
Abstract
COVID-19 is a serious virus that can have a lot of effects, one of which is a secondary bacterial infection that can be more life-threatening and even lethal than the initial viral infection. Hence a fast and sensitive HPLC/UV method was developed and validated for the first estimation of a binary mixture of molnupiravir (MOL) and ertapenem (ERT) as a co-administrated medicine for the management of COVID-19 in pharmaceutical dosage forms, and human plasma samples. The drug combination was separated within 5 min via RP-ODS column using isocratic elution with a mobile phase of 0.05 M phosphate buffer (pH 3.5): acetonitrile with a 76: 24% ratio v/v. The presented method provided a linear response ranging from 0.03 to 17.0 and 0.05–20 µg mL−1 with LOD values of 0.009 and 0.008 µg mL−1 for MOL and ERT respectively. The good separation and high sensitivity of the HPLC method provide the determination of the cited drugs in human plasma without matrix interference with a percent of recovery ranging from 94.97 ± 2.05 to 98.44 ± 1.92. Based on the results, this method could be utilized to monitor cited drugs in quality control and therapeutic laboratories.
Similar content being viewed by others


Introduction
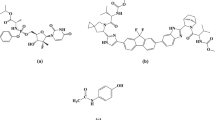
At the end of 2019, numerous cases of patients suffering from mild to severe symptoms that are similar to bacterial pneumonia without any known cause were reported in Wuhan, Hubei province, China [1]. An outbreak of the infection resulted in thousands and millions of cases worldwide. The unidentified pneumonia was defined to be caused by a novel coronavirus (CoV) named 2019-nCoV [2, 3]. On February 11, 2020, the International Committee of Taxonomy of Viruses (ICTV) named this novel coronavirus as SARS-CoV-2 [4]. In March 2020, the World Health Organization (WHO) declared the COVID-19 virus a global pandemic [5]. Plenty of efforts around the world have been directed toward developing therapeutic strategies and instructions to decrease the spread of infection and disease symptoms [6]. Although the reported total COVID-19 deaths for the two most recent years were around 5.94 million worldwide, an estimated 18.2 million people died from COVID-19 disease during this period. This could be due to poor reporting, insufficient testing facilities, reduced access to healthcare services, and even political considerations [7]. For control of the health hazards of COVID-19 disease, an antiviral drug directly acting on coronavirus was urgently needed. MOL (Fig. 1) chemically is N-Hydroxy-5’-O-isobutyryl-3,4-dihydrocytidine [(2R,3 S,4R,5R)-3,4-Dihydroxy-5-[4-(hydroxyamino)-2-oxopyrimidin-1-yl] oxolan-2-yl] methyl 2-methylpropanoate. MOL is a broad-spectrum antiviral prodrug that is rapidly metabolized in vivo into the active triphosphate form, inhibiting viral RNA polymerase, which is necessary for viral replication [8]. MOL is the first oral, direct-acting antiviral that is highly effective at reducing nasopharyngeal SARS-CoV-2 infectious virus and viral RNA with a favorable tolerability and safety profile [9]. The severity of COVID-19 disease is not only attributed to the viral invasion but also to the secondary bacterial infection that can be more life-threatening and even lethal than the initial viral infection [10]. So, the need for a wide-spectrum antibiotic oriented to face the bacterial infection that may have emerged after or just before the COVID-19 infection is essential.

Chemical structure of studied drugs (a) MOL and (b) ERT.
Ertapenem sodium (Fig. 1) is the monosodium salt of (4R,5 S,6 S)-3-[(3 S,5 S)-5-[(3-carboxyphenyl) carbamoyl] pyrrolidin-3-yl] sulfanyl-6-(1-hydroxyethyl)-4- methyl-7-oxo-1- azabicyclo [3.2.0] hept-2-ene-2-carboxylic acid. It is a β-lactam antibiotic of the carbapenem class with an exceptionally broad spectrum of activity [11]. ERT has the advantage of being a broader spectrum of activity than other beta-lactams like penicillins and cephalosporins and is more resistant to the enzyme β-lactamase which is the main cause of resistance of many bacteria [12]. In the covid-19 pandemic period, daily inpatient ERT therapy can be an alternative to hospitalization for the treatment of complicated urinary tract infections, which is safe and cost-effective [13]. The need for an effective antibiotic in covid-19 patients for treating a complication of the covid-19 disease concomitant with an antiviral is so essential. ERT combined with cefazolin resulted in successful sterilization of blood cultures within 24 h of administration in a covid-19 patient previously bacteremic for more than 10 days [14]. Co-administration of MOL as an antiviral drug with ERT as an antibacterial drug for secondary bacterial infection for a covid-19 patient is crucial. Till the time of writing this manuscript, there is no reported method for the determination of both MOL and ERT as possible co-administered drugs in some cases related to covid-19 patients.
