
Abstract
Background
Acyclovir is an anti-viral medication given to treat herpes simplex and herpes zoster infection. In some severe conditions such as herpes encephalitis, acyclovir is administered intravenously. However, high acyclovir doses may cause acute kidney injury and low acyclovir dose may predispose the patient to inadequate exposure to acyclovir which could be fatal in some conditions. In such cases, the acyclovir plasma concentrations will potentially guide the diagnosis and management of the kidney injury. In this study, we provide a simple and time-efficient method for analyzing acyclovir in human plasma using high-performance liquid chromatography (HPLC).
Results
The process starts with a single protein precipitation step by adding acetonitrile to deproteinize 300 µL of plasma. The chromatographic separation conditions consist of a mobile phase of water: methanol (97:3, v/v), a flow rate of 1 mL/min, a run time of 17 min, and a detection wavelength of 254 nm. The calibration curve was linear over the range of (0.70–60 mg/L) (r2 ˃ 0.99). The retention times of acyclovir and the internal standard were around 15 and 12 min, respectively. The intra-day and inter-day analysis of acyclovir in plasma using this method exhibited accuracy and precision of less than 7%, which lies within the acceptable range. Different greenness assessment tools confirmed that the proposed method is eco-friendly.
Conclusion
The proposed method of analysis of acyclovir in the plasma using HPLC is simple, green and accurate method. This method could be applied in clinical settings where monitoring acyclovir concentrations is essential as it has wide range of the concentrations that could be detected.
Similar content being viewed by others

Background
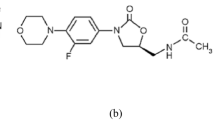
Acyclovir (9-[2-hydroxyethoxymethyl]-9H-guanine) (Fig. 1) is a synthetic nucleoside analog that has an anti-viral activity against herpes simplex and varicella zoster virus [1]. Patients with severe infections such as herpes encephalitis may require careful monitoring of the plasma acyclovir level. High acyclovir concentrations could predispose the patient to acyclovir nephrotoxicity and neurotoxicity. On the other hand, low acyclovir concentration that is below the 50% inhibitory concentration (0.56 mg/L for herpes simplex virus and 1.125 mg/L for varicella zoster virus) could lead to treatment failure and worsen the patient’s condition [2,3,4].

Acyclovir chemical structure
Acyclovir follows a two-compartment model with first-order elimination kinetics [5]. The primary elimination pathway for acyclovir is through the renal route, making it significantly influenced by kidney function [6]. Impaired kidney function could result in acyclovir accumulation in the body and the development of either nephrotoxicity, neurotoxicity, or both, while enhanced kidney function could lead to subtherapeutic concentrations of acyclovir and subsequently treatment failure [7, 8].
The most common reported methods for the detection of acyclovir in the plasma were high-performance liquid chromatography (HPLC) and liquid chromatography–mass spectrometry (LC–MS) [9,10,11,12]. Most of these methods used a mobile phase with very low acidic pH [13, 14], which by time could degrade the stationary phase of the column.
Green analytical chemistry (GAC) is an area of activity that ensures that the analytical practices are environmentally friendly [15]. The assessment of greenness is crucial to ensure that analytical methods produce the least threats to the environment [16]. There are different developed tools to assess the greenness of the analytical methods such as analytical method GREEnness score (AMGS) [17], analytical eco-scale [18], and analytical GREEnness (AGREE) [19].
This method aimed to develop a simple, sensitive, and time-efficient method for analyzing acyclovir in plasma. The advantages of our method over the previously developed method are that it utilizes a single protein denaturation step, does not include buffers for the preparation of the mobile phase and it provides higher range of concentrations that could detect acyclovir toxicity. The development of buffer-free HPLC method offers a greener, more efficient, and more versatile approach to chemical analysis which protects the column for a longer time and avoids time-consuming cleaning process and waste generated. Furthermore, the absence of buffers allows for a simplified and more cost-effective analytical process. To our knowledge, this is the first acyclovir analysis method using the HPLC that used the greenness assessment tools to ensure that the method is environmentally friendly.
