phosphatase A (PtpA), an effector secreted by Mtb, inhibits GPX4 expression by targeting protein arginine methyltransferase 6 (PRMT6), ultimately precipitating ferroptosis and bolstering the pathogenicity and dispersion of Mtb [692]. In an in vivo context, GPX4-deficient mice infected with Mtb displayed a significant upsurge in lung necrosis and bacterial load, meanwhile, an outcome relieved by the lipid peroxidation inhibitor, ferrostatin-1 [700]. These findings support the role of iron-induced death in Mtb-triggered necrosis. Further research revealed that the knockdown of heme oxygenase 1 (Hmox1) by siRNA resulted in a diminution of antioxidant factors GPX4 and FSP1 [32], along with an increased release of intracellular bacteria in Bacillus Calmette-Guérin (BCG)-infected macrophages [743]. These observations suggest that Mtb propagation occurs through ferroptosis, thereby inspiring us to search for promising strategies to manage pulmonary tuberculosis by focusing on ferroptosis-induced necrosis.
Ferroptosis also has a bearing on the evolution and progression of sepsis [744]. Sepsis is a severe medical condition characterized by dysfunctional organ function resulting from the host's inadequate response to infection [745]. The immune response, initiated by the invading pathogen, fails to restore normal balance, leading to a pathological syndrome characterized by sustained inflammation and immunosuppression [746]. Intense stress during sepsis can disrupt the metabolic processes of ions, lipids, and energy in organisms [747]. Numerous studies have increasingly demonstrated the significant role of ferroptosis in modulating inflammation and sepsis [748, 749]. The interplay between Stimulator of Interferon Response cGAMP Interactor 1 (STING) and Nuclear Receptor Coactivator 4 (NCOA4) triggers ferritin-phagocytosis mediated ferroptosis, culminating in an amplified inflammatory response and impacting the transcription factor [722]. Subsequent studies revealed that HET0016, a selective 20-HETE synthase inhibitor, could reverse this mechanism [722]. Moreover, our body can also alleviate sepsis by modulating ferroptosis. Sestrin 2 (Sesn2), a stress-responsive protein, inhibits ferroptosis in septic Dendritic Cells (DC) by downregulating the ATF4-CHOP-CHAC1 signaling pathway [694]. Yes-associated protein 1 (YAP1), a crucial regulator of the Hippo signaling pathway, can disrupt the interaction between NCOA4 and ferritin heavy chain 1 (FTH1) to inhibit lipid peroxidation and ferroptosis [21]. Another investigation found that adipose-derived stem cells (ADSCs) exosomes augment the expression of NRF2 and GPX4, resulting in a relief of oxidative stress injury and ferroptosis in lung tissue [696]. These studies collectively suggest that interference with ferroptosis can to some extent govern the progression of sepsis. However, further relevant research in this domain is still required to provide promising target insights and effective therapeutic agents for sepsis.
Viral infection
Viral hepatitis, a collection of infectious diseases primarily evidenced by liver inflammation and necrotic lesions, results from the influence of an array of hepatitis viruses [750]. Several different viruses cause hepatitis, including hepatitis A, B, C, D, and E. The hepatitis A and E viruses typically cause acute infections. The hepatitis B, C, and D viruses can cause acute and chronic infections [751]. The transmission of viral hepatitis poses a public health concern, and chronic infection can negatively impact a person's quality of life, leading to symptoms and long-term complications [752]. Studies have shown that ferroptosis may be involved in the development of inflammatory responses, hepatocyte damage, and liver fibrosis in the liver tissues of patients with viral hepatitis [753]. The human hepatitis A virus 3C protease (3Cpro) has been recently identified as the instigator of caspase-independent cell death, with affected cells demonstrating plasma membrane rupture, depletion of mitochondrial potential, and mitochondrial and nuclear engorgement [708]. Subsequent investigations unveiled that cell death orchestrated by 3Cpro was proficiently obviated by ferroptosis inhibitors [708]. These findings infer that 3Cpro expression provokes ferroptosis in human cells. HBV X protein (HBx), a crucial HBV regulatory protein, bears associations with oxidative stress and lipid peroxidation [754]. In vitro and in vivo examinations revealed that HBx curbed the expression of solute carrier family 3 member 2 (SLC3A2), amplifying liver toxicity and ferroptosis induced by d-galactosamine/lipopolysaccharide (D-GalN) [649]. Nevertheless, the host also possesses the capacity to curtail infection via ferroptosis induction. Oxidative degradation of cellular lipids drastically impedes hepatitis C virus (HCV) replication [691]. Yamane and colleagues posited that fatty acid desaturase 2 (FADS2) operates as a rate-limiting factor for ferroptosis, with the escalated expression of FADS2 inhibiting HCV replication9. Moreover, BWA4C, a 5-lipoxygenase inhibitor, can endorse LPO restriction to limit HCV replication [691]. Despite the multitude of studies forging connections between viral hepatitis and ferroptosis, additional research is mandated to cultivate pharmaceuticals for these associated targets.
The COVID-19 pandemic in 2019 jolted the global populace. The observation that augmented ferroptosis transpires in various tissues and cells impacted by COVID-19 warrants significant attention. Han and his team discovered that SARS-CoV-2 infection initiates dysfunction in the human sinoatrial node (SAN)-like pacemaker cells and induces ferroptosis [755]. Vitamin K diminishes the levels of ROS by managing the expression of antioxidant enzymes, proven to curtail lipid peroxidation and inhibit ferroptosis, contributing to its therapeutic efficacy in COVID-19 patients [756]. Han and his group, conducting drug screening utilizing Hesc-SAN-like pacemaker cells, recognized imatinib and deferoxamine as potential candidates for safeguarding pacemaker cells against SARS-CoV-2 infection and ferroptosis [756].
One more instance pertains to AIDS. AIDS, caused by the human immunodeficiency virus (HIV), is a chronic infectious disease that primarily attacks the immune system, specifically CD4+ T cells [757]. As the virus replicates and weakens the immune system, it significantly compromises the body's ability to defend against infections and diseases [758]. In the context of HIV, it has been observed that key markers of ferroptosis, such as iron accumulation and lipid peroxidation, play a significant role [759, 760]. In further research, Xiao and colleagues documented that classical indicators of ferroptosis were discernible in CD4+ T cells of HIV immune non-responders, inclusive of increased lipid peroxidation in mitochondria and destruction of mitochondrial structure [720]. Furthermore, Kannan and colleagues declared that the HIV-1 Tat protein can upregulate ACSL4 expression, escalating lipid peroxidation, which results in the discharge of proinflammatory cytokines and the activation of microglia730. Further studies disclosed that miR-204 functions as an upstream regulator of ACSL4 and inhibits both HIV-1 TAT-mediated ferroptosis and pro-inflammatory cytokine release [721]. These results suggest that the HIV-1 Tat protein and miR-204 might represent potential therapeutic targets against HIV infection. Consequently, the regulation of ferroptosis emerges as a promising therapeutic target and strategy for combating HIV. Nevertheless, further research is necessary to elucidate this and provide treatment strategies targeting ferroptosis for AIDS patients.
Other infection
In recent years, there has been growing evidence implicating the involvement of ferroptosis in the pathogenesis of malaria [761], a persistent public health challenge in economically disadvantaged regions, posing a grave threat to the well-being and lives of local populations [762]. Heather S Kain discerned that impeding GPX4 or SLC7A11 precipitates a substantial reduction in malaria liver-stage parasite infection [733]. Further, Erastin and Sorafenib, inhibitors of SLC7A11, exhibit inhibitory actions on malaria [733]. Another study has shown that desferriamine and lipstatin-1 can stimulate cellular lipid peroxidation and the accumulation of unstable iron associated with dead iron [763]. However, the role of ferroptosis in malaria remains murky. Further research is required to elucidate the intricate interactions between factors related to iron toxicity, malarial parasites, and host immune response in order to combat this devastating infectious disease.
Regardless, extant studies of ferroptosis-pathogen interactions remain relatively rudimentary. Moreover, the understanding of the mechanism underlying the pathogenic regulation of ferroptosis is still deficient in certain areas. Particular pathogen infections necessitate more rigorous investigation to provide novel therapeutic strategies for the evolution of antiviral pharmaceuticals or vaccines. A question that arises is how current medications ameliorate disease symptoms via iron-induced death. For instance, the ferroptosis inhibitor, ferrostatin-1, demonstrates a more pronounced inhibitory effect on Mtb-induced ferroptosis than the reactivation of GPX4 [700], intimating those other mechanisms may participate in Mtb-induced ferroptosis. Recognizing which cellular constituents are involved in the regulation of pathogen-associated ferroptosis may likewise lay the groundwork for drug screening initiatives designed to treat infectious diseases.
Ferroptosis in iron-overload diseases
Iron-overload diseases represent a cluster of disorders distinguished by the excessive accumulation of iron within the body. Several circumstances such as genetic aberrations, blood transfusions, or extended intake of iron supplements can initiate an iron load that outstrips the capacity of iron-binding proteins, precipitating tissue damage [764, 765]. Common iron-overload maladies encompass hereditary hemochromatosis, alcoholic liver disease (ALD), chronic liver disease, and aplastic anemia [766,767,768]. Left untreated, these diseases can cause serious health problems, including liver disease, heart disease, diabetes, and arthritis [767,768,769]
Iron-overload diseases can be divided into two categories: primary and secondary overload [770]. The primary iteration, exemplified by hereditary hemochromatosis and juvenile haemochromatosis, predominantly arises from genetic perturbations, which attenuate hepcidin levels and enhance bodily iron absorption, instigating excessive iron deposition within internal organs [767, 770,771,772,773]. Conversely, the pathogenesis of secondary iron overload predominantly originates from ineffective hematopoiesis, induced by auto-anemic disorders, leading to diminished secretion levels of hepcidin and an upsurge in intestinal iron absorption, thereby engendering iron overload [774, 775]. Concurrently, human blood transfusions liberate substantial quantities of iron, culminating in an iron overburden [774].
Chronic liver disease
The liver, due to its inherent predisposition to oxidative detriment, frequently presents with excessive iron accretion, a quintessential characteristic pervading a plethora of severe hepatic afflictions [776]. Studies have delineated iron overload as a seminal feature of ALD [777, 778], postulating that ethanol may engender liver iron overburden through assorted mechanisms. These encompass the activation of iron-regulatory proteins, thereby elevating transferrin receptor expression, and the repression of the transcription factor CCAAT/enhancer-binding protein α(C/EBPα) or bone morphogenetic protein (BMP)-mediated Smad signaling pathways, resulting in attenuated hepcidin expression [779,780,781].
This hepcidin downregulation fosters increased expression of the divalent metal transporter 1 and ferroportin within the duodenum, culminating in enhanced intestinal iron absorption [782, 783]. Hence, strategies aiming to augment hepcidin or activate the transferrin receptor may exhibit therapeutic potential [20, 778, 784]. Utilization of specific antioxidants, such as vitamin E and N-acetylcysteine (NAC), may relieve alcohol-mediated C/EBPα inhibition in the liver, reduce hepcidin expression, and enhance DMT1. Simultaneously, targeting hepatic sirtuin 1 and cytochrome P450 2E1(CYP2E1) may also provide therapeutic benefits in ALD, principally through lipid peroxidation reduction [20, 785, 786].
Non-alcoholic fatty liver disease and steatohepatitis, conversely, are characterized by iron-deficient hepatocytes and iron overload in hepatic stellate cells [787]. This phenomenon occurs due to iron deficiency intensifying hepatocyte lipogenesis and insulin resistance through HIF2α-ATF4 signaling, while the accumulation of iron engenders excess reactive oxygen species production, thereby exacerbating liver fibrosis [787,788,789]. Iron chelators such as DFO can efficaciously reverse abnormal lipid metabolism and hepatic damage induced by high-fat, high-iron diets [790,791,792]. Therapeutic approaches that aim to enhance NRF2 activity have been shown to facilitate ubiquitination and proteasomal degradation of target proteins, which are mediated by Kelch-like ECH-associated protein 1, β-transducin repeat-containing protein, and/or HMG-CoA reductase degradation protein 1 [793]. Preventative measures such as Vitamin C, quercetin, mitochondrial ROS scavenger Mito-TEMPO, and curcumol to thwart steatosis, and the utilization of iron chelators or ferroptosis inhibitor liproxstatin-1 to maintain iron homeostasis, are favorable approaches to relieve nonalcoholic fatty liver disease (NAFLD) [794,795,796,797].
In conclusion, iron overload-induced chronic liver disease caused by iron overload is a complex condition that requires multidisciplinary approaches for effective management. While current therapies aim to reduce iron burden and relieve the harmful effects of oxidative stress and inflammation, emerging therapies targeting specific pathways involved in disease pathogenesis offer hope for improved treatment outcomes in the future.
Brain iron accumulation
Cerebral iron accumulation, a prevalent comorbidity in a multitude of cognitive and motor function disorders such as AD, PD, multiple system atrophy, and multiple sclerosis, is often evidenced by heightened iron deposition in the brain [469, 503, 798,799,800,801,802,803]. Yet, the mechanistic comprehension of the correlation between this accumulation and neurodegenerative disorders remains insufficient [802].
Friedrich's ataxia (FRDA), a monogenic recessive neurodegenerative condition, is characterized by progressive cerebellar and sensory ataxia, precipitated by the amplification of GAA repeats within the frataxin (FXN) gene, which encodes for the mitochondrial protein frataxin involved in iron-sulfur cluster biogenesis [804,805,806]. Frataxin deficiency can influence iron-sulfur cluster-containing proteins, culminating in iron accumulation within the brains and hearts of afflicted individuals [807]. The primary drivers of FRDA encompass aberrant iron metabolism, mitochondrial dysfunction, and subsequent oxidative damage [808].
Frataxin deficiency will curtail the availability of coenzyme A for TfR1 palmitoylation, while compounds such as artesunate, coenzyme A, and dichloroacetate may ameliorate iron overload through the enhancement of TfR1 palmitoylation [804]. In FRDA, NRF2 is typically downregulated, however, treating afflicted fibroblasts with NRF2 inducers like EPI-743 and sulforaphane could rectify iron deficiency and redox imbalance by targeting NRF2-mediated iron homeostasis [809, 810]. Concurrently, the utilization of leriglitazone and targeting of peroxisome-proliferator-activated receptor gamma may serve as efficacious strategies to improve mitochondrial function, thus offering a therapeutic avenue for FRDA [808, 811].
At present, an absolute remedy for FRDA remains elusive, with extant treatment modalities merely offering symptomatic relief. However, recent years have seen the advent of innovative therapeutic stratagems encompassing gene therapy, small molecule pharmaceuticals, and cell-based interpositions, all of which imbue optimism for the development of more efficacious treatment alternatives for afflicted patients.
IO-associated endocrine diseases
Accumulating evidence implicates iron dysregulation as a pivotal factor in the progression of an array of endocrine disorders, including those of the pancreas and kidneys [812, 813]. Within the pancreas, iron deficiency in β cells can result in diminished insulin secretion [813, 814]. Similarly, iron deficiency in the liver, adipose tissue, and muscles can induce insulin resistance, thereby mediating the onset and advancement of type 2 diabetes mellitus (T2DM) [769, 813].
Research has revealed that iron overload inhibits insulin secretion and compromises islet β cell function through the downregulation of synaptotagmin 7 (SYT7), both in vivo and in vitro models [815]. This suggests SYT7might present a potential therapeutic target for T2DM. Furthermore, free fatty acids, hyperglycemia, and inflammatory cytokines are principal mediators of β-cell toxicity inT2DM, impairing mitochondrial metabolism [816]. Preservation of mitochondrial homeostasis through glutaredoxin 5, caveolin-1, and mitochondrial electron transport can relieve the impacts of T2DM [512, 816, 817].
In summation, iron overload-induced T2DM embodies a multifaceted condition encompassing an array of mechanisms, inclusive of oxidative stress, inflammation, and mitochondrial dysfunction. Existing therapeutic options remain limited, necessitating innovative therapeutic strategies such as antioxidant therapy and targeted interventions. To develop more efficacious treatments, an intricate understanding of the underlying pathophysiological mechanisms remains paramount. At present, phlebotomy, iron modulator supplementation, and iron chelation therapy stand as the primary modalities to mitigate excessive iron accumulation. Clarifying the root cause of iron overload could indeed enhance clinical therapeutics. Notably, iron overload within the body can precipitate a wide array of organ complications, with numerous specific mechanisms still awaiting elucidation. The future trajectory of iron overload disease management may well lie in employing targeted pharmaceuticals and synthesizing drug complexes from these specific materials. Potential therapeutic targets and compounds are duly summarized in Tables 13 & 14.
Conclusion and perspective
Ferroptosis, a form of regulated cell death characterized by iron-dependent accumulation of lipid hydroperoxides, has emerged as a significant area of study in cell biology and disease research. It is distinct from other forms of cell death such as apoptosis, necrosis, and autophagy, and is tightly linked to numerous biological processes, including amino acid, iron, and polyunsaturated fatty acid metabolism, and the biosynthesis of glutathione, phospholipids, NADPH, and CoQ10 [9, 857].
The role of ferroptosis in pathological cell death associated with degenerative diseases, carcinogenesis, stroke, intracerebral hemorrhage, traumatic brain injury, ischemia–reperfusion injury, and kidney degeneration is increasingly being recognized [10, 858,859,860,861]. Moreover, the potential of ferroptosis as a tumor-suppressor function that could be harnessed for cancer therapy is an exciting development.
However, the strategies for tumor suppression and organ injury are fundamentally incongruous. Additional elucidation of the mechanisms of iron-dependent cellular death at every disease stage can equip us with more precise preventative and therapeutic strategies. In addition, other forms of cellular death, such as cuproptosis, have been discovered. The roles that these various forms of cell death play in disease processes warrant further exploration.
Therefore, Future investigations in the field of ferroptosis should focus on further elucidating the molecular mechanisms underlying this form of cell death. This includes understanding the roles of key regulators such as GPX4, FSP1, NRF2, NADPH oxidase, and p53 in ferroptosis.
Hitherto, an array of compounds targeting essential proteins have been deployed to either promote or inhibit ferroptosis, though little has been found in clinical application. Hence, the development of effective therapeutic strategies to modulate ferroptosis could have significant implications for the treatment of a wide range of diseases, including cancer and neurodegenerative disorders. The potential of ferroptosis inhibitors in protecting against pathological conditions such as acute kidney injury also warrants further exploration. Whether multi-target therapy will also seize a prominent position in this field remains a topic of ongoing research.
Moreover, the identification of ferroptosis markers is crucial in differentiating them from cell death induced by oxidative stress and in guiding the development and evaluation of ferroptosis-specific drugs. Assuring safety, efficacy, minimizing off-target effects, and ensuring effective drug delivery present formidable challenges to ongoing research.
In general, a deeper understanding of the specific mechanisms of ferroptosis in different diseases and interventions targeting ferroptosis at various stages of disease progression will provide valuable insights and inform more accurate prevention and treatment strategies for patients.
Availability of data and materials
Not applicable.
References
Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–21. https://doi.org/10.1038/s41423-020-00630-3.
Das S, Shukla N, Singh SS, Kushwaha S, Shrivastava R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis. 2021;26(9–10):512–33. https://doi.org/10.1007/s10495-021-01687-9.
Raj S, Jaiswal SK, DePamphilis ML. Cell death and the p53 enigma during mammalian embryonic development. Stem Cells. 2022;40(3):227–38. https://doi.org/10.1093/stmcls/sxac003.
Kist M, Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J. 2021;40(5):e106700. https://doi.org/10.15252/embj.2020106700.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–96. https://doi.org/10.1016/s1535-6108(03)00050-3.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–45. https://doi.org/10.1016/j.chembiol.2008.02.010.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. https://doi.org/10.1038/nature14344.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82. https://doi.org/10.1038/s41580-020-00324-8.
Yan H-F, Zou T, Tuo Q-Z, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):1–16. https://doi.org/10.1038/s41392-020-00428-9.
Stockwell SJDaBR. The hallmarks of ferroptosis. Annu Rev Cancer Biol. 2019; 2019. 3:35–54. doi:https://doi.org/10.1146/annurev-.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34. https://doi.org/10.1186/s13045-019-0720-y.
Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411-23 e7. https://doi.org/10.1016/j.celrep.2020.02.049.
Schiavi A, Salveridou E, Brinkmann V, Shaik A, Menzel R, Kalyanasundaram S, et al. Mitochondria hormesis delays aging and associated diseases in Caenorhabditis elegans impacting on key ferroptosis players. iScience. 2023;26(4):106448. https://doi.org/10.1016/j.isci.2023.106448.
Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-78.e6. https://doi.org/10.1016/j.ccell.2022.02.003.
Muller S, Sindikubwabo F, Caneque T, Lafon A, Versini A, Lombard B, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat Chem. 2020;12(10):929–38. https://doi.org/10.1038/s41557-020-0513-5.
Wang Y, Zhang M, Bi R, Su Y, Quan F, Lin Y, et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022;51:102262. https://doi.org/10.1016/j.redox.2022.102262.
Schreiber R, Buchholz B, Kraus A, Schley G, Scholz J, Ousingsawat J, et al. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J Am Soc Nephrol. 2019;30(2):228–42. https://doi.org/10.1681/ASN.2018010039.
Maser RL, Vassmer D, Magenheimer BS, Calvet JP. Oxidant stress and reduced antioxidant enzyme protection in polycystic kidney disease. J Am Soc Nephrol. 2002;13(4):991–9. https://doi.org/10.1681/ASN.V134991.
Zhou Z, Ye TJ, DeCaro E, Buehler B, Stahl Z, Bonavita G, et al. Intestinal SIRT1 deficiency protects mice from ethanol-induced liver injury by mitigating ferroptosis. Am J Pathol. 2020;190(1):82–92. https://doi.org/10.1016/j.ajpath.2019.09.012.
Wang J, Zhu Q, Li R, Zhang J, Ye X, Li X. YAP1 protects against septic liver injury via ferroptosis resistance. Cell Biosci. 2022;12(1):163. https://doi.org/10.1186/s13578-022-00902-7.
Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10(6):449. https://doi.org/10.1038/s41419-019-1678-y.
Belaidi AA, Gunn AP, Wong BX, Ayton S, Appukuttan AT, Roberts BR, et al. Marked age-related changes in brain iron homeostasis in amyloid protein precursor knockout mice. Neurotherapeutics. 2018;15(4):1055–62. https://doi.org/10.1007/s13311-018-0656-x.
Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262-79 e25. https://doi.org/10.1016/j.cell.2019.03.032.
Granata S, Votrico V, Spadaccino F, Catalano V, Netti GS, Ranieri E et al. Oxidative stress and ischemia/reperfusion injury in kidney transplantation: focus on ferroptosis, mitophagy and new antioxidants. Antioxidants (Basel). 2022;11(4). https://doi.org/10.3390/antiox11040769.
Yu X, Ma X, Lyu J, Jiang N, Lu Y, Liao Y, et al. Ferroptosis involved in sevoflurane-aggravated young rats brain injury induced by liver transplantation. NeuroReport. 2022;33(16):705–13. https://doi.org/10.1097/wnr.0000000000001836.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–80. https://doi.org/10.1073/pnas.1821022116.
Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314(3):H659–68. https://doi.org/10.1152/ajpheart.00452.2017.
Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-CoA thioesterase 1 prevents cardiomyocytes from Doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. 2020;11(9):756. https://doi.org/10.1038/s41419-020-02948-2.
Zang J, Cui M, Xiao L, Zhang J, Jing R. Overexpression of ferroptosis-related genes FSP1 and CISD1 is related to prognosis and tumor immune infiltration in gastric cancer. Clin Transl Oncol. 2023. https://doi.org/10.1007/s12094-023-03136-2.
Panda SK, Peng V, Sudan R, Ulezko Antonova A, Di Luccia B, Ohara TE, et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity. 2023;56(4):797-812.e4. https://doi.org/10.1016/j.immuni.2023.01.023.
Yan Q, Zheng W, Jiang Y, Zhou P, Lai Y, Liu C, et al. Transcriptomic reveals the ferroptosis features of host response in a mouse model of Zika virus infection. J Med Virol. 2023;95(1):e28386. https://doi.org/10.1002/jmv.28386.
Sawicki KT, De Jesus A, Ardehali H. Iron metabolism in cardiovascular disease: physiology, mechanisms, and therapeutic targets. Circ Res. 2023;132(3):379–96. https://doi.org/10.1161/circresaha.122.321667.
Zeng F, Ye L, Zhou Q, He Y, Zhang Y, Deng G, et al. Inhibiting SCD expression by IGF1R during lorlatinib therapy sensitizes melanoma to ferroptosis. Redox Biol. 2023;61:102653. https://doi.org/10.1016/j.redox.2023.102653.
Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao W, et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the anti-PD-1/L1 response. Adv Sci (Weinh). 2023;10(2):e2203973. https://doi.org/10.1002/advs.202203973.
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341–55. https://doi.org/10.1172/JCI99032.
Yan R, Xie E, Li Y, Li J, Zhang Y, Chi X, et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 2022;32(7):687–90. https://doi.org/10.1038/s41422-022-00642-w.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226. https://doi.org/10.3389/fcell.2020.590226.
Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. 2023;20(1):7–23. https://doi.org/10.1038/s41569-022-00735-4.
Gan B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct Target Ther. 2022;7(1):128. https://doi.org/10.1038/s41392-022-01004-z.
Chen X, Kang R, Tang D. Ferroptosis by lipid peroxidation: the tip of the iceberg? Front Cell Dev Biol. 2021;9:646890. https://doi.org/10.3389/fcell.2021.646890.
Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32(9–10):602–19. https://doi.org/10.1101/gad.314674.118.
Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biology (Basel). 2021;10(3). https://doi.org/10.3390/biology10030184.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–75. https://doi.org/10.1073/pnas.1603244113.
Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. https://doi.org/10.1038/nchembio.2238.
Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–76. https://doi.org/10.1016/j.tcb.2015.10.014.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. https://doi.org/10.3389/fcell.2020.586578.
Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021;7(1):193. https://doi.org/10.1038/s41420-021-00579-w.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–25. https://doi.org/10.1038/s41422-020-00441-1.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85. https://doi.org/10.1016/j.freeradbiomed.2020.02.027.
Zhang LL, Tang RJ, Yang YJ. The underlying pathological mechanism of ferroptosis in the development of cardiovascular disease. Front Cardiovasc Med. 2022;9:964034. https://doi.org/10.3389/fcvm.2022.964034.
Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12(7):706. https://doi.org/10.1038/s41419-021-04008-9.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32(6):920–37. https://doi.org/10.1016/j.cmet.2020.10.011.
Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5(1):108. https://doi.org/10.1038/s41392-020-00216-5.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85. https://doi.org/10.1016/j.cell.2017.09.021.
Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. https://doi.org/10.1016/j.redox.2019.101107.
Xu Z, Xie Y, Mao Y, Huang J, Mei X, Song J, et al. Ferroptosis-related gene signature predicts the prognosis of skin cutaneous melanoma and response to immunotherapy. Front Genet. 2021;12:758981. https://doi.org/10.3389/fgene.2021.758981.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388(6641):482–8. https://doi.org/10.1038/41343.
Wang C-Y, Knutson MD. Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology. 2013;58(2):788–98. https://doi.org/10.1002/hep.26401.
Ems T, St Lucia K, Huecker MR. Biochemistry, iron absorption. Treasure Island (FL): StatPearls; 2023.
Zhang DL, Ghosh MC, Ollivierre H, Li Y, Rouault TA. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood. 2018;132(19):2078–87. https://doi.org/10.1182/blood-2018-04-842997.
Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab. 2015;22(5):777–87. https://doi.org/10.1016/j.cmet.2015.09.006.
Fillebeen C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load. Blood. 2019;133(4):344–55. https://doi.org/10.1182/blood-2018-05-850404.
Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55–63. https://doi.org/10.1016/j.freeradbiomed.2018.07.020.
Chutvanichkul B, Vattanaviboon P, Mas-Oodi S, Y UP, Wanachiwanawin W. Labile iron pool as a parameter to monitor iron overload and oxidative stress status in β-thalassemic erythrocytes. Cytometry B Clin Cytom. 2018;94(4):631–6. https://doi.org/10.1002/cyto.b.21633.
Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic Biol Med. 2002;33(8):1037–46. https://doi.org/10.1016/s0891-5849(02)01006-7.
Ali MY, Oliva CR, Flor S, Griguer CE. Mitoferrin, cellular and mitochondrial iron homeostasis. Cells. 2022;11(21). https://doi.org/10.3390/cells11213464.
Kotla NK, Dutta P, Parimi S, Das NK. The role of ferritin in health and disease: recent advances and understandings. Metabolites. 2022;12(7). https://doi.org/10.3390/metabo12070609.
Acoba MG, Alpergin ESS, Renuse S, Fernández-Del-Río L, Lu YW, Khalimonchuk O, et al. The mitochondrial carrier SFXN1 is critical for complex III integrity and cellular metabolism. Cell Rep. 2021;34(11):108869. https://doi.org/10.1016/j.celrep.2021.108869.
Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. 2017;106(Suppl 6):1559S-S1566. https://doi.org/10.3945/ajcn.117.155804.
Coffey R, Ganz T. Iron homeostasis: An anthropocentric perspective. J Biol Chem. 2017;292(31):12727–34. https://doi.org/10.1074/jbc.R117.781823.
Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61(6):933–52. https://doi.org/10.1136/gut.2010.214312.
Zhang S, Xin W, Anderson GJ, Li R, Gao L, Chen S, et al. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022;13(1):40. https://doi.org/10.1038/s41419-021-04490-1.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. https://doi.org/10.1038/nchembio.1416.
Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69(6):423–34. https://doi.org/10.1002/iub.1616.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–9. https://doi.org/10.1038/nature13148.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575-86.e4. https://doi.org/10.1016/j.devcel.2019.10.007.
Su L-J, Zhang J-H, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843. https://doi.org/10.1155/2019/5080843.
Stadtman ER, Berlett BS. Fenton chemistry. Amino acid oxidation J Biol Chem. 1991;266(26):17201–11.
Prousek J. Fenton chemistry in biology and medicine. Pure Appl Chem. 2007;79(12):2325–38. https://doi.org/10.1351/pac200779122325.
Waldvogel-Abramowski S, Waeber G, Gassner C, Buser A, Frey BM, Favrat B, et al. Physiology of iron metabolism. Transfus Med Hemother. 2014;41(3):213–21. https://doi.org/10.1159/000362888.
Huang J, Simcox J, Mitchell TC, Jones D, Cox J, Luo B, et al. Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013;27(7):2845–54. https://doi.org/10.1096/fj.12-216929.
Jahng JWS, Alsaadi RM, Palanivel R, Song E, Hipolito VEB, Sung HK, et al. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019;20(10):e47911. https://doi.org/10.15252/embr.201947911.
Davis MR, Rendina E, Peterson SK, Lucas EA, Smith BJ, Clarke SL. Enhanced expression of lipogenic genes may contribute to hyperglycemia and alterations in plasma lipids in response to dietary iron deficiency. Genes Nutr. 2012;7(3):415–25. https://doi.org/10.1007/s12263-011-0278-y.
Potashnik R, Kozlovsky N, Ben-Ezra S, Rudich A, Bashan N. Regulation of glucose transport and GLUT-1 expression by iron chelators in muscle cells in culture. Am J Physiol. 1995;269(6):E1052–8. https://doi.org/10.1152/ajpendo.1995.269.6.E1052.
Backe MB, Moen IW, Ellervik C, Hansen JB, Mandrup-Poulsen T. Iron regulation of pancreatic beta-cell functions and oxidative stress. Annu Rev Nutr. 2016;36:241–73. https://doi.org/10.1146/annurev-nutr-071715-050939.
Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, et al. Metabolic adaptation establishes disease tolerance to sepsis. Cell. 2017;169(7):1263-75.e14. https://doi.org/10.1016/j.cell.2017.05.031.
Hay SM, McArdle HJ, Hayes HE, Stevens VJ, Rees WD. The effect of iron deficiency on the temporal changes in the expression of genes associated with fat metabolism in the pregnant rat. Physiol Rep. 2016;4(21). https://doi.org/10.14814/phy2.12908.
Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad Sci U S A. 2018;115(23):5839–48. https://doi.org/10.1073/pnas.1804932115.
Pereira M, Chen T-D, Buang N, Olona A, Ko J-H, Prendecki M, et al. Acute iron deprivation reprograms human macrophage metabolism and reduces inflammation in vivo. Cell Rep. 2019;28(2):498-511.e5. https://doi.org/10.1016/j.celrep.2019.06.039.
Sherman AR. Lipogenesis in iron-deficient adult rats. Lipids. 1978;13(7):473–8. https://doi.org/10.1007/BF02533616.
Myllyharju J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003;22(1):15–24. https://doi.org/10.1016/s0945-053x(03)00006-4.
Buongiorno D, Straganz GD. Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord Chem Rev. 2013;257(2):541–63. https://doi.org/10.1016/j.ccr.2012.04.028.
Yu Q, Tai Y-Y, Tang Y, Zhao J, Negi V, Culley MK, et al. BOLA (BolA Family Member 3) deficiency controls endothelial metabolism and glycine homeostasis in pulmonary hypertension. Circulation. 2019;139(19):2238–55. https://doi.org/10.1161/CIRCULATIONAHA.118.035889.
Góger S, Bogáth D, Baráth G, Simaan AJ, Speier G, Kaizer J. Bio-inspired amino acid oxidation by a non-heme iron catalyst. J Inorg Biochem. 2013;123:46–52. https://doi.org/10.1016/j.jinorgbio.2013.02.007.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–8. https://doi.org/10.1080/15548627.2016.1187366.
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–18. https://doi.org/10.1016/j.freeradbiomed.2020.08.009.
Tang M, Huang Z, Luo X, Liu M, Wang L, Qi Z, et al. Ferritinophagy activation and sideroflexin1-dependent mitochondria iron overload is involved in apelin-13-induced cardiomyocytes hypertrophy. Free Radic Biol Med. 2019;134:445–57. https://doi.org/10.1016/j.freeradbiomed.2019.01.052.
Wolff NA, Garrick MD, Zhao L, Garrick LM, Ghio AJ, Thévenod F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci Rep. 2018;8(1):211. https://doi.org/10.1038/s41598-017-18584-4.
Nigam S, Schewe T. Phospholipase A(2)s and lipid peroxidation. Biochim Biophys Acta. 2000;1488(1–2):167–81. https://doi.org/10.1016/s1388-1981(00)00119-0.
Muralikrishna Adibhatla R, Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40(3):376–87. https://doi.org/10.1016/j.freeradbiomed.2005.08.044.
Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, et al. Pathological aspects of lipid peroxidation. Free Radic Res. 2010;44(10):1125–71. https://doi.org/10.3109/10715762.2010.498478.
Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. 2009;47(5):469–84. https://doi.org/10.1016/j.freeradbiomed.2009.05.032.
H Y, L X, Na P. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10). https://doi.org/10.1021/cr200084z.
Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res. 1998;39(8):1529–42.
Kanner J, German JB, Kinsella JE. Initiation of lipid peroxidation in biological systems. Crit Rev Food Sci Nutr. 1987;25(4):317–64. https://doi.org/10.1080/10408398709527457.
A A, Mf M, S A. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014. https://doi.org/10.1155/2014/360438.
Bayir H, Dixon SJ, Tyurina YY, Kellum JA, Kagan VE. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol. 2023;19(5):315–36. https://doi.org/10.1038/s41581-023-00689-x.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8. https://doi.org/10.1038/nchembio.2239.
Yuan ZH, Liu T, Wang H, Xue LX, Wang JJ. Fatty acids metabolism: the bridge between ferroptosis and ionizing radiation. Front Cell Dev Biol. 2021;9:675617. https://doi.org/10.3389/fcell.2021.675617.
White B. Dietary fatty acids. Am Fam Physician. 2009;80(4):345–50.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–34. https://doi.org/10.1038/s41556-020-0461-8.
Definition of polyunsaturated fatty acid - NCI Drug Dictionary - NCI. 2011. https://www.cancer.gov/publications/dictionaries/cancer-drug/def/polyunsaturated-fatty-acid.
Bentsen H. Dietary polyunsaturated fatty acids, brain function and mental health. Microb Ecol Health Dis. 2017;28(sup1):1281916. https://doi.org/10.1080/16512235.2017.1281916.
Wiktorowska-Owczarek A, Berezinska M, Nowak JZ. PUFAs: Structures, metabolism and functions. Adv Clin Exp Med. 2015;24(6):931–41. https://doi.org/10.17219/acem/31243.
Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem Rev. 2003;103(6):2239–304. https://doi.org/10.1021/cr000068x.
Xiao M, Zhong H, Xia L, Tao Y, Yin H. Pathophysiology of mitochondrial lipid oxidation: role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic Biol Med. 2017;111:316–27. https://doi.org/10.1016/j.freeradbiomed.2017.04.363.
Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420-32.e9. https://doi.org/10.1016/j.chembiol.2018.11.016.
Klasson TD, LaGory EL, Zhao H, Huynh SK, Papandreou I, Moon EJ, et al. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 2022;10(1):14. https://doi.org/10.1186/s40170-022-00290-z.
Pope LE, Dixon S. The role of monounsaturated fatty acids in ferroptosis. The FASEB Journal. 2022;36(S1). https://doi.org/10.1096/fasebj.2022.36.S1.0R871.
Tanaka A, Yamamoto A, Murota K, Tsujiuchi T, Iwamori M, Fukushima N. Polyunsaturated fatty acids induce ovarian cancer cell death through ROS-dependent MAP kinase activation. Biochem Biophys Res Commun. 2017;493(1):468–73. https://doi.org/10.1016/j.bbrc.2017.08.168.
Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012;2012:137289. https://doi.org/10.5402/2012/137289.
Cui W, Liu D, Gu W, Chu B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021;28(8):2536–51. https://doi.org/10.1038/s41418-021-00769-0.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603–8. https://doi.org/10.1038/s41586-020-2732-8.
Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111(10):6130–85. https://doi.org/10.1021/cr200085w.
Bourdillon MT, Ford BA, Knulty AT, Gray CN, Zhang M, Ford D, et al. Oxidation of plasmalogen, low-density lipoprotein and RAW 264.7 cells by photoactivatable atomic oxygen precursors. Photochem Photobiol. 2014;90(2):386–93. https://doi.org/10.1111/php.12201.
Dean JM, Lodhi IJ. Structural and functional roles of ether lipids. Protein Cell. 2018;9(2):196–206. https://doi.org/10.1007/s13238-017-0423-5.
Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary lipids induce ferroptosis in caenorhabditiselegans and human cancer cells. Dev Cell. 2020;54(4):447-54 e4. https://doi.org/10.1016/j.devcel.2020.06.019.
Balgoma D, Hedeland M. Etherglycerophospholipids and ferroptosis: structure, regulation, and location. Trends Endocrinol Metab. 2021;32(12):960–2. https://doi.org/10.1016/j.tem.2021.08.005.
Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(13):2748-64 e22. https://doi.org/10.1016/j.cell.2023.05.003.
Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U S A. 2008;105(8):2830–5. https://doi.org/10.1073/pnas.0712245105.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–9. https://doi.org/10.1021/acschembio.5b00245.
Küch E-M, Vellaramkalayil R, Zhang I, Lehnen D, Brügger B, Sreemmel W, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. 2014;1841(2):227–39. https://doi.org/10.1016/j.bbalip.2013.10.018.
Grube J, Woitok MM, Mohs A, Erschfeld S, Lynen C, Trautwein C, et al. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022;13(8):1–13. https://doi.org/10.1038/s41419-022-05137-5.
Reed A, Ichu T-A, Milosevich N, Melillo B, Schafroth MA, Otsuka Y, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. 2022;17(6):1607–18. https://doi.org/10.1021/acschembio.2c00317.
Mbah NE, Lyssiotis CA. Metabolic regulation of ferroptosis in the tumor microenvironment. J Biol Chem. 2022;298(3). https://doi.org/10.1016/j.jbc.2022.101617.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628-41.e26. https://doi.org/10.1016/j.cell.2017.09.044.
Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15(12):1137–47. https://doi.org/10.1038/s41589-019-0408-1.
Fenton HJH. LXXIII.—Oxidation of tartaric acid in presence of iron. J Chem Soc. 1894;65(0):899–910.
A R, Cm S. Chemistry of phospholipid oxidation. Biochim Biophys Acta. 2012;1818(10). https://doi.org/10.1016/j.bbamem.2012.02.002.
Kuhn H, Saam J, Eibach S, Holzhutter HG, Ivanov I, Walther M. Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem Biophys Res Commun. 2005;338(1):93–101. https://doi.org/10.1016/j.bbrc.2005.08.238.
Xu S, Mueser TC, Marnett LJ, Funk MO. Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure. 2012;20(9):1490–7. https://doi.org/10.1016/j.str.2012.06.00.
Newcomer ME, Brash AR. The structural basis for specificity in lipoxygenase catalysis. Protein Sci. 2015;24(3):298–309. https://doi.org/10.1002/pro.2626.
Ou Y, Wang S-J, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–12. https://doi.org/10.1073/pnas.1607152113.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107(16):7455–60. https://doi.org/10.1073/pnas.1001006107.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. https://doi.org/10.1016/j.molcel.2015.06.011.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–704. https://doi.org/10.1016/j.celrep.2017.07.055.
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–75. https://doi.org/10.1016/j.celrep.2017.12.077.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–91. https://doi.org/10.1038/s41556-019-0305-6.
Zhao J, O’Donnell VB, Balzar S, St Croix CM, Trudeau JB, Wenzel SE. 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc Natl Acad Sci U S A. 2011;108(34):14246–51. https://doi.org/10.1073/pnas.1018075108.
Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401(6749):173–7. https://doi.org/10.1038/43686.
Sheng XH, Cui CC, Shan C, Li YZ, Sheng DH, Sun B, et al. O-Phenylenediamine: a privileged pharmacophore of ferrostatins for radical-trapping reactivity in blocking ferroptosis. Org Biomol Chem. 2018;16(21):3952–60. https://doi.org/10.1039/c8ob00546j.
Anthonymuthu TS, Tyurina YY, Sun W-Y, Mikulska-Ruminska K, Shrivastava IH, Tyurin VA, et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021;38:101744. https://doi.org/10.1016/j.redox.2020.101744.
Vermot A, Petit-Hartlein I, Smith SME, Fieschi F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants (Basel). 2021;10(6). https://doi.org/10.3390/antiox10060890.
Yang W-H, Huang Z, Wu J, Ding C-KC, Murphy SK, Chi J-T. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. 2020;18(1):79–90. https://doi.org/10.1158/1541-7786.MCR-19-0691.
Yang W-H, Ding C-KC, Sun T, Rupprecht G, Lin C-C, Hsu D, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28(10):2501-8.e4. https://doi.org/10.1016/j.celrep.2019.07.107.
Augsburger F, Filippova A, Rasti D, Seredenina T, Lam M, Maghzal G, et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019;26:101272. https://doi.org/10.1016/j.redox.2019.101272.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–9. https://doi.org/10.1038/s41589-020-0472-6.
Riddick DS, Ding X, Wolf CR, Porter TD, Pandey AV, Zhang Q-Y, et al. NADPH-cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metab Dispos. 2013;41(1):12–23. https://doi.org/10.1124/dmd.112.048991.
Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81(2):355-69.e10. https://doi.org/10.1016/j.molcel.2020.11.024.
Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30(1–2):1–12. https://doi.org/10.1016/j.mam.2008.08.006.
Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57(3–4):145–55. https://doi.org/10.1016/s0753-3322(03)00043-x.
Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:736837. https://doi.org/10.1155/2012/736837.
Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J, et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4(6):1247–53. https://doi.org/10.3892/ol.2012.931.
Angeli JPF, Proneth B, Hammond VJ, Tyurina YY, Tyurin VA, O`Donnell VB et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in a Therapeutically Relevant Mechanism. Nat Cell Biol. 2014;Supplement 1(76):S77-S8. https://doi.org/10.1038/ncb3064.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–31. https://doi.org/10.1016/j.cell.2013.12.010.
Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255(6):2372–6.
Kobayashi S, Sato M, Kasakoshi T, Tsutsui T, Sugimoto M, Osaki M, et al. Cystathionine is a novel substrate of cystine/glutamate transporter: IMPLICATIONS FOR IMMUNE FUNCTION*. J Biol Chem. 2015;290(14):8778–88. https://doi.org/10.1074/jbc.M114.625053.
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins*. J Biol Chem. 1999;274(17):11455–8. https://doi.org/10.1074/jbc.274.17.11455.
Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34(18):3421–34. https://doi.org/10.1128/MCB.00221-14.
Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30(8):918–30. https://doi.org/10.1101/gad.275891.115.
Mandal PK, Seiler A, Perisic T, Kölle P, Banjac Canak A, Förster H, et al. System xc− and thioredoxin reductase 1 cooperatively rescue glutathione deficiency*. J Biol Chem. 2010;285(29):22244–53. https://doi.org/10.1074/jbc.M110.121327.
Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–8. https://doi.org/10.1038/cdd.2015.93.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: cystine supplier and beyond. Amino Acids. 2012;42(1):231–46. https://doi.org/10.1007/s00726-011-0867-5.
Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830(5):3143–53. https://doi.org/10.1016/j.bbagen.2012.09.008.
Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30(1–2):42–59. https://doi.org/10.1016/j.mam.2008.05.005.
Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry. 2017;22(3):328–35. https://doi.org/10.1038/mp.2016.196.
Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021;35(11):109235. https://doi.org/10.1016/j.celrep.2021.109235.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. https://doi.org/10.1038/s41467-019-09277-9.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–92. https://doi.org/10.1038/s41586-019-1705-2.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–8. https://doi.org/10.1038/s41586-019-1707-0.
Gong M, Hay S, Marshall KR, Munro AW, Scrutton NS. DNA binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J Biol Chem. 2007;282(41):30331–40. https://doi.org/10.1074/jbc.M703713200.
Wu M, Xu L-G, Li X, Zhai Z, Shu H-B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem. 2002;277(28):25617–23. https://doi.org/10.1074/jbc.M202285200.
Li J, Cao F, Yin H-L, Huang Z-J, Lin Z-T, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):1–13. https://doi.org/10.1038/s41419-020-2298-2.
Nyquist SE, Barr R, Morré DJ. Ubiquinone from rat liver Golgi apparatus fractions. Biochim Biophys Acta. 1970;208(3):532–4. https://doi.org/10.1016/0304-4165(70)90228-x.
Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K, et al. Aifm2, a NADH oxidase, supports robust glycolysis and Is required for cold- and diet-induced thermogenesis. Mol Cell. 2020;77(3):600-17.e4. https://doi.org/10.1016/j.molcel.2019.12.002.
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778–83. https://doi.org/10.1038/s41586-022-05022-3.
Jin DY, Chen X, Liu Y, Williams CM, Pedersen LC, Stafford DW, et al. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat Commun. 2023;14(1):828. https://doi.org/10.1038/s41467-023-36446-8.
Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13(1):2206. https://doi.org/10.1038/s41467-022-29905-1.
Muller F, Lim JKM, Bebber CM, Seidel E, Tishina S, Dahlhaus A, et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023;30(2):442–56. https://doi.org/10.1038/s41418-022-01096-8.
Hendricks JM, Doubravsky CE, Wehri E, Li Z, Roberts MA, Deol KK, et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem Biol. 2023. https://doi.org/10.1016/j.chembiol.2023.04.007.
Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–48. https://doi.org/10.1016/j.cmet.2008.07.005.
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–6. https://doi.org/10.1038/s41586-019-1426-6.
Schneider M, Wortmann M, Mandal PK, Arpornchayanon W, Jannasch K, Alves F, et al. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia. 2010;12(3):254–63. https://doi.org/10.1593/neo.91782.
Zhao B, Lei Q-Y, Guan K-L. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20(6):638–46. https://doi.org/10.1016/j.ceb.2008.10.001.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. https://doi.org/10.1016/j.devcel.2010.09.011.
Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54(2):281–8. https://doi.org/10.1016/j.molcel.2014.03.030.
Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345(6203):1250256. https://doi.org/10.1126/science.1250256.
Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16(10):635–49. https://doi.org/10.1038/nrc.2016.77.
Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther. 2020;5(1):187. https://doi.org/10.1038/s41392-020-00297-2.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. https://doi.org/10.7554/eLife.02523.
Henning Y, Blind US, Larafa S, Matschke J, Fandrey J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022;13(7):662. https://doi.org/10.1038/s41419-022-05121-z.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61. https://doi.org/10.1126/science.abf0529.
Zeng F, Nijiati S, Tang L, Ye J, Zhou Z, Chen X. Ferroptosis detection: from approaches to applications. Angew Chem Int Ed Engl. 2023:e202300379. https://doi.org/10.1002/anie.202300379.
Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162. https://doi.org/10.3389/fcell.2021.637162.
Zeng C, Lin J, Zhang K, Ou H, Shen K, Liu Q, et al. SHARPIN promotes cell proliferation of cholangiocarcinoma and inhibits ferroptosis via p53/SLC7A11/GPX4 signaling. Cancer Sci. 2022;113(11):3766–75. https://doi.org/10.1111/cas.15531.
He C, Wang C, Liu H, Shan B. Kayadiol exerted anticancer effects through p53-mediated ferroptosis in NKTCL cells. BMC Cancer. 2022;22(1):724. https://doi.org/10.1186/s12885-022-09825-5.
Chen X, Zhang T, Su W, Dou Z, Zhao D, Jin X, et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 2022;13(11):974. https://doi.org/10.1038/s41419-022-05408-1.
Xia Z, Kon N, Gu AP, Tavana O, Gu W. Deciphering the acetylation code of p53 in transcription regulation and tumor suppression. Oncogene. 2022;41(22):3039–50. https://doi.org/10.1038/s41388-022-02331-9.
Yang J, Wei X, Hu F, Dong W, Sun L. Development and validation of a novel 3-gene prognostic model for pancreatic adenocarcinoma based on ferroptosis-related genes. Cancer Cell Int. 2022;22(1):21. https://doi.org/10.1186/s12935-021-02431-8.
Wang D, Tang L, Zhang Y, Ge G, Jiang X, Mo Y, et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022;13(6):544. https://doi.org/10.1038/s41419-022-04927-1.
Leu JI, Murphy ME, George DL. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A. 2019;116(17):8390–6. https://doi.org/10.1073/pnas.1821277116.
Wang X, Chen Y, Yang X, Cheng L, He Z, Xin Y, et al. Activation of ALOX12 by a multi-organelle-orienting photosensitizer drives ACSL4-independent cell ferroptosis. Cell Death Dis. 2022;13(12):1040. https://doi.org/10.1038/s41419-022-05462-9.
Egolf S, Zou J, Anderson A, Simpson CL, Aubert Y, Prouty S, et al. MLL4 mediates differentiation and tumor suppression through ferroptosis. Sci Adv. 2021;7(50):eabj9141. https://doi.org/10.1126/sciadv.abj9141.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–92. https://doi.org/10.1038/s41556-018-0178-0.
Zhang Y, Zhuang L, Gan B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol. 2019;6(1):1536845. https://doi.org/10.1080/23723556.2018.1536845.
Zhang Y, Koppula P, Gan B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle. 2019;18(8):773–83. https://doi.org/10.1080/15384101.2019.1597506.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639–43. https://doi.org/10.1038/nature24637.
Terzi EM, Sviderskiy VO, Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv. 2021;7(22):eabg4302. https://doi.org/10.1126/sciadv.abg4302.
Lee J, Shin D, Roh JL. Lipid metabolism alterations and ferroptosis in cancer: Paving the way for solving cancer resistance. Eur J Pharmacol. 2023;941:175497. https://doi.org/10.1016/j.ejphar.2023.175497.
Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L, et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021;33(8):1701-15.e5. https://doi.org/10.1016/j.cmet.2021.05.016.
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33(5):1001-12.e5. https://doi.org/10.1016/j.cmet.2021.02.015.
Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–4. https://doi.org/10.1038/s41586-019-1170-y.
Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021;22(9):1127–39. https://doi.org/10.1038/s41590-021-00996-0.
Zheng H, Jiang L, Tsuduki T, Conrad M, Toyokuni S. Embryonal erythropoiesis and aging exploit ferroptosis. Redox Biol. 2021;48:102175. https://doi.org/10.1016/j.redox.2021.102175.
Mi Y, Wei C, Sun L, Liu H, Zhang J, Luo J, et al. Melatonin inhibits ferroptosis and delays age-related cataract by regulating SIRT6/p-Nrf2/GPX4 and SIRT6/NCOA4/FTH1 pathways. Biomed Pharmacother. 2023;157:114048. https://doi.org/10.1016/j.biopha.2022.114048.
Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. Elife. 2020;9. https://doi.org/10.7554/eLife.56580.
Shen Q, Liang M, Yang F, Deng YZ, Naqvi NI. Ferroptosis contributes to developmental cell death in rice blast. New Phytol. 2020;227(6):1831–46. https://doi.org/10.1111/nph.16636.