The literature review revealed only a few methods for determining the cited drugs alone or in combination with other drugs, such as HPLC methods [15,16,17,18,19,20,21,22,23,24,25,26], HPTLC methods [27], electro-analytical methods [28,29,30], spectrophotometric [31,32,33,34,35,36,37], and spectrofluorimetric [3, 38,39,40].
The presented method describes a simple, rapid, sensitive, reliable, and cost-effective HPLC method for simultaneous estimation of MOL and ERT in their pharmaceutical formulations and human plasma samples which could be helpful in clinical and therapeutic laboratories.
Experimental
Instrumentation and software
The HPLC separation of the MOL and ERT mixture was carried out using Waters 717 Instrument, the instrument connected with an autosampler with a sample thermostat that contains Alltech, 426 LC pump, and UV/VIS detector (Waters Millipore, USA). The results were obtained using Kromex (Estonia) software.
Chemicals and standard solutions
MOL (analytical standard, purity 99.96%) was mercifully given by the Egyptian International Pharmaceutical Industries Co. (EIPICo., Egypt). Molcovir® capsules (containing 200 mg MOL per capsule; batch number: MOLCD1003D) were obtained from Optimus Pharma, India.
ERT (analytical standard, purity 99.89%) and Invanz® vial (containing 1000 mg ERT per vial; batch number: W011981) were obtained from Merck Sharp & Dohme, USA. All the solvents used were of HPLC grade. Acetonitrile (ACN), sodium di-hydrogen phosphate, and 85% orthophosphoric acid were HPLC grade obtained from EL-Nasr Co., Egypt.
Standard drug solutions preparation
The standard stock solutions of concentration 1.0 mg mL− 1 for each drug were made by transferring accurately weighed 25 mg of authentic powder into a 25-mL calibrated flask then diluted with about 15 ml double distilled water and sonicated for about 5 min. The flask was completed to 25 ml by double distilled water to obtain a stock solution of a concentration of 1.0 mg mL− 1, then stored in the refrigerator at 4ºC. The working solutions of MOL and ERT were prepared by diluting the standard stock solution (1.0 mg mL− 1) by the mobile phase using volumetric flasks to obtain working solutions within the concentration range of 0.03–20 µg mL− 1.
The chromatographic conditions
Twenty microliters of working solution for the specified drugs in the concentration range of 0.03–20 µg mL− 1, were injected into the HPLC-UV system. The separation and quantitation were carried out using a GL Science RP-ODS column (25 cm x 4.6 mm id, 5 μm particle size) (Japan) as a stationary phase. The mobile phase consists of 0.05 M phosphate buffer (pH 3.5 adjusted by 85% ortho-phosphoric acid) and acetonitrile in the ratio of 76: 24% (v/v). The flow rate of the mobile phase in this method was set to 1 mL min− 1 and the eluted drugs were detected using a UV detector at 230 nm. The separation and quantitation were performed in this method at ambient temperature.
Pharmaceutical dosage form preparation
In order to analyze the studied drugs, ten capsules of Molcovir® were evacuated and mixed well then, an equivalent amount of 25 mg of the studied drug was transferred into a 25 mL volumetric flask containing ultra-pure water. The solution was filtrated through a 0.45-µm cellulose acetate membrane and diluted using the mobile phase for the preparation of working solutions in the linearity range of the calibration.
Invanz® vial was evacuated and mixed well and then an equivalent amount of 25 mg of the studied drug was put into a 25 mL volumetric flask containing ultra-pure water, filtered, and diluted in the same way previously mentioned to obtain working solutions with concentrations in the linearity range of the calibration.