Methods
Reagents and chemicals
Acyclovir with a purity of (95.8% ± 0.9) and the internal standard (IS) ganciclovir with a purity of (99.3% ± 0.2) were obtained from Sigma-Aldrich (Oakville, ON, Canada). HPLC-grade acetonitrile, water, and methanol were purchased from Fisher Scientific (Edmonton, AB, Canada). Drug-free human plasma was acquired from Cedarlane Laboratories (Burlington, ON, Canada).
Instruments
HPLC–UV system (Shimadzu, Kyoto, Japan) was used to perform the analysis. It consisted of a system controller (SCL-10Avp), an autosampler (SIL-HTC), two pumps (LC-10 AD), and a UV–vis detector (SPD-10AV). The chromatographic separation was achieved by using a C18 reverse phase Supleco Discovery® C18 column (5 μm, 250 × 4.6 mm) (Supleco Inc., Mississauga, ON, Canada) protected by a Discovery® C18 Supelguard™ guard column (5 μm, 20 × 4 mm) (Supleco Inc., Mississauga, ON, Canada). Clarity® software version 8.7 (DataApex, Prague, The Czech Republic) was utilized for data collection and analysis.
Chromatographic conditions
The chromatographic separation was conducted through isocratic elution of a mobile phase mixture consisting of water and methanol (97:3, v/v). The flow rate was maintained at 1 mL/min for a total run time of 17 min for elution, and the detection wavelength was set at 254 nm. All the steps were conducted at room temperature.
System suitability
The system suitability was tested through the calculation of the tailing factor (symmetry factor), resolution, capacity factor and the height of theoretical plate (HETP) of five replicates. The results were compared with the guidelines to ensure that the chromatographic condition in optimal conditions, and the method is suitable for its intended purpose.
Preparation of stock and working solutions
Acyclovir and ganciclovir were dissolved in HPLC-grade water to prepare stock solutions of 400 mg/L and 500 mg/L, respectively. Working solutions of 50 mg/L and 100 mg/L of acyclovir as well as 100 mg/L ganciclovir were prepared by further diluting the stock solutions. All solutions were prepared fresh daily.
Preparation of calibration concentrations and quality control samples
Serial dilutions of acyclovir in blank human plasma were prepared to obtain the calibration concentrations of (0.7, 2, 5, 15, 25, 60 mg/L) of acyclovir. Four quality control (QC) concentrations were prepared for the method validation. The quality control samples were the lower limit of quantification (LLOQ, 0.7 mg/L), low-level QC (2 mg/L), middle-level QC (25 mg/L), and high-level QC (45 mg/L).
Sample preparation
Sixty µL of 100 mg/L IS were added to 300 µL of blank plasma spiked with acyclovir and vortex mixed for 30 s. Then, 2 mL of acetonitrile were added to the prepared plasma spiked with acyclovir and ganciclovir and vortex mixed for 1 min for the purpose of the plasma proteins denaturation. Then, the prepared samples were centrifuged (Eppendorf centrifuge 5804, Eppendorf SE, Barkhausenweg, Hamburg, Germany) at 5000 rpm for 20 min. The obtained supernatant was then transferred to clean tubes and concentrated using SpeedVac® Vacuum Concentrator (Thermo Fisher Scientific, Waltham, MA, USA). Reconstitution was performed by the addition of 200 µL of the mobile phase (water: methanol, 97:3, v/v) and vortex mixed for 15 s. A volume of 50 µL of the prepared samples was injected into the HPLC for the chromatographic separation.
Method validation
The validation was done following the Guideline on bioanalytical method validation guidelines developed by the European Medicines Agency (EMA, 2011) [20]. The method validation included linearity, selectivity and sensitivity, precision and accuracy, carry-over, stability, and recovery.
Linearity
The linearity of the method was determined by plotting the calibration curves of the peak height ratios (acyclovir /ganciclovir) vs. the calibration concentrations. Linear regression was performed to obtain the slope, intercept, and coefficient of determination (r2) of the calibration curve.
Selectivity and sensitivity
The selectivity of the developed method was assured by the absence of any plasma peaks interfering with acyclovir and ganciclovir peaks when comparing chromatograms obtained from blank plasma with those obtained from acyclovir-containing samples. The sensitivity was determined in terms of the LLOQ, in which its response must be at least 5 times higher than the plasma response.