Zhang L, Li XM, Shi XH, Ye K, Fu XL, Wang X, et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin. 2023;44(3):622–34. https://doi.org/10.1038/s41401-022-00981-9.
Qiu S, Zhong X, Meng X, Li S, Qian X, Lu H, et al. Mitochondria-localized cGAS suppresses ferroptosis to promote cancer progression. Cell Res. 2023;33(4):299–311. https://doi.org/10.1038/s41422-023-00788-1.
Wu K, Yan M, Liu T, Wang Z, Duan Y, Xia Y, et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat Cell Biol. 2023;25(5):714–25. https://doi.org/10.1038/s41556-023-01133-9.
Cui X, Yun X, Sun M, Li R, Lyu X, Lao Y, et al. HMGCL-induced β-hydroxybutyrate production attenuates hepatocellular carcinoma via DPP4-mediated ferroptosis susceptibility. Hepatol Int. 2023;17(2):377–92. https://doi.org/10.1007/s12072-022-10459-9.
Liu D, Liang CH, Huang B, Zhuang X, Cui W, Yang L, et al. Tryptophan metabolism acts as a new anti-ferroptotic pathway to mediate tumor growth. Adv Sci. 2023;10(6):e2204006. https://doi.org/10.1002/advs.202204006.
Xu FL, Wu XH, Chen C, Wang K, Huang LY, Xia J, et al. SLC27A5 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by downregulating glutathione reductase. Cell Death Dis. 2023;14(1):22. https://doi.org/10.1038/s41419-023-05558-w.
Zhang T, Sun L, Hao Y, Suo C, Shen S, Wei H, et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat Cancer. 2022;3(1):75–89. https://doi.org/10.1038/s43018-021-00299-1.
Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J, et al. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer. 2022;21(1):11. https://doi.org/10.1186/s12943-021-01466-9.
Zhang X, Zheng Q, Yue X, Yuan Z, Ling J, Yuan Y, et al. ZNF498 promotes hepatocellular carcinogenesis by suppressing p53-mediated apoptosis and ferroptosis via the attenuation of p53 Ser46 phosphorylation. J Exp Clin Cancer Res. 2022;41(1):79. https://doi.org/10.1186/s13046-022-02288-3.
Wang Y, Zhao M, Zhao L, Geng Y, Li G, Chen L, et al. HBx-induced HSPA8 stimulates HBV replication and suppresses ferroptosis to support liver cancer progression. Cancer Res. 2023;83(7):1048–61. https://doi.org/10.1158/0008-5472.CAN-22-3169.
Liu J, Liu Y, Wang Y, Li C, Xie Y, Klionsky DJ, et al. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy. 2023;19(3):945–56. https://doi.org/10.1080/15548627.2022.2111635.
Li D, Wang Y, Dong C, Chen T, Dong A, Ren J, et al. CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene. 2023;42(2):83–98. https://doi.org/10.1038/s41388-022-02537-x.
Guo S, Deng J, Wang P, Kou F, Wu Z, Zhang N, et al. The malignancy suppression and ferroptosis facilitation of BCL6 in gastric cancer mediated by FZD7 repression are strengthened by RNF180/RhoC pathway. Cell Biosci. 2023;13(1):73. https://doi.org/10.1186/s13578-023-01020-8.
Qu X, Liu B, Wang L, Liu L, Zhao W, Liu C, et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist Updat. 2023;68:100936. https://doi.org/10.1016/j.drup.2023.100936.
Chen C, Yang Y, Guo Y, He J, Chen Z, Qiu S, et al. CYP1B1 inhibits ferroptosis and induces anti-PD-1 resistance by degrading ACSL4 in colorectal cancer. Cell Death Dis. 2023;14(4):271. https://doi.org/10.1038/s41419-023-05803-2.
Liu MY, Li HM, Wang XY, Xia R, Li X, Ma YJ, et al. TIGAR drives colorectal cancer ferroptosis resistance through ROS/AMPK/SCD1 pathway. Free Radic Biol Med. 2022;182:219–31. https://doi.org/10.1016/j.freeradbiomed.2022.03.002.
Chen L, Cai Q, Yang R, Wang H, Ling H, Li T, et al. GINS4 suppresses ferroptosis by antagonizing p53 acetylation with Snail. Proc Natl Acad Sci U S A. 2023;120(15):e2219585120. https://doi.org/10.1073/pnas.2219585120.
Xu X, Cui J, Wang H, Ma L, Zhang X, Guo W, et al. IGF2BP3 is an essential N(6)-methyladenosine biotarget for suppressing ferroptosis in lung adenocarcinoma cells. Mater Today Bio. 2022;17:100503. https://doi.org/10.1016/j.mtbio.2022.100503.
Wang Q, Gao S, Shou Y, Jia Y, Wei Z, Liu Y, et al. AIM2 promotes renal cell carcinoma progression and sunitinib resistance through FOXO3a-ACSL4 axis-regulated ferroptosis. Int J Biol Sci. 2023;19(4):1266–83. https://doi.org/10.7150/ijbs.79853.
Zheng J, Zhang Q, Zhao Z, Qiu Y, Zhou Y, Wu Z, et al. Epigenetically silenced lncRNA SNAI3-AS1 promotes ferroptosis in glioma via perturbing the m(6)A-dependent recognition of Nrf2 mRNA mediated by SND1. J Exp Clin Cancer Res. 2023;42(1):127. https://doi.org/10.1186/s13046-023-02684-3.
Liu Y, Chou FJ, Lang F, Zhang M, Song H, Zhang W, et al. Protein kinase B (PKB/AKT) protects IDH-Mutated glioma from ferroptosis via Nrf2. Clin Cancer Res. 2023;29(7):1305–16. https://doi.org/10.1158/1078-0432.Ccr-22-3179.
Wen RJ, Dong X, Zhuang HW, Pang FX, Ding SC, Li N, et al. Baicalin induces ferroptosis in osteosarcomas through a novel Nrf2/xCT/GPX4 regulatory axis. Phytomedicine. 2023;116:154881. https://doi.org/10.1016/j.phymed.2023.154881.
Ding Z, Liang X, Wang J, Song Z, Guo Q, Schafer MKE, et al. Inhibition of spinal ferroptosis-like cell death alleviates hyperalgesia and spontaneous pain in a mouse model of bone cancer pain. Redox Biol. 2023;62:102700. https://doi.org/10.1016/j.redox.2023.102700.
Su Z, Kon N, Yi J, Zhao H, Zhang W, Tang Q, et al. Specific regulation of BACH1 by the hotspot mutant p53(R175H) reveals a distinct gain-of-function mechanism. Nat Cancer. 2023;4(4):564–81. https://doi.org/10.1038/s43018-023-00532-z.
Asif K, Adeel M, Rahman MM, Caligiuri I, Perin T, Cemazar M, et al. Iron nitroprusside as a chemodynamic agent and inducer of ferroptosis for ovarian cancer therapy. J Mater Chem B. 2023;11(14):3124–35. https://doi.org/10.1039/d2tb02691k.
Zhang X, Zheng X, Ying X, Xie W, Yin Y, Wang X. CEBPG suppresses ferroptosis through transcriptional control of SLC7A11 in ovarian cancer. J Transl Med. 2023;21(1):334. https://doi.org/10.1186/s12967-023-04136-0.
Anandhan A, Dodson M, Shakya A, Chen J, Liu P, Wei Y, et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv. 2023;9(5):eade9585. https://doi.org/10.1126/sciadv.ade9585.
Wang CK, Chen TJ, Tan GYT, Chang FP, Sridharan S, Yu CA, et al. MEX3A mediates p53 degradation to suppress ferroptosis and facilitate ovarian cancer tumorigenesis. Cancer Res. 2023;83(2):251–63. https://doi.org/10.1158/0008-5472.Can-22-1159.
Wang ME, Chen J, Lu Y, Bawcom AR, Wu J, Ou J et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J Clin Invest. 2023;133(10). https://doi.org/10.1172/JCI166647.
Cheng L, He Q, Liu B, Chen L, Lv F, Li X, et al. SGK2 promotes prostate cancer metastasis by inhibiting ferroptosis via upregulating GPX4. Cell Death Dis. 2023;14(1):74. https://doi.org/10.1038/s41419-023-05614-5.
Jiang K, Yin X, Zhang Q, Yin J, Tang Q, Xu M, et al. STC2 activates PRMT5 to induce radioresistance through DNA damage repair and ferroptosis pathways in esophageal squamous cell carcinoma. Redox Biol. 2023;60:102626. https://doi.org/10.1016/j.redox.2023.102626.
Wang S, Yi X, Wu Z, Guo S, Dai W, Wang H, et al. CAMKK2 Defines Ferroptosis Sensitivity of Melanoma Cells by Regulating AMPK-NRF2 Pathway. J Invest Dermatol. 2022;142(1):189-200.e8. https://doi.org/10.1016/j.jid.2021.05.025.
Zhang H-L, Hu B-X, Li Z-L, Du T, Shan J-L, Ye Z-P, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24(1):88–98. https://doi.org/10.1038/s41556-021-00818-3.
Wang S, Wang Y, Li Q, Zeng K, Li X, Feng X. RUNX1-IT1 favors breast cancer carcinogenesis through regulation of IGF2BP1/GPX4 axis. Discov Oncol. 2023;14(1):42. https://doi.org/10.1007/s12672-023-00652-z.
Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y, et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol. 2022;15(1):2. https://doi.org/10.1186/s13045-021-01223-x.
Wang YF, Feng JY, Zhao LN, Zhao M, Wei XF, Geng Y et al. Aspirin triggers ferroptosis in hepatocellular carcinoma cells through restricting NF-κB p65-activated SLC7A11 transcription. Acta Pharmacol Sin. 2023. https://doi.org/10.1038/s41401-023-01062-1.
Zhu ZH, Xu XT, Shen CJ, Yuan JT, Lou SY, Ma XL, et al. A novel sesquiterpene lactone fraction from Eupatorium chinense L. suppresses hepatocellular carcinoma growth by triggering ferritinophagy and mitochondrial damage. Phytomedicine. 2023;112:154671. https://doi.org/10.1016/j.phymed.2023.154671.
Xi J, Tian LL, Xi J, Girimpuhwe D, Huang C, Ma R, et al. Alterperylenol as a novel thioredoxin reductase inhibitor induces liver cancer cell apoptosis and ferroptosis. J Agric Food Chem. 2022;70(50):15763–75. https://doi.org/10.1021/acs.jafc.2c05339.
Liu X, Peng X, Cen S, Yang C, Ma Z, Shi X. Wogonin induces ferroptosis in pancreatic cancer cells by inhibiting the Nrf2/GPX4 axis. Front Pharmacol. 2023;14:1129662. https://doi.org/10.3389/fphar.2023.1129662.
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023:1–15. https://doi.org/10.1080/15548627.2023.2165323.
Cui W, Zhang J, Wu D, Zhang J, Zhou H, Rong Y, et al. Ponicidin suppresses pancreatic cancer growth by inducing ferroptosis: Insight gained by mass spectrometry-based metabolomics. Phytomedicine. 2022;98:153943. https://doi.org/10.1016/j.phymed.2022.153943.
Zheng F, Wang Y, Zhang Q, Chen Q, Liang CL, Liu H, et al. Polyphyllin I suppresses the gastric cancer growth by promoting cancer cell ferroptosis. Front Pharmacol. 2023;14:1145407. https://doi.org/10.3389/fphar.2023.1145407.
Xu X, Li Y, Wu Y, Wang M, Lu Y, Fang Z, et al. Increased ATF2 expression predicts poor prognosis and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox Biol. 2023;59:102564. https://doi.org/10.1016/j.redox.2022.102564.
Bian Z, Sun X, Liu L, Qin Y, Zhang Q, Liu H et al. Sodium butyrate induces CRC cell ferroptosis via the CD44/SLC7A11 pathway and exhibits a synergistic therapeutic effect with erastin. Cancers (Basel). 2023;15(2). https://doi.org/10.3390/cancers15020423.
Miao Q, Deng WQ, Lyu WY, Sun ZT, Fan SR, Qi M et al. Erianin inhibits the growth and metastasis through autophagy-dependent ferroptosis in KRAS(G13D) colorectal cancer. Free Radic Biol Med. 2023. https://doi.org/10.1016/j.freeradbiomed.2023.05.008.
Zhu JF, Liu Y, Li WT, Li MH, Zhen CH, Sun PW, et al. Ibrutinib facilitates the sensitivity of colorectal cancer cells to ferroptosis through BTK/NRF2 pathway. Cell Death Dis. 2023;14(2):151. https://doi.org/10.1038/s41419-023-05664-9.
Guo S, Zhao W, Zhang W, Li S, Teng G, Liu L. Vitamin D promotes ferroptosis in colorectal cancer stem cells via SLC7A11 downregulation. Oxid Med Cell Longev. 2023;2023:4772134. https://doi.org/10.1155/2023/4772134.
Hao J, Chen Q, Feng Y, Jiang Q, Sun H, Deng B, et al. Combination treatment with FAAH inhibitors/URB597 and ferroptosis inducers significantly decreases the growth and metastasis of renal cell carcinoma cells via the PI3K-AKT signaling pathway. Cell Death Dis. 2023;14(4):247. https://doi.org/10.1038/s41419-023-05779-z.
Kang L, Wang D, Shen T, Liu X, Dai B, Zhou D, et al. PDIA4 confers resistance to ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma. Cell Death Dis. 2023;14(3):193. https://doi.org/10.1038/s41419-023-05719-x.
Xu C, Jiang ZB, Shao L, Zhao ZM, Fan XX, Sui X, et al. β-Elemene enhances erlotinib sensitivity through induction of ferroptosis by upregulating lncRNA H19 in EGFR-mutant non-small cell lung cancer. Pharmacol Res. 2023;191:106739. https://doi.org/10.1016/j.phrs.2023.106739.
Zhou C, Yu T, Zhu R, Lu J, Ouyang X, Zhang Z, et al. Timosaponin aIII promotes non-small-cell lung cancer ferroptosis through targeting and facilitating HSP90 mediated GPX4 ubiquitination and degradation. Int J Biol Sci. 2023;19(5):1471–89. https://doi.org/10.7150/ijbs.77979.
Zhang W, Jiang B, Liu Y, Xu L, Wan M. Bufotalin induces ferroptosis in non-small cell lung cancer cells by facilitating the ubiquitination and degradation of GPX4. Free Radic Biol Med. 2022;180:75–84. https://doi.org/10.1016/j.freeradbiomed.2022.01.009.
Han N, Yang ZY, Xie ZX, Xu HZ, Yu TT, Li QR, et al. Dihydroartemisinin elicits immunogenic death through ferroptosis-triggered ER stress and DNA damage for lung cancer immunotherapy. Phytomedicine. 2023;112:154682. https://doi.org/10.1016/j.phymed.2023.154682.
Hu CY, Wu HT, Shan YS, Wang CT, Shieh GS, Wu CL et al. Evodiamine exhibits anti-bladder cancer activity by suppression of glutathione peroxidase 4 and induction of ferroptosis. Int J Mol Sci. 2023;24(7). https://doi.org/10.3390/ijms24076021.
Cai J, Ye Z, Hu Y, Ye L, Gao L, Wang Y, et al. Fatostatin induces ferroptosis through inhibition of the AKT/mTORC1/GPX4 signaling pathway in glioblastoma. Cell Death Dis. 2023;14(3):211. https://doi.org/10.1038/s41419-023-05738-8.
Ni M, Zhou J, Zhu Z, Xu Q, Yin Z, Wang Y, et al. Shikonin and cisplatin synergistically overcome cisplatin resistance of ovarian cancer by inducing ferroptosis via upregulation of HMOX1 to promote Fe(2+) accumulation. Phytomedicine. 2023;112:154701. https://doi.org/10.1016/j.phymed.2023.154701.
Mao G, Xin D, Wang Q, Lai D. Sodium molybdate inhibits the growth of ovarian cancer cells via inducing both ferroptosis and apoptosis. Free Radic Biol Med. 2022;182:79–92. https://doi.org/10.1016/j.freeradbiomed.2022.02.023.
Cao YY, Chen YY, Wang MS, Tong JJ, Xu M, Zhao C, et al. A catalase inhibitor: Targeting the NADPH-binding site for castration-resistant prostate cancer therapy. Redox Biol. 2023;63:102751. https://doi.org/10.1016/j.redox.2023.102751.
Zhao P, Song H, Gao F, Chen L, Qiu J, Jin J et al. A novel derivative of curcumol, HCL-23, inhibits the malignant phenotype of triple-negative breast cancer and induces apoptosis and HO-1-dependent ferroptosis. Molecules. 2023;28(8). https://doi.org/10.3390/molecules28083389.
Zhang B, Hou Q, Zhang X, Ma Y, Yuan J, Li S, et al. Anesthetic propofol inhibits ferroptosis and aggravates distant cancer metastasis via Nrf2 upregulation. Free Radic Biol Med. 2023;195:298–308. https://doi.org/10.1016/j.freeradbiomed.2022.12.092.
Chen H, Li Z, Xu J, Zhang N, Chen J, Wang G, et al. Curcumin induces ferroptosis in follicular thyroid cancer by upregulating HO-1 expression. Oxid Med Cell Longev. 2023;2023:6896790. https://doi.org/10.1155/2023/6896790.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. https://doi.org/10.1158/2159-8290.Cd-21-1059.
Advancing Cancer Therapy. Nature Cancer. 2021;2(3):245-6. https://doi.org/10.1038/s43018-021-00192-x.
Saito A, Toyoda H, Kobayashi M, Koiwa Y, Fujii H, Fujita K, et al. Prediction of early recurrence of hepatocellular carcinoma after resection using digital pathology images assessed by machine learning. Mod Pathol. 2021;34(2):417–25. https://doi.org/10.1038/s41379-020-00671-z.
Poh AR, Ernst M. Functional roles of SRC signaling in pancreatic cancer: Recent insights provide novel therapeutic opportunities. Oncogene. 2023;42(22):1786–801. https://doi.org/10.1038/s41388-023-02701-x.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21(1):47. https://doi.org/10.1186/s12943-022-01530-y.
Spangler B, Morgan CW, Fontaine SD, Vander Wal MN, Chang CJ, Wells JA, et al. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat Chem Biol. 2016;12(9):680–5. https://doi.org/10.1038/nchembio.2116.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–96. https://doi.org/10.1038/s41571-020-00462-0.
Kim E, Han DJ, Kim BH, Yoo J, Kim HJ, Wu HG, et al. Whole-genome sequencing reveals mutational signatures related to radiation-induced sarcomas and DNA-damage-repair pathways. Mod Pathol. 2023;36(1):100004. https://doi.org/10.1016/j.modpat.2022.100004.
Meng Z, Lu J, Ge G, Wang G, Zhang R, Li Y, et al. Ginsenoside Rb1 induces autophagic lipid degradation via miR-128 targeting TFEB. Food Funct. 2023;14(1):240–9. https://doi.org/10.1039/d2fo02719d.
Truong H, Breen K, Nandakumar S, Sjoberg DD, Kemel Y, Mehta N, et al. Gene-based confirmatory germline testing following tumor-only sequencing of prostate cancer. Eur Urol. 2023;83(1):29–38. https://doi.org/10.1016/j.eururo.2022.08.028.
Pennington KL, McEwan CM, Woods J, Muir CM, Pramoda Sahankumari AG, Eastmond R, et al. SGK2, 14–3-3, and HUWE1 cooperate to control the localization, stability, and function of the oncoprotein PTOV1. Mol Cancer Res. 2022;20(2):231–43. https://doi.org/10.1158/1541-7786.Mcr-20-1076.
Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061. https://doi.org/10.1038/nrdp.2016.61.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309. https://doi.org/10.1038/s41586-019-1730-1.
Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. 2019;12(1):134. https://doi.org/10.1186/s13045-019-0818-2.
Sharifi-Rad J, Herrera-Bravo J, Salazar LA, Shaheen S, Abdulmajid Ayatollahi S, Kobarfard F, et al. The therapeutic potential of wogonin observed in preclinical studies. Evid Based Complement Alternat Med. 2021;2021:9935451. https://doi.org/10.1155/2021/9935451.
Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–54. https://doi.org/10.1016/S0140-6736(21)00312-3.
Fan G, Wei X, Xu X. Is the era of sorafenib over? A review of the literature. Ther Adv Med Oncol. 2020;12:1758835920927602. https://doi.org/10.1177/1758835920927602.
Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2017;8:992. https://doi.org/10.3389/fphar.2017.00992.
Li B, Yang L, Peng X, Fan Q, Wei S, Yang S, et al. Emerging mechanisms and applications of ferroptosis in the treatment of resistant cancers. Biomed Pharmacother. 2020;130:110710. https://doi.org/10.1016/j.biopha.2020.110710.
Mikubo M, Inoue Y, Liu G, Tsao MS. Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J Thorac Oncol. 2021;16(11):1798–809. https://doi.org/10.1016/j.jtho.2021.07.017.
Zhang Z, Tan Y, Huang C, Wei X. Redox signaling in drug-tolerant persister cells as an emerging therapeutic target. EBioMedicine. 2023;89:104483. https://doi.org/10.1016/j.ebiom.2023.104483.
Qi R, Bai Y, Li K, Liu N, Xu Y, Dal E, et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting miRNAs. Drug Resist Updat. 2023;68:100960. https://doi.org/10.1016/j.drup.2023.100960.
Xiong K, Wang Z, Hounye AH, Peng L, Zhang J, Qi M. Development and validation of ferroptosis-related lncRNA signature and immune-related gene signature for predicting the prognosis of cutaneous melanoma patients. Apoptosis. 2023;28(5–6):840–59. https://doi.org/10.1007/s10495-023-01831-7.
Yan H, Talty R, Jain A, Cai Y, Zheng J, Shen X, et al. Discovery of decreased ferroptosis in male colorectal cancer patients with KRAS mutations. Redox Biol. 2023;62:102699. https://doi.org/10.1016/j.redox.2023.102699.
Chen H, Yin L, Yang J, Ren N, Chen J, Lu Q, et al. Genetic polymorphisms in genes regulating cell death and prognosis of patients with rectal cancer receiving postoperative chemoradiotherapy. Cancer Biol Med. 2023;20(4):297–316. https://doi.org/10.20892/j.issn.2095-3941.2022.0711.
Liu S, Zhang HL, Li J, Ye ZP, Du T, Li LC, et al. Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis. Redox Biol. 2023;62:102677. https://doi.org/10.1016/j.redox.2023.102677.
Wang C, Zheng C, Wang H, Shui S, Jin H, Liu G, et al. Dual degradation mechanism of GPX4 degrader in induction of ferroptosis exerting anti-resistant tumor effect. Eur J Med Chem. 2023;247:115072. https://doi.org/10.1016/j.ejmech.2022.115072.
Gu Y, Li Y, Wang J, Zhang L, Zhang J, Wang Y. Targeting ferroptosis: Paving new roads for drug design and discovery. Eur J Med Chem. 2023;247:115015. https://doi.org/10.1016/j.ejmech.2022.115015.
Zhang X, Xu X, Liu H, Ni N, Liu S, Gong Y et al. CCR2-overexpressing biomimetic carrier-free nanoplatform for enhanced cascade ferroptosis tumor therapy. Acta Biomater. 2023. https://doi.org/10.1016/j.actbio.2023.05.006.
Zhang Z, Lo H, Zhao X, Li W, Wu K, Zeng F, et al. Mild photothermal/radiation therapy potentiates ferroptosis effect for ablation of breast cancer via MRI/PA imaging guided all-in-one strategy. J Nanobiotechnology. 2023;21(1):150. https://doi.org/10.1186/s12951-023-01910-6.
Chen Y, Yao Z, Liu P, Hu Q, Huang Y, Ping L, et al. A self-assembly nano-prodrug for triple-negative breast cancer combined treatment by ferroptosis therapy and chemotherapy. Acta Biomater. 2023;159:275–88. https://doi.org/10.1016/j.actbio.2023.01.050.
Soares ROS, Losada DM, Jordani MC, Évora P, Castro-E-Silva O. Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. Int J Mol Sci. 2019;20(20):5034. https://doi.org/10.3390/ijms20205034.
Heusch G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17(12):773–89. https://doi.org/10.1038/s41569-020-0403-y.
Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190(3):255–66. https://doi.org/10.1002/(SICI)1096-9896(200002)190:3%3c255::AID-PATH526%3e3.0.CO;2-6.
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46(4):1650–67. https://doi.org/10.1159/000489241.
Chen C, Zheng M, Hou H, Fang S, Chen L, Yang J, et al. Cellular senescence in ischemia/reperfusion injury. Cell Death Discov. 2022;8(1):420. https://doi.org/10.1038/s41420-022-01205-z.
Verma S, Fedak PW, Weisel RD, Butany J, Rao V, Maitland A, et al. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation. 2002;105(20):2332–6. https://doi.org/10.1161/01.cir.0000016602.96363.36.
Mandalaneni K, Rayi A, Jillella DV. Stroke reperfusion injury. Treasure Island (FL): StatPearls; 2023.
Wu L, Xiong X, Wu X, Ye Y, Jian Z, Zhi Z, et al. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci. 2020;13:28. https://doi.org/10.3389/fnmol.2020.00028.
Slegtenhorst BR, Dor FJ, Rodriguez H, Voskuil FJ, Tullius SG. Ischemia/reperfusion injury and its consequences on immunity and inflammation. Curr Transplant Rep. 2014;1(3):147–54. https://doi.org/10.1007/s40472-014-0017-6.
Sanchez-Hernandez CD, Torres-Alarcon LA, Gonzalez-Cortes A, Peon AN. Ischemia/reperfusion injury: pathophysiology, current clinical management, and potential preventive approaches. Mediators Inflamm. 2020;2020:8405370. https://doi.org/10.1155/2020/8405370.
Zhou Y, He Y, Yan S, Chen L, Zhang R, Xu J, et al. Reperfusion injury is associated with poor outcome in patients with recanalization after thrombectomy. Stroke. 2023;54(1):96–104. https://doi.org/10.1161/strokeaha.122.039337.
Nieuwenhuijs-Moeke GJ, Pischke SE, Berger SP, Sanders JSF, Pol RA, Struys M et al. Ischemia and reperfusion injury in kidney transplantation: Relevant mechanisms in injury and repair. J Clin Med. 2020;9(1). https://doi.org/10.3390/jcm9010253.
Fernandez AR, Sanchez-Tarjuelo R, Cravedi P, Ochando J, Lopez-Hoyos M. Review: Ischemia Reperfusion Injury-A Translational Perspective in Organ Transplantation. Int J Mol Sci. 2020;21(22). https://doi.org/10.3390/ijms21228549.
Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. 2021;166:105466. https://doi.org/10.1016/j.phrs.2021.105466.
Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, et al. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61(3):414–26. https://doi.org/10.1016/j.cardiores.2003.12.023.
Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. https://doi.org/10.1016/B978-0-12-394309-5.00006-7.
Zhou L, Han S, Guo J, Qiu T, Zhou J, Shen L. Ferroptosis-a new dawn in the treatment of organ ischemia-reperfusion injury. Cells. 2022;11(22). https://doi.org/10.3390/cells11223653.
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26(11):2284–99. https://doi.org/10.1038/s41418-019-0299-4.
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang X, et al. Alox15/15-HpETE aggravates myocardial ischemia-reperfusion injury by promoting cardiomyocyte ferroptosis. Circulation. 2023;147(19):1444–60. https://doi.org/10.1161/CIRCULATIONAHA.122.060257.
Jiang YQ, Yang XY, Duan DQ, Zhang YY, Li NS, Tang LJ, et al. Inhibition of MALT1 reduces ferroptosis in rat hearts following ischemia/reperfusion via enhancing the Nrf2/SLC7A11 pathway. Eur J Pharmacol. 2023;950:175774. https://doi.org/10.1016/j.ejphar.2023.175774.
Ji JJ, Chen SY, Yang ZW, Zhang R, Qian LL, Jiang Y, et al. Delivery of Mir-196c-3p with NIR-II light-triggered gel attenuates cardiomyocyte ferroptosis in cardiac ischemia-reperfusion injury. Nanomedicine. 2023;47:102618. https://doi.org/10.1016/j.nano.2022.102618.
Zhang JK, Zhang Z, Guo ZA, Fu Y, Chen XJ, Chen WJ, et al. The BMSC-derived exosomal lncRNA Mir9-3hg suppresses cardiomyocyte ferroptosis in ischemia-reperfusion mice via the Pum2/PRDX6 axis. Nutr Metab Cardiovasc Dis. 2022;32(2):515–27. https://doi.org/10.1016/j.numecd.2021.10.017.
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang JJ, Luo XJ, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic Biol Med. 2021;162:339–52. https://doi.org/10.1016/j.freeradbiomed.2020.10.307.
Liu H, Mo H, Yang C, Mei X, Song X, Lu W, et al. A novel function of ATF3 in suppression of ferroptosis in mouse heart suffered ischemia/reperfusion. Free Radic Biol Med. 2022;189:122–35. https://doi.org/10.1016/j.freeradbiomed.2022.07.006.
Zhuang Y, Yang D, Shi S, Wang L, Yu M, Meng X, et al. MiR-375-3p promotes cardiac fibrosis by regulating the ferroptosis mediated by GPX4. Comput Intell Neurosci. 2022;2022:9629158. https://doi.org/10.1155/2022/9629158.
Wu T, Shi G, Ji Z, Wang S, Geng L, Guo Z. Circulating small extracellular vesicle-encapsulated SEMA5A-IT1 attenuates myocardial ischemia-reperfusion injury after cardiac surgery with cardiopulmonary bypass. Cell Mol Biol Lett. 2022;27(1):95. https://doi.org/10.1186/s11658-022-00395-9.
Zhang W, Qiao W, Zuo L. A(1) and A(2b) adenosine receptors regulate GPX4 against ferroptosis of cardiomyocytes in myocardial infarction rat model and in vitro. Tissue Cell. 2022;77:101828. https://doi.org/10.1016/j.tice.2022.101828.
Zhang GY, Gao Y, Guo XY, Wang GH, Guo CX. MiR-199a-5p promotes ferroptosis-induced cardiomyocyte death responding to oxygen-glucose deprivation/reperfusion injury via inhibiting Akt/eNOS signaling pathway. Kaohsiung J Med Sci. 2022;38(11):1093–102. https://doi.org/10.1002/kjm2.12605.
Ma S, Sun L, Wu W, Wu J, Sun Z, Ren J. USP22 protects against myocardial ischemia-reperfusion injury via the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Front Physiol. 2020;11:551318. https://doi.org/10.3389/fphys.2020.551318.
Tian H, Xiong Y, Zhang Y, Leng Y, Tao J, Li L, et al. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones. 2021;27(2):149–64. https://doi.org/10.1007/s12192-022-01257-1.
Li W, Li W, Wang Y, Leng Y, Xia Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Dis. 2021;7(1):267. https://doi.org/10.1038/s41420-021-00656-0.
Stamenkovic A, O’Hara KA, Nelson DC, Maddaford TG, Edel AL, Maddaford G, et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2021;320(3):H1170–84. https://doi.org/10.1152/ajpheart.00237.2020.
Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P, Zheng SY. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. 2021;27(1):14. https://doi.org/10.1186/s10020-021-00271-w.
Sun W, Shi R, Guo J, Wang H, Shen L, Shi H, et al. miR-135b-3p promotes cardiomyocyte ferroptosis by targeting GPX4 and aggravates myocardial ischemia/reperfusion injury. Front Cardiovasc Med. 2021;8:663832. https://doi.org/10.3389/fcvm.2021.663832.
Sun W, Wu X, Yu P, Zhang Q, Shen L, Chen J, et al. LncAABR07025387.1 enhances myocardial ischemia/reperfusion injury via miR-205/ACSL4-mediated ferroptosis. Front Cell Dev Biol. 2022;10:672391. https://doi.org/10.3389/fcell.2022.672391.
Hu M, Huang J, Chen L, Sun XR, Yao ZM, Tong XH, et al. Upregulation of CDGSH iron sulfur domain 2 attenuates cerebral ischemia/reperfusion injury. Neural Regen Res. 2023;18(7):1512–20. https://doi.org/10.4103/1673-5374.355766.
Liu Q, Liu Y, Li Y, Hong Z, Li S, Liu C. PUM2 aggravates the neuroinflammation and brain damage induced by ischemia-reperfusion through the SLC7A11-dependent inhibition of ferroptosis via suppressing the SIRT1. Mol Cell Biochem. 2023;478(3):609–20. https://doi.org/10.1007/s11010-022-04534-w.
Xiong L, Zhang J, Shi H, Zhu G, Ji X, Li M, et al. Downregulation of TNFAIP1 alleviates OGD/R-induced neuronal damage by suppressing Nrf2/GPX4-mediated ferroptosis. Exp Ther Med. 2023;25(1):25. https://doi.org/10.3892/etm.2022.11724.
Yang J, Guo Q, Wang L, Yu S. POU domain class 2 transcription factor 2 inhibits ferroptosis in cerebral ischemia reperfusion injury by activating sestrin2. Neurochem Res. 2023;48(2):658–70. https://doi.org/10.1007/s11064-022-03791-x.
Zhang T, Yang M, Ma C, Wei X, Zhang Z. BACH1 encourages ferroptosis by activating KDM4C-mediated COX2 demethylation after cerebral ischemia-reperfusion injury. Eur J Neurosci. 2023. https://doi.org/10.1111/ejn.16035.
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22(11):1520–30. https://doi.org/10.1038/mp.2017.171.
Li C, Sun G, Chen B, Xu L, Ye Y, He J, et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol Res. 2021;174:105933. https://doi.org/10.1016/j.phrs.2021.105933.
Chen W, Jiang L, Hu Y, Tang N, Liang N, Li XF, et al. Ferritin reduction is essential for cerebral ischemia-induced hippocampal neuronal death through p53/SLC7A11-mediated ferroptosis. Brain Res. 2021;1752:147216. https://doi.org/10.1016/j.brainres.2020.147216.
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021;12(5):447. https://doi.org/10.1038/s41419-021-03725-5.
Huang Y, Liu J, He J, Hu Z, Tan F, Zhu X, et al. UBIAD1 alleviates ferroptotic neuronal death by enhancing antioxidative capacity by cooperatively restoring impaired mitochondria and Golgi apparatus upon cerebral ischemic/reperfusion insult. Cell Biosci. 2022;12(1):42. https://doi.org/10.1186/s13578-022-00776-9.
Xu Y, Liu Y, Li K, Yuan D, Yang S, Zhou L, et al. COX-2/PGE2 pathway inhibits the ferroptosis induced by cerebral ischemia reperfusion. Mol Neurobiol. 2022;59(3):1619–31. https://doi.org/10.1007/s12035-021-02706-1.
Zhao J, Wu Y, Liang S, Piao X. Activation of SSAT1/ALOX15 Axis aggravates cerebral ischemia/reperfusion injury via triggering neuronal ferroptosis. Neuroscience. 2022;485:78–90. https://doi.org/10.1016/j.neuroscience.2022.01.017.
Tuo QZ, Liu Y, Xiang Z, Yan HF, Zou T, Shu Y, et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct Target Ther. 2022;7(1):59. https://doi.org/10.1038/s41392-022-00917-z.
Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020;260:118305. https://doi.org/10.1016/j.lfs.2020.118305.
Zou S, Sun H, Peng Y, Liang C, Zheng C, Wang L, et al. Mu-opioid receptor alleviated ferroptosis in hepatic ischemia-reperfusion injury via the HIF-1alpha/KCNQ1OT1 axis. Am J Physiol Cell Physiol. 2023;324(4):C927–40. https://doi.org/10.1152/ajpcell.00394.2022.
Li X, Wu L, Tian X, Zheng W, Yuan M, Tian X, et al. miR-29a-3p in exosomes from heme oxygenase-1 modified bone marrow mesenchymal stem cells alleviates steatotic liver ischemia-reperfusion injury in rats by suppressing ferroptosis via iron responsive element binding protein 2. Oxid Med Cell Longev. 2022;2022:6520789. https://doi.org/10.1155/2022/6520789.
Wu S, Yang J, Sun G, Hu J, Zhang Q, Cai J, et al. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br J Pharmacol. 2021;178(18):3783–96. https://doi.org/10.1111/bph.15518.
Wu Y, Jiao H, Yue Y, He K, Jin Y, Zhang J, et al. Ubiquitin ligase E3 HUWE1/MULE targets transferrin receptor for degradation and suppresses ferroptosis in acute liver injury. Cell Death Differ. 2022;29(9):1705–18. https://doi.org/10.1038/s41418-022-00957-6.
Shi L, Song Z, Li Y, Huang J, Zhao F, Luo Y, et al. MiR-20a-5p alleviates kidney ischemia/reperfusion injury by targeting ACSL4-dependent ferroptosis. Am J Transplant. 2023;23(1):11–25. https://doi.org/10.1016/j.ajt.2022.09.003.
Sun X, Huang N, Li P, Dong X, Yang J, Zhang X, et al. TRIM21 ubiquitylates GPX4 and promotes ferroptosis to aggravate ischemia/reperfusion-induced acute kidney injury. Life Sci. 2023;321:121608. https://doi.org/10.1016/j.lfs.2023.121608.
Sun Z, Wu J, Bi Q, Wang W. Exosomal lncRNA TUG1 derived from human urine-derived stem cells attenuates renal ischemia/reperfusion injury by interacting with SRSF1 to regulate ASCL4-mediated ferroptosis. Stem Cell Res Ther. 2022;13(1):297. https://doi.org/10.1186/s13287-022-02986-x.
Huang LL, Liao XH, Sun H, Jiang X, Liu Q, Zhang L. Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J Cell Mol Med. 2019;23(6):4153–64. https://doi.org/10.1111/jcmm.14302.
Su L, Jiang X, Yang C, Zhang J, Chen B, Li Y, et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J Biol Chem. 2019;294(50):19395–404. https://doi.org/10.1074/jbc.RA119.010949.
Sui M, Xu D, Zhao W, Lu H, Chen R, Duan Y, et al. CIRBP promotes ferroptosis by interacting with ELAVL1 and activating ferritinophagy during renal ischaemia-reperfusion injury. J Cell Mol Med. 2021;25(13):6203–16. https://doi.org/10.1111/jcmm.16567.
Chen C, Wang D, Yu Y, Zhao T, Min N, Wu Y, et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021;12(1):65. https://doi.org/10.1038/s41419-020-03362-4.
Eleftheriadis T, Pissas G, Golfinopoulos S, Liakopoulos V, Stefanidis I. Role of indoleamine 2,3-dioxygenase in ischemia-reperfusion injury of renal tubular epithelial cells. Mol Med Rep. 2021;23(6). https://doi.org/10.3892/mmr.2021.12111.
Feng R, Xiong Y, Lei Y, Huang Q, Liu H, Zhao X, et al. Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J Cell Mol Med. 2022;26(15):4254–67. https://doi.org/10.1111/jcmm.17444.
Ding C, Ding X, Zheng J, Wang B, Li Y, Xiang H, et al. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 2020;11(10):929. https://doi.org/10.1038/s41419-020-03135-z.
Tao W, Liu F, Zhang J, Fu S, Zhan H, Qian K. miR-3587 inhibitor attenuates ferroptosis following renal ischemia-reperfusion through HO-1. Front Mol Biosci. 2021;8:789927. https://doi.org/10.3389/fmolb.2021.789927.
Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY). 2020;12(13):12943–59. https://doi.org/10.18632/aging.103378.
Qiang Z, Dong H, Xia Y, Chai D, Hu R, Jiang H. Nrf2 and STAT3 alleviates ferroptosis-mediated IIR-ALI by regulating SLC7A11. Oxid Med Cell Longev. 2020;2020:5146982. https://doi.org/10.1155/2020/5146982.
Dong H, Xia Y, Jin S, Xue C, Wang Y, Hu R, et al. Nrf2 attenuates ferroptosis-mediated IIR-ALI by modulating TERT and SLC7A11. Cell Death Dis. 2021;12(11):1027. https://doi.org/10.1038/s41419-021-04307-1.
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27(9):2635–50. https://doi.org/10.1038/s41418-020-0528-x.
Rong Y, Fan J, Ji C, Wang Z, Ge X, Wang J, et al. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2022;29(6):1164–75. https://doi.org/10.1038/s41418-021-00907-8.
Gerber Y, Weston SA, Jiang R, Roger VL. The changing epidemiology of myocardial infarction in Olmsted County, Minnesota, 1995–2012. Am J Med. 2015;128(2):144–51. https://doi.org/10.1016/j.amjmed.2014.09.012.
Correction: Twenty-five-year trends in myocardial infarction attack and mortality rates, and case-fatality, in six European populations. Heart. 2018;104(16):e2. https://doi.org/10.1136/heartjnl-2014-307310corr1.
Degano IR, Salomaa V, Veronesi G, Ferrieres J, Kirchberger I, Laks T, et al. Twenty-five-year trends in myocardial infarction attack and mortality rates, and case-fatality, in six European populations. Heart. 2015;101(17):1413–21. https://doi.org/10.1136/heartjnl-2014-307310.
Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D’Agostino RB, et al. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118(20):2057–62. https://doi.org/10.1161/CIRCULATIONAHA.108.784215.
Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JM, Warnica JW, et al. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol. 2003;42(8):1446–53. https://doi.org/10.1016/s0735-1097(03)01057-x.
Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, et al. Innate immune receptors, key actors in cardiovascular diseases. JACC Basic Transl Sci. 2020;5(7):735–49. https://doi.org/10.1016/j.jacbts.2020.03.015.
Lillo-Moya J, Rojas-Solé C, Muñoz-Salamanca D, Panieri E, Saso L, Rodrigo R. Targeting ferroptosis against ischemia/reperfusion cardiac injury. Antioxidants (Basel). 2021;10(5). https://doi.org/10.3390/antiox10050667.
Miyamoto HD, Ikeda M, Ide T, Tadokoro T, Furusawa S, Abe K, et al. Iron overload via heme degradation in the endoplasmic reticulum triggers ferroptosis in myocardial ischemia-reperfusion injury. JACC Basic Transl Sci. 2022;7(8):800–19. https://doi.org/10.1016/j.jacbts.2022.03.012.
Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16(3):123–32. https://doi.org/10.1177/1089253211436350.
Huang C, Zhou S, Chen C, Wang X, Ding R, Xu Y, et al. Biodegradable redox-responsive AIEgen-based-covalent organic framework nanocarriers for long-term treatment of myocardial ischemia/reperfusion injury. Small. 2022;18(47):e2205062. https://doi.org/10.1002/smll.202205062.
Li JY, Liu SQ, Yao RQ, Tian YP, Yao YM. A novel insight into the fate of cardiomyocytes in ischemia-reperfusion injury: from iron metabolism to ferroptosis. Front Cell Dev Biol. 2021;9:799499. https://doi.org/10.3389/fcell.2021.799499.
Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ, Wang G, et al. ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct Target Ther. 2022;7(1):288. https://doi.org/10.1038/s41392-022-01090-z.
Lin JH, Yang KT, Lee WS, Ting PC, Luo YP, Lin DJ, et al. Xanthohumol protects the rat myocardium against ischemia/reperfusion injury-induced ferroptosis. Oxid Med Cell Longev. 2022;2022:9523491. https://doi.org/10.1155/2022/9523491.
Chen W, Zhang Y, Wang Z, Tan M, Lin J, Qian X, et al. Dapagliflozin alleviates myocardial ischemia/reperfusion injury by reducing ferroptosis via MAPK signaling inhibition. Front Pharmacol. 2023;14:1078205. https://doi.org/10.3389/fphar.2023.1078205.
Zhao Y, Zhang X, Chen X, Wei Y. Neuronal injuries in cerebral infarction and ischemic stroke: From mechanisms to treatment (Review). Int J Mol Med. 2022;49(2). https://doi.org/10.3892/ijmm.2021.5070.
Cetin N, Dasdelen D, Mogulkoc R, Menevse E, Baltaci AK. Role of exogenous putrescine in the status of energy, DNA damage, inflammation, and spermidine/spermine-n(1)- acetyltransferase in brain ischemia-reperfusion in rats. Iran J Basic Med Sci. 2022;25(5):597–603. https://doi.org/10.22038/IJBMS.2022.63733.14046.
Bu ZQ, Yu HY, Wang J, He X, Cui YR, Feng JC, et al. Emerging role of ferroptosis in the pathogenesis of ischemic stroke: a new therapeutic target? ASN Neuro. 2021;13:17590914211037504. https://doi.org/10.1177/17590914211037505.
Yang L, Du B, Zhang S, Wang M. RXRγ attenuates cerebral ischemia-reperfusion induced ferroptosis in neurons in mice through transcriptionally promoting the expression of GPX4. Metab Brain Dis. 2022;37(5):1351–63. https://doi.org/10.1007/s11011-022-00988-5.
Chen Y, He W, Wei H, Chang C, Yang L, Meng J, et al. Srs11-92, a ferrostatin-1 analog, improves oxidative stress and neuroinflammation via Nrf2 signal following cerebral ischemia/reperfusion injury. CNS Neurosci Ther. 2023;29(6):1667–77. https://doi.org/10.1111/cns.14130.
Xu S, Li X, Li Y, Li X, Lv E, Zhang X, et al. Neuroprotective effect of Dl-3-n-butylphthalide against ischemia-reperfusion injury is mediated by ferroptosis regulation via the SLC7A11/GSH/GPX4 pathway and the attenuation of blood-brain barrier disruption. Front Aging Neurosci. 2023;15:1028178. https://doi.org/10.3389/fnagi.2023.1028178.
Chen L, Huang J, Yao ZM, Sun XR, Tong XH, Hu M et al. Procyanidins alleviated cerebral ischemia/reperfusion injury by inhibiting ferroptosis via the Nrf2/HO-1 signaling pathway. Molecules. 2023;28(8). https://doi.org/10.3390/molecules28083582.
Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59(5):1094–106. https://doi.org/10.1016/j.jhep.2013.06.017.
Liu J, Zhang W, Zhou C, Li M, Wang X, Zhang W, et al. Precision navigation of hepatic ischemia-reperfusion injury guided by lysosomal viscosity-activatable NIR-II fluorescence. J Am Chem Soc. 2022;144(30):13586–99. https://doi.org/10.1021/jacs.2c03832.
Gao F, Qiu X, Wang K, Shao C, Jin W, Zhang Z, et al. Targeting the hepatic microenvironment to improve ischemia/reperfusion injury: New insights into the immune and metabolic compartments. Aging Dis. 2022;13(4):1196–214. https://doi.org/10.14336/ad.2022.0109.
Mao L, Zhao T, Song Y, Lin L, Fan X, Cui B, et al. The emerging role of ferroptosis in non-cancer liver diseases: hype or increasing hope? Cell Death Dis. 2020;11(7):518. https://doi.org/10.1038/s41419-020-2732-5.
Ye J, Peng J, Liu K, Zhang T, Huang W. MCTR1 inhibits ferroptosis by promoting NRF2 expression to attenuate hepatic ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2022;323(3):G283–93. https://doi.org/10.1152/ajpgi.00354.2021.
Guo J, Song Z, Yu J, Li C, Jin C, Duan W, et al. Hepatocyte-specific TMEM16A deficiency alleviates hepatic ischemia/reperfusion injury via suppressing GPX4-mediated ferroptosis. Cell Death Dis. 2022;13(12):1072. https://doi.org/10.1038/s41419-022-05518-w.
Qiu S, Li X, Zhang J, Shi P, Cao Y, Zhuang Y, et al. Neutrophil membrane-coated taurine nanoparticles protect against hepatic ischemia-reperfusion injury. Eur J Pharmacol. 2023;949:175712. https://doi.org/10.1016/j.ejphar.2023.175712.
Qi D, Chen P, Bao H, Zhang L, Sun K, Song S, et al. Dimethyl fumarate protects against hepatic ischemia-reperfusion injury by alleviating ferroptosis via the NRF2/SLC7A11/HO-1 axis. Cell Cycle. 2023;22(7):818–28. https://doi.org/10.1080/15384101.2022.2155016.
Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. https://doi.org/10.1038/nrneph.2011.16.
Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–21. https://doi.org/10.1172/jci45161.
Reuter S, Mrowka R. Acute kidney injury. Acta Physiol (Oxf). 2015;215(2):73–5. https://doi.org/10.1111/apha.12555.
Han SJ, Lee HT. Mechanisms and therapeutic targets of ischemic acute kidney injury. Kidney Res Clin Pract. 2019;38(4):427–40. https://doi.org/10.23876/j.krcp.19.062.
Thapa K, Singh TG, Kaur A. Targeting ferroptosis in ischemia/reperfusion renal injury. Naunyn Schmiedebergs Arch Pharmacol. 2022;395(11):1331–41. https://doi.org/10.1007/s00210-022-02277-5.
Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2021;28:231–43. https://doi.org/10.1016/j.jare.2020.07.007.
Du YW, Li XK, Wang TT, Zhou L, Li HR, Feng L, et al. Cyanidin-3-glucoside inhibits ferroptosis in renal tubular cells after ischemia/reperfusion injury via the AMPK pathway. Mol Med. 2023;29(1):42. https://doi.org/10.1186/s10020-023-00642-5.
Qi Y, Hu M, Qiu Y, Zhang L, Yan Y, Feng Y, et al. Mitoglitazone ameliorates renal ischemia/reperfusion injury by inhibiting ferroptosis via targeting mitoNEET. Toxicol Appl Pharmacol. 2023;465:116440. https://doi.org/10.1016/j.taap.2023.116440.
Zhang Y, Ren X, Wang Y, Chen D, Jiang L, Li X, et al. Targeting ferroptosis by polydopamine nanoparticles protects heart against ischemia/reperfusion injury. ACS Appl Mater Interfaces. 2021;13(45):53671–82. https://doi.org/10.1021/acsami.1c18061.
He P, Zhang M, Zhao M, Zhang M, Ma B, Lv H, et al. A novel polysaccharide from chuanminshen violaceum and its protective effect against myocardial injury. Front Nutr. 2022;9:961182. https://doi.org/10.3389/fnut.2022.961182.
Peng Y, Liao B, Zhou Y, Zeng W, Zeng ZY. Atorvastatin inhibits ferroptosis of H9C2 cells by regulatingSMAD7/Hepcidin expression to improve ischemia-reperfusion injury. Cardiol Res Pract. 2022;2022:3972829. https://doi.org/10.1155/2022/3972829.
Wang Z, Yao M, Jiang L, Wang L, Yang Y, Wang Q, et al. Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3beta/Nrf2 axis. Biomed Pharmacother. 2022;154:113572. https://doi.org/10.1016/j.biopha.2022.113572.
Ma X, Xu J, Gao N, Tian J, Song T. Dexmedetomidine attenuates myocardial ischemia-reperfusion injury via inhibiting ferroptosis by the cAMP/PKA/CREB pathway. Mol Cell Probes. 2023;68:101899. https://doi.org/10.1016/j.mcp.2023.101899.
Li T, Tan Y, Ouyang S, He J, Liu L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene. 2022;808:145968. https://doi.org/10.1016/j.gene.2021.145968.
Mei SL, Xia ZY, Qiu Z, Jia YF, Zhou JJ, Zhou B. Shenmai injection attenuates myocardial ischemia/reperfusion injury by targeting Nrf2/GPX4 signalling-mediated ferroptosis. Chin J Integr Med. 2022;28(11):983–91. https://doi.org/10.1007/s11655-022-3620-x.
Zhang F, Li Z, Gao P, Zou J, Cui Y, Qian Y, et al. HJ11 decoction restrains development of myocardial ischemia-reperfusion injury in rats by suppressing ACSL4-mediated ferroptosis. Front Pharmacol. 2022;13:1024292. https://doi.org/10.3389/fphar.2022.1024292.
Ding Y, Li W, Peng S, Zhou G, Chen S, Wei Y, et al. Puerarin protects against myocardial ischemia/reperfusion injury by inhibiting ferroptosis. Biol Pharm Bull. 2023;46(4):524–32. https://doi.org/10.1248/bpb.b22-00174.
Guo L, Shi L. Vitexin Improves Cerebral ischemia-reperfusion Injury by Attenuating Oxidative Injury and Ferroptosis via Keap1/Nrf2/HO-1signaling. Neurochem Res. 2023;48(3):980–95. https://doi.org/10.1007/s11064-022-03829-0.
Liu J, Zhou Y, Xie C, Li C, Ma L, Zhang Y. Anti-Ferroptotic Effects of bone Marrow Mesenchymal Stem Cell-Derived Extracellular Vesicles Loaded with Ferrostatin-1 in Cerebral ischemia-reperfusion Injury Associate with the GPX4/COX-2 Axis. Neurochem Res. 2023;48(2):502–18. https://doi.org/10.1007/s11064-022-03770-2.
Zhao J, Ma M, Li L, Fang G. Oxysophoridine protects against cerebral ischemia/reperfusion injury via inhibition of TLR4/p38MAPK‑mediated ferroptosis. Mol Med Rep. 2023;27(2). https://doi.org/10.3892/mmr.2023.12931.
