
Abstract
Ferroptosis is a novel type of cell death with distinct properties and recognizing functions involved in physical conditions or various diseases including cancers. The fast-growing studies of ferroptosis in cancer have boosted a perspective for its usage in cancer therapeutics. Here, we review the current findings of ferroptosis regulation and especially focus on the function of ncRNAs in mediating the process of cell ferroptotic death and on how ferroptosis was in relation to other regulated cell deaths. Aberrant ferroptosis in diverse cancer types and tissues were summarized, and we elaborated recent data about the novel actors of some “conventional” drugs or natural compounds as ferroptosis inducers in cancer. Finally, we deliberate future orientation for ferroptosis in cancer cells and current unsettled issues, which may forward the speed of clinical use of ferroptosis induction in cancer treatment.
Similar content being viewed by others

Background
Ferroptosis was first proposed by Dixon as a novel cell death in 2012 [1]. Unlike autophagy and apoptosis, ferroptosis is an iron-dependent and reactive oxygen species (ROS)-reliant cell death with characteristics mainly of cytological changes, including decreased or vanished mitochondria cristae, a ruptured outer mitochondrial membrane, and a condensed mitochondrial membrane [2,3,4,5,6]. These cell abnormalities resulted from the loss of selective permeability of plasma membrane due to intense membrane lipid peroxidation and the occurrence of oxidative stress (Table 1) [7].
Researches indicated that ferroptosis could be triggered by diverse physiological conditions and pathological stresses in humans and animals [8]. Ferroptosis is gradually accepted as an adaptive feature to eliminate the malignant cells. It plays a pivotal role in the depression of tumorigenesis by removing the cells that are deficient in key nutrients in the environment or damaged by infection or ambient stress [9]. Studies have shown that the classic oxidative stress pathway was an important causative factor to induce ferroptosis. Although cancer cells are under continuous oxidative stress with an exquisite balance between thiols and catalytic iron, ferroptosis does not often happen in the cancer development [10]. The underlying molecular mechanisms remain poorly understood. We herein review the occurrence and regulation of ferroptosis in various cancer cells. The opportunity and challenge of cancer treatment based on ferroptosis will be detailed, which was desired to prosper new strategies for cancer therapy of clinical value.
Ferroptosis, from a cancer perspective: an overview
Ferroptosis was first observed in oncogenic Ras-expressing human foreskin fibroblast cell line by a battery of small compounds considered as ferroptosis-inducing agents (FIN), including erastin and Ras-selective lethal small molecule 3 (RSL 3). With the following studies, the relationship of Ras oncoprotein with ferroptosis becomes agnostic. Some Ras WT cells including fibrosarcoma cells, kidney tubule cells, and T cells are vulnerable to erastin, but the RMS13 rhabdomyosarcoma cells with Ras mutation were resistant to erastin and RLS3. Indeed, the ferroptosis inducer artesunate/erastin can promote ferroptosis in a Ras-reliant way in pancreatic cancer or transformed fibroblastic cells, while in a Ras-independent manner in leukemia cells [6, 11].
Emerging evidence implicated that ferroptosis may be an adaptive process which was critical for eradicating the carcinogenic cells [1]. More clues for this role of ferroptosis can be derived from recent researches of the tumor suppressor P53 (TP53). The acetylation-defective mutant TP533KR lost the ability to induce cell senescence, apoptosis, and cell-cycle arrest, which were the main functions of TP53 in tumor suppression. Impressively, TP533KR can still hold the capacity of inhibiting tumorigenesis due to its ferroptosis induction [12,13,14]. An argument does emerge that P53 expression may promote, limit, or detain the outset of ferroptosis in certain cells or conditions (Fig. 1). These opposite jobs of p53 in operating the process of ferroptotic cell death were executed by different mechanisms, including effects on metabolic genes transcription, post-translational regulation or by virtue of P53-P21 axis [15, 16]. The bidirectional regulation of ferroptosis by P53 in a cell-specific or context-dependent manner needs to be further investigated. Moreover, it is still obscure that what kind of role P53-target genes take part in manipulating of the ferroptotic cell death [17]?

P53 regulate ferroptosis. P533KR cannot elicit apoptosis activity, it retains the ability to promote ferroptosis. GLS2 and SAT1 contribute, at least in part, to P533KR-mediated ferroptosis. P533KR completely retains the ability to regulate the expression of SLC7A11. Interestingly, WTP53 inhibits ferroptosis via blocking the DPP4 activity. And WTP53 can delay ferroptosis by promoting the expression of P53 transcriptional target CDKN1A. However, P533KR is unable to induce P21. Other P53 mutants, P534KR, P53P47S, and TP53KO, cannot induce ferroptosis and weaken the blocking of cell growth
Ferroptosis is programmed necrosis mainly triggered by extra-mitochondrial lipid peroxidation arising from an iron-dependent ROS accretion. Excessive iron originally from aberrant iron metabolism or maladjustment of two major redox systems (lipid peroxidation and thiols) was the main incentive factors of ROS production. Glutathione (GSH), a thiol-containing tripeptide, synthesis is determined by the constant import of cysteine (Cys2) by the cell surface Cys2/glutamate antiporter xCT (Fig. 2).