Precision and accuracy
The intra-day and inter-day precision and accuracy of the developed method were tested by injecting five replicates of each of the four QC samples mentioned earlier on three consecutive days. The method’s precision was presented as the coefficient of variation (CV, %), and the accuracy was expressed as a percentage error.
Carry-over
Carry-over was assessed by injecting drug-free plasma after the injection of the upper limit of quantification (60 mg/L). Based on the EMA guidelines, the blank plasma response must not exceed ± 20% of acyclovir LLOQ response and 5% of the internal standard response.
Stability
The stability of the method was assessed in either plasma spiked with acyclovir or final prepared samples for HPLC injection (e.g., concentrated and reconstituted) over two weeks of three replicates of two QC samples (2 and 55 mg/L). The stability of the plasma spiked with acyclovir was determined at the preparation time and after three hours and 24 h at room temperature. Furthermore, the stability after 1 and 2 weeks stored at 4–8, − 20, and – 80 °C was also assessed. Moreover, the stability of the final prepared samples was examined over one week and stored at room temperature (autosampler), 4–8, − 20, and − 80 °C. Also, the freeze and thaw stability of acyclovir in plasma was assessed by initially freezing the three replicates of the two QC samples at − 80 °C for 24 h followed by thawing them at room temperature. This cycle was repeated for three days before preparing the samples to be injected into the HPLC. In addition, the stability of stock and working solutions kept in the fridge were tested after 2 months by preparing a working solution from the stock solution stored at 2-8֯ C and comparing the results of samples prepared from these working solutions.
Recovery
The average extraction recovery of acyclovir was measured by injecting three replicates of three QC samples (5, 15, 25 mg/L) and comparing their chromatographic peaks with those of plasma samples spiked with equivalent concentrations of acyclovir after protein precipitation and sample concentration.
Assessment of greenness
The eco-friendliness of the proposed method was tested using different greenness assessment tools. Three were used to evaluate the greenness which are analytical method GREEnness score (AMGS) [17], analytical eco-scale [18], and analytical GREEnness (AGREE) [19].
Results
Method development
Preliminary experiments were done to optimize the chromatographic conditions of acyclovir analysis. Various solvents and different compositions were tested to obtain the optimum mobile phase composition to run the analysis. Different compositions of acetonitrile and water as well as different compositions of methanol and water were tested as mobile phases, and it was found that the composition of 97% water and 3% methanol gave the best chromatograms of acyclovir samples. The selected mobile phase composition resulted in increasing the retention time compared to more methanol percentage in the mobile phase. Nevertheless, it had the advantages of better peak separation, decreasing the cost of the analysis and reducing the environmental impact of methanol. Moreover, different wavelengths over the range of 200–800 nm were tested to select the wavelength that provides maximum acyclovir UV absorbance and less plasma absorbance which was found to be 254 nm. The effect of different flow rates was studied, and a flow rate of 1 mL/min was chosen for the method. Using flow rates of more than 1 mL/min resulted in increasing the pressure, while using flow rate of less than 1 mL/min resulted in increasing the retention time and hence the run time. Furthermore, acetonitrile and methanol were tested as protein precipitation solvents, and acetonitrile gave better results. The retention times of acyclovir and ganciclovir using the optimum conditions were around 15 and 12 min, respectively. The ratios of the peak heights of acyclovir to ganciclovir were used in all the calculations as they gave more accurate results than the peak area ratios. Although column temperature affects the resolution of the samples, it was not used to make the study applicable to different systems, and the measurements were all in the room temperature.
System suitability
The results of the system suitability (Table 1) were obtained and compared to the reference ranges. All of the obtained values are within the acceptable ranges.
Method validation
Linearity
The calibration curves of the plasma samples of acyclovir were done to test the linearity of the developed method. The peak height ratios of acyclovir to ganciclovir showed linearity over the range of 0.7–60 mg/L with r2 > 0.99 (Fig. 2).

Calibration curve of peak height ratios of acyclovir to the internal standard versus acyclovir concentrations
Selectivity and sensitivity
There were no interfering plasma peaks with the acyclovir or the ganciclovir as shown in Fig. 3. The lower limit of quantification of acyclovir that gives accurate and precise results was 0.70 mg/L.