Tuo Q-Z, Masaldan S, Southon A, Mawal C, Ayton S, Bush AI, et al. Characterization of selenium compounds for anti-ferroptotic activity in neuronal cells and after cerebral ischemia-reperfusion injury. Neurotherapeutics. 2021;18(4):2682–91. https://doi.org/10.1007/s13311-021-01111-9.
Shi Y, Han L, Zhang X, Xie L, Pan P, Chen F. Selenium alleviates cerebral ischemia/reperfusion injury by regulating oxidative stress, mitochondrial fusion and ferroptosis. Neurochem Res. 2022;47(10):2992–3002. https://doi.org/10.1007/s11064-022-03643-8.
Guan X, Li X, Yang X, Yan J, Shi P, Ba L, et al. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019;235:116795. https://doi.org/10.1016/j.lfs.2019.116795.
Fu C, Wu Y, Liu S, Luo C, Lu Y, Liu M, et al. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J Ethnopharmacol. 2022;289:115021. https://doi.org/10.1016/j.jep.2022.115021.
Guan X, Li Z, Zhu S, Cheng M, Ju Y, Ren L, et al. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci. 2021;264:118660. https://doi.org/10.1016/j.lfs.2020.118660.
Guo H, Zhu L, Tang P, Chen D, Li Y, Li J et al. Carthamin yellow improves cerebral ischemia‑reperfusion injury by attenuating inflammation and ferroptosis in rats. Int J Mol Med. 2021;47(4). https://doi.org/10.3892/ijmm.2021.4885.
Yuan Y, Zhai Y, Chen J, Xu X, Wang H. Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 axis. Biomolecules. 2021;11(7). doi:https://doi.org/10.3390/biom11070923.
Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520(3):606–11. https://doi.org/10.1016/j.bbrc.2019.10.006.
Xu Y, Li X, Cheng Y, Yang M, Wang R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 2020;34(12):16262–75. https://doi.org/10.1096/fj.202001758R.
Yamada N, Karasawa T, Wakiya T, Sadatomo A, Ito H, Kamata R, et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am J Transplant. 2020;20(6):1606–18. https://doi.org/10.1111/ajt.15773.
Ma L, Liu X, Zhang M, Zhou L, Jiang L, Gao L, et al. Paeoniflorin alleviates ischemia/reperfusion induced acute kidney injury by inhibiting Slc7a11-mediated ferroptosis. Int Immunopharmacol. 2023;116:109754. https://doi.org/10.1016/j.intimp.2023.109754.
Kar F, Yildiz F, Hacioglu C, Kar E, Donmez DB, Senturk H, et al. LoxBlock-1 or Curcumin attenuates liver, pancreas and cardiac ferroptosis, oxidative stress and injury in Ischemia/reperfusion-damaged rats by facilitating ACSL/GPx4 signaling. Tissue Cell. 2023;82:102114. https://doi.org/10.1016/j.tice.2023.102114.
Kolbrink B, von Samson-Himmelstjerna FA, Messtorff ML, Riebeling T, Nische R, Schmitz J, et al. Vitamin K1 inhibits ferroptosis and counteracts a detrimental effect of phenprocoumon in experimental acute kidney injury. Cell Mol Life Sci. 2022;79(7):387. https://doi.org/10.1007/s00018-022-04416-w.
Jiang GP, Liao YJ, Huang LL, Zeng XJ, Liao XH. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Mol Med Rep. 2021;23(1). https://doi.org/10.3892/mmr.2020.11704.
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836–41. https://doi.org/10.1073/pnas.1415518111.
Zhao Z, Wu J, Xu H, Zhou C, Han B, Zhu H, et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020;11(8):629. https://doi.org/10.1038/s41419-020-02871-6.
Tonnus W, Meyer C, Steinebach C, Belavgeni A, von Massenhausen A, Gonzalez NZ, et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1):4402. https://doi.org/10.1038/s41467-021-24712-6.
Yang J, Sun X, Huang N, Li P, He J, Jiang L, et al. Entacapone alleviates acute kidney injury by inhibiting ferroptosis. FASEB J. 2022;36(7):e22399. https://doi.org/10.1096/fj.202200241RR.
Wang Y, Chen Z, Luo J, Zhang J, Sang AM, Cheng ZS, et al. Salidroside postconditioning attenuates ferroptosis-mediated lung ischemia-reperfusion injury by activating the Nrf2/SLC7A11 signaling axis. Int Immunopharmacol. 2023;115:109731. https://doi.org/10.1016/j.intimp.2023.109731.
Zhongyin Z, Wei W, Juan X, Guohua F. Isoliquiritin apioside relieves intestinal ischemia/reperfusion-induced acute lung injury by blocking Hif-1alpha-mediated ferroptosis. Int Immunopharmacol. 2022;108:108852. https://doi.org/10.1016/j.intimp.2022.108852.
Ma X, Yan W, He N. Lidocaine attenuates hypoxia/reoxygenation‑induced inflammation, apoptosis and ferroptosis in lung epithelial cells by regulating the p38 MAPK pathway. Mol Med Rep. 2022;25(5). https://doi.org/10.3892/mmr.2022.12666.
Chen K, Xu Z, Liu Y, Wang Z, Li Y, Xu X et al. Irisin protects mitochondria function during pulmonary ischemia/reperfusion injury. Sci Transl Med. 2017;9(418). https://doi.org/10.1126/scitranslmed.aao6298.
Zhang J, Bi J, Ren Y, Du Z, Li T, Wang T, et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. J Cell Physiol. 2021;236(2):931–45. https://doi.org/10.1002/jcp.29903.
Kang Y, Tiziani S, Park G, Kaul M, Paternostro G. Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun. 2014;5:3672. https://doi.org/10.1038/ncomms4672.
Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, et al. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 2013;4(1):e470. https://doi.org/10.1038/cddis.2012.216.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91. https://doi.org/10.1038/ncb3064.
Bartzokis G, Tishler TA, Shin I-S, Lu PH, Cummings JL. Brain ferritin iron as a risk factor for age at onset in neurodegenerative diseases. Ann N Y Acad Sci. 2004;1012:224–36. https://doi.org/10.1196/annals.1306.019.
Connor JR, Menzies SL, St Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J Neurosci Res. 1992;31(1):75–83. https://doi.org/10.1002/jnr.490310111.
Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect. 1990;2(4):327–40. https://doi.org/10.1007/BF02252926.
Quintana C, Bellefqih S, Laval JY, Guerquin-Kern JL, Wu TD, Avila J, et al. Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol. 2006;153(1):42–54.
Belaidi AA, Masaldan S, Southon A, Kalinowski P, Acevedo K, Appukuttan AT, et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-022-01568-w.
Ayton S, Faux NG, Bush AI. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun. 2015;6:6760. https://doi.org/10.1038/ncomms7760.
Wu Y, Torabi S-F, Lake RJ, Hong S, Yu Z, Wu P, et al. Simultaneous Fe2+/Fe3+ imaging shows Fe3+ over Fe2+ enrichment in Alzheimer’s disease mouse brain. Sci Adv. 2023;9(16):eade7622. https://doi.org/10.1126/sciadv.ade7622.
Greenough MA, Lane DJR, Balez R, Anastacio HTD, Zeng Z, Ganio K, et al. Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differ. 2022;29(11):2123–36. https://doi.org/10.1038/s41418-022-01003-1.
Zhu Z-Y, Liu Y-D, Gong Y, Jin W, Topchiy E, Turdi S, et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol Sin. 2022;43(1):39–49. https://doi.org/10.1038/s41401-021-00635-2.
Chen L, Dar NJ, Na R, McLane KD, Yoo K, Han X et al. Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic Biol Med. 2022;180. https://doi.org/10.1016/j.freeradbiomed.2022.01.002.
Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947. https://doi.org/10.1016/j.redox.2021.101947.
Wang S, Jiang Y, Liu Y, Liu Q, Sun H, Mei M, et al. Ferroptosis promotes microtubule-associated protein tau aggregation via GSK-3β activation and proteasome inhibition. Mol Neurobiol. 2022;59(3):1486–501. https://doi.org/10.1007/s12035-022-02731-8.
Sun J, Lin X-M, Lu D-H, Wang M, Li K, Li S-R et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J Clin Invest. 2023;133(10). https://doi.org/10.1172/JCI165228.
Bouchaoui H, Mahoney-Sanchez L, Garçon G, Berdeaux O, Alleman LY, Devos D, et al. ACSL4 and the lipoxygenases 15/15B are pivotal for ferroptosis induced by iron and PUFA dyshomeostasis in dopaminergic neurons. Free Radic Biol Med. 2023;195:145–57. https://doi.org/10.1016/j.freeradbiomed.2022.12.086.
Ryan SK, Zelic M, Han Y, Teeple E, Chen L, Sadeghi M, et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci. 2023;26(1):12–26. https://doi.org/10.1038/s41593-022-01221-3.
Oun A, Soliman A, Trombetta-Lima M, Tzepapadaki A, Tsagkari D, Kortholt A, et al. LRRK2 protects immune cells against erastin-induced ferroptosis. Neurobiol Dis. 2022;175:105917. https://doi.org/10.1016/j.nbd.2022.105917.
Mahoney-Sanchez L, Bouchaoui H, Boussaad I, Jonneaux A, Timmerman K, Berdeaux O, et al. Alpha synuclein determines ferroptosis sensitivity in dopaminergic neurons via modulation of ether-phospholipid membrane composition. Cell Rep. 2022;40(8):111231. https://doi.org/10.1016/j.celrep.2022.111231.
Huang Z, Han J, Wu P, Wu C, Fan Y, Zhao L, et al. Sorting nexin 5 plays an important role in promoting ferroptosis in Parkinson’s Disease. Oxid Med Cell Longev. 2022;2022:5463134. https://doi.org/10.1155/2022/5463134.
Lee WJ, Lee HG, Hur J, Lee GH, Won JP, Kim E et al. PPARδ activation mitigates 6-OHDA-induced neuronal damage by regulating intracellular iron levels. Antioxidants (Basel). 2022;11(5). https://doi.org/10.3390/antiox11050810.
Han K, Jin X, Guo X, Cao G, Tian S, Song Y, et al. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic Biol Med. 2021;162:592–602. https://doi.org/10.1016/j.freeradbiomed.2020.11.019.
Kambey PA, Liu W-Y, Wu J, Bosco B, Nadeem I, Kanwore K, et al. Single-nuclei RNA sequencing uncovers heterogenous transcriptional signatures in Parkinson’s disease associated with nuclear receptor-related factor 1 defect. Neural Regen Res. 2023;18(9):2037–46. https://doi.org/10.4103/1673-5374.366493.
Zhang N, Yu X, Song L, Xiao Z, Xie J, Xu H. Ferritin confers protection against iron-mediated neurotoxicity and ferroptosis through iron chelating mechanisms in MPP(+)-induced MES23.5 dopaminergic cells. Free Radic Biol Med. 2022;193(Pt 2):751–63. https://doi.org/10.1016/j.freeradbiomed.2022.11.018.
Bai L, Yan F, Deng R, Gu R, Zhang X, Bai J. Thioredoxin-1 rescues MPP+/MPTP-induced ferroptosis by increasing glutathione peroxidase 4. Mol Neurobiol. 2021;58(7):3187–97. https://doi.org/10.1007/s12035-021-02320-1.
Wang D, Liang W, Huo D, Wang H, Wang Y, Cong C, et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ. 2023;30(2):369–82. https://doi.org/10.1038/s41418-022-01089-7.
Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, et al. Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death Differ. 2022;29(6):1187–98. https://doi.org/10.1038/s41418-021-00910-z.
Peng J, Pan J, Mo J, Peng Y. MPO/HOCl facilitates apoptosis and ferroptosis in the SOD1G93A motor neuron of Amyotrophic Lateral Sclerosis. Oxid Med Cell Longev. 2022;2022:8217663. https://doi.org/10.1155/2022/8217663.
Song S, Su Z, Kon N, Chu B, Li H, Jiang X, et al. ALOX5-mediated ferroptosis acts as a distinct cell death pathway upon oxidative stress in Huntington’s disease. Genes Dev. 2023;37(5–6):204–17. https://doi.org/10.1101/gad.350211.122.
Ralhan I, Chang J, Moulton MJ, Goodman LD, Lee NYJ, Plummer G et al. Autolysosomal exocytosis of lipids protect neurons from ferroptosis. J Cell Biol. 2023;222(6). https://doi.org/10.1083/jcb.202207130.
Santambrogio P, Ripamonti M, Cozzi A, Raimondi M, Cavestro C, Di Meo I, et al. Massive iron accumulation in PKAN-derived neurons and astrocytes: light on the human pathological phenotype. Cell Death Dis. 2022;13(2):185. https://doi.org/10.1038/s41419-022-04626-x.
Musheshe N, Oun A, Sabogal-Guáqueta AM, Trombetta-Lima M, Mitchel SC, Adzemovic A et al. Pharmacological inhibition of epac1 averts ferroptosis cell death by preserving mitochondrial integrity. Antioxidants (Basel, Switzerland). 2022;11(2). https://doi.org/10.3390/antiox11020314.
Hoffmann L, Waclawczyk MS, Tang S, Hanschmann EM, Gellert M, Rust MB, et al. Cofilin1 oxidation links oxidative distress to mitochondrial demise and neuronal cell death. Cell Death Dis. 2021;12(11):953. https://doi.org/10.1038/s41419-021-04242-1.
Fernández-Mendívil C, Luengo E, Trigo-Alonso P, García-Magro N, Negredo P, López MG. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 2021;38:101789. https://doi.org/10.1016/j.redox.2020.101789.
Diaw SH, Ganos C, Zittel S, Plötze-Martin K, Kulikovskaja L, Vos M et al. Mutant WDR45 leads to altered ferritinophagy and ferroptosis in β-propeller protein-associated neurodegeneration. Int J Mol Sci. 2022;23(17). https://doi.org/10.3390/ijms23179524.
Zhang H, Zhou W, Li J, Qiu Z, Wang X, Xu H, et al. Senegenin rescues PC12 cells with oxidative damage through inhibition of ferroptosis. Mol Neurobiol. 2022;59(11):6983–92. https://doi.org/10.1007/s12035-022-03014-y.
Li L, Li W-J, Zheng X-R, Liu Q-L, Du Q, Lai Y-J, et al. Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol Med. 2022;28(1):11. https://doi.org/10.1186/s10020-022-00442-3.
Wang C, Chen S, Guo H, Jiang H, Liu H, Fu H, et al. Forsythoside A Mitigates Alzheimer’s-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. Int J Biol Sci. 2022;18(5):2075–90. https://doi.org/10.7150/ijbs.69714.
Liu Y, Han C, Dai R, Li B. Rational design, synthesis and activities of hydroxylated chalcones as highly potent dual functional agents against Alzheimer’s disease. Bioorg Chem. 2022;122:105662. https://doi.org/10.1016/j.bioorg.2022.105662.
Yang S, Wang L, Zeng Y, Wang Y, Pei T, Xie Z, et al. Salidroside alleviates cognitive impairment by inhibiting ferroptosis via activation of the Nrf2/GPX4 axis in SAMP8 mice. Phytomedicine. 2023;114:154762. https://doi.org/10.1016/j.phymed.2023.154762.
Gao Y, Li J, Wu Q, Wang S, Yang S, Li X, et al. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int Immunopharmacol. 2021;99:108002. https://doi.org/10.1016/j.intimp.2021.108002.
Lin Z-H, Liu Y, Xue N-J, Zheng R, Yan Y-Q, Wang Z-X, et al. Quercetin protects against MPP+/MPTP-induced dopaminergic neuron death in Parkinson’s disease by inhibiting ferroptosis. Oxid Med Cell Longev. 2022;2022:7769355. https://doi.org/10.1155/2022/7769355.
Li K, Wang M, Huang Z-H, Wang M, Sun W-Y, Kurihara H et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol Res. 2023:106779. https://doi.org/10.1016/j.phrs.2023.106779.
Xi J, Zhang Z, Wang Z, Wu Q, He Y, Xu Y, et al. Hinokitiol functions as a ferroptosis inhibitor to confer neuroprotection. Free Radic Biol Med. 2022;190:202–15. https://doi.org/10.1016/j.freeradbiomed.2022.08.011.
Sun W-Y, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai Y-J, et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17(4):465–76. https://doi.org/10.1038/s41589-020-00734-x.
Cao Z, Wang X, Zhang T, Fu X, Zhang F, Zhu J. Discovery of novel 2-(4-(benzyloxy)-5-(hydroxyl) phenyl) benzothiazole derivatives as multifunctional MAO-B inhibitors for the treatment of Parkinson’s disease. J Enzyme Inhib Med Chem. 2023;38(1):2159957. https://doi.org/10.1080/14756366.2022.2159957.
Shen Z-B, Meng H-W, Meng X-S, Lv Z-K, Fang M-Y, Zhang L-L, et al. Design, synthesis, and SAR study of novel flavone 1,2,4-oxadiazole derivatives with anti-inflammatory activities for the treatment of Parkinson’s disease. Eur J Med Chem. 2023;255:115417. https://doi.org/10.1016/j.ejmech.2023.115417.
Sun Y, He L, Wang W, Xie Z, Zhang X, Wang P, et al. Activation of Atg7-dependent autophagy by a novel inhibitor of the Keap1-Nrf2 protein-protein interaction from Penthorum chinense Pursh. attenuates 6-hydroxydopamine-induced ferroptosis in zebrafish and dopaminergic neurons. Food Funct. 2022;13(14):7885–900. https://doi.org/10.1039/d2fo00357k.
Liang Z, Soriano-Castell D, Kepchia D, Duggan BM, Currais A, Schubert D, et al. Cannabinol inhibits oxytosis/ferroptosis by directly targeting mitochondria independently of cannabinoid receptors. Free Radic Biol Med. 2022;180:33–51. https://doi.org/10.1016/j.freeradbiomed.2022.01.001.
Tang W, Li Y, He S, Jiang T, Wang N, Du M, et al. Caveolin-1 alleviates diabetes-associated cognitive dysfunction through modulating neuronal ferroptosis-mediated mitochondrial homeostasis. Antioxid Redox Signal. 2022;37(13–15):867–86. https://doi.org/10.1089/ars.2021.0233.
Li Y, Sun M, Cao F, Chen Y, Zhang L, Li H et al. The ferroptosis inhibitor liproxstatin-1 ameliorates LPS-induced cognitive impairment in mice. Nutrients. 2022;14(21). https://doi.org/10.3390/nu14214599.
Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. https://doi.org/10.1038/s41572-021-00269-y.
Ayton S, Wang Y, Diouf I, Schneider JA, Brockman J, Morris MC, et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry. 2020;25(11):2932–41. https://doi.org/10.1038/s41380-019-0375-7.
Huang L, McClatchy DB, Maher P, Liang Z, Diedrich JK, Soriano-Castell D, et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 2020;11(10):828. https://doi.org/10.1038/s41419-020-03020-9.
Yang Y, Wang X, Xiao A, Han J, Wang Z, Wen M. Ketogenic diet prevents chronic sleep deprivation-induced Alzheimer’s disease by inhibiting iron dyshomeostasis and promoting repair via Sirt1/Nrf2 pathway. Front Aging Neurosci. 2022;14:998292. https://doi.org/10.3389/fnagi.2022.998292.
Feng J, Song G, Wu Y, Chen X, Pang J, Xu Y, et al. Plasmalogens improve swimming performance by modulating the expression of genes involved in amino acid and lipid metabolism, oxidative stress, and ferroptosis in an Alzheimer’s disease zebrafish model. Food Funct. 2021;12(23):12087–97. https://doi.org/10.1039/d1fo01471d.
Faucheux BA, Martin M-E, Beaumont C, Hunot S, Hauw J-J, Agid Y, et al. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J Neurochem. 2002;83(2):320–30. https://doi.org/10.1046/j.1471-4159.2002.01118.x.
Ayton S, Lei P, Duce JA, Wong BXW, Sedjahtera A, Adlard PA, et al. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol. 2013;73(4):554–9. https://doi.org/10.1002/ana.23817.
Bi M, Du X, Jiao Q, Liu Z, Jiang H. α-synuclein regulates iron homeostasis via preventing Parkin-mediated DMT1 ubiquitylation in Parkinson’s Disease models. ACS Chem Neurosci. 2020;11(11):1682–91. https://doi.org/10.1021/acschemneuro.0c00196.
Zhang Y, Li S, Hou L, Wu M, Liu J, Wang R, et al. NLRP3 mediates the neuroprotective effects of SVHRSP derived from scorpion venom in rotenone-induced experimental Parkinson’s disease model. J Ethnopharmacol. 2023;312:116497. https://doi.org/10.1016/j.jep.2023.116497.
Desouky MA, George MY, Michel HE, Elsherbiny DA. Roflumilast escalates α-synuclein aggregate degradation in rotenone-induced Parkinson’s disease in rats: Modulation of the ubiquitin-proteasome system and endoplasmic reticulum stress. Chem Biol Interact. 2023;379:110491. https://doi.org/10.1016/j.cbi.2023.110491.
Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918–29. https://doi.org/10.1111/ene.14393.
Akçimen F, Lopez ER, Landers JE, Nath A, Chiò A, Chia R, et al. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet. 2023. https://doi.org/10.1038/s41576-023-00592-y.
Matsuo T, Adachi-Tominari K, Sano O, Kamei T, Nogami M, Ogi K, et al. Involvement of ferroptosis in human motor neuron cell death. Biochem Biophys Res Commun. 2021;566:24–9. https://doi.org/10.1016/j.bbrc.2021.05.095.
Moreau C, Danel V, Devedjian JC, Grolez G, Timmerman K, Laloux C, et al. Could conservative iron chelation lead to neuroprotection in amyotrophic lateral sclerosis? Antioxid Redox Signal. 2018;29(8):742–8. https://doi.org/10.1089/ars.2017.7493.
Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. 2020;16(10):529–46. https://doi.org/10.1038/s41582-020-0389-4.
Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects : a cross-sectional study. J Neurol. 2007;254(12):1676–83.
Rosas HD, Chen YI, Doros G, Salat DH, Chen N-K, Kwong KK, et al. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol. 2012;69(7):887–93.
Amini M, Zayeri F, Salehi M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: results from global burden of disease study 2017. BMC Public Health. 2021;21(1):401. https://doi.org/10.1186/s12889-021-10429-0.
Wang K, Chen XZ, Wang YH, Cheng XL, Zhao Y, Zhou LY, et al. Emerging roles of ferroptosis in cardiovascular diseases. Cell Death Discov. 2022;8(1):394. https://doi.org/10.1038/s41420-022-01183-2.
Li D, Liu X, Pi W, Zhang Y, Yu L, Xu C, et al. Fisetin Attenuates Doxorubicin-Induced Cardiomyopathy In Vivo and In Vitro by Inhibiting Ferroptosis Through SIRT1/Nrf2 Signaling Pathway Activation. Front Pharmacol. 2021;12:808480. https://doi.org/10.3389/fphar.2021.808480.
Wang R, Xu Y, Fang Y, Wang C, Xue Y, Wang F, et al. Pathogenetic mechanisms of septic cardiomyopathy. J Cell Physiol. 2022;237(1):49–58. https://doi.org/10.1002/jcp.30527.
Izzy M, Oh J, Watt KD. Cirrhotic cardiomyopathy after transplantation: neither the transient nor innocent bystander. Hepatology. 2018;68(5):2008–15. https://doi.org/10.1002/hep.30040.
Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381(9862):242–55. https://doi.org/10.1016/s0140-6736(12)60397-3.
Wang Y, Lu Y, Chen W, Xie X. Inhibition of ferroptosis alleviates high-power microwave-induced myocardial injury. Front Cardiovasc Med. 2023;10:1157752. https://doi.org/10.3389/fcvm.2023.1157752.
Liu F, Jiang LJ, Zhang YX, Xu ST, Liu SL, Ye JT, et al. Inhibition of miR-214-3p attenuates ferroptosis in myocardial infarction via regulating ME2. Biochem Biophys Res Commun. 2023;661:64–74. https://doi.org/10.1016/j.bbrc.2023.04.031.
Jiang Y, Qiao Y, He D, Tian A, Li Z. Adaptor protein HIP-55-mediated signalosome protects against ferroptosis in myocardial infarction. Cell Death Differ. 2023;30(3):825–38. https://doi.org/10.1038/s41418-022-01110-z.
Cao G, Yang C, Jin Z, Wei H, Xin C, Zheng C, et al. FNDC5/irisin reduces ferroptosis and improves mitochondrial dysfunction in hypoxic cardiomyocytes by Nrf2/HO-1 axis. Cell Biol Int. 2022;46(5):723–36. https://doi.org/10.1002/cbin.11763.
Gao F, Zhao Y, Zhang B, Xiao C, Sun Z, Gao Y, et al. Suppression of lncRNA Gm47283 attenuates myocardial infarction via miR-706/ Ptgs2/ferroptosis axis. Bioengineered. 2022;13(4):10786–802. https://doi.org/10.1080/21655979.2022.2065743.
Wu Y, Huang T, Li X, Shen C, Ren H, Wang H, et al. Retinol dehydrogenase 10 reduction mediated retinol metabolism disorder promotes diabetic cardiomyopathy in male mice. Nat Commun. 2023;14(1):1181. https://doi.org/10.1038/s41467-023-36837-x.
Zhang J, Qiu Q, Wang H, Chen C, Luo D. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp Cell Res. 2021;407(2):112800. https://doi.org/10.1016/j.yexcr.2021.112800.
Wang N, Ma H, Li J, Meng C, Zou J, Wang H, et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol. 2021;150:65–76. https://doi.org/10.1016/j.yjmcc.2020.10.010.
Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12(2):708–22. https://doi.org/10.1016/j.apsb.2021.10.005.
Chen L, Yin Z, Qin X, Zhu X, Chen X, Ding G, et al. CD74 ablation rescues type 2 diabetes mellitus-induced cardiac remodeling and contractile dysfunction through pyroptosis-evoked regulation of ferroptosis. Pharmacol Res. 2022;176:106086. https://doi.org/10.1016/j.phrs.2022.106086.
Leng B, Deng L, Tan J, Lee WT, Cao CR, Wang ZP, et al. Targeting the Na(+)/K(+) ATPase DR-region with DR-Ab improves doxorubicin-induced cardiotoxicity. Free Radic Biol Med. 2023;204:38–53. https://doi.org/10.1016/j.freeradbiomed.2023.04.008.
Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, et al. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022;29(10):1982–95. https://doi.org/10.1038/s41418-022-00990-5.
Ta N, Qu C, Wu H, Zhang D, Sun T, Li Y, et al. Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2022;119(36):e2117396119. https://doi.org/10.1073/pnas.2117396119.