Mechanism of ferroptotic cell death. System xc- transports intracellular Glu to the extracellular space and extracellular Cys2 into the cell, which is then transformed into Cys for GSH synthesis. GPX4 reduces the endogenous neutralization of PUFAs-OOH to PUFAs-OH, ultimately reducing ROS accumulation. Excess irons are the basis for ferroptosis execution. Circulated iron was combined with transferrin in the form of Fe3+, and then it entered into cells by TFR1. Iron in Fe3+ form was deoxidized to iron in Fe2+ by iron oxide reductase STEAP3. Ultimately, Fe2+ was released into a labile iron pool in the cytoplasm from the endosome mediated by DMT1
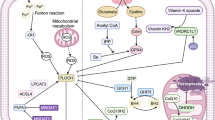
The activation of Ras-mitogen-activated protein kinase (MEK) signaling can attribute to the sensitivity of cancer cells to ferroptosis, resulting from its promoting iron abundance in cancer by governing the expression levels of the transferrin receptor and ferritin [2, 7, 8]. And the overactive Ras-MEK pathway may enhance ROS generation via inhibiting cystine (Cys2) uptake or mitochondrial voltage-dependent anion channel 2/3 (VDAC 2/3) and consequently sensitize cancer cells to ferroptosis [18, 19]. But scientist and skeptics argued that the conclusion is unreliable because the MEK inhibitor U0126 was used in these above studies. Compared with U0126, the special MEK1/2 inhibitor, PD0325901 cannot halt cell ferroptotic cell death induced by FIN. So MEK activity is not indispensable for ferroptosis [20, 21]. Other signal pathways were also pinpointed to regulate the process of cell ferroptotic death, e.g., Keleh-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2), lymphoid-specific helicase (LSH), Egl nine homolog 1 (EGLN1)/cellular myelocytomatosis oncogene (c-Myc), mevalonate (MVA), sulfur-transfer, mucin 1 C-terminal (MUC1-C)/system xc- (xCT) and heat shock factor-1 (HSF1)-heat shock protein beta-1 (HSPB1) pathway [5]. RNAi against Fms-like tyrosine kinase 3 (Flt3) saves cells from ferroptosis by limiting lipid peroxidation and inactivating p22phox which back the role for Flt3 kinase in ferroptosis. A new study indicates that adenosine 5′-monophosphate-activated protein kinase (AMPK)-mediated the phosphorylation of Beclin1 (BECN1) directly blocks the activity of system xc- and thus results in the occurrence of ferroptosis [22, 23]. Recent investigations have unraveled a perplexing network in the ferroptosis regulation which was shown in Fig. 3.

The regulatory network of ferroptosis. PHKG2, IREB2, and CISD1 play an important role in ferroptosis by their function in iron metabolism balance. The phosphorylation of HSP27 induces ferroptosis resistance through blocking cytoskeleton-mediated iron absorption. EGLN1 can upregulate LSH expression by limiting HIF 1α. LSH inhibits ferroptosis by affecting metabolism-associated genes including SCD1, GLUT1, and FADS2. Also, protein kinase C-mediated HSPB1 is a negative regulator of ferroptosis by inhibiting ROS production and reducing iron uptake. The p62-Keap1-Nrf2 pathway plays a vital role against ferroptosis by regulating Nrf2-targeted genes HO-1, FTH1, and NQO1. AMPK-mediated BECN1 phosphorylation and BAP1 directly represses system xc- activity, leading to the elevated ROS level and ferroptosis. MUC1-C binding with CD44v promotes the stability of system xc-. The inhibition of CDO1 restores the levels of GSH and increases ROS. Methionine can be converted to S-adenosylhomocysteine and Cys through the sulfur transfer pathway, which is essential for GPX4 biosynthesis. IPP and CoQ10 are the important products of the MVA pathway, which promotes GPX4 synthesis. FIN56 treatment also reduces CoQ10 by modulating SQS. VDAC2/3 and CARS are positive regulators of ferroptosis. ROS accumulation requires the activation of PUFAs by ACSL4 and LPCAT3. And LOX directly catalyzes the peroxidation of phospholipid PUFAs
Interestingly, microRNA and long non-coding RNA (lncRNA) are increasingly recognized as the crucial mediators in the regulation of ferroptosis (Table 2). The cytosolic lncRNA P53RRA can promote ferroptosis via nuclear sequestration of P53. P53RRA interplays with the RNA recognition motif (RRM) domain of Ras GTPase-activating protein-binding protein 1 (G3BP1), resulting in the combination of G3BP1 with P53, which cause more P53 custody in the nucleus and less sequestration of p53 in the cytoplasm [24]. P53RRA increased the intracellular concentrations of iron and lipid ROS by means of the augmented P53 pathway. Taken together, the cytosolic P53RRA-G3BP1 is a novel mechanism for inducing ferroptosis in lung adenocarcinoma.
In melanoma cell lines G-361 and A375, miR-9 can inhibit ferroptosis via targeting glutamic-oxaloacetic transaminase 1 (GOT1), an enzyme via glutaminolysis converting glutamine (Gln) ultimately to α-ketoglutarate (α-KG), which can promote ROS accumulation and thus irritate ferroptosis [25]. Knockdown miR-9 elevated the level of GOT1 and a-KG, which subsequently increased the sensitivity of cells to erastin- and RSL3-induced ferroptosis. Intriguingly, the other study showed that miR-137 impedes ferroptosis through directly suppressing solute carrier family 1 member 5 (SLC1A5), which is a major receptor for Gln uptake [26]. Knockdown of miR-137 can enhance the antitumor activity of erastin by enhancing ferroptosis.
As a specific light-chain submission of the Cys2/glutamate (Glu) antiporter, solute carrier family 7 member 11 (SLC7A11) plays a critical role in the negative regulation of ferroptosis. Researches showed that miR-375, miR-27a, miR-26b, and As-SLC7A11 (antisense lncRNAs) could suppress the transcription of SLC7A11 mRNA and impair the strength of its protein [27,28,29,30]. So, it is plausible that these miRNAs can promote ferroptosis by targeting SLC7A11.
The Nrf2 is the vital inhibitor of ferroptosis due to its ability to inhibit cellular iron uptake, limiting ROS production, and upregulating SLC7A11. First, miR-7 and miR-200a readily induce the activation of the Nrf2 pathway by repressing Keap1 expression [31, 32]. Contrastingly, miR-28 suppresses Nrf2 expression in a Keap1-independent way [33]. Second, both miR-101 and miR-455 can promote Nrf2 nuclear accumulation by targeting Cullin-3 (Cul3) [34, 35]. Finally, miR-153, miR-142-5p, miR-27a [36], miR-144 [37], miR-93 [38], miR-34a [39], miR-365-1, miR-193b, and miR-29-b1 [40] can decrease Nrf2 level through different mechanisms. These results indicated that miRNA might modify ferroptosis by means of regulating the expression of Nrf2.
It was confirmed that iron overload could contribute to ferroptosis in cancer. The iron metabolism-related genes, such as transferrin (TF), transferrin receptor 1 (TFR1), ferroportin (FPN), divalent metal transporter 1 (DMT1), ferritin heavy chain 1 (FTH1), and ferritin light chain (FTL), were the critical mediators in the ferroptosis procedure. Based on the existing research findings, miRNAs were also involved in the regulation of iron export, storage, utilization, and uptake. MiR-20a and miR-485-3p can reduce iron output by targeting FPN genes [41, 42]. MiR-210 and miR-152 inhibit the expression level of TFR, thereby reducing the uptake of TF [43, 44]. Concurrently, the expressions of miR-200b and miR-Let-7d effectively reduce iron accumulation by inhibiting the expression of FTH and DMT1-iron-responsive element (IRE), respectively [45, 46].