The mixture of working solutions for the binary mixture was prepared using the mobile phase.
Analysis of the studied drugs in human plasma
Human plasma samples were collected from five healthy volunteers aged 22–35 years into heparinized tubes, in accordance with the responsible committee on human experimentation’s (institutional and national) ethical guidelines and the Helsinki Declaration of 1975, as revised in 2008. The method and the study were approved by the Egyptian Network of Research Ethics Committees (ENREC). After collecting plasma samples, 1.0 mL of plasma samples were spiked with different concentrations of the working standard solution (5, 50, 100, 150, and 170 µg mL− 1). For protein precipitation, 1.0 mL of acetonitrile was added [3]. After vortex mixing, the supernatant solution was separated and diluted in 10 ml volumetric flasks with the mobile phase to obtain final concentrations (0.5, 5.0, 10, 15.0, and 17.0 µg mL− 1). The final solution was centrifuged afterward for 20 min at 4000 rpm. A 0.45 μm cellulose acetate membrane was used for filtration of the supernatant solution, then 20 µL of supernatant was injected into the HPLC-UV system.
Results and discussion
The goal of this study is to create a rapid, simple, and sensitive HPLC method for the simultaneous quantification of MOL and ERT as a binary mixture in pure form and human plasma for the first time (Figure 2).

HPLC Chromatogram for separation of studied drugs (5 µg mL− 1 for each)
Various system suitability factors including capacity factor (k’), retention time (tR), resolution (Rs), separation factor (α), tailing factor (T), and number of theoretical plates (N) were investigated to assess the system performance and the method repeatability for separation of cited drugs. MOL and ERT were separated simultaneously after 3.2 and 4.86 min, respectively (Figure 2).
The summarized results in Table 1 revealed a good separation between MOL and ERT, where MOL and ERT were separated at 3.2 and 4.86 min respectively having tailing factor values of 1.09 and 1.05 for MOL and ERT respectively. The capacity factor (K’) was found to be 7 and 11.15 for MOL and ERT respectively. The column efficiency was studied by calculating the resolution (RS). It was found that Rs between MOL and ERT was equal to 2.3 with a selectivity factor equal to 1.53 which refers to a good separation of the two studied drugs (Table 1).
Optimization of HPLC variables
For achieving the most suitable drug separation, different factors which influence separation time and peak symmetry were investigated to choose the most appropriate conditions. These factors include mobile phase composition, flow rate, and buffer concentration. Each factor was assessed individually while others remain unchanged.
Mobile phase composition
To obtain the most optimum drugs separation in a suitable time giving reliable peak area, various mobile phases composition was tried as acetonitrile (ACN): methanol, water: methanol, and ACN: water with varying percentages, and it was observed that no good separation of studied peaks was achieved with these trials. Therefore, the mobile phase that consists of a mixture of 0.05 M phosphate buffer (pH 3.5 adjusted by 85% ortho-phosphoric acid), acetonitrile (76: 24% v/v), was found to be the optimum mobile phase for separation as in Fig. 2. As it is observed from Fig. 1, MOL contains four hydrogen bond donors and seven hydrogen bond acceptors, enabling it to form four hydrogen bonds at least with orthophosphoric acid. Also, ERT contains five hydrogen bond donors and nine hydrogen bond acceptors, which easily bind with orthophosphoric acid. This makes the forcing power for both drugs elution.
ERT has a higher retention time than MOL due to the presence of lipophilic phenyl moiety in ERT which decreases polarity in comparison to MOL as well as log p of ERT (-1.8) is more than the log p of MOL (−0.8) [41, 42].
Besides, the effect of the flow rate was further studied to achieve sharp symmetric peaks of the cited drugs within a reasonable time. It was found that flow rates from 0.9 to 1.1 ml/min showed good separation between the MOL and ERT with sharp symmetric peaks, so 1 mL/min was the suitable flow rate.
Buffer PH optimization
The buffer PH of 0.05 M phosphate buffer was checked in the range from 2.5 to 4.1 to obtain good separation for MOL and ERT (Fig. 3a), after comparing it was observed that pH 3.5 was selected as the optimum pH for the separation of the studied drugs.