Wang AJ, Tang Y, Zhang J, Wang BJ, Xiao M, Lu G, et al. Cardiac SIRT1 ameliorates doxorubicin-induced cardiotoxicity by targeting sestrin 2. Redox Biol. 2022;52:102310. https://doi.org/10.1016/j.redox.2022.102310.
Kawano I, Adamcova M. MicroRNAs in doxorubicin-induced cardiotoxicity: the DNA damage response. Front Pharmacol. 2022;13:1055911. https://doi.org/10.3389/fphar.2022.1055911.
Pan J, Xiong W, Zhang A, Zhang H, Lin H, Gao L, et al. The imbalance of p53-Park7 signaling axis induces iron homeostasis dysfunction in doxorubicin-challenged cardiomyocytes. Adv Sci (Weinh). 2023;10(15):e2206007. https://doi.org/10.1002/advs.202206007.
Yu Y, Wu T, Lu Y, Zhao W, Zhang J, Chen Q, et al. Exosomal thioredoxin-1 from hypoxic human umbilical cord mesenchymal stem cells inhibits ferroptosis in doxorubicin-induced cardiotoxicity via mTORC1 signaling. Free Radic Biol Med. 2022;193(Pt 1):108–21. https://doi.org/10.1016/j.freeradbiomed.2022.10.268.
Hu C, Zhang X, Song P, Yuan YP, Kong CY, Wu HM, et al. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020;37:101747. https://doi.org/10.1016/j.redox.2020.101747.
Wu X, Wang L, Wang K, Li J, Chen R, Wu X, et al. ADAR2 increases in exercised heart and protects against myocardial infarction and doxorubicin-induced cardiotoxicity. Mol Ther. 2022;30(1):400–14. https://doi.org/10.1016/j.ymthe.2021.07.004.
Yu W, Chen C, Xu C, Xie D, Wang Q, Liu W, et al. Activation of p62-NRF2 axis protects against doxorubicin-induced ferroptosis in cardiomyocytes: a novel Role and molecular mechanism of resveratrol. Am J Chin Med. 2022;50(8):2103–23. https://doi.org/10.1142/S0192415X22500902.
Peng H, Fu S, Wang S, Xu H, Dhanasekaran M, Chen H, et al. Ablation of FUNDC1-dependent mitophagy renders myocardium resistant to paraquat-induced ferroptosis and contractile dysfunction. Biochim Biophys Acta Mol Basis Dis. 2022;1868(9):166448. https://doi.org/10.1016/j.bbadis.2022.166448.
Kong C, Ni X, Wang Y, Zhang A, Zhang Y, Lin F, et al. ICA69 aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Dis. 2022;8(1):187. https://doi.org/10.1038/s41420-022-00957-y.
Chen Z, Cao Z, Gui F, Zhang M, Wu X, Peng H et al. TMEM43 protects against sepsis-induced cardiac injury via inhibiting ferroptosis in mice. Cells. 2022;11(19). https://doi.org/10.3390/cells11192992.
Shen H, Xie K, Tian Y, Wang X. N6-methyladenosine writer METTL3 accelerates the sepsis-induced myocardial injury by regulating m6A-dependent ferroptosis. Apoptosis. 2023;28(3–4):514–24. https://doi.org/10.1007/s10495-022-01808-y.
Wang JX, Zhao Y, Chen MS, Zhang H, Cui JG, Li JL. Heme-oxygenase-1 as a target for phthalate-induced cardiomyocytes ferroptosis. Environ Pollut. 2023;317:120717. https://doi.org/10.1016/j.envpol.2022.120717.
Chen D, Geng Y, Deng Z, Li P, Xue S, Xu T et al. Inhibition of TLR4 alleviates heat stroke-Induced cardiomyocyte injury by down-regulating inflammation and ferroptosis. Molecules. 2023;28(5). https://doi.org/10.3390/molecules28052297.
Zhang X, Zheng C, Gao Z, Chen H, Li K, Wang L, et al. SLC7A11/xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs Ther. 2022;36(3):437–47. https://doi.org/10.1007/s10557-021-07220-z.
Ahola S, Rivera Mejías P, Hermans S, Chandragiri S, Giavalisco P, Nolte H, et al. OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell Metab. 2022;34(11):1875-91.e7. https://doi.org/10.1016/j.cmet.2022.08.017.
Liao J, Xie SS, Deng Y, Wu DD, Meng H, Lan WF, et al. PRDX6-mediated pulmonary artery endothelial cell ferroptosis contributes to monocrotaline-induced pulmonary hypertension. Microvasc Res. 2023;146:104471. https://doi.org/10.1016/j.mvr.2022.104471.
Li H, Ding J, Liu W, Wang X, Feng Y, Guan H, et al. Plasma exosomes from patients with acute myocardial infarction alleviate myocardial injury by inhibiting ferroptosis through miR-26b-5p/SLC7A11 axis. Life Sci. 2023;322:121649. https://doi.org/10.1016/j.lfs.2023.121649.
Li D, Zhang G, Wang Z, Guo J, Liu Y, Lu Y, et al. Idebenone attenuates ferroptosis by inhibiting excessive autophagy via the ROS-AMPK-mTOR pathway to preserve cardiac function after myocardial infarction. Eur J Pharmacol. 2023;943:175569. https://doi.org/10.1016/j.ejphar.2023.175569.
Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther. 2022;2022:3159717. https://doi.org/10.1155/2022/3159717.
Chen Y, Li S, Yin M, Li Y, Chen C, Zhang J, et al. Isorhapontigenin attenuates cardiac microvascular injury in diabetes via the inhibition of mitochondria-associated ferroptosis through PRDX2-MFN2-ACSL4 pathways. Diabetes. 2023;72(3):389–404. https://doi.org/10.2337/db22-0553.
Xu C, Ou E, Li Z, Chen Z, Jia Q, Xu X, et al. Synthesis and in vivo evaluation of new steviol derivatives that protect against cardiomyopathy by inhibiting ferroptosis. Bioorg Chem. 2022;129:106142. https://doi.org/10.1016/j.bioorg.2022.106142.
Tadokoro T, Ikeda M, Abe K, Ide T, Miyamoto HD, Furusawa S, et al. Ethoxyquin is a competent radical-trapping antioxidant for preventing ferroptosis in doxorubicin cardiotoxicity. J Cardiovasc Pharmacol. 2022;80(5):690–9. https://doi.org/10.1097/fjc.0000000000001328.
Zhu X, Wang X, Zhu B, Ding S, Shi H, Yang X. Disruption of histamine/H(1)R-STAT3-SLC7A11 axis exacerbates doxorubicin-induced cardiac ferroptosis. Free Radic Biol Med. 2022;192:98–114. https://doi.org/10.1016/j.freeradbiomed.2022.09.012.
Luo LF, Guan P, Qin LY, Wang JX, Wang N, Ji ES. Astragaloside IV inhibits adriamycin-induced cardiac ferroptosis by enhancing Nrf2 signaling. Mol Cell Biochem. 2021;476(7):2603–11. https://doi.org/10.1007/s11010-021-04112-6.
Abe K, Ikeda M, Ide T, Tadokoro T, Miyamoto HD, Furusawa S, et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci Signal. 2022;15(758):eabn8017. https://doi.org/10.1126/scisignal.abn8017.
Zhang Y, Liu S, Peng J, Cheng S, Zhang Q, Zhang N et al. Biomimetic nanozymes suppressed ferroptosis to ameliorate doxorubicin-induced cardiotoxicity via synergetic effect of antioxidant stress and GPX4 restoration. Nutrients. 2023;15(5). https://doi.org/10.3390/nu15051090.
Liu X, Li D, Pi W, Wang B, Xu S, Yu L, et al. LCZ696 protects against doxorubicin-induced cardiotoxicity by inhibiting ferroptosis via AKT/SIRT3/SOD2 signaling pathway activation. Int Immunopharmacol. 2022;113(Pt A):109379. https://doi.org/10.1016/j.intimp.2022.109379.
Wang M, Liu M, Tang L, Shen L, Xiao J, Li R. Liquiritin reduces ferroptosis in doxorubicin-induced cardiotoxicity through targeting SLC7A11/GPX4 pathway. Naunyn Schmiedebergs Arch Pharmacol. 2023. https://doi.org/10.1007/s00210-023-02515-4.
Sun L, Wang H, Xu D, Yu S, Zhang L, Li X. Lapatinib induces mitochondrial dysfunction to enhance oxidative stress and ferroptosis in doxorubicin-induced cardiomyocytes via inhibition of PI3K/AKT signaling pathway. Bioengineered. 2022;13(1):48–60. https://doi.org/10.1080/21655979.2021.2004980.
Cheah IK, Tang RMY, Wang X, Sachaphibulkij K, Chong SY, Lim LHK et al. Protection against doxorubicin-induced cardiotoxicity by ergothioneine. Antioxidants. 2023;12(2). https://doi.org/10.3390/antiox12020320.
Chen H, Zhu J, Le Y, Pan J, Liu Y, Liu Z, et al. Salidroside inhibits doxorubicin-induced cardiomyopathy by modulating a ferroptosis-dependent pathway. Phytomedicine. 2022;99:153964. https://doi.org/10.1016/j.phymed.2022.153964.
He H, Wang L, Qiao Y, Yang B, Yin D, He M. Epigallocatechin-3-gallate pretreatment alleviates doxorubicin-induced ferroptosis and cardiotoxicity by upregulating AMPKα2 and activating adaptive autophagy. Redox Biol. 2021;48:102185. https://doi.org/10.1016/j.redox.2021.102185.
Shubhra QTH, Guo K, Liu Y, Razzak M, Serajum Manir M, Moshiul Alam AKM. Dual targeting smart drug delivery system for multimodal synergistic combination cancer therapy with reduced cardiotoxicity. Acta Biomater. 2021;131:493–507. https://doi.org/10.1016/j.actbio.2021.06.016.
Fu X, Eggert M, Yoo S, Patel N, Zhong J, Steinke I, et al. The cardioprotective mechanism of phenylaminoethyl selenides (PAESe) against doxorubicin-induced cardiotoxicity involves frataxin. Front Pharmacol. 2020;11:574656. https://doi.org/10.3389/fphar.2020.574656.
Song C, Li D, Zhang J, Zhao X. Berberine hydrochloride alleviates imatinib mesylate - induced cardiotoxicity through the inhibition of Nrf2-dependent ferroptosis. Food Funct. 2023;14(2):1087–98. https://doi.org/10.1039/d2fo03331c.
Ma S, He LL, Zhang GR, Zuo QJ, Wang ZL, Zhai JL, et al. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn Schmiedebergs Arch Pharmacol. 2022;395(8):945–62. https://doi.org/10.1007/s00210-022-02243-1.
Zhang W, Lu J, Wang Y, Sun P, Gao T, Xu N et al. Canagliflozin attenuates lipotoxicity in cardiomyocytes by inhibiting inflammation and ferroptosis through activating AMPK pathway. Int J Mol Sci. 2023;24(1). https://doi.org/10.3390/ijms24010858.
Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res. 2021;2021:9999612. https://doi.org/10.1155/2021/9999612.
Li D, Song C, Zhang J, Zhao X. Resveratrol alleviated 5-FU-induced cardiotoxicity by attenuating GPX4 dependent ferroptosis. J Nutr Biochem. 2023;112:109241. https://doi.org/10.1016/j.jnutbio.2022.109241.
Wang X, Simayi A, Fu J, Zhao X, Xu G. Resveratrol mediates the miR-149/HMGB1 axis and regulates the ferroptosis pathway to protect myocardium in endotoxemia mice. Am J Physiol Endocrinol Metab. 2022;323(1):E21-e32. https://doi.org/10.1152/ajpendo.00227.2021.
Min J, Wu L, Liu Y, Song G, Deng Q, Jin W, et al. Empagliflozin attenuates trastuzumab-induced cardiotoxicity through suppression of DNA damage and ferroptosis. Life Sci. 2023;312:121207. https://doi.org/10.1016/j.lfs.2022.121207.
Bian J, Ding Y, Wang S, Jiang Y, Wang M, Wei K, et al. Celastrol confers ferroptosis resistance via AKT/GSK3β signaling in high-fat diet-induced cardiac injury. Free Radic Biol Med. 2023;200:36–46. https://doi.org/10.1016/j.freeradbiomed.2023.03.004.
Liu T, Shu J, Liu Y, Xie J, Li T, Li H, et al. Atorvastatin attenuates ferroptosis-dependent myocardial injury and inflammation following coronary microembolization via the Hif1a/Ptgs2 pathway. Front Pharmacol. 2022;13:1057583. https://doi.org/10.3389/fphar.2022.1057583.
Zhou B, Zhang J, Chen Y, Liu Y, Tang X, Xia P, et al. Puerarin protects against sepsis-induced myocardial injury through AMPK-mediated ferroptosis signaling. Aging (Albany NY). 2022;14(8):3617–32. https://doi.org/10.18632/aging.204033.
Zhang J, Wang X, Guan B, Wang X, An X, Wang T, et al. Qing-Xin-Jie-Yu Granule inhibits ferroptosis and stabilizes atherosclerotic plaques by regulating the GPX4/xCT signaling pathway. J Ethnopharmacol. 2023;301:115852. https://doi.org/10.1016/j.jep.2022.115852.
Yu LM, Dong X, Huang T, Zhao JK, Zhou ZJ, Huang YT, et al. Inhibition of ferroptosis by icariin treatment attenuates excessive ethanol consumption-induced atrial remodeling and susceptibility to atrial fibrillation, role of SIRT1. Apoptosis. 2023;28(3–4):607–26. https://doi.org/10.1007/s10495-023-01814-8.
Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76(25):2982–3021. https://doi.org/10.1016/j.jacc.2020.11.010.
Li RL, Fan CH, Gong SY, Kang S. Effect and mechanism of LRP6 on cardiac myocyte ferroptosis in myocardial infarction. Oxid Med Cell Longev. 2021;2021:8963987. https://doi.org/10.1155/2021/8963987.
Liu K, Chen S, Lu R. Identification of important genes related to ferroptosis and hypoxia in acute myocardial infarction based on WGCNA. Bioengineered. 2021;12(1):7950–63. https://doi.org/10.1080/21655979.2021.1984004.
Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524–33. https://doi.org/10.1038/s41586-021-03392-8.
Ouyang S, You J, Zhi C, Li P, Lin X, Tan X, et al. Ferroptosis: the potential value target in atherosclerosis. Cell Death Dis. 2021;12(8):782. https://doi.org/10.1038/s41419-021-04054-3.
Vinchi F, Porto G, Simmelbauer A, Altamura S, Passos ST, Garbowski M, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. 2020;41(28):2681–95. https://doi.org/10.1093/eurheartj/ehz112.
Guo Z, Ran Q, Roberts LJ 2nd, Zhou L, Richardson A, Sharan C, et al. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic Biol Med. 2008;44(3):343–52. https://doi.org/10.1016/j.freeradbiomed.2007.09.009.
Poch D, Mandel J. Pulmonary hypertension. Ann Intern Med. 2021;174(4):Itc49-itc64. https://doi.org/10.7326/aitc202104200.
Kamada H, Kaneyama J, Inoue YY, Noda T, Ueda N, Nakajima K, et al. Long term prognosis in patients with pulmonary hypertension undergoing catheter ablation for supraventricular tachycardia. Sci Rep. 2021;11(1):16176. https://doi.org/10.1038/s41598-021-95508-3.
Liu SF, Nambiar Veetil N, Li Q, Kucherenko MM, Knosalla C, Kuebler WM. Pulmonary hypertension: linking inflammation and pulmonary arterial stiffening. Front Immunol. 2022;13:959209. https://doi.org/10.3389/fimmu.2022.959209.
Xu D, Hu YH, Gou X, Li FY, Yang XY, Li YM et al. Oxidative stress and antioxidative therapy in pulmonary arterial hypertension. Molecules. 2022;27(12). doi:https://doi.org/10.3390/molecules27123724.
Zou HX, Qiu BQ, Lai SQ, Zhou XL, Gong CW, Wang LJ, et al. Iron metabolism and idiopathic pulmonary arterial hypertension: new insights from bioinformatic analysis. Biomed Res Int. 2021;2021:5669412. https://doi.org/10.1155/2021/5669412.
Xie SS, Deng Y, Guo SL, Li JQ, Zhou YC, Liao J, et al. Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep. 2022;12(1):3056. https://doi.org/10.1038/s41598-022-06848-7.
Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic cardiomyopathy - a comprehensive updated review. Prog Cardiovasc Dis. 2019;62(4):315–26. https://doi.org/10.1016/j.pcad.2019.03.003.
Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Fail Clin. 2019;15(3):341–7. https://doi.org/10.1016/j.hfc.2019.02.003.
Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB et al. Pathophysiology of T\type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17). https://doi.org/10.3390/ijms21176275.
Yang P, Feng J, Peng Q, Liu X, Fan Z. Advanced glycation end products: potential mechanism and therapeutic target in cardiovascular complications under diabetes. Oxid Med Cell Longev. 2019;2019:9570616. https://doi.org/10.1155/2019/9570616.
Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. 2018;104(4):293–9. https://doi.org/10.1136/heartjnl-2017-311448.
Yang S, Wong KH, Hua P, He C, Yu H, Shao D, et al. ROS-responsive fluorinated polyethyleneimine vector to co-deliver shMTHFD2 and shGPX4 plasmids induces ferroptosis and apoptosis for cancer therapy. Acta Biomater. 2022;140:492–505. https://doi.org/10.1016/j.actbio.2021.11.042.
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79(4):390–414. https://doi.org/10.1016/j.jacc.2021.11.021.
Wang Z, Xia Q, Su W, Cao M, Sun Y, Zhang M, et al. Exploring the communal pathogenesis, ferroptosis mechanism, and potential therapeutic targets of dilated cardiomyopathy and hypertrophic cardiomyopathy via a microarray data analysis. Front Cardiovasc Med. 2022;9:824756. https://doi.org/10.3389/fcvm.2022.824756.
Khalili-Tanha G, Moghbeli M. Long non-coding RNAs as the critical regulators of doxorubicin resistance in tumor cells. Cell Mol Biol Lett. 2021;26(1):39. https://doi.org/10.1186/s11658-021-00282-9.
Wu BB, Leung KT, Poon EN. Mitochondrial-targeted therapy for doxorubicin-induced cardiotoxicity. Int J Mol Sci. 2022;23(3). https://doi.org/10.3390/ijms23031912.
Ozcan M, Guo Z, Valenzuela Ripoll C, Diab A, Picataggi A, Rawnsley D, et al. Sustained alternate-day fasting potentiates doxorubicin cardiotoxicity. Cell Metab. 2023. https://doi.org/10.1016/j.cmet.2023.02.006.
Chen Y, Shi S, Dai Y. Research progress of therapeutic drugs for doxorubicin-induced cardiomyopathy. Biomed Pharmacother. 2022;156:113903. https://doi.org/10.1016/j.biopha.2022.113903.
Kitakata H, Endo J, Ikura H, Moriyama H, Shirakawa K, Katsumata Y et al. Therapeutic targets for DOX-induced cardiomyopathy: Role of apoptosis vs. ferroptosis. Int J Mol Sci. 2022;23(3). https://doi.org/10.3390/ijms23031414.
Wang Q, Jiang F, Zhao C, Song J, Hu M, Lv Y, et al. miR-21–5p prevents doxorubicin-induced cardiomyopathy by downregulating BTG2. Heliyon. 2023;9(5):e15451. https://doi.org/10.1016/j.heliyon.2023.e15451.
Zhang D, Li Y, Du C, Sang L, Liu L, Li Y, et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J Transl Med. 2022;20(1):363. https://doi.org/10.1186/s12967-022-03566-6.
Fan J, Jiang T, He D. Emerging insights into the role of ferroptosis in the pathogenesis of autoimmune diseases. Front Immunol. 2023;14:1120519. https://doi.org/10.3389/fimmu.2023.1120519.
Lai B, Wu CH, Wu CY, Luo SF, Lai JH. Ferroptosis and autoimmune diseases. Front Immunol. 2022;13:916664. https://doi.org/10.3389/fimmu.2022.916664.
Chen Q, Wang J, Xiang M, Wang Y, Zhang Z, Liang J, et al. The potential role of ferroptosis in systemic lupus erythematosus. Front Immunol. 2022;13:855622. https://doi.org/10.3389/fimmu.2022.855622.
Zhao T, Yang Q, Xi Y, Xie Z, Shen J, Li Z, et al. Ferroptosis in rheumatoid arthritis: a potential therapeutic strategy. Front Immunol. 2022;13:779585. https://doi.org/10.3389/fimmu.2022.779585.
Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4–regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. 2021;22(9):1107–17. https://doi.org/10.1038/s41590-021-00993-3.
López-Pedrera C, Villalba JM, Patiño-Trives AM, Luque-Tévar M, Barbarroja N, Aguirre M et al. Therapeutic potential and immunomodulatory role of coenzyme Q(10) and its analogues in systemic autoimmune diseases. Antioxidants (Basel). 2021;10(4). https://doi.org/10.3390/antiox10040600.
Vats K, Kruglov O, Mizes A, Samovich SN, Amoscato AA, Tyurin VA, et al. Keratinocyte death by ferroptosis initiates skin inflammation after UVB exposure. Redox Biol. 2021;47:102143. https://doi.org/10.1016/j.redox.2021.102143.
Song X, Zhang H, Zhao Y, Lin Y, Tang Q, Zhou X, et al. HMGB1 activates myeloid dendritic cells by up-regulating mTOR pathway in systemic lupus erythematosus. Front Med. 2021;8:636188. https://doi.org/10.3389/fmed.2021.636188.
Ling H, Li M, Yang C, Sun S, Zhang W, Zhao L, et al. Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis. Rheumatology. 2022;61(11):4521–34. https://doi.org/10.1093/rheumatology/keac069.
Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun. 2022;13(1):676. https://doi.org/10.1038/s41467-021-27948-4.
Xie M, Zhu C, Ye Y. Ferroptosis-related molecular clusters and diagnostic model in rheumatoid arthritis. Int J Mol Sci. 2023;24(8). https://doi.org/10.3390/ijms24087342.
Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation. 2021;44(1):35–47. https://doi.org/10.1007/s10753-020-01338-2.
Li RL, Duan HX, Liang Q, Huang YL, Wang LY, Zhang Q, et al. Targeting matrix metalloproteases: a promising strategy for herbal medicines to treat rheumatoid arthritis. Front Immunol. 2022;13:1046810. https://doi.org/10.3389/fimmu.2022.1046810.
Homma T, Kobayashi S, Fujii J. Methionine deprivation reveals the pivotal roles of cell cycle progression in ferroptosis that is induced by cysteine starvation. Cells. 2022;11(10). https://doi.org/10.3390/cells11101603.
Hu J, Zhang R, Chang Q, Ji M, Zhang H, Geng R, et al. p53: a regulator of ferroptosis induced by galectin-1 derived peptide 3 in MH7A cells. Front Genet. 2022;13:920273. https://doi.org/10.3389/fgene.2022.920273.
Zhan Y, Yang Z, Zhan F, Huang Y, Lin S. SIRT1 is transcriptionally repressed by YY1 and suppresses ferroptosis in rheumatoid arthritis. Adv Rheumatol. 2023;63(1):9. https://doi.org/10.1186/s42358-023-00289-0.
Yang Z, Lin SD, Zhan F, Liu Y, Zhan YW. LncRNA GAS5 alleviates rheumatoid arthritis through regulating miR-222-3p/Sirt1 signalling axis. Autoimmunity. 2021;54(1):13–22. https://doi.org/10.1080/08916934.2020.1846183.
Shen P, Deng X, Chen Z, Ba X, Qin K, Huang Y, et al. SIRT1: A potential therapeutic target in autoimmune diseases. Front Immunol. 2021;12:779177. https://doi.org/10.3389/fimmu.2021.779177.
Wu YJ, Fang WJ, Pan S, Zhang SS, Li DF, Wang ZF, et al. Regulation of Sirt1 on energy metabolism and immune response in rheumatoid arthritis. Int Immunopharmacol. 2021;101(Pt A):108175. https://doi.org/10.1016/j.intimp.2021.108175.
Chen Y, Wang J, Li J, Zhu J, Wang R, Xi Q, et al. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur J Pharmacol. 2021;911:174518. https://doi.org/10.1016/j.ejphar.2021.174518.
Gu SC, Yuan CX, Gu C. Identification of ferroptosis-related gene signatures associated with multiple sclerosis using weighted gene co-expression network analysis. Medicine. 2022;101(51):e31802. https://doi.org/10.1097/md.0000000000031802.
Cui Y, Zhang Z, Zhou X, Zhao Z, Zhao R, Xu X, et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression. J Neuroinflammation. 2021;18(1):249. https://doi.org/10.1186/s12974-021-02231-x.
Jhee JH, Nam BY, Park JT, Kim HW, Chang TI, Kang EW, et al. CD71 mesangial IgA1 receptor and the progression of IgA nephropathy. Transl Res. 2021;230:34–43. https://doi.org/10.1016/j.trsl.2020.10.007.
Mohammad G, Matakidou A, Robbins PA, Lakhal-Littleton S. The kidney hepcidin/ferroportin axis controls iron reabsorption and determines the magnitude of kidney and systemic iron overload. Kidney Int. 2021;100(3):559–69. https://doi.org/10.1016/j.kint.2021.04.034.
Fan J, Han Y, Sun H, Sun S, Wang Y, Guo R, et al. Mesenchymal stem cell-derived exosomal microRNA-367–3p alleviates experimental autoimmune encephalomyelitis via inhibition of microglial ferroptosis by targeting EZH2. Biomed Pharmacother. 2023;162:114593. https://doi.org/10.1016/j.biopha.2023.114593.
Liu GZ, Xu XW, Tao SH, Gao MJ, Hou ZH. HBx facilitates ferroptosis in acute liver failure via EZH2 mediated SLC7A11 suppression. J Biomed Sci. 2021;28(1):67. https://doi.org/10.1186/s12929-021-00762-2.
Li H, Zeng Y, Luo S, Li Z, Huang F, Liu Z. GPX4 aggravates experimental autoimmune encephalomyelitis by inhibiting the functions of CD4+ T cells. Biochem Biophys Res Commun. 2023;642:57–65. https://doi.org/10.1016/j.bbrc.2022.12.034.
Luoqian J, Yang W, Ding X, Tuo QZ, Xiang Z, Zheng Z, et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol Immunol. 2022;19(8):913–24. https://doi.org/10.1038/s41423-022-00883-0.
Li Q, Chen Z, Yang C, Wang L, Ma J, He T, et al. Role of ferroptosis-associated genes in ankylosing spondylitis and immune cell infiltration. Front Genet. 2022;13:948290. https://doi.org/10.3389/fgene.2022.948290.