Beyond that, previous studies have confirmed that ROS generation requires the activation of polyunsaturated fatty acids (PUFAs) by Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). MiR-3595 [47], miR-205 [48], miR-224-5P [49], miR-19b-3p, miR-130a-3p, miR-150-5p, miR-144-3p, miR-16-5p, miR-7a-5p, and miR-17-5p [50] can decrease the expression of ACSL4. It is conceivable but not yet demonstrated that these miRNAs can regulate ferroptosis by targeting ACSL4.
Ultimately, ROS is the indispensable molecule in the process of ferroptosis. Previous studies have confirmed that miRNAs are closely related to redox signaling and ROS production, and we can further speculate that miRNAs can regulate ferroptosis by regulating the expression of ROS. MiR-206 significantly induces ROS accumulation by binding to the mRNA of superoxide dismutase 1 (SOD1) [51]. MiR-155 increases the generation of ROS by inducing Foxo3a deficiency [52]. Also, miR-25 and miR-448-3p have been proved to reduce ROS level by targeting the nicotinamide adenine dinucleotide phosphate-oxidase (NOX) [53, 54].
Ferroptosis, as an expanding network of programmed cell death in cancer
Apart from apoptosis and autophagy, other programmed cell death such as programmed necrosis was discovered. Ferroptosis, as well as necroptosis, parthanatos, and pyroptosis, are all belonging to the programmed necrosis, which was carried out by a specific program of genetically encoded cellular machinery demolishing the cell in an ordered fashion [9]. Over the latest 5 years, an astonishing boost in our perception of ferroptosis has been seen, and it was linked with apoptosis, autophagy, and other programmed necrosis which are tentatively investigated.
Ferroptosis and apoptosis: switch, synergism, or antagonism?
Recently, a growing research suggested the interconnection of ferroptosis and apoptosis. Besides preventing tumorigenesis by cell-cycle arrest and cell apoptosis induction, the canonical tumor suppressor protein P53 can also induce ferroptosis in certain conditions. Zheng et al. designed a novel type of P53 complex named metal organic network-P53 (MON-P53), which is tannic acid integrated with ferric ions forming MON on the external of the P53 plasmid [55]. When MON-P53 was internalized, ferric ions can induce Fenton reaction which will cause ROS generation. In vivo and vitro experiment, the dominant ferroptotic cell death, apart from cell apoptosis, was observed in the MON-P53-treated cells. The tumor growth was suppressed, and the life span of tumor-bearing mice was also prolonged. Therefore, this approach will direct a ferroptosis/apoptosis hybrid anti-cancer therapy [9].
Conversely, ferroptotic agents such as artesunate and erastin can induce the unfolded protein response (UPR) which sequentially promotes the expression of P53 upregulated modulator of apoptosis (PUMA) via C/EBP-homologous protein (CHOP) in the P53-independent way [56]. PUMA can enhance the function of apoptotic agent tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in promoting cell apoptosis (Fig. 4). This observation of ferroptotic agent-mediated sensitization to TRAIL-induced apoptosis implies ferroptosis inducer combined with TRAIL can strongly augment the tumoricidal efficacy. Another opinion is that metabolic or other alterations related to the irritation of ferroptosis biochemically block apoptosis occurrence. Cells that were subjected to ferroptosis due to cysteine (Cys) deprivation have about 10% of the normal level of intracellular glutathione (GSH). The power of reduced GSH may be necessary for the cascade activation of caspases-3 and-8. Therefore, cells’ lack of GSH cannot activate caspases correctly [10, 57].

Cross-talk between ferroptosis, apoptotic, and autophagy in cancer. Autophagy can modulate cell sensitivity to ferroptosis through various pathways. Erastin, artesunate, MON-P53, and Cys closely link ferroptosis with apoptosis. ELAVL1 promoted autophagy by binding to the AU-rich elements within the F3 of the 3′-untranslated regions of BECN1 mRNA. BECN1 can promote ferroptosis via directly blocking system xc-
Autophagy degrades ferritin to facilitate ferroptosis
Autophagy is a lysosome-dependent degradation pathway. The study provided data that the activation of autophagy pathway can degrade ferritin and then trigger ferroptosis in cancer cells [58]. Ferritinophagy, the autophagic turnover of ferritin, is critical to induce ferroptosis [59]. Further study indicated that BECN1 generation from the stimulation of upregulated embryonic lethal, abnormal vision, Drosophila-like 1 (ELAVL1) was responsible for the activation of autophagy in erastin- or sorafenib-irritated ferroptosis [60].
Autophagy can contribute to ferroptosis via the generation of lysosomal ROS and providing available labile iron via NCOA4-mediated ferritinophagy. And pharmacological blockage of autophagy weakens drug-induced ferroptosis in cancer cells. Genetic knockout of autophagy-related 5 (Atg5) and Atg7 limited erastin-induced ferroptosis by decreasing lipid peroxidation and intracellular ferrous iron levels. Significantly, blockage of nuclear receptor coactivator 4 (NCOA4), which was a choosy cargo receptor for ferritinophgay, suppresses ferritin degradation and inhibits ferroptosis. Contrarily, NCOA4 overexpression reinforces ferritin degradation and then drives ferroptosis. Autophagy supplies available labile iron via NCOA4-mediated ferritinophagy to the process of ferroptosis [61,62,63]. These results unraveled the molecular interactions between ferroptosis and apoptosis, and ferroptosis is an autophagic cell death process [64]. But in the absence of ferroptosis, prolonged iron mediates ROS accumulation and triggers autophagy and then results in autosis. Ferroptosis and autophagy can motivate cell death occurrence separately in breast cancer cells with siramesine and lapatinib treatment [58]. But contrastly, Buccarelli et al. reported that the blockage of autophagy using quinacrine can enhance glioblastoma stem cells (GSC) sensitivity to temozolomide by means of ferroptotic cell death [65].