Different chromatograms of a Blank plasma; b Plasma containing internal standard (20 mg/L)-retention time 12.1 min; c Plasma containing acyclovir (25 mg/L) and internal standard (20 mg/L) retention times 15 and 12.2 min, respectively
Precision and accuracy
To assess the intra-day and inter-day precision and accuracy, we prepared five replicates of four quality control samples and analyzed their concentrations in three different days. The method is precise and accurate as shown in Table 2. The intra-day coefficient of variation was less than 2.2%, and the percentage of error was less than 7%. On the other hand, the inter-day coefficient of variation was less than 4.6%, and the percentage of error was less than 6%.
Carry-over
There were no peaks appearing in the blank plasma chromatogram after injecting the upper limit of quantification either in the retention time of acyclovir or ganciclovir. This indicates that there were no carry-over effects of high acyclovir concentrations.
Stability
Plasma samples spiked with acyclovir were stable for 2 weeks at different temperatures. Also, the finally prepared samples for injection showed stability for one week stored at different temperatures. Acyclovir showed stability after three freeze and thaw cycles. All the % remaining of the acyclovir in samples compared with the initial samples (zero time) ranges from 88 to 113% (Table 3) which lies within ± 15% indicating the stability of acyclovir samples. Moreover, the stock and working solutions were stable for 2 months.
Recovery
The mean percentages recovery of the three QC of 5, 15, 25 mg/L (n = 3) concentrations is in Table 4. It ranged from 88 to 90%.
Assessment of greenness
The web application version of the AGREE tool provided the result based on the 12 criteria of green analytical chemistry. The score was 0.64 which indicates that the method is green. The AMGS score, which calculates the instrument energy score, solvent energy score and solvent EHS score were 216.34 which also indicates the greenness of the method. The analytical eco-scale resulted in a score of (100-16) = 84 of which means that the method is green.
Discussion
Acyclovir is the anti-viral nucleoside analog drug used for the treatment of herpes infections. Acyclovir is given intravenously to patients suffering from herpes encephalitis at a dose of 10 mg/kg/dose every 8 h [23]. High concentrations of acyclovir in plasma above 25 mg/L could highly predispose the patient to nephrotoxicity and neurotoxicity [24]. This paper describes a simple, rapid, accurate, and precise method for the quantifying of acyclovir in human plasma.
This method utilizes a small volume of the plasma 0.3 mL compared to other methods which use larger volumes (0.5–1 mL) [9, 13, 14, 25]. The run time was 17 min which could aid in the analysis of large numbers of samples in a short time. The wavelength that showed the best chromatograms was 254 nm after scanning the UV range (200–800 nm). Although the mobile phase composition was 97% water and 3% methanol which resulted in increasing the retention time compared to more methanol percentage in the mobile phase, it has the advantages of better peak separation, decreasing the cost of the analysis and reducing the environmental impact of methanol. The mobile phase used to elute acyclovir did not contain buffers, which provides the advantages of the simplicity of the method and protection of the column without affecting the results’ selectivity and sensitivity.
The greenness of the developed method was assessed using different tools. The AGREE tool was first developed in 2020 by Pena-Pereira, et al. [19]. It involves the 12 concepts of green analytical chemistry in its evaluation [15]. The analytical eco-scale is also an interesting tool to assess if the method is environmentally safe, and it has a score of hundred and good results expected to be > 75 [18]. Analytical method GREEnness score (AMGS) is a semi-quantitative user-friendly tool to evaluate the greenness of the analytical methods [17]. The proposed study is determined as green and environmentally safe based on the results obtained from this tool. This was the first acyclovir HPLC analysis study that evaluated the greenness of the proposed method.
It has the advantage of a single protein precipitation step which also reinforces the uncomplicatedness of the developed method compared to other methods [14]. Moreover, the stability of the developed method was assessed, and acyclovir showed stability in stock and working solutions kept for two months in the fridge. In addition, it showed stability in the plasma over two weeks and finally prepared samples over one week in different storage conditions. The validation of the developed method showed precision and accuracy results within the acceptable range (± 15%) as reported by the EMA guidelines [20].