(a) effect of pH range for separation MOL and ERT mixture (5 µg mL− 1 for each), (b) UV spectrum for studied drugs
PH 3.5 was chosen according to the pKa of both drugs which is 2.2, 10.2, and 12 for MOL, 3.2 and 9 for ERT, so the use of buffer to maintain the best ionization and solubility of both drugs above pKa 2 and below 10.
Also, the wavelength for the cited drugs was estimated using a Shimadzu spectrophotometer, and the UV spectra of the cited drugs were recorded at 230 nm (Fig. 3b).
Validation of the presented HPLC method
The HPLC method was validated based on International Conference on Harmonization (ICH) guidelines [39].
Linearity and calibration curve
To obtain the calibration curve for each drug, the peak area for each concentration in the range of (0.03–17.0 µg mL− 1) for MOL and (0.05–20.0 µg mL− 1) for ERT was plotted against the corresponding peak area, and the range specified for each drug was observed to produce a linear relationship with high correlation coefficient.
The linearity range and the sensitivity parameters were summarized in Table 2.
Accuracy
The accuracy of the proposed approach was evaluated using five distinct concentration levels (0.5,5.0, 10.0, 15, and 17.0 µg mL-1) by injecting each concentration three times on the HPLC-UV system, for the drugs under investigation. The results were summarized in Table 3 which refers to the high accuracy of the proposed method.
Repeatability and intermediate precision
The intra-day repeatability was evaluated as RSD using 5 different concentrations for each drug, each analyzed three replicates (n = 3) on the same day, and the intermediate precision was checked by analyzing the same 5 different concentrations on three consecutive days. RSD was found to range from 0.19 to 2.16 which shows acceptable repeatability and intermediate precision.
The limit of detection (LOD), the limit of quantitation (LOQ), and the sensitivity
According to ICH guidelines recommendations [43], the lower limit of detection (LOD) and lower limit of quantitation (LOQ) were calculated using the following equations:
Where ϭ is the standard deviation of the response and S is the slope.
The calculated results were summarized in Table 2, which refers to the high sensitivity of the HPLC method compared to other reported methods [15,16,17,18,19,20,21,22,23,24]. For the reason of high sensitivity, the proposed HPLC method gives great value in the determination of the studied drugs in plasma samples.
Robustness
To examine the robustness of the proposed HPLC method, one experimental variable was varied independently while the others were kept constant. The variables examined were mobile phase composition ratio, buffer concentration, value of pH, mobile phase flow rate, and detection wavelength. To check the effect of mobile phase ratio change, 74/26 v/v, and 78/22 v/v ratios were checked. The pH of the buffer solution was checked at 3.4 and 3.6 pH values and the buffer concentration was changed to 0.04 and 0.06 M. As shown in Table 4 these slight modifications of the separation system parameters have no significant effect on the results of the method revealing method robustness.
Linearity, accuracy, and precision in human plasma
The proposed method was applied for the determination of the studied drugs in human plasma samples, and it was observed to give a linear relationship between spiked drug concentration and the corresponding peak area in the range of (0.03–17.0 µg mL− 1) for MOL and (0.05–20.0 µg mL− 1) for ERT. The value of both LOD and LOQ were calculated using the equations previously mentioned in Sect. 3.2.4. The LOQ for MOL and ERT was found to be 0.04 and 0.05 µg mL− 1 respectively and the LOD was found to be 0.013 and 0.015 µg mL− 1 for MOL and ERT respectively.
According to US-FDA criteria [44], the explored approach underwent bio-analytical validation, where the accuracy and precision were examined in human plasma. Using low-quality control sample (LQC), medium-quality control sample (MQC), and high-quality control samples (HQC) for MOL and ERT, the three concentration points were analyzed at the same day (intra-day) where n = 6, and inter-daily (n = 9). According to the results listed in Table S1, the application of the proposed method for analysis of the studied drugs in human plasma exhibits good repeatability precision, as the percent RSD ranges from 1.32 to 2.50 and from 1.44 to 2.20 for MOL and ERT respectively, and the percent recovery ranging from 95.01 to 97.10%.