Dong S, Lu Y, Peng G, Li J, Li W, Li M, et al. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Dig Liver Dis. 2021;53(10):1276–85. https://doi.org/10.1016/j.dld.2021.02.011.
Jiang H, Fang Y, Wang Y, Li T, Lin H, Lin J, et al. FGF4 improves hepatocytes ferroptosis in autoimmune hepatitis mice via activation of CISD3. Int Immunopharmacol. 2023;116:109762. https://doi.org/10.1016/j.intimp.2023.109762.
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33–43. https://doi.org/10.1016/j.jot.2020.09.006.
Chang S, Tang M, Zhang B, Xiang D, Li F. Ferroptosis in inflammatory arthritis: a promising future. Front Immunol. 2022;13:955069. https://doi.org/10.3389/fimmu.2022.955069.
Tang B, Zhu J, Fang S, Wang Y, Vinothkumar R, Li M, et al. Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radic Biol Med. 2021;172:312–29. https://doi.org/10.1016/j.freeradbiomed.2021.06.012.
Hou L, Gong F, Liu B, Yang X, Chen L, Li G, et al. Orally administered titanium carbide nanosheets as anti-inflammatory therapy for colitis. Theranostics. 2022;12(8):3834–46. https://doi.org/10.7150/thno.70668.
Li X, Chu Y, Ma R, Dou M, Li S, Song Y, et al. Ferroptosis as a mechanism of oligodendrocyte loss and demyelination in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2022;373:577995. https://doi.org/10.1016/j.jneuroim.2022.577995.
Zhu L, Chen D, Zhu Y, Pan T, Xia D, Cai T, et al. GPX4-regulated ferroptosis mediates s100-induced experimental autoimmune hepatitis associated with the Nrf2/HO-1 signaling pathway. Oxid Med Cell Longev. 2021;2021:6551069. https://doi.org/10.1155/2021/6551069.
Luo H, Zhang R. Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the Xc-/GPX4 axis. Exp Ther Med. 2021;21(1):72. https://doi.org/10.3892/etm.2020.9504.
Zhao C, Chen J, Ye J, Li Z, Su L, Wang J, et al. Structural transformative antioxidants for dual-responsive anti-inflammatory delivery and photoacoustic inflammation imaging. Angew Chem Int Ed Engl. 2021;60(26):14458–66. https://doi.org/10.1002/anie.202100873.
Lin D, Zhang M, Luo C, Wei P, Cui K, Chen Z. Targeting ferroptosis attenuates inflammation, fibrosis, and mast cell activation in chronic prostatitis. J Immunol Res. 2022;2022:6833867. https://doi.org/10.1155/2022/6833867.
Alli AA, Desai D, Elshika A, Conrad M, Proneth B, Clapp W, et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin Immunol. 2023;248:109213. https://doi.org/10.1016/j.clim.2022.109213.
Pons-Estel GJ, Alarcón GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39(4):257–68. https://doi.org/10.1016/j.semarthrit.2008.10.007.
Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. 2016;7:35. https://doi.org/10.3389/fimmu.2016.00035.
Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, et al. Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol. 2021;22(9):1107–17. https://doi.org/10.1038/s41590-021-00993-3.
Kim SJ, Lee K, Diamond B. Follicular helper T cells in systemic lupus erythematosus. Front Immunol. 2018;9:1793. https://doi.org/10.3389/fimmu.2018.01793.
Gao X, Song Y, Wu J, Lu S, Min X, Liu L et al. Iron-dependent epigenetic modulation promotes pathogenic T cell differentiation in lupus. J Clin Invest. 2022;132(9). https://doi.org/10.1172/jci152345.
Philippou E, Petersson SD, Rodomar C, Nikiphorou E. Rheumatoid arthritis and dietary interventions: systematic review of clinical trials. Nutr Rev. 2021;79(4):410–28. https://doi.org/10.1093/nutrit/nuaa033.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38. https://doi.org/10.1016/s0140-6736(16)30173-8.
Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–33. https://doi.org/10.1038/s41584-020-0413-5.
Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet. 2017;389(10080):1741–55. https://doi.org/10.1016/s0140-6736(16)31711-1.
Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606–19. https://doi.org/10.1016/s0140-6736(12)60150-0.
Zhong W, Xia Z, Hinrichs D, Rosenbaum JT, Wegmann KW, Meyrowitz J, et al. Hemin exerts multiple protective mechanisms and attenuates dextran sulfate sodium-induced colitis. J Pediatr Gastroenterol Nutr. 2010;50(2):132–9. https://doi.org/10.1097/MPG.0b013e3181c61591.
Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9(11):778–88. https://doi.org/10.1038/nri2655.
Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27(3):1008–22. https://doi.org/10.1038/s41418-019-0393-7.
Schmitz ML, Shaban MS, Albert BV, Gökçen A, Kracht M. The crosstalk of endoplasmic reticulum (ER) stress pathways with NF-κB: complex mechanisms relevant for cancer, inflammation and infection. Biomedicines. 2018;6(2). https://doi.org/10.3390/biomedicines6020058.
Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang J, et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020;11(2):86. https://doi.org/10.1038/s41419-020-2299-1.
Tan S, Xu J, Lai A, Cui R, Bai R, Li S, et al. Curculigoside exerts significant anti-arthritic effects in vivo and in vitro via regulation of the JAK/STAT/NF-κB signaling pathway. Mol Med Rep. 2019;19(3):2057–64. https://doi.org/10.3892/mmr.2019.9854.
Wang S, Liu W, Wang J, Bai X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 2020;259:118356. https://doi.org/10.1016/j.lfs.2020.118356.
Chen Y, Zhang P, Chen W, Chen G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol Lett. 2020;225:9–15. https://doi.org/10.1016/j.imlet.2020.06.005.
Rodríguez Murúa S, Farez MF, Quintana FJ. The immune response in multiple sclerosis. Annu Rev Pathol. 2022;17:121–39. https://doi.org/10.1146/annurev-pathol-052920-040318.
Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. 2019;25(2):112–23. https://doi.org/10.1016/j.molmed.2018.11.005.
Gao J, Wang Q, Tang YD, Zhai J, Hu W, Zheng C. When ferroptosis meets pathogenic infections. Trends Microbiol. 2023;31(5):468–79. https://doi.org/10.1016/j.tim.2022.11.006.
Lopez-Jimenez AT, Mostowy S. Emerging technologies and infection models in cellular microbiology. Nat Commun. 2021;12(1):6764. https://doi.org/10.1038/s41467-021-26641-w.
Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6). https://doi.org/10.1084/jem.20210518.
Wang MP, Joshua B, Jin NY, Du SW, Li C. Ferroptosis in viral infection: the unexplored possibility. Acta Pharmacol Sin. 2022;43(8):1905–15. https://doi.org/10.1038/s41401-021-00814-1.
Newton HJ, Ang DK, van Driel IR, Hartland EL. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev. 2010;23(2):274–98. https://doi.org/10.1128/cmr.00052-09.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–64. https://doi.org/10.1038/s41422-019-0164-5.
Yamane D, Hayashi Y, Matsumoto M, Nakanishi H, Imagawa H, Kohara M, et al. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem Biol. 2022;29(5):799-810.e4. https://doi.org/10.1016/j.chembiol.2021.07.022.
Qiang L, Zhang Y, Lei Z, Lu Z, Tan S, Ge P, et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat Commun. 2023;14(1):1430. https://doi.org/10.1038/s41467-023-37148-x.
Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556–70. https://doi.org/10.1084/jem.20181776.
Li JY, Ren C, Wang LX, Yao RQ, Dong N, Wu Y, et al. Sestrin2 protects dendrite cells against ferroptosis induced by sepsis. Cell Death Dis. 2021;12(9):834. https://doi.org/10.1038/s41419-021-04122-8.
Wang Y, Chen D, Xie H, Jia M, Sun X, Peng F, et al. AUF1 protects against ferroptosis to alleviate sepsis-induced acute lung injury by regulating NRF2 and ATF3. Cell Mol Life Sci. 2022;79(5):228. https://doi.org/10.1007/s00018-022-04248-8.
Shen K, Wang X, Wang Y, Jia Y, Zhang Y, Wang K, et al. miR-125b-5p in adipose derived stem cells exosome alleviates pulmonary microvascular endothelial cells ferroptosis via Keap1/Nrf2/GPX4 in sepsis lung injury. Redox Biol. 2023;62:102655. https://doi.org/10.1016/j.redox.2023.102655.
Wang YM, Gong FC, Qi X, Zheng YJ, Zheng XT, Chen Y, et al. Mucin 1 inhibits ferroptosis and sensitizes vitamin e to alleviate sepsis-induced acute lung injury through GSK3β/Keap1-Nrf2-GPX4 pathway. Oxid Med Cell Longev. 2022;2022:2405943. https://doi.org/10.1155/2022/2405943.
Shimizu J, Murao A, Nofi C, Wang P, Aziz M. Extracellular CIRP promotes GPX4-mediated ferroptosis in sepsis. Front Immunol. 2022;13:903859. https://doi.org/10.3389/fimmu.2022.903859.
Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci. 2022;18(8):3337–57. https://doi.org/10.7150/ijbs.69141.
Amaral EP, Foreman TW, Namasivayam S, Hilligan KL, Kauffman KD, Barbosa Bomfim CC et al. GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. J Exp Med. 2022;219(11). https://doi.org/10.1084/jem.20220504.
Ma C, Wu X, Zhang X, Liu X, Deng G. Corrigendum: Heme oxygenase-1 modulates ferroptosis by fine-tuning levels of intracellular iron and reactive oxygen species of macrophages in response to Bacillus Calmette-Guerin infection. Front Cell Infect Microbiol. 2022;12:1105336. https://doi.org/10.3389/fcimb.2022.1105336.
Shi X, Li C, Cheng L, Ullah H, Sha S, Kang J, et al. Mycobacterium tuberculosis Rv1324 protein contributes to mycobacterial persistence and causes pathological lung injury in mice by inducing ferroptosis. Microbiol Spectr. 2023;11(1):e0252622. https://doi.org/10.1128/spectrum.02526-22.
Liu Y, Chen W, Cen Y, Zhao X, Chen Z, Liang Y, et al. Hepatocyte ferroptosis contributes to anti-tuberculosis drug-induced liver injury: Involvement of the HIF-1α/SLC7A11/GPx4 axis. Chem Biol Interact. 2023;376:110439. https://doi.org/10.1016/j.cbi.2023.110439.
Dar HH, Epperly MW, Tyurin VA, Amoscato AA, Anthonymuthu TS, Souryavong AB et al. P. aeruginosa augments irradiation injury via 15-lipoxygenase-catalyzed generation of 15-HpETE-PE and induction of theft-ferroptosis. JCI Insight. 2022;7(4). https://doi.org/10.1172/jci.insight.156013.
Dar HH, Anthonymuthu TS, Ponomareva LA, Souryavong AB, Shurin GV, Kapralov AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO(•) sabotage of theft-ferroptosis. Redox Biol. 2021;45:102045. https://doi.org/10.1016/j.redox.2021.102045.
Vaillancourt M, Galdino ACM, Limsuwannarot SP, Celedonio D, Dimitrova E, Broerman M, et al. A compensatory RNase E variation increases Iron Piracy and Virulence in multidrug-resistant Pseudomonas aeruginosa during Macrophage infection. PLoS Pathog. 2023;19(4):e1010942. https://doi.org/10.1371/journal.ppat.1010942.
Wang Y, Wang L, Sun T, Shen S, Li Z, Ma X, et al. Study of the inflammatory activating process in the early stage of Fusobacterium nucleatum infected PDLSCs. Int J Oral Sci. 2023;15(1):8. https://doi.org/10.1038/s41368-022-00213-0.
Komissarov AA, Karaseva MA, Roschina MP, Shubin AV, Lunina NA, Kostrov SV et al. Individual expression of hepatitis a virus 3C protease induces ferroptosis in human cells In vitro. Int J Mol Sci. 2021;22(15). https://doi.org/10.3390/ijms22157906.
Zhang Q, Qu Y, Zhang Q, Li F, Li B, Li Z, et al. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol Toxicol. 2022. https://doi.org/10.1007/s10565-021-09684-z.
Liu L, Lv Z, Wang M, Zhang D, Liu D, Zhu F. HBV enhances sorafenib resistance in hepatocellular carcinoma by reducing ferroptosis via SRSF2-mediated abnormal PCLAF splicing. Int J Mol Sci. 2023;24(4). https://doi.org/10.3390/ijms24043263.
Miao X, Yin Y, Chen Y, Bi W, Yin Y, Chen S et al. Bidirectionally regulating viral and cellular ferroptosis with metastable iron sulfide against influenza virus. Adv Sci (Weinh). 2023:e2206869. https://doi.org/10.1002/advs.202206869.
Liu C, Wu X, Bing X, Qi W, Zhu F, Guo N, et al. H1N1 influenza virus infection through NRF2-KEAP1-GCLC pathway induces ferroptosis in nasal mucosal epithelial cells. Free Radic Biol Med. 2023;204:226–42. https://doi.org/10.1016/j.freeradbiomed.2023.05.004.
Kung YA, Chiang HJ, Li ML, Gong YN, Chiu HP, Hung CT, et al. Acyl-coenzyme a synthetase long-chain family member 4 Is involved in viral replication organelle formation and facilitates virus replication via ferroptosis. mBio. 2022;13(1):e0271721. https://doi.org/10.1128/mbio.02717-21.
Wen Y, Wang Y, Chen S, Zhou X, Zhang Y, Yang D, et al. Dysregulation of Cytosolic c-di-GMP in Edwardsiella piscicida Promotes Cellular Non-Canonical Ferroptosis. Front Cell Infect Microbiol. 2022;12:825824. https://doi.org/10.3389/fcimb.2022.825824.
Xu XQ, Xu T, Ji W, Wang C, Ren Y, Xiong X, et al. Herpes simplex virus 1-induced ferroptosis contributes to viral encephalitis. mBio. 2023;14(1):e0237022. https://doi.org/10.1128/mbio.02370-22.
Cao L, Qin R, Liu J. Farnesoid X receptor protects against lipopolysaccharide-induced endometritis by inhibiting ferroptosis and inflammatory response. Int Immunopharmacol. 2023;118:110080. https://doi.org/10.1016/j.intimp.2023.110080.
Burton EM, Voyer J, Gewurz BE. Epstein-Barr virus latency programs dynamically sensitize B cells to ferroptosis. Proc Natl Acad Sci U S A. 2022;119(11):e2118300119. https://doi.org/10.1073/pnas.2118300119.
Yi L, Hu Y, Wu Z, Li Y, Kong M, Kang Z, et al. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022;13(7):592. https://doi.org/10.1038/s41419-022-05027-w.
Wang Y, Tian Q, Hao Y, Yao W, Lu J, Chen C, et al. The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4(+) T cells by preventing ferroptosis. Nat Immunol. 2022;23(2):303–17. https://doi.org/10.1038/s41590-021-01090-1.
Xiao Q, Yan L, Han J, Yang S, Tang Y, Li Q, et al. Metabolism-dependent ferroptosis promotes mitochondrial dysfunction and inflammation in CD4(+) T lymphocytes in HIV-infected immune non-responders. EBioMedicine. 2022;86:104382. https://doi.org/10.1016/j.ebiom.2022.104382.
Kannan M, Sil S, Oladapo A, Thangaraj A, Periyasamy P, Buch S. HIV-1 Tat-mediated microglial ferroptosis involves the miR-204-ACSL4 signaling axis. Redox Biol. 2023;62:102689. https://doi.org/10.1016/j.redox.2023.102689.
Wu J, Liu Q, Zhang X, Tan M, Li X, Liu P, et al. The interaction between STING and NCOA4 exacerbates lethal sepsis by orchestrating ferroptosis and inflammatory responses in macrophages. Cell Death Dis. 2022;13(7):653. https://doi.org/10.1038/s41419-022-05115-x.
Lai K, Song C, Gao M, Deng Y, Lu Z, Li N et al. Uridine alleviates sepsis-induced acute lung injury by inhibiting ferroptosis of macrophage. Int J Mol Sci. 2023;24(6). https://doi.org/10.3390/ijms24065093.
Lv YW, Du Y, Ma SS, Shi YC, Xu HC, Deng L, et al. Proanthocyanidins attenuates ferroptosis against influenza-induced acute lung injury in mice by reducing IFN-γ. Life Sci. 2023;314:121279. https://doi.org/10.1016/j.lfs.2022.121279.
He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 2022;8(1):43. https://doi.org/10.1038/s41420-021-00807-3.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24(1):97-108.e4. https://doi.org/10.1016/j.chom.2018.05.009.
Geng S, Hao P, Wang D, Zhong P, Tian F, Zhang R, et al. Zinc oxide nanoparticles have biphasic roles on Mycobacterium-induced inflammation by activating autophagy and ferroptosis mechanisms in infected macrophages. Microb Pathog. 2023;180:106132. https://doi.org/10.1016/j.micpath.2023.106132.
Huang M, Wang Z, Yao L, Zhang L, Gou X, Mo H, et al. Ferric chloride induces ferroptosis in Pseudomonas aeruginosa and heals wound infection in a mouse model. Int J Antimicrob Agents. 2023;61(5):106794. https://doi.org/10.1016/j.ijantimicag.2023.106794.
Ousingsawat J, Schreiber R, Gulbins E, Kamler M, Kunzelmann KP. aeruginosa induced lipid peroxidation causes ferroptotic Cell death in airways. Cell Physiol Biochem. 2021;55(5):590–604. https://doi.org/10.33594/000000437.
Nishiga M, Jahng JWS, Wu JC. Ferroptosis of pacemaker cells in COVID-19. Circ Res. 2022;130(7):978–80. https://doi.org/10.1161/circresaha.122.320951.
Carota G, Ronsisvalle S, Panarello F, Tibullo D, Nicolosi A, Li Volti G. Role of iron chelation and protease inhibition of natural products on COVID-19 infection. J Clin Med. 2021;10(11). https://doi.org/10.3390/jcm10112306.
Chavarría AP, Vázquez RRV, Cherit JGD, Bello HH, Suastegui HC, Moreno-Castañeda L, et al. Antioxidants and pentoxifylline as coadjuvant measures to standard therapy to improve prognosis of patients with pneumonia by COVID-19. Comput Struct Biotechnol J. 2021;19:1379–90. https://doi.org/10.1016/j.csbj.2021.02.009.
Li S, Xu W, Wang H, Tang T, Ma J, Cui Z, et al. Ferroptosis plays an essential role in the antimalarial mechanism of low-dose dihydroartemisinin. Biomed Pharmacother. 2022;148:112742. https://doi.org/10.1016/j.biopha.2022.112742.
Parkins MD, Somayaji R, Waters VJ. Epidemiology, Biology, and Impact of Clonal Pseudomonas aeruginosa Infections in Cystic Fibrosis. Clin Microbiol Rev. 2018;31(4). https://doi.org/10.1128/cmr.00019-18.
de Sousa T, Hébraud M, Dapkevicius M, Maltez L, Pereira JE, Capita R et al. Genomic and metabolic characteristics of the pathogenicity in pseudomonas aeruginosa. Int J Mol Sci. 2021;22(23). https://doi.org/10.3390/ijms222312892.
Langton Hewer SC, Smith S, Rowbotham NJ, Yule A, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst Rev. 2023;6(6):Cd004197. https://doi.org/10.1002/14651858.CD004197.pub6.
Albin OR, Kaye KS, McCreary EK, Pogue JM. Less is more? antibiotic treatment duration in pseudomonas aeruginosa ventilator-associated pneumonia. Clin Infect Dis. 2023;76(4):745–9. https://doi.org/10.1093/cid/ciac784.
Jean SS, Liu IM, Hsieh PC, Kuo DH, Liu YL, Hsueh PR. Off-label use versus formal recommendations of conventional and novel antibiotics for the treatment of infections caused by multidrug-resistant bacteria. Int J Antimicrob Agents. 2023;61(5):106763. https://doi.org/10.1016/j.ijantimicag.2023.106763.
Kanj SS, Bassetti M, Kiratisin P, Rodrigues C, Villegas MV, Yu Y, et al. Clinical data from studies involving novel antibiotics to treat multidrug-resistant Gram-negative bacterial infections. Int J Antimicrob Agents. 2022;60(3):106633. https://doi.org/10.1016/j.ijantimicag.2022.106633.
Jeong GJ, Khan F, Khan S, Tabassum N, Mehta S, Kim YM. Pseudomonas aeruginosa virulence attenuation by inhibiting siderophore functions. Appl Microbiol Biotechnol. 2023;107(4):1019–38. https://doi.org/10.1007/s00253-022-12347-6.
Rada B, Leto TL. Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol. 2013;21(2):73–81. https://doi.org/10.1016/j.tim.2012.10.004.
Bourzac K. Infectious disease: beating the big three. Nature. 2014;507(7490):S4-7. https://doi.org/10.1038/507s4a.
Ma C, Wu X, Zhang X, Liu X, Deng G. Heme oxygenase-1 modulates ferroptosis by fine-tuning levels of intracellular iron and reactive oxygen species of macrophages in response to Bacillus Calmette-Guerin infection. Front Cell Infect Microbiol. 2022;12:1004148. https://doi.org/10.3389/fcimb.2022.1004148.
Xl L, Gy Z, R G, N C. Ferroptosis in sepsis: The mechanism, the role and the therapeutic potential. Front Immunol. 2022;13:956361. https://doi.org/10.3389/fimmu.2022.956361.
Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol. 2017;2(2):135–43. https://doi.org/10.1016/s2468-1253(16)30119-4.
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–20. https://doi.org/10.1038/nri.2017.36.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555–68. https://doi.org/10.1084/jem.20140857.
Xie Z, Xu M, Xie J, Liu T, Xu X, Gao W, et al. Inhibition of ferroptosis attenuates glutamate excitotoxicity and nuclear autophagy in a CLP septic mouse model. Shock. 2022;57(5):694–702. https://doi.org/10.1097/shk.0000000000001893.
Gao N, Tang AL, Liu XY, Chen J, Zhang GQ. p53-Dependent ferroptosis pathways in sepsis. Int Immunopharmacol. 2023;118:110083. https://doi.org/10.1016/j.intimp.2023.110083.
Terrault NA, Levy MT, Cheung KW, Jourdain G. Viral hepatitis and pregnancy. Nat Rev Gastroenterol Hepatol. 2021;18(2):117–30. https://doi.org/10.1038/s41575-020-00361-w.
Castaneda D, Gonzalez AJ, Alomari M, Tandon K, Zervos XB. From hepatitis A to E: a critical review of viral hepatitis. World J Gastroenterol. 2021;27(16):1691–715. https://doi.org/10.3748/wjg.v27.i16.1691.
Odenwald MA, Paul S. Viral hepatitis: past, present, and future. World J Gastroenterol. 2022;28(14):1405–29. https://doi.org/10.3748/wjg.v28.i14.1405.
Shojaie L, Iorga A, Dara L. Cell death in liver diseases: a review. Int J Mol Sci. 2020;21(24). https://doi.org/10.3390/ijms21249682.
Liu C, Zhao K, Chen Y, Yao Y, Tang J, Wang J, et al. Mitochondrial glycerol-3-phosphate dehydrogenase restricts HBV replication via the TRIM28-mediated degradation of HBx. J Virol. 2023;97(5):e0058023. https://doi.org/10.1128/jvi.00580-23.
Han Y, Zhu J, Yang L, Nilsson-Payant BE, Hurtado R, Lacko LA, et al. SARS-CoV-2 Infection Induces Ferroptosis of Sinoatrial Node Pacemaker Cells. Circ Res. 2022;130(7):963–77. https://doi.org/10.1161/circresaha.121.320518.
Nuszkiewicz J, Sutkowy P, Wróblewski M, Pawłowska M, Wesołowski R, Wróblewska J et al. Links between vitamin K, ferroptosis and SARS-CoV-2 infection. Antioxidants. 2023;12(3). https://doi.org/10.3390/antiox12030733.
Peterlin BM, Trono D. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat Rev Immunol. 2003;3(2):97–107. https://doi.org/10.1038/nri998.
Redd AD, Quinn TC, Tobian AA. Frequency and implications of HIV superinfection. Lancet Infect Dis. 2013;13(7):622–8. https://doi.org/10.1016/s1473-3099(13)70066-5.
Subashini D, Dinesha TR, Srirama RB, Boobalan J, Poongulali S, Chitra DA, et al. Mitochondrial DNA content of peripheral blood mononuclear cells in ART untreated & stavudine/zidovudine treated HIV-1-infected patients. Indian J Med Res. 2018;148(2):207–14. https://doi.org/10.4103/ijmr.IJMR_1144_16.
Perrin S, Cremer J, Roll P, Faucher O, Ménard A, Reynes J, et al. HIV-1 infection and first line ART induced differential responses in mitochondria from blood lymphocytes and monocytes: the ANRS EP45 “Aging” study. PLoS One. 2012;7(7):e41129. https://doi.org/10.1371/journal.pone.0041129.
Sena-Dos-Santos C, Braga-da-Silva C, Marques D, Azevedo Dos Santos Pinheiro J, Ribeiro-Dos-Santos Â, Cavalcante GC. Unraveling cell death pathways during malaria infection: What do we know so far? Cells. 2021;10(2). https://doi.org/10.3390/cells10020479.
Malaria's WHO moment. Nat Biotechnol. 2021;39(11):1322. https://doi.org/10.1038/s41587-021-01129-6.
Chen S, Fan F, Zhang Y, Zeng J, Li Y, Xu N, et al. Metabolites from scutellarin alleviating deferoxamine-induced hypoxia injury in BV2 cells cultured on microfluidic chip combined with a mass spectrometer. Talanta. 2023;259:124478. https://doi.org/10.1016/j.talanta.2023.124478.
Gattermann N, Muckenthaler MU, Kulozik AE, Metzgeroth G, Hastka J. The evaluation of iron deficiency and iron overload. Dtsch Arztebl Int. 2021;118(49):847–56. https://doi.org/10.3238/arztebl.m2021.0290.
Loughnan R, Ahern J, Tompkins C, Palmer CE, Iversen J, Thompson WK, et al. Association of genetic variant linked to hemochromatosis with brain magnetic resonance imaging measures of iron and movement disorders. JAMA Neurol. 2022;79(9):919–28. https://doi.org/10.1001/jamaneurol.2022.2030.
Crichton R. Iron metabolism : from molecular mechanisms to clinical consequences. other: other. 2018.
Griffiths WJH, Besser M, Bowden DJ, Kelly DA. Juvenile haemochromatosis. Lancet Child Adolesc Health. 2021;5(7):524–30. https://doi.org/10.1016/S2352-4642(20)30392-8.