The linearity range of the method (0.70–60 mg/L) covers a wider range than previous study, including high acyclovir concentrations, which are known to be associated with potential adverse effects [14, 26, 27]. Acyclovir levels in herpes encephalitis should be maintained above the 50% inhibitory concentration which is 0.56 mg/L for herpes simplex virus and 1.125 mg/L for varicella zoster virus. On the other hand, high acyclovir concentrations above 25 mg/L could cause acyclovir adverse effects [2, 4, 28]. These concentrations could be readily measured in patients experiencing adverse effects, such as acute kidney failure, neurotoxicity, or worsening symptoms, using this method.
Conclusion
This article represents a quantitative method that is fully validated based on the EMA guidelines for the analysis of acyclovir in the plasma. It has a linearity range of (0.7–60 mg/L) which make it applicable in the clinical practice. The method has undergone evaluation for its eco-friendliness using diverse green assessment tools, positioning it as a novel method of acyclovir detection in the plasma assigned to be green. The developed method is simple and utilizes a single protein precipitation step and excludes the use of the buffered mobile phase. Overall, it is a rapid, selective, accurate, and precise method that could be used widely in clinical settings.
Availability of data and materials
The data used to support the findings of this study are available from the corresponding author upon request.
Abbreviations
- HPLC:
-
High-performance liquid chromatography
- LC-MS:
-
Liquid chromatography- mass spectrometry
- AMGS:
-
Analytical method GREEnness score
- AGREE:
-
Analytical GREEnness
- IS:
-
Internal standard
- QC:
-
Quality control concentration
- LLOQ:
-
Lower limit of quantification
- EMA:
-
European Medicines Agency
- r 2 :
-
Coefficient of determination
- CV:
-
Coefficient of variation
- Rs:
-
Resolution
- T :
-
Tailing factor
- HETP:
-
Height of theoretical plate
- S.D:
-
Standard deviation
References
Schaeffer HJ, Beauchamp L, de Miranda P, Elion GB, Bauer DJ, Collins P (1978) 9-(2-hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature 272:583–585
McGrath N, Anderson NE, Croxson MC, Powell KF (1997) Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 63:321–326
Whitley RJ, Alford CA, Hirsch MS, Schooley RT, Luby JP, Aoki FY, Hanley D, Nahmias AJ, Soong SJ (1986) Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med 314:144–149
Abdalla S, Briand C, Oualha M, Bendavid M, Beranger A, Benaboud S, Treluyer JM, Zheng Y, Capito C, Demir Z, Foissac F, Winter S, Gana I, Boujaafar S, Bouazza N, Hirt D (2020) Population pharmacokinetics of intravenous and oral acyclovir and oral valacyclovir in pediatric population to optimize dosing regimens. Antimicrob Agents Chemother 64
Whitley RJ, Blum MR, Barton N, de Miranda P (1982) Pharmacokinetics of acyclovir in humans following intravenous administration. A model for the development of parenteral antivirals. Am J Med 73:165–171
de Miranda P, Whitley RJ, Blum MR, Keeney RE, Barton N, Cocchetto DM, Good S, Hemstreet GP 3rd, Kirk LE, Page DA, Elion GB (1979) Acyclovir kinetics after intravenous infusion. Clin Pharmacol Ther 26:718–728
Gurgel Assis MS, FernandesPedrosa TC, de Moraes FS, Caldeira TG, Pereira GR, de Souza J, Ruela ALM (2021) Novel insights to enhance therapeutics with acyclovir in the management of herpes simplex encephalitis. J Pharm Sci 110:1557–1571
Mahmoud SH, Shen C (2017) Augmented renal clearance in critical illness: an important consideration in drug dosing. Pharmaceutics 9
Boulieu R, Gallant C, Silberstein N (1997) Determination of acyclovir in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 693:233–236
Sharma M, Nautiyal P, Jain S, Jain D (2010) Simple and rapid RP-HPLC method for simultaneous determination of acyclovir and zidovudine in human plasma. J AOAC Int 93:1462–1467
Arlemalm A, Hellden A, Karlsson L, Carlsson B (2022) Rapid determination of acyclovir, its main metabolite 9-carboxymethoxymethylguanine, ganciclovir, and penciclovir in human serum using LC-MS/MS. Biomed Chromatogr 36:e5315