Matrix effect and selectivity
For evaluation of the method selectivity, three points of quality control samples (low-quality control sample (LQC), medium quality sample (MQC), and high-quality control sample (HQC) were examined. Those concentrations were (0.5, 5.0, and 15.0 µg mL− 1) for both MOL and ERT that were used to examine the possible plasma matrix effect in human plasma samples on the determination of medications under investigation. The recovery was discovered to range from 95.55 ± 1.86 to 97.40 ± 2.44 as in Fig. 4. The results refer to the absence of a significant plasma matrix with the tested regimen as a binary mixture for treatment of COVID-19, which validates the excellent selectivity of the suggested technique as depicted in Fig. 4.

3D Chromatogram for matrix effect using the synthetic mixture in human plasma (0.5, 5.0, and 15.0 µg mL− 1 for each)
Stability
The stability of the studied analytes was assessed under different experimental conditions, resembling plasma sample storage and preparation until HPLC analysis. QC samples at three concentration levels (LQC, MQC, and HQC) were assessed under the following conditions: (1) Short-term stability for 12 h at -20 °C, (2) Long-term stability for 15 days at -20 °C, (3) post-preparative stability for 6 h at room temperature 25 °C, (4) Three Freeze–thaw cycle stability at -20 °C. The results obtained from the tested samples at these different conditions revealed that the percentage of recovery was between 85 and 115% which indicates the accepted stability of the studied drugs under the studied different conditions (Table 5).
Applications of the chromatographic method
Estimation of MOL and ERT in their pharmaceutical forms
The developed HPLC method was utilized for the determination of the studied drugs in their dosage forms (Molcovir capsules® and Invanz vial®). The percentage of recovery obtained by the proposed method was determined to be 101.50 ± 0.92 and 101.33 ± 0.53 respectively and was compared with that of the reported method [3, 23]. All the results summarized in Table 6 refer to the high accuracy of the proposed HPLC method.
Applications of HPLC in spiked human plasma
The new method’s high sensitivity enables the detection of MOL and ERT medicines in spiked human plasma without matrix interference as a synthetic mixture (Fig. S1). For the tested procedures at five different concentration levels applied, the recovery percentage was discovered to be between 94.97 and 98.44%, as indicated in Table 7. For the examined medicines, the percent RSD values fall between 1.55 and 2.70 respectively. The data obtained revealed that the studied drugs can be determined in plasma samples without the interference of the matrix effect.
Comparison study between the presented method and reported methods
Comparing the results in our work with other reported as in Table 8. It was found the presented work can serve as a probe for the first simultaneous estimation of a binary mixture of MOL and ERT with low concentration with higher sensitivity and reliability than other reported methods.
Conclusion
This study aims to the first simultaneous estimation of a binary mixture of MOL and ERT as a possible treatment regimen in COVID-19 infection. It has been successively applied to the analysis of the studied drugs in their commercial dosage forms and human plasma samples. The proposed method appears to be sensitive (LOD is 0.009 µg mL− 1 and 0.008 µg mL− 1 for MOL and ERT respectively), selective, rapid, and relatively low cost which facilitates its application in quality control units. Also, the high sensitivity and selectivity obtained after application of the proposed method on analysis of the studied drugs in human plasma samples gives the advantage of being applied for drug analysis in clinical units.
Declaration section.
Data Availability
All data generated or analyzed during this study are included in this published article [and its additional files].
References
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2001316.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Phil D, Tan W. A novel coronavirus from patients with pneumonia in China, 2019. New England journal of medicine 2020. https://doi.org/10.1056/NEJMoa2001017.
Salman BI, Ibrahim AE, El Deeb S, Saraya RE. Fabrication of novel quantum dots for the estimation of COVID-19 antiviral drug using green chemistry: application to real human plasma. RSC Adv. 2022;12(26):16624–31. https://doi.org/10.1039/D2RA02241A.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Caraco Y, Crofoot GE, Moncada PA, Galustyan AN, Musungaie DB, Payne B, Kovalchuk E, Gonzalez A, Brown ML, Williams-Diaz A, Gao W, Strizki JM, Grobler J, Du J, Assaid CA, Paschke A, Butterton JR, Johnson MG, De Anda C. Phase 2/3 trial of molnupiravir for treatment of Covid-19 in nonhospitalized adults. NEJM Evid. 2022;1(2):EVIDoa2100043. https://doi.org/10.1056/EVIDoa2100043.