Recent research shows that SOCS1 is required for p53 activation and the regulation of cellular senescence. SOCS1 can regulate the expression of p53 target genes such as reducing the expression of the cystine transporter SLC7A11 and the levels of glutathione, and it therefore sensitize cells to ferroptosis [66].
Although the mechanism among diverse types of regulated cell death has distinctive morphological and biochemical traits, some crosstalk still prevails between regulators and components of these various processes [67, 68]. The pathway of ferroptosis was linked with that of oxytosis by transactivating BH3-interacting domain death agonist (BID), the pro-apoptotic member of Bcl-2 family proteins, which converged to mitochondrial damage [69]. MiR137 was identified as a central mediator among apoptosis, autophagy, and ferroptosis [26]. HSPB1 is a small heat-shock protein that is crucial in controlling autophagy and ferroptosis [70]. Erastin can simultaneously induce the ferroptosis and necroptosis in HL-60 cell line [6]. Therefore, explicating how these pathways of regulated cell death are interplayed at the molecular level and how these pathways could be mapped and integrated will advance new ways to systematical research on this field. Discerning the critical factors such as ncRNAs should enable these processes to be therapeutically targeted and would be highly desired.
Aberrant ferroptosis in diverse cancer types and tissues
The susceptibility of different types of cancer cells to ferroptosis was significantly different. NCI-60, a panel of different cancer cell lines from eight various tissue types, recommended by the US National Cancer Institute Developmental Therapeutics Program. Among them, diffuse large B cell lymphomas and Renal cell carcinoma are more susceptible to erastin-induced ferroptosis than other cancer cells from the six tissues (the breast, lung, colon, melanocytes, central nervous system, and ovary) [6]. Some argue that the sensitivity of different cell lines to ferroptosis is different because of the difference of their basic metabolic state. Numerous studies have confirmed the pivotal role of ferroptosis in killing cancer cells and suppressing cancer growth. Further investigations showed that chemotherapeutic drugs such as cytarabine/ara-C, cisplatin, doxorubicin/Adriamycin, and temozolomide combining with the ferroptosis inducer erastin gained a remarkable synergistic effect on their anti-tumor activity [3]. The prognosis is better than traditional chemotherapy alone. Here, we summarize the possible mechanism of ferroptosis in various cancer types and putative indictor of ferroptosis for clinical application.
Hepatocellular carcinoma
Ferroptosis was one of the underlying mechanisms in sorafenib treating HCC. HCC cells with the retinoblastoma (RB) protein deficiency had 2–3 times higher death rate more than that of cells with a normal level of RB protein [71]. This susceptibility of HCC with deactivated RB protein to ferroptosis was due to the augment of oxidative stress response in cells from increased reactive oxygen concentration in mitochondria. Metallothionein-1g (MT-1G) is a novel negative regulator of ferroptosis in HCC. MT-1G knockdown contributed to sorafenib-induced ferroptosis by increasing lipid peroxidation and GSH depletion. CDGSH iron sulfur domain 1 (CISD1) and ACSL4 inhibition promote erastin-induced ferroptosis in HCC. Low-density lipoprotein (LDL)–docosahexaenoic acid (DHA) nanoparticles cause cell death in HCC cells through the ferroptosis pathway. The p62-Keap1-Nrf2 pathway plays a vital role in saving HCC cells from ferroptosis, and Ras/Raf/MEK pathway is reported to be a critically important target for ferroptosis in treating HCC [72].
Colorectal cancer
Xie et al. reported that colorectal cancer was resistant to ferroptosis resulting from the inhibition of dipeptidyl-peptidase-4 (DDP4) activity by TP53 in a transcription-independent way [12]. P53 loss promotes DDP4 gathering to plasma-membrane and thus augments DDP4-dependent lipid peroxidation, eventually causing ferroptotic cell death.
Gastric cancer
Hao et al. found that erastin irritates ferroptosis in GC cells [73]. Ferrostatin-1 and liproxstatin-1 can reverse this effect. C-Myc increases the expression of cysteine dioxygenase type 1 (CDO1), facilitating ferroptosis occurrence. Mechanistically, CDO1 suppression leads to the resistance of GC cells to erastin-induced ferroptosis by restoring cellular glutathione peroxidase 4 (GPX4) expression and GSH levels, and also by decreasing ROS generation. Another study reported that the CD44 variant (CD44v) stabilizes xCT at the plasma membrane and increases Cys2 uptake for GSH synthesis, blocking the ROS-induced stress signaling, and thus confer to ferroptosis resistance in GC cell [74, 75].
Ovarian cancer
Ovarian cancer cells are characteristics of ferroptosis susceptibility because of excess iron overload by its tumor-initiating cells (TICs), which have overexpressed TFR1 and decreased iron efflux pump FPN level [76]. Artesunate (ART) can induce ferroptosis in a ROS-dependent way in ovarian cancer. Ferrostatin-1 can significantly reverse ART-induced cell ferroptosis, but transferrin pretreatment augments the ferroptosis of ovarian cancer cells induced by ART via enhancing cellular iron level [77].
Prostate adenocarcinoma
Erastin induces ferroptosis in Ras-carrying human prostate adenocarcinoma cells. The phosphorylation of HSF1-dependent HSPB1 contributes to the ferroptosis resistance to erastin through inhibiting lipid ROS accumulation and iron uptake [70]. HSPB1 inhibition specifically increased erastin-induced ferroptosis by facilitating iron accumulation from the upregulation of TFR1 and slight reduction of FTH1 expression.
Breast cancer
Ma et al. reported that siramesine and lapatinib induce ferroptosis by increasing iron-dependent ROS productions, and CDO1 overexpression can exacerbate ferroptotic cell death by the further accumulation of high-level ROS result from the decreased GSH levels in breast cancer cells [58]. In contrast, MUC1-C can upregulate the GSH expression by its formation of a complex with CD44v, which cause the cripple of ferroptosis in breast cancer cells [74].
Lung cancer
Ferroptosis of lung cancer cell was first induced by erastin in the K-ras mutated A549 cells [2]. And the following report shows that erastin sensitizes lung cancer cells to cisplatin in ferroptosis manner by GSH reduction and GPXs inactivation [19]. Cysteine desulfurase (NFS1), as an iron-sulfur cluster biosynthetic enzyme, can protect cells from ferroptosis under the high-oxygen tension by sustaining the iron-sulfur cofactors. Coinhibition of NFS1 and Cys transport can evoke ferroptosis in vitro and suppress tumor growth [20].