Mancardi D, Mezzanotte M, Arrigo E, Barinotti A, Roetto A. Iron overload, oxidative stress, and ferroptosis in the failing heart and liver. Antioxidants. 2021;10(12). https://doi.org/10.3390/antiox10121864.
Harrison AV, Lorenzo FR, McClain DA. Iron and the pathophysiology of diabetes. Annu Rev Physiol. 2023;85:339–62. https://doi.org/10.1146/annurev-physiol-022522-102832.
Hsu CC, Senussi NH, Fertrin KY, Kowdley KV. Iron overload disorders. Hepatol Commun. 2022;6(8):1842–54. https://doi.org/10.1002/hep4.2012.
Baas FS, Rishi G, Swinkels DW, Subramaniam VN. Genetic diagnosis in hereditary hemochromatosis: discovering and understanding the biological relevance of variants. Clin Chem. 2021;67(10):1324–41. https://doi.org/10.1093/clinchem/hvab130.
Baschant U, Altamura S, Steele-Perkins P, Muckenthaler MU, Spasi MV, Hofbauer LC, et al. Iron effects versus metabolic alterations in hereditary hemochromatosis driven bone loss. Trends Endocrinol Metab. 2022;33(9):652–63. https://doi.org/10.1016/j.tem.2022.06.004.
Cartella I, Tavecchia GA, Quattrocchi G, Giannattasio C, Volpato E, Palazzini M, et al. A heart of iron: juvenile haemochromatosis presents with cardiac failure. Lancet. 2022;400(10352):616. https://doi.org/10.1016/S0140-6736(22)01285-5.
Palmer WC, Vishnu P, Sanchez W, Aqel B, Riegert-Johnson D, Seaman LAK, et al. Diagnosis and management of genetic iron overload disorders. J Gen Intern Med. 2018;33(12):2230–6. https://doi.org/10.1007/s11606-018-4669-2.
Srole DN, Ganz T. Erythroferrone structure, function, and physiology: iron homeostasis and beyond. J Cell Physio. 2021;236(7):4888–901. https://doi.org/10.1002/jcp.30247.
Wu J, Wang Y, Jiang R, Xue R, Yin X, Wu M, et al. Ferroptosis in liver disease: new insights into disease mechanisms. Cell Death Discov. 2021;7(1):276. https://doi.org/10.1038/s41420-021-00660-4.
Kim E, Jean J, Kearns C, Ballard DH. Perivenous hepatic iron deposition in alcoholic cirrhosis. Radiographics. 2021;41(6):1570–1. https://doi.org/10.1148/rg.2021210190.
Li Y, Qin M, Zhong W, Liu C, Deng G, Yang M, et al. RAGE promotes dysregulation of iron and lipid metabolism in alcoholic liver disease. Redox Biol. 2023;59:102559. https://doi.org/10.1016/j.redox.2022.102559.
Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281(32):22974–82. https://doi.org/10.1074/jbc.M602098200.
Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132(14):1473–7. https://doi.org/10.1182/blood-2018-06-857995.
Rosato BE, Marra R, D'Onofrio V, Del Giudice F, Della Monica S, Iolascon A et al. SEC23B loss-of-function suppresses hepcidin expression by impairing glycosylation pathway in human hepatic cells. Int J Mol Sci. 2022;23(3). https://doi.org/10.3390/ijms23031304.
Marketa D-C, Kamila B, Karolina K, Jitka C, Vaclav H, Jan H, et al. Role of duodenal iron transporters and hepcidin in patients with alcoholic liver disease. J Cell Mol Med. 2014;18(9):1840–50. https://doi.org/10.1111/jcmm.12310.
Jiang L, Wang J, Wang K, Wang H, Wu Q, Yang C, et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021;138(8):689–705. https://doi.org/10.1182/blood.2020008986.
Xue M, Tian Y, Sui Y, Zhao H, Gao H, Liang H, et al. Protective effect of fucoidan against iron overload and ferroptosis-induced liver injury in rats exposed to alcohol. Biomed Pharmacother. 2022;153:113402. https://doi.org/10.1016/j.biopha.2022.113402.
Yuan R, Tao X, Liang S, Pan Y, He L, Sun J, et al. Protective effect of acidic polysaccharide from Schisandra chinensis on acute ethanol-induced liver injury through reducing CYP2E1-dependent oxidative stress. Biomed Pharmacother. 2018;99:537–42. https://doi.org/10.1016/j.biopha.2018.01.079.
Harjum?ki R, Pridgeon CS, Ingelman-Sundberg M. CYP2E1 in alcoholic and non-alcoholic liver injury. roles of ROS, reactive intermediates and lipid overload. International journal of molecular sciences. 2021;22(15). https://doi.org/10.3390/ijms22158221.
Corradini E, Buzzetti E, Dongiovanni P, Scarlini S, Caleffi A, Pelusi S, et al. Ceruloplasmin gene variants are associated with hyperferritinemia and increased liver iron in patients with NAFLD. J Hepatol. 2021;75(3):506–13. https://doi.org/10.1016/j.jhep.2021.03.014.
Gao H, Jin Z, Bandyopadhyay G, Wang G, Zhang D, Rocha KCE, et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022;34(8):1201-13.e5. https://doi.org/10.1016/j.cmet.2022.07.006.
Feng H, Liu Q, Deng Z, Li H, Zhang H, Song J, et al. Human umbilical cord mesenchymal stem cells ameliorate erectile dysfunction in rats with diabetes mellitus through the attenuation of ferroptosis. Stem Cell Res Ther. 2022;13(1):450. https://doi.org/10.1186/s13287-022-03147-w.
Duygu Dee H-F, Elizabeth K, Callie C, John E, Nikolai T, John G. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology. 2007;46(6):1979–85. https://doi.org/10.1002/hep.21895.
Yu L, He M, Liu S, Dou X, Li L, Gu N, et al. Fluorescent egg white-based carbon dots as a high-sensitivity iron chelator for the therapy of nonalcoholic fatty liver disease by iron overload in zebrafish. ACS Appl Mater Interfaces. 2021;13(46):54677–89. https://doi.org/10.1021/acsami.1c14674.
Zhang L, Dai X, Wang L, Cai J, Shen J, Shen Y, et al. Iron overload accelerated lipid metabolism disorder and liver injury in rats with non-alcoholic fatty liver disease. Front Nutr. 2022;9:961892. https://doi.org/10.3389/fnut.2022.961892.
Bathish B, Robertson H, Dillon JF, Dinkova-Kostova AT, Hayes JD. Nonalcoholic steatohepatitis and mechanisms by which it is ameliorated by activation of the CNC-bZIP transcription factor Nrf2. Free Radic Biol Med. 2022;188:221–61. https://doi.org/10.1016/j.freeradbiomed.2022.06.226.
Qi X, Song A, Ma M, Wang P, Zhang X, Lu C, et al. Curcumol inhibits ferritinophagy to restrain hepatocyte senescence through YAP/NCOA4 in non-alcoholic fatty liver disease. Cell Prolif. 2021;54(9):e13107. https://doi.org/10.1111/cpr.13107.
Hu Z, Li Y, Ma B, Lei S, Wang X. Iron metabolism mediates the relationship between Vitamin C and hepatic steatosis and fibrosis in NAFLD. Front Nutr. 2022;9:952056. https://doi.org/10.3389/fnut.2022.952056.
Jiang JJ, Zhang GF, Zheng JY, Sun JH, Ding SB. Targeting mitochondrial ROS-mediated ferroptosis by quercetin alleviates high-fat diet-induced hepatic lipotoxicity. Front Pharmacol. 2022;13:876550. https://doi.org/10.3389/fphar.2022.876550.
Tong J, Lan XT, Zhang Z, Liu Y, Sun DY, Wang XJ, et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2023;44(5):1014–28. https://doi.org/10.1038/s41401-022-01010-5.
Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, et al. Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PloS one. 2013;8(10):e77023. https://doi.org/10.1371/journal.pone.0077023.
Domínguez JF, Ng AC, Poudel G, Stout JC, Churchyard A, Chua P, et al. Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry. 2016;87(5):545–9. https://doi.org/10.1136/jnnp-2014-310183.
Ayton S, Portbury S, Kalinowski P, Agarwal P, Diouf I, Schneider JA, et al. Regional brain iron associated with deterioration in Alzheimer’s disease: a large cohort study and theoretical significance. Alzheimers Dement. 2021;17(7):1244–56. https://doi.org/10.1002/alz.12282.
Peng Y, Chang X, Lang M. Iron homeostasis disorder and Alzheimer's Disease. Int J Mol Sci. 2021;22(22). https://doi.org/10.3390/ijms222212442.
Rosenblum SL, Kosman DJ. Aberrant cerebral iron trafficking co-morbid with chronic inflammation: molecular mechanisms and pharmacologic intervention. Front Neurol. 2022;13:855751. https://doi.org/10.3389/fneur.2022.855751.
Spagnoli C, Fusco C, Pisani F. Pediatric-onset epilepsy and developmental epileptic encephalopathies followed by early-onset parkinsonism. Int J Mol Sci. 2023;24(4). https://doi.org/10.3390/ijms24043796.
Petit F, Drecourt A, Dussiot M, Zangarelli C, Hermine O, Munnich A, et al. Defective palmitoylation of transferrin receptor triggers iron overload in Friedreich ataxia fibroblasts. Blood. 2021;137(15):2090–102. https://doi.org/10.1182/blood.2020006987.
Payne RM. Cardiovascular research in friedreich ataxia: unmet needs and opportunities. JACC Basic Transl Sci. 2022;7(12):1267–83. https://doi.org/10.1016/j.jacbts.2022.04.005.
Pellerin D, Danzi MC, Wilke C, Renaud M, Fazal S, Dicaire MJ, et al. Deep intronic FGF14 GAA repeat expansion in late-onset cerebellar ataxia. N Engl J Med. 2023;388(2):128–41. https://doi.org/10.1056/NEJMoa2207406.
Rodrigues AV, Batelu S, Hinton TV, Rotondo J, Thompson L, Brunzelle JS, et al. Drosophila melanogaster frataxin: protein crystal and predicted solution structure with identification of the iron-binding regions. Acta crystallographica Section D, Structural Biology. 2023;79(Pt 1):22–30. https://doi.org/10.1107/S2059798322011639.
Rodríguez-Pascau L, Britti E, Calap-Quintana P, Dong YN, Vergara C, Delaspre F, et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol Dis. 2021;148:105162. https://doi.org/10.1016/j.nbd.2020.105162.
La Rosa P, Petrillo S, Turchi R, Berardinelli F, Schirinzi T, Vasco G, et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021;38:101791. https://doi.org/10.1016/j.redox.2020.101791.
Xu L, Sun Z, Xing Z, Liu Y, Zhao H, Tang Z, et al. Cur@SF NPs alleviate Friedreich’s ataxia in a mouse model through synergistic iron chelation and antioxidation. J Nanobiotechnology. 2022;20(1):118. https://doi.org/10.1186/s12951-022-01333-9.
Villa C, Legato M, Umbach A, Riganti C, Jones R, Martini B et al. Treatment with ROS detoxifying gold quantum clusters alleviates the functional decline in a mouse model of Friedreich ataxia. Sci Transl Med. 2021;13(607). https://doi.org/10.1126/scitranslmed.abe1633.
Zhang X, Li LX, Ding H, Torres VE, Yu C, Li X. Ferroptosis promotes cyst growth in autosomal dominant polycystic kidney disease mouse models. J Am Soc Nephrol. 2021;32(11):2759–76. https://doi.org/10.1681/asn.2021040460.
Miao R, Fang X, Zhang Y, Wei J, Zhang Y, Tian J. Iron metabolism and ferroptosis in type 2 diabetes mellitus and complications: mechanisms and therapeutic opportunities. Cell Death Dis. 2023;14(3):186. https://doi.org/10.1038/s41419-023-05708-0.
Karmi O, Sohn YS, Marjault HB, Israeli T, Leibowitz G, Ioannidis K et al. A combined drug treatment that reduces mitochondrial iron and reactive oxygen levels recovers insulin secretion in NAF-1-deficient pancreatic cells. Antioxidants. 2021;10(8). https://doi.org/10.3390/antiox10081160.
Zhao X, Ma Y, Shi M, Huang M, Xin J, Ci S, et al. Excessive iron inhibits insulin secretion via perturbing transcriptional regulation of SYT7 by OGG1. Cell Mol Life Sci. 2023;80(6):159. https://doi.org/10.1007/s00018-023-04802-y.
Petry SF, A Rm, Rawat D, Brunner L, Lerch N, Zhou M et al. Loss and recovery of glutaredoxin 5 is inducible by diet in a murine model of diabesity and mediated by free fatty acids in vitro. Antioxidants. 2022;11(4). https://doi.org/10.3390/antiox11040788.
Tasnim H, Ding H. Electron transfer activity of the nanodisc-bound mitochondrial outer membrane protein mitoNEET. Free Radic Biol Med. 2022;187:50–8. https://doi.org/10.1016/j.freeradbiomed.2022.05.011.
You Y, Liu C, Liu T, Tian M, Wu N, Yu Z, et al. FNDC3B protects steatosis and ferroptosis via the AMPK pathway in alcoholic fatty liver disease. Free Radic Biol Med. 2022;193(Pt 2):808–19. https://doi.org/10.1016/j.freeradbiomed.2022.10.322.
Xiong H, Zhang C, Han L, Xu T, Saeed K, Han J, et al. Suppressed farnesoid X receptor by iron overload in mice and humans potentiates iron-induced hepatotoxicity. Hepatology. 2022;76(2):387–403. https://doi.org/10.1002/hep.32270.
Chakraborty S, Andrieux G, Kastl P, Adlung L, Altamura S, Boehm ME, et al. Erythropoietin-driven dynamic proteome adaptations during erythropoiesis prevent iron overload in the developing embryo. Cell Rep. 2022;40(12):111360. https://doi.org/10.1016/j.celrep.2022.111360.
Jadhav S, Protchenko O, Li F, Baratz E, Shakoury-Elizeh M, Maschek A, et al. Mitochondrial dysfunction in mouse livers depleted of iron chaperone PCBP1. Free Radical Biol Med. 2021;175:18–27. https://doi.org/10.1016/j.freeradbiomed.2021.08.232.
Federti E, Vinchi F, Iatcenko I, Ghigo A, Matte A, Toya SCM, et al. Duality of Nrf2 in iron-overload cardiomyopathy. Haematologica. 2023;108(5):1335–48. https://doi.org/10.3324/haematol.2022.281995.
Culley MK, Zhao J, Tai YY, Tang Y, Perk D, Negi V et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J Clin Invest. 2021;131(11). https://doi.org/10.1172/JCI136459.
Medina-Carbonero M, Sanz-Alcázar A, Britti E, Delaspre F, Cabiscol E, Ros J, et al. Mice harboring the FXN I151F pathological point mutation present decreased frataxin levels, a Friedreich ataxia-like phenotype, and mitochondrial alterations. Cell Mol Life Sci. 2022;79(2):74. https://doi.org/10.1007/s00018-021-04100-5.
Cotticelli MG, Xia S, Truitt R, Doliba NM, Rozo AV, Tobias JW et al. Acute frataxin knockdown in induced pluripotent stem cell-derived cardiomyocytes activates a type I interferon response. Dis Model Mech. 2023;16(5). https://doi.org/10.1242/dmm.049497.
Culley MK, Rao RJ, Mehta M, Zhao J, El Khoury W, Harvey LD, et al. Frataxin deficiency disrupts mitochondrial respiration and pulmonary endothelial cell function. Vascul Pharmacol. 2023;151:107181. https://doi.org/10.1016/j.vph.2023.107181.
Guo T, Yu Y, Yan W, Zhang M, Yi X, Liu N, et al. Erythropoietin ameliorates cognitive dysfunction in mice with type 2 diabetes mellitus via inhibiting iron overload and ferroptosis. Exp Neurol. 2023;365:114414. https://doi.org/10.1016/j.expneurol.2023.114414.
Lv Z, Han J, Li J, Guo H, Fei Y, Sun Z, et al. Single cell RNA-seq analysis identifies ferroptotic chondrocyte cluster and reveals TRPV1 as an anti-ferroptotic target in osteoarthritis. EBioMedicine. 2022;84:104258. https://doi.org/10.1016/j.ebiom.2022.104258.
Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao J, et al. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine. 2022;76:103847. https://doi.org/10.1016/j.ebiom.2022.103847.
Noonan ML, Ni P, Solis E, Marambio YG, Agoro R, Chu X, et al. Osteocyte Egln1/Phd2 links oxygen sensing and biomineralization via FGF23. Bone Res. 2023;11(1):7. https://doi.org/10.1038/s41413-022-00241-w.
Sun K, Hou L, Guo Z, Wang G, Guo J, Xu J, et al. JNK-JUN-NCOA4 axis contributes to chondrocyte ferroptosis and aggravates osteoarthritis via ferritinophagy. Free Radic Biol Med. 2023;200:87–101. https://doi.org/10.1016/j.freeradbiomed.2023.03.008.
Di Modica SM, Tanzi E, Olivari V, Lidonnici MR, Pettinato M, Pagani A, et al. Transferrin receptor 2 (Tfr2) genetic deletion makes transfusion-independent a murine model of transfusion-dependent β-thalassemia. Am J Hematol. 2022;97(10):1324–36. https://doi.org/10.1002/ajh.26673.
Ledesma-Colunga MG, Baschant U, Weidner H, Alves TC, Mirtschink P, Hofbauer LC, et al. Transferrin receptor 2 deficiency promotes macrophage polarization and inflammatory arthritis. Redox Biol. 2023;60:102616. https://doi.org/10.1016/j.redox.2023.102616.
Xiao X, Moschetta GA, Xu Y, Fisher AL, Alfaro-Magallanes VM, Dev S, et al. Regulation of iron homeostasis by hepatocyte TfR1 requires HFE and contributes to hepcidin suppression in β-thalassemia. Blood. 2023;141(4):422–32. https://doi.org/10.1182/blood.2022017811.
Wang Y, Bi R, Quan F, Cao Q, Lin Y, Yue C, et al. Ferroptosis involves in renal tubular cell death in diabetic nephropathy. Eur J Pharmacol. 2020;888:173574. https://doi.org/10.1016/j.ejphar.2020.173574.
Chiang S, Kalinowski DS, Dharmasivam M, Braidy N, Richardson DR, Huang MLH. The potential of the novel NAD(+) supplementing agent, SNH6, as a therapeutic strategy for the treatment of Friedreich’s ataxia. Pharmacol Res. 2020;155:104680. https://doi.org/10.1016/j.phrs.2020.104680.
Abdul Y, Li W, Ward R, Abdelsaid M, Hafez S, Dong G, et al. Deferoxamine treatment prevents post-stroke vasoregression and neurovascular unit remodeling leading to improved functional outcomes in yype 2 male diabetic rats: role of endothelial ferroptosis. Transl Stroke Res. 2021;12(4):615–30. https://doi.org/10.1007/s12975-020-00844-7.
Pan Z, He Q, Zeng J, Li S, Li M, Chen B, et al. Naringenin protects against iron overload-induced osteoarthritis by suppressing oxidative stress. Phytomedicine. 2022;105:154330. https://doi.org/10.1016/j.phymed.2022.154330.
Mattè A, Kosinski PA, Federti E, Dang L, Recchiuti A, Russo R et al. Mitapivat, a pyruvate kinase activator, improves transfusion burden and reduces iron overload in β-thalassemic mice. Haematologica. 2023. https://doi.org/10.3324/haematol.2022.282614.
Sripetchwandee J, Khamseekaew J, Svasti S, Srichairatanakool S, Fucharoen S, Chattipakorn N et al. Corrigendum to "Deferiprone and efonidipine mitigated iron-overload induced neurotoxicity in wild-type and thalassemic mice" [Life Sci. 239 (2019) 116878]. Life Sci. 2023;315:121401. https://doi.org/10.1016/j.lfs.2023.121401.
Adramerina A, Printza N, Hatzipantelis E, Symeonidis S, Tarazi L, Teli A et al. Use of deferasirox film-coated tablets in pediatric patients with transfusion dependent thalassemia: a single center experience. Biology. 2022;11(2). https://doi.org/10.3390/biology11020247.
Sriwichaiin S, Thiennimitr P, Thonusin C, Sarichai P, Buddhasiri S, Kumfu S, et al. Deferiprone has less benefits on gut microbiota and metabolites in high iron-diet induced iron overload thalassemic mice than in iron overload wild-type mice: a preclinical study. Life Sci. 2022;307:120871. https://doi.org/10.1016/j.lfs.2022.120871.
Abbina S, Abbasi U, Gill A, Leitch H, Kizhakkedathu JN. Active transport nanochelators for the reduction of liver iron burden in iron overload. J Control Release. 2022;350:857–69. https://doi.org/10.1016/j.jconrel.2022.08.056.
Burton LH, Afzali MF, Radakovich LB, Campbell MA, Culver LA, Olver CS, et al. Systemic administration of a pharmacologic iron chelator reduces cartilage lesion development in the Dunkin-Hartley model of primary osteoarthritis. Free Radic Biol Med. 2022;179:47–58. https://doi.org/10.1016/j.freeradbiomed.2021.12.257.
Feng W, Xiao Y, Zhao C, Zhang Z, Liu W, Ma J, et al. New deferric amine compounds efficiently chelate excess iron to treat iron overload disorders and to prevent ferroptosis. Adv Sci. 2022;9(29):e2202679. https://doi.org/10.1002/advs.202202679.
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–6. https://doi.org/10.1021/ja411006a.
Mao XY, Zhou HH, Jin WL. Ferroptosis induction in pentylenetetrazole kindling and pilocarpine-induced epileptic seizures in mice. Front Neurosci. 2019;13:721. https://doi.org/10.3389/fnins.2019.00721.
Chen KN, Guan QW, Yin XX, Wang ZJ, Zhou HH, Mao XY. Ferrostatin-1 obviates seizures and associated cognitive deficits in ferric chloride-induced posttraumatic epilepsy via suppressing ferroptosis. Free Radic Biol Med. 2022;179:109–18. https://doi.org/10.1016/j.freeradbiomed.2021.12.268.
van Vuren A, Kerkhoffs JL, Schols S, Rijneveld A, Nur E, Peereboom D, et al. Proton pump inhibition for secondary hemochromatosis in hereditary anemia: a phase III placebo-controlled randomized cross-over clinical trial. Am J Hematol. 2022;97(7):924–32. https://doi.org/10.1002/ajh.26581.
Prasad S, DuBourdieu D, Srivastava A, Kumar P, Lall R. Metal-curcumin complexes in therapeutics: an approach to enhance pharmacological effects of curcumin. Int J Mol Sci. 2021;22(13). https://doi.org/10.3390/ijms22137094.
Sindone AP, De Pasquale C, Amerena J, Burdeniuk C, Chan A, Coats A, et al. Consensus statement on the current pharmacological prevention and management of heart failure. Med J Aust. 2022;217(4):212–7. https://doi.org/10.5694/mja2.51656.
Fuchs Andersen C, Omar M, Glenth?j A, El Fassi D, HJ Ml, Lindholm Kurtzhals JA et al. Effects of empagliflozin on erythropoiesis in heart failure: data from the Empire HF trial. Eur J Heart Fail. 2023;25(2):226–34. https://doi.org/10.1002/ejhf.2735.
Chifotides HT, Bose P, Verstovsek S. Momelotinib: an emerging treatment for myelofibrosis patients with anemia. J Hematol Oncol. 2022;15(1):7. https://doi.org/10.1186/s13045-021-01157-4.
Radadiya PS, Thornton MM, Puri RV, Yerrathota S, Dinh-Phan J, Magenheimer B et al. Ciclopirox olamine induces ferritinophagy and reduces cyst burden in polycystic kidney disease. JCI insight. 2021;6(8). https://doi.org/10.1172/jci.insight.141299.
He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2023;157:113915. https://doi.org/10.1016/j.biopha.2022.113915.
Du K, Zhou Q, Wang Z, Mo C, Dong W, Wei N et al. Polydatin ameliorates inflammation and oxidative stress associated with MSU-induced gouty arthritis in mice by regulating PPAR-γ and ferritin activation. Life Sci. 2023:121766. https://doi.org/10.1016/j.lfs.2023.121766.
Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–21. https://doi.org/10.1016/j.cell.2022.06.003.
Wu H, Li D, Zhang T, Zhao G. Novel mechanisms of perioperative neurocognitive disorders: ferroptosis and pyroptosis. Neurochem Res. 2023. https://doi.org/10.1007/s11064-023-03963-3.
Li P, Yu J, Huang F, Zhu YY, Chen DD, Zhang ZX, et al. SLC7A11-associated ferroptosis in acute injury diseases: mechanisms and strategies. Eur Rev Med Pharmacol Sci. 2023;27(10):4386–98. https://doi.org/10.26355/eurrev_202305_32444.
Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 in cell death, autophagy, and disease. Autophagy. 2023:1–18. https://doi.org/10.1080/15548627.2023.2218764.
Yang M, Luo H, Yi X, Wei X, Jiang DS. The epigenetic regulatory mechanisms of ferroptosis and its implications for biological processes and diseases. MedComm. 2023;4(3):e267. https://doi.org/10.1002/mco2.267.
Acknowledgements
We acknowledge https://smart.servier.com for the assistance of producing the schematic diagram figures. This work was supported by the NSFC grants 32071214 (Q. S.), the Natural Science Foundation of Sichuan, China 2022NSFSC0049 (Q. S.), 32201025 (D. T.), and the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University [ZYYC20016 (S. Q.), and ZYJC18015 (K. W.)], and the Fundamental Research Funds for the Central Universities 20826041F4141 (D. T.).
Institutional review board statement and informed consent statement
Not applicable.
Funding
This work was supported by the NSFC grants 32071214 (Q. S.) and 32201025 (D. T.), the Natural Science Foundation of Sichuan, China 2022NSFSC0049 (Q. S.), and the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University [ZYYC20016 (S. Q.), and ZYJC18015 (K. W.)], and the Fundamental Research Funds for the Central Universities 20826041F4141 (D. T.).
Author information
Authors and Affiliations
Contributions
SF, DT, YW, and XL drafted the manuscript. HB, CT, XD, XL, QY, YY, ZY, TS, KZ, XH, and ZW edited the manuscript. KW and SQ supervised the work and edited the manuscript. All authors contributed to the article. And all authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, S., Tang, D., Wang, Y. et al. The mechanism of ferroptosis and its related diseases. Mol Biomed 4, 33 (2023). https://doi.org/10.1186/s43556-023-00142-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-023-00142-2