Cevik M, Tate M, Lloyd O, Maraolo AE, Schafers J, Ho A. SARS-CoV-2 SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. The Lancet Microbe. 2021;2(1):e13–e22. https://doi.org/10.1016/S2666-5247(20)30172-5.
Wang H, Paulson KR, Pease SA, Watson S. Estimating excess mortality due to the COVID- 19 pandemic: a systematic analysis of COVID-19-related mortality, 2020–21. The Lancet. 2022;399(10334):1513–36. https://doi.org/10.1016/S0140-6736(21)02796-3.
Painter GR, Natchus MG, Cohen O, Holman W, Painter WP. Developing a direct-acting, orally available antiviral agent in a pandemic: the evolution of molnupiravir as a potential treatment for COVID-19. Current opinion in virology 2021;17–22. https://doi.org/10.1016/j.coviro.2021.06.003.
Fischer W, Eron JJ Jr, Holman W, Cohen MS, Fang L, Szewczyk LJ, Sheahan TP, Baric R, Mollan KR, Wolfe CR, Duke ER, Azizad MM, Borroto-Esoda K, Wohl DA, Loftis AJ, Alabanza P, Lipansky F, Painter WP. Molnupiravir, an oral antiviral treatment for COVID-19. MedRxiv 2021. https://doi.org/10.1101/2021.06.17.21258639.
Gonzalez-Zorn B. Antibiotic use in the COVID-19 crisis in Spain. Clin Microbiol Infect. 2021;27(4):646–7. https://doi.org/10.1016/j.cmi.2020.09.055.
Zhanel GG, Wiebe R, Dilay L, Thomson K, Rubinstein E, Hoban DJ, Noreddin AM, Karlowsky AJ. Comparative review of the carbapenems. Drugs. 2007;67(7):1027–52.
Nicolau DP. Carbapenems: a potent class of antibiotics. Expert opinion on pharmacotherapy 2008;9(1):23–37. https://doi.org/10.1517/14656566.9.1.23.
Zeka AN, Avkan-Oguz V, Irmak C, Kutsoylu OE, Cavus SA, Kuruüzüm Z, Ergon MC. Daily inpatient ertapenem therapy can be an alternative to hospitalization for the treatment of complicated urinary tract infections during the COVID-19 pandemic. Int J Clin Pract. 2021;75(7):e14230. https://doi.org/10.1111/ijcp.14230.
Ilges D, Krishnan G, Geng E. Persistent methicillin-susceptible bacteremia rapidly cleared with cefazolin and Ertapenem Combination Therapy in a patient with COVID-19. Case Reports in Infectious Diseases. 2022. https://doi.org/10.1155/2022/6828538.
Bindu M, Gandla K, Vemireddy S, Samuel S, Praharsha Y. A validated stability indicating RP-HPLC method for the determination of molnupiravir in pharmaceutical dosage form. World J Adv Res Reviews. 2022;15(1):580–90. https://doi.org/10.30574/wjarr.2022.15.1.0720.
Reçber T, Timur SS, Kablan SE, Yalcin F, Karabulut TC, Gursoy RN, Eroglu H, KIt S, Nemutlu E. Stability indicating RP-HPLC method for determination of the COVID-19 drug molnupiravir applied using nanoformulations in permeability studies. J Pharm Biomed Anal. 2022;214:114693. https://doi.org/10.1016/j.jpba.2022.114693.
Gouda AS, Marzouk HM, Rezk MR, Salem AM, Morsi MI, Nouman EG, Abdallah YM, Hassan AY, Abdel-Megied AM. A validated LC-MS/MS method for determination of antiviral prodrug molnupiravir in human plasma and its application for a pharmacokinetic modeling study in healthy egyptian volunteers. J Chromatogr B. 2022;1206:123363. https://doi.org/10.1016/j.jchromb.2022.123363.
Zajac M, Cielecka-Piontek J, Jelinska A. Development and validation of UV spectrophotometric and RP-HPLC methods for determination of ertapenem during stability studies. Chem Anal (Warsaw). 2006;51(5):761.