Rhabdomyosarcoma
High level of GSH biosynthesis is essential for RMS cells to grow and become multidrug resistant. GPX4 inhibition using RSL3 and erastin can induce ferroptosis in RMS13 cells by lessening GSH level [21]. Another possible reason for high susceptibility of RMS13 cell lines to erastin and RSL3 was related to its higher activity of intrinsic Ras/extracellular signal-regulated kinase (ERK). But contrastingly, the RMS13 cells with oncogenic Ras mutation were resistant to the oxidative stress-induced ferroptosis.
Hematological malignancies
Diffuse large B-cell lymphoma (DLBCL) cells were notably one of the most sensitive to ferroptosis inducer in the eight cell lines harvested from various tissues [78]. It is proved that the enhanced sensitivity might be due to its weakness in the sulfur transfer pathways, which causes more extracellular Cys and Cys2 required for cells survival [79]. Yu et al. reported low-dose erastin could remarkably increase the ability of cytarabine and doxorubicin to kill non-APL acute myeloid leukemia (AML) cells by irritating both necroptosis and ferroptosis [80].
Ferroptosis-induction cancer therapy
Since erastin, a novel compound is found in human tumor cells in 2003 [81]. It was first identified as a ferroptosis inducer in 2012 [8], several clinical drugs have also been found to hold a capacity of inducing ferroptosis in cancer cells.
Chemotherapeutic agents
Sulfasalazine
SSZ is recently recognized as a system xc- inhibitor [41]. xCT expression has circadian rhythm and the expression of TFR1 was affected by the circadian organization of molecular clock. Bmal1 and the clock regulate the circadian rhythm of xCT expression. The clock-controlled gene c-Myc rhythmically activated the transcription of the TfR1 gene. SSZ has been reported to disrupt the circadian rhythm of transferrin receptor 1 gene expression and thus it was plausible that SSZ may affect iron metabolism [82,83,84]. Toyokuni et al. report that sulfasalazine inhibits Cys2 uptake via system xc-, resulting in ferroptosis in glioma cells [85]. Based on the circadian rhythm of xCT, SSZ has different effects on inducing ferroptosis at various times. But some argued that ferroptosis was not observed in the mouse embryonic fibroblasts treated with sulfasalazine [86]. One reasonable interpretation was the discrepancy of different cell lines on the sensitivity to ferroptosis.
Artesunate
ART and its derivatives can produce ROS and cause oxidative stress in cancer cells. In pancreatic ductal adenocarcinoma, head and neck cancers (HNCs), and ovarian cancer cells, the mechanism underlying the antitumor effect of ART was ferroptosis-induction [87]. However, because of the activation of Nrf2-antioxidant response element signaling pathway, the ferroptosis induction of artesunate can be partially attenuated in some cisplatin-resistant HNCs [88]. So Nrf2 inhibition via silencing Keap1 helps the reversal of ferroptosis resistance to artesunate in HNC cells.
Temozolomide
TMZ markedly induces system xc- expression via the activation of activating transcription factor 4 (ATF4) and Nrf2 pathway in glioblastoma multiforme (GBM) cells [89]. Cystathionine γ-lyase (CTH), an enzyme in the transsulfuration pathway, is induced after temozolomide treatment, which can supply Cys when system xc- is blocked. Based on the finding of erastin facilitating ferroptotic cell death to temozolomide, thus, the combination of TMZ and erastin maybe a promising therapy in GMB treatment [90].
Cisplatin
From the screening among five chemotherapeutic drugs, cisplatin was found as a ferroptosis-inducer. Cisplatin exerts its cytotoxic effects on A549 and HCT116 cells to undergo ferroptosis by reduced GSH depletion together with GPXs inactivation [19].
Targeted agents
Sorafenib
Sorafenib was first identified as a ferroptosis-inducer in HCC cell lines [91]. System xc- inhibition and GSH depletion, the accomplices for ROS accumulation, were the main mechanism for sorafenib-induced ferroptosis. Haloperidol, as a sigma receptor 1 antagonist, can bolster erastin and sorafenib-induced ferroptosis by stimulating cellular iron accumulation, GSH depletion, and lipid peroxidation [92]. But the overactive p62-keap1-Nrf2 pathway will weaken the ferroptosis process, owing to the target genes of Nrf2 including heme oxygenase-1 (HO-1), FTH1, and quinone oxidoreductase-1 (NQO1) which can directly inhibit ROS accretion. Nrf2 inhibition using genetic tools or drugs could remarkably reinforce the anti-tumor effect of sorafenib [93].
Lapatinib and BAY87–2243
Lapatinib is a tyrosine kinase inhibitor. It can incite ferroptosis in breast cancer cells when it was used together with siramesine [58]. BAY87-2243, a robust inhibitor of NADH-coenzyme Q oxidoreductase, can promote ferroptosis in a dose-dependent manner on a series of BRaf (V600E) melanoma cell lines [94].
Others
Lanperisone
Lanperisone promotes ROS production to kill K-Ras-mutant mouse embryonic fibroblasts in ferroptotic ways. And it also induces lung cancer cell ferroptotic death by inhibiting Cys2 uptake in the mouse model [95].
Besides the clinically approved drugs, two antibiotics such as salinomycin and ironomycin can promote ferroptosis in colon cancer cells via interfering with iron metabolism allowing for ROS production [65]. Other natural compounds such as bromelain [50], baicalein, artenimol, artemisinin, cotylenin A (CN-A) [11], and various vitamins can regulate cell ferroptotic death by acting on the lipid peroxidation and ROS occurrence (Table 3). Numerous nanomaterials have also been prospered for ferroptosis-based cancer therapy. Most of them are iron-based nanomaterials, which can be used as carriers of certain genes, such as P53 and ACSL4, to inhibit or promote the expression of certain critical molecules correlated with ferroptosis [55, 94, 96].
Challenges
How does ferroptosis interact with other cell death at the molecular level and how could these pathways be mapped and integrated into the cellular events?
How does ncRNA regulate the process of ferroptotic cell death?
Can ferroptosis enhance the cell immunogenicity to the host and thereby evoke an adaptive immune response, as shown in necroptotic cell death?