Huidobro AL, Ruperez FJ, Barbas C (2005) LC methods for acyclovir and related impurities determination. J Pharm Biomed Anal 37:687–694
Bangaru RA, Bansal YK, Rao AR, Gandhi TP (2000) Rapid, simple and sensitive high-performance liquid chromatographic method for detection and determination of acyclovir in human plasma and its use in bioavailability studies. J Chromatogr B Biomed Sci Appl 739:231–237
Fernandez M, Sepulveda J, Aranguiz T, von Plessing C (2003) Technique validation by liquid chromatography for the determination of acyclovir in plasma. J Chromatogr B Analyt Technol Biomed Life Sci 791:357–363
Galuszka A, Migaszewski Z, Namiesnik J (2013) The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. Trac-Trends Anal Chem 50:78–84
Kannaiah KP, Sugumaran A, Chanduluru HK, Rathinam S (2021) Environmental impact of greenness assessment tools in liquid chromatography: a review. Microchem J 170
Hicks MB, Farrell W, Aurigemma C, Lehmann L, Weisel L, Nadeau K, Lee H, Moraff C, Wong ML, Huang Y, Ferguson P (2019) Making the move towards modernized greener separations: introduction of the analytical method greenness score (AMGS) calculator. Green Chem 21:1816–1826
Galuszka A, Konieczka P, Migaszewski ZM, Namiesnik J (2012) Analytical Eco-Scale for assessing the greenness of analytical procedures. Trac-Trends Anal Chem 37:61–72
Pena-Pereira F, Wojnowski W, Tobiszewski M (2020) AGREE-analytical GREEnness metric approach and software. Anal Chem 92:10076–10082
Guideline on Bioanalytical Method Validation. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 1 Aug 2023
Pharmacopia U.S <621> Chromatography. https://www.usp.org/harmonization-standards/pdg/excipients/chromatography (2022). Accessed 15 March 2024
Food and Drug Administration (1994) Reviewer Guidance: Validation of chromatographic methods. Cited November
Tunkel AR, Glaser CA, Bloch KC, Sejvar JJ, Marra CM, Roos KL, Hartman BJ, Kaplan SL, Scheld WM, Whitley RJ, Infectious Diseases Society of A (2008) The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 47:303–327
Bean B, Aeppli D (1985) Adverse effects of high-dose intravenous acyclovir in ambulatory patients with acute herpes zoster. J Infect Dis 151:362–365
Muralidharan S, Kalaimani J, Parasuraman S, Dhanaraj SA (2014) Development and validation of acyclovir HPLC external standard method in human plasma: application to pharmacokinetic studies. Adv Pharm 2014:1–5
Teshima D, Otsubo K, Yoshida T, Itoh Y, Oishi R (2003) A simple and simultaneous determination of acyclovir and ganciclovir in human plasma by high-performance liquid chromatography. Biomed Chromatogr 17:500–503
Zendelovska D, Simeska S, Atanasovska E, Georgievska K, Kikerkov I, Labachevski N, Jakovski K, Balkanov T (2015) Determination of acyclovir in human plasma samples by HPLC method with UV detection: application to single-dose pharmacokinetic study. Open Access Maced J Med Sci 3:32–36
Aboelezz A, Mahmoud SH (2024) Acyclovir dosing in herpes encephalitis: a scoping review. J Am Pharm Assoc 2003:102040
Acknowledgements
Not applicable.
Funding
This research received funding from the University Hospital Foundation (UHF), and the Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada.
Author information
Authors and Affiliations
Contributions
Conceptualization was done by S.H.M.; methodology was done by S.H.M., A.A and M.K.; validation was done by A.A and M.K.; formal analysis was done by S.H.M. A.A and M.K.; writing—original draft preparation was done by A.A.; writing—review and editing was done by S.H.M., A.A and M.K.; supervision was done by S.H.M.; funding acquisition was done by S.H.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no any competing interests.
Studies involving plants
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aboelezz, A., Kharouba, M. & Mahmoud, S.H. A simple method for the determination of acyclovir concentrations in human plasma using high-performance liquid chromatography. Futur J Pharm Sci 10, 74 (2024). https://doi.org/10.1186/s43094-024-00649-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-024-00649-7