Pedroso TM, Medeiros AC, Salgado HR. RP-HPLC× HILIC chromatography for quantifying ertapenem sodium with a look at green chemistry, Talanta 2016;160:745–53. https://doi.org/10.1016/j.talanta.2016.08.016.
Abdel-Moety EM, Elragehy NA, Hassan NY, Rezk MR. Selective determination of ertapenem and imipenem in the presence of their degradants. J Chromatogr Sci. 2010;48(8):624–30. https://doi.org/10.1093/chromsci/48.8.624.
Mundkowski RG, Majcher-Peszynska J, Burkhardt O, Welte T, Drewelow B. A new simple HPLC assay for the quantification of ertapenem in human plasma, lung tissue, and broncho-alveolar lavage fluid. J Chromatogr B. 2006;832(2):231–5. https://doi.org/10.1016/j.jchromb.2006.01.005.
Pickering M, Brown S. Quantification and validation of HPLC-UV and LC‐MS assays for therapeutic drug monitoring of ertapenem in human plasma. Biomed Chromatogr. 2013;27(5):568–74. https://doi.org/10.1002/bmc.2829.
Ali MFB, Marzouq MA, Hussein SA, Salman BI. A bio-analytically validated HPLC-UV method for simultaneous determination of doripenem and ertapenem in pharmaceutical dosage forms and human plasma: a dual carbapenem regimen for treatment of drug-resistant strain of Klebsiella pneumoniae. RSC Adv. 2021;11(5):3125–33. https://doi.org/10.1039/D0RA10466C.
Musson DG, Kitchen CJ, Hsieh JY-K, Birk KL. Modified high-performance liquid chromatographic method for the determination of ertapenem in human urine: enhanced selectivity and automation. J Chromatogr B. 2002;779(2):341–6. https://doi.org/10.1016/S1570-0232(02)00377-X.
Sravanthi G, Gandla KS, Repudi L. New analytical method development and validation for estimation of molnupiravir in bulk and tablet dosage form by RP-HPLC method. Cell Mol Biomedical Rep. 2023;3(3):130–6. https://doi.org/10.55705/cmbr.2023.375093.1087.
Hafez HM, El Deeb S, Swaif MM, Ibrahim RI, Kamil RA, Abdelwahed AS, Ibrahim AE. Micellar Organic-solvent free HPLC design of experiment for the determination of Ertapenem and meropenem; assessment using GAPI, AGREE and analytical eco-scale models. Microchem J. 2023;185:108262. https://doi.org/10.1016/j.microc.2022.108262.
Saraya RE, El Deeb S, Salman BI, Ibrahim AE. Highly sensitive high-performance thin‐layer chromatography method for the simultaneous determination of Molnupiravir, Favipiravir, and Ritonavir in pure forms and pharmaceutical formulations. J Sep Sci. 2022. https://doi.org/10.1002/jssc.202200178.
Kablam SE, Reçber T, Tezel G, Timur SS, karabulut C, Karabulut TC, Eroğlu H, Kır S, Nemutlu E. Voltammetric sensor for COVID-19 drug Molnupiravir on modified glassy carbon electrode with electrochemically reduced graphene oxide. J Electroanal Chem. 2022;920:116579. https://doi.org/10.1016/j.jelechem.2022.116579.
Fayed AS, Youssif RM, Salama NN, Elzanfaly ES, Hendawy HAM. Ultra-sensitive stripping SWV for determination of ertapenem via ZnONPs/MWCNT/CP sensor: greenness assessment. Microchem J. 2021;162:105752. https://doi.org/10.1016/j.microc.2020.105752.
Gumustas M, Karadas N, Ozkan SA. Validated electroanalytical and RP-LC assay of ertapenem in its pharmaceutical dosage form. Rev Roum Chim. 2013;58:7–8. http://web.icf.ro/rrch/.
Hassan NY, Abdel-Moety EM, Elragehy NA, Rezk MR. Selective determination of ertapenem in the presence of its degradation product. Spectrochim Acta Part A Mol Biomol Spectrosc. 2009;72(5):915–21. https://doi.org/10.1016/j.saa.2008.12.025.