Are the ferroptosis inducers effective in killing the cancer cells in pre-clinical or clinical trials?
Conclusion & perspective
Collectively, ferroptosis has taken a full expectation from us to provide a new approach in anti-tumor therapies. Current researches have mainly focused on the eradication of residual or resistant cancer cells, where ferroptotic cell death emerges to be a new cell death for this purpose (Fig. 5). Conspicuously, obtaining a mesenchymal cell state (e.g., epithelial-mesenchymal transition (EMT) or cancer stem cells) has been suggested to determine metastatic dissemination and chemo-resistance [97]. More recently, the cancer cells with the high-mesenchymal state have arisen as a vital mechanism of both acquired and de novo resistance to targeted therapies [98, 99]. This therapy-resistant mesenchymal cancer cells have bred a state of non-oncogene addition to GPX4, which inhibition will intuitively result in ferroptosis. Consistently, persistent cancer cells which are nominated to escape from conventional cytotoxic treatment via a dormant state tumor showed an identically selective dependency on the GPX4 pathway [100, 101]. Therefore, ferroptosis might be considered a viable therapeutic strategy to reverse therapy-resistance in cancer strategy.

Perspectives of ferroptosis in cancer therapeutics. Ferroptosis can be as a novel cell death for killing mesenchymal-state cells or as an important synergist for immunotherapy and chemotherapy. P53 regulate ferroptosis sensitivity in a cell-type-special way. A large number of small molecules and drugs regulate ferroptosis in a Ras-dependent or -independent manner
Ferroptosis is a kind of programmed necrosis, which is accepted to be more immunogenic than apoptosis. Due to damage associated with molecular patterns (DAMPs) (e.g., HMGB1) release, ferroptosis was considered as a pro-inflammatory process [102]. By deliverying chemoattractant signals, ferroptosis hold the ability to recruit and activate immune cells at tumor sites, which provide the possibility of ferroptosis inducer as a suitable enhancer for anti-tumor immunotherapy treatment such as checkpoint-inhibitor [103]. Indeed, a large number of immune cells were observed inside the tumor mass when the mouse tumor xenografts underwent cell ferroptotic death induced by ultrasmall nanoparticles [104]. However, scientist skeptics argued that ferroptosis and necroinflammation did not have an unequivocal relationship [102].
Several studies of drug re-position suggest that “conventional” agents (i.e., SASP and artesunate) have antitumor therapeutic effects by activating ferroptosis [105]. As a chemo-sensitizer by ferroptosis-induction, erastin can be used with various drugs such as cisplatin, temozolomide, doxorubicin/adriamycin, and cytarabine/ara-C in different type cancers. Albeit there is limited knowledge of the elaborated mechanism in the ferroptosis pathway that is engaged by current ferroptosis inducers, ferroptosis may provide a new form of cell death for approaching the reversal of drug-resistance and boosting the host immune system. Although it was promising from the advantages of ferroptosis in cancer therapeutics, ferroptosis is still waiting for formal addressing in a pre-clinical setting and clinical achievability, partially due to the complexity of it observed in different contexts such as P53 or Ras-mutant cancer cells. Another challenge is that ferroptosis induction such as GPX4 inhibitor affects the development and function of nervous system and kidney, by causing GPX4 gene which is fundamental for embryonic development and some adult tissue homeostasis in mice [106]. In addition, another noticeable issue is that the occurrence of ferroptotic resistance, which was originally observed in the Hela cells with the erastin treatment. The resistance mechanism was the HSP27 overactivation by suppressing cytoskeleton-mediated iron absorption [107].
Abbreviations
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- AML:
-
Acute myeloid leukemia
- AMPK:
-
Adenosine 5′-monophosphate-activated protein kinase
- ART:
-
Artesunate
- As-SLC7A11:
-
Antisense lncRNAs
- ATF4:
-
Activating transcription factor 4
- Atg5:
-
Autophagy-related 5
- BAP1:
-
Breast cancer susceptibility gene 1-associated protein 1
- BECN1:
-
Beclin1
- BID:
-
BH3 interacting domain death agonist
- CARS :
-
Cysteinyl-tRNA synthetase
- CD44v:
-
CD44 variant
- CDO1:
-
Cysteine dioxygenase type 1
- CHOP:
-
C/EBP-homologous protein
- CISD1:
-
CDGSH iron sulfur domain 1
- c-Myb:
-
Cellular homolog
- C-Myc:
-
Cellular myelocytomatosis oncogene
- CN-A:
-
Cotylenin A
- CoQ10:
-
Coenzyme Q10
- CTH:
-
Cystathionine γ-lyase
- Cul3:
-
Cullin-3
- Cys:
-
Cysteine
- Cys2:
-
Cystine
- DAMPs:
-
Damage-associated molecular patterns
- DDP4:
-
Dipeptidyl-peptidase-4
- DHA:
-
Docosahexaenoic acid
- DLBCL:
-
Diffuse large B-cell lymphoma
- DMT1:
-
Divalent metal transporter 1
- EGLN1:
-
Egl nine homolog 1
- ELAVL1:
-
Embryonic lethal, abnormal vision, Drosophila-like 1
- EMT:
-
Epithelial-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- FADS2:
-
Fatty acid desaturase 2
- FIN:
-
Ferroptosis inducing agents
- Flt3:
-
Fms-like tyrosine kinase 3
- FPN:
-
Ferroportin
- FTH1:
-
Ferritin heavy chain 1
- FTL:
-
Ferritin light chain
- G3BP1:
-
Ras GTPase-activating protein-binding protein 1
- GBM:
-
Glioblastoma multiforme
- Gln:
-
Glutamine
- GLS2:
-
Glutaminase 2
- Glu:
-
Glutamate
- GLUT1:
-
Glucose transporter 1
- GOT1:
-
Glutamic-oxaloacetic transaminase 1
- GPX4:
-
Glutathione peroxidase 4
- GSC:
-
Glioblastoma stem cells
- GSH:
-
Glutathione
- HIF:
-
Hypoxia-inducible transcriptional factor
- HNCs:
-
Head and neck cancers
- HO-1:
-
Heme oxygenase-1
- HSF1:
-
Heat shock factor-1
- HSPB1:
-
Heat shock protein beta-1
- IPP:
-
Isopentenyl pyrophosphate
- IRE:
-
Iron-responsive element
- IREB2:
-
Iron response element binding protein 2
- Keap1 :
-
Keleh-like ECH-associated protein 1
- LDL:
-
Low-density lipoprotein
- LncRNA:
-
Long non-coding RNA
- LPCAT3:
-
Lysophosphatidylcholine acyltransferase 3
- LSH:
-
Lymphoid-specific helicase
- MEK:
-
Ras-mitogen-activated protein kinase kinase
- MON-P53:
-
Metal organic network-P53
- MT-1G:
-
Metallothionein-1 g
- MUC1-C:
-
Mucin 1 C-terminal
- MVA:
-
Mevalonate
- NCOA4:
-
Nuclear receptor coactivator 4
- NFS1:
-
Cysteine desulfurase
- NOX:
-
Nicotinamide adenine dinucleotide phosphate-oxidase
- NQO1:
-
Quinone oxidoreductase-1
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- PHKG2 :
-
Phosphorylase kinase gamma 2
- PIK3C3:
-
Phosphatidylinositol 3-kinase catalytic subunit type 3
- PL-PUFAs:
-
Phospholipid-polyunsaturated fatty acids
- PtdIns3K :
-
Phosphatidylinositol 3-kinase
- PUFAs:
-
Polyunsaturated fatty acids
- PUMA:
-
P53 upregulated modulator of apoptosis
- RB:
-
Retinoblastoma
- ROS:
-
Reactive oxygen species
- RRM:
-
RNA recognition motif
- RSL3:
-
Ras-selective lethal small molecule 3
- SAT1:
-
Spermidine/spermine N1-acetyltransferase 1
- SCD1:
-
Stearoyl-CoA desaturase 1
- Se:
-
Selenium
- SLC1A5:
-
Solute carrier family 1 member 5
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- SOD1:
-
Superoxide dismutase 1
- SQS:
-
Squalene synthase
- STEAP3:
-
Six-transmembrane epithelial antigen of the prostate 3
- TF:
-
Transferrin
- TFR1:
-
Transferrin receptor 1
- TICs:
-
Tumor-initiating cells
- TP53:
-
Tumor suppressor P53
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- UPR:
-
Unfolded protein response
- VDAC2/3:
-
Voltage-dependent anion channel 2/3
- xCT:
-
System xc-
- α-KG:
-
α-ketoglutarate
References
Dixon SJ. Ferroptosis: bug or feature? Immunol Rev. 2017;277:150–7.
Yagoda N, Von RM, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648–57.
Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta. 2017;1861(8):1893.
Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–209.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Fearnhead HO, Vandenabeele P, Vanden TB. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017;24:1991–8.
Ueda S, Nakamura H, Masutani H, Sasada T, Yonehara S, Takabayashi A, et al. Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J Immunol. 1998;161:6689–95.
Ye J, Zhang R, Wu F, Zhai L, Wang K, Xiao M, et al. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018;420:210–27.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93–103.
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–75.
Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2017;25:104–13.
Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front Endocrinol. 2018;9:124.
Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–60.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–43.
Fanzani A, Poli M. Iron, oxidative damage and ferroptosis in rhabdomyosarcoma. Int J Mol Sci. 2017;18:1718.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system xc(−) activity. Curr Biol. 2018;28:2388–99.e5.
Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14:2173–5.
Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–96.
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinog. 2018;57:1566–76.
Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457–72.
Wu Y, Sun X, Song B, Qiu X, Zhao J. MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017;6:1686–97.
Drayton RM, Dudziec E, Peter S, Bertz S, Hartmann A, Bryant HE, et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res. 2014;20:1990–2000.
Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett. 2011;585:1363–7.
Yuan J, Liu Z, Song R. Antisense lncRNA As-SLC7A11 suppresses epithelial ovarian cancer progression mainly by targeting SLC7A11. Pharmazie. 2017;72:402–7.
Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015;89:548–56.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725–33.
Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat. 2011;129:983–91.
Kim JH, Lee KS, Lee DK, Kim J, Kwak SN, Ha KS, et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid Redox Signal. 2014;21:2469–82.
Xu D, Zhu H, Wang C, Zhu X, Liu G, Chen C, et al. microRNA-455 targets cullin 3 to activate Nrf2 signaling and protect human osteoblasts from hydrogen peroxide. Oncotarget. 2017;8:59225–34.
Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS One. 2012;7:e51111.
Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–48.
Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis. 2013;34:1165–72.
Zhang Y, Tao X, Yin L, Xu L, Xu Y, Qi Y, et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br J Pharmacol. 2017;174:2512–27.
Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40:7416–29.
Babu KR, Muckenthaler MU. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J Mol Med (Berl). 2016;94:347–59.
Sangokoya C, Doss JF, Chi JT. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013;9:e1003408.
Yoshioka Y, Kosaka N, Ochiya T, Kato T. Micromanaging iron homeostasis: hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J Biol Chem. 2012;287:34110–9.
Kindrat I, Tryndyak V, de Conti A, Shpyleva S, Mudalige TK, Kobets T, et al. MicroRNA-152-mediated dysregulation of hepatic transferrin receptor 1 in liver carcinogenesis. Oncotarget. 2016;7:1276–87.
Shpyleva SI, Tryndyak VP, Kovalchuk O, Starlarddavenport A, Chekhun VF, Beland FA, et al. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res Treat. 2011;126:63–71.
Andolfo I, De Falco L, Asci R, Russo R, Colucci S, Gorrese M, et al. Regulation of divalent metal transporter 1 (DMT1) non-IRE isoform by the microRNA let-7d in erythroid cells. Haematologica. 2010;95:1244–52.
Wu X, Zhi F, Lun W, Deng Q, Zhang W. Baicalin inhibits PDGF-BB-induced hepatic stellate cell proliferation, apoptosis, invasion, migration and activation via the miR-3595/ACSL4 axis. Int J Mol Med. 2018;41:1992–2002.
Cui M, Xiao ZL, Sun BD, Wang Y, Zheng MY, Ye LH, et al. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochem Biophys Res Commun. 2014;445:651–5.
Peng Y, Xiang H, Chen C, Zheng R, Chai J, Peng J, et al. MiR-224 impairs adipocyte early differentiation and regulates fatty acid metabolism. Int J Biochem Cell Biol. 2013;45:1585–93.
Park S, Oh J, Kim M, Jin E-J. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst. 2018;22:334–40.
Zhang Y, Zheng S, Geng Y, Xue J, Wang Z, Xie X, et al. MicroRNA profiling of atrial fibrillation in canines: miR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD1. PLoS One. 2015;10:e0122674.
Peng W, Zhu CF, Ma MZ, Gang C, Ming S, Zeng ZL, et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget. 2015;6:21148–58.
Varga ZV, Kupai K, Szucs G, Gaspar R, Paloczi J, Farago N, et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J Mol Cell Cardiol. 2013;62:111–21.
Kyrychenko S, Kyrychenko V, Badr MA, Ikeda Y, Sadoshima J, Shirokova N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc Res. 2015;108:324–34.
Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C, et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17:284–91.
Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: cross-talk between ferroptosis and apoptosis. Mol Cancer Res. 2018;16:1073–6.
Hentze H, Schmitz I, Latta M, Krueger A, Krammer PH, Wendel A. Glutathione dependence of caspase-8 activation at the death-inducing signaling complex. J Biol Chem. 2002;277:5588–95.
Ma S, Dielschneider RF, Henson ES, Xiao W, Choquette TR, Blankstein AR, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12:e0182921.
Tang M, Chen Z, Wu D, Chen L. Ferritinophagy/ferroptosis: iron-related newcomers in human diseases. J Cell Physiol. 2018;233:9179–90.
Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–103.
Abdelmonsif DA, Sultan AS, El-Hadidy WF, Abdallah DM. Targeting AMPK, mTOR and β-catenin by combined metformin and aspirin therapy in HCC: an appraisal in Egyptian HCC patients. Mol Diagn Ther. 2017;22(5):1–13.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10(1):67.
Lu BCX, Ying MD, He QJ, Cao J, Yang B. The role of ferroptosis in cancer development and treatment response. Front Pharmacol. 2018;8:992.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021.
Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D'Alessandris QG, Martini M, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018;9:841.
Saintgermain E, Lian M, Vernier M. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging. 2017;9(10):2137–62.
Cotto-Rios XM, Gavathiotis E. Chemical genetics: unraveling cell death mysteries. Nat Chem Biol. 2016;12:470–1.
Dong T, Liao D, Liu X, Lei X. Using small molecules to dissect non-apoptotic programmed cell death: necroptosis, Ferroptosis, and Pyroptosis. Chembiochem. 2015;16:2557–61.
Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–70.
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–25.
Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971–7.
Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144:2329–37.
Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. 2017;19:1022–32.
Masanori H, Hidekazu T, Hasan R, Maroof A, Yozo S, Li Y, et al. Functional interactions of the cystine/glutamate antiporter, CD44v and MUC1-C oncoprotein in triple-negative breast cancer cells. Oncotarget. 2016;7:11756–69.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400.
Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36:4089–99.
Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2016;56:75–93.
Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Kinowaki Y, Kurata M, Ishibashi S, Ikeda M, Tatsuzawa A, Yamamoto M, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab Investig. 2018;98:609–19.
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–96.
Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34(1):1–10.
Okazaki F, Matsunaga N, Hamamura K, Suzuki K, Nakao T, Okazaki H, et al. Administering xCT inhibitors based on circadian clock improves antitumor effects. Cancer Res. 2017;77(23):0720.2017.
Yoshida GJ. Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res. 2018;37(1):173.
Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis. Free Radic Biol Med. 2017;108:610–26.
Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52(3):1011–22.
Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381:96–103.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep. 2015;33:1465–74.
Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyupoglu IY, Savaskan NE. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget. 2016;7:74630–47.
Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34:6417–22.
Bai T, Wang S, Zhao Y, Zhu R, Wang W, Sun Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2017;491:919–25.
Sauzay C, Louandre C, Bodeau S, Anglade F, Godin C, Saidak Z, et al. Protein biosynthesis, a target of sorafenib, interferes with the unfolded protein response (UPR) and ferroptosis in hepatocellular carcinoma cells. Oncotarget. 2018;9:8400–14.
Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X. Emerging strategies of cancer therapy based on ferroptosis. Adv Mater. 2018;30:e1704007.
Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A. 2011;108:8773–8.
Bazak R, Houri M, Achy SE, Hussein W, Refaat T. Passive targeting of nanoparticles to cancer: a comprehensive review of the literature. Mol Clin Oncol. 2014;2:904–8.
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512.
Salt MB, Bandyopadhyay S, Mccormick F. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discovery. 2014;4:186–99.
Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Ahern TP, Lash TL, Damkier P, Christiansen PM, Cronin-Fenton DP. Statins and breast cancer prognosis: evidence and opportunities. Lancet Oncol. 2014;15:e461–8.
Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019;26:14–24.
Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280:126–48.
Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11:977–85.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10:67.
Liu H, Schreiber SL, Stockwell BR. Targeting dependency on the GPX4 lipid peroxide repair pathway for cancer therapy. Biochemistry. 2018;57:2059–60.
Razaghi A, Heimann K, Schaeffer PM, Gibson SB. Negative regulators of cell death pathways in cancer: perspective on biomarkers and targeted therapies. Apoptosis. 2018;23:93–112.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523.
Eling N, Reuter L, Hazin J, Hamacherbrady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–32.
Zhou L, Zhao B, Zhang L, Wang S, Dong D, Lv H, et al. Alterations in cellular iron metabolism provide more therapeutic opportunities for cancer. Int J Mol Sci. 2018;19:1545.
Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, et al. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci Rep. 2018;8:968.
Hinman A, Holst CR, Latham JC, Bruegger JJ, Ulas G, Mccusker KP, et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One. 2018;13:e0201369.
Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7:3293–305.
Acknowledgements
Not applicable
Funding
This work was supported by grants from the National Natural Science Foundation of China (81200366,81572281,81702278,81171841) and Province Natural Science Foundation of Hunan (No.14JJ6004) and the Key Subject Education Department of Hunan ([2012]594).
Availability of data and materials
The materials supporting the conclusion of this review have been included within the article.
Author information
Authors and Affiliations
Contributions
YHM and BL drafted the manuscript and prepared the figures and tables. JW, JCW, DH, CFZ, and CJD collected the references and participated in the discussion. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mou, Y., Wang, J., Wu, J. et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 12, 34 (2019). https://doi.org/10.1186/s13045-019-0720-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-019-0720-y