Babu KR, Kumari NA, Lakshmi RV. Spectrophotometric determination of doripenem, ertapenem in bulk and injection formulations by FC reagent. Int J Pharm Sci Drug Res. 2013;5(4):184–6.
Fayed AS, Youssif RM, Salama NN, Hendawy HA, Elzanfaly ES. Two-wavelength manipulation stability-indicating spectrophotometric methods for determination of meropenem and ertapenem: greenness consolidation and pharmaceutical product application. Chem Pap. 2019;73(11):2723–36. https://doi.org/10.1007/s11696-019-00824-8.
Cielecka-Piontek J, Jelińska A. The UV-derivative spectrophotometry for the determination of doripenem in the presence of its degradation products. Spectrochim Acta Part A Mol Biomol Spectrosc. 2010;77(2):554–7. https://doi.org/10.1016/j.saa.2010.06.019.
Moneim MA, Kamal M, Hamdy M. Simple Green Spectrophotometric & Chromatographic Assay of the oral antiviral treatment of COVID-19: Molnupiravir-EIDD-2801. Egypt J Chem. 2022. https://doi.org/10.21608/ejchem.2022.135659.5976.
Alekhya K, Bhoomika S, Aaliya P, Dharani S, Faizal AS, Ilango K. Estimation of Molnupiravir in bulk and formulation using green UV-Spectrophotometric method. Eur Chem Bull 2023;12(Special Issue 4), 14874–85.
Abdelazim AH, Abourehab MA, Abd Elhalim LM, Almrasy AA, Ramzy S. Green adherent spectrophotometric determination of molnupiravir based on computational calculations; application to a recently FDA-approved pharmaceutical dosage form. Spectrochim Acta Part A Mol Biomol Spectrosc. 2023;285:121911. https://doi.org/10.1016/j.saa.2022.121911.
Fayed AS, Youssif RM, Salama NN, Elzanfaly ES, Hendawy HAM. Utility of silver-nanoparticles for Nano Spectrofluorimetric determination of Meropenem and Ertapenem: bio-analytical validation. Spectrochim Acta Part A Mol Biomol Spectrosc. 2021;262:120077. https://doi.org/10.1016/j.saa.2021.120077.
Salman BI, Saraya RE. Bio-analytically fluorimetric method for estimation of ertapenem in real human plasma and commercial samples; application to pharmacokinetics study. Luminescence. 2022;37(5):769–802. https://doi.org/10.1002/bio.4223.
Elzanfaly ES, Youssif RM, Salama NN, Fayed AS, Hendawy HAM, Salem MY. Zero and second-derivative synchronous fluorescence spectroscopy for the quantification of two non‐classical β‐lactams in pharmaceutical vials: application to stability studies. Luminescence. 2017;32(8):1517–27. https://doi.org/10.1002/bio.3353.
National Center for Biotechnology Information. (2023). PubChem Compound Summary for CID 145996610, Molnupiravir. https://pubchem.ncbi.nlm.nih.gov/compound/Molnupiravir.
National Center for Biotechnology Information. (2023). PubChem Compound Summary for CID 150610, Ertapenem. https://pubchem.ncbi.nlm.nih.gov/compound/Ertapenem.
International Conference on Harmonization, Topic Q2 (R1) Validation of analytical procedures: text and methodology. 2005.
Zimmer D. New US FDA draft guidance on bioanalytical method validation versus current FDA and EMA guidelines: chromatographic methods and ISR. Bioanalysis. 2014;6(1):13–9. https://doi.org/10.4155/bio.13.298.
Funding
Open access funding is provided by The Science, The Technology, and Innovation Funding Authority (STF) in cooperation with The Egyptian Knowledge Bank (EKB).
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
KA and RA conceived and designed the experiments. KA, ME, and MN conducted the experiment and interpreted the results. All the authors analyzed the data and wrote the paper. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Human plasma samples were collected from healthy volunteers in accordance with the responsible committee on human experimentation’s (institutional and national) ethical guidelines and the Helsinki Declaration of 1975, as revised in 2008. The method and the study were approved by the Egyptian Network of Research Ethics Committees (ENREC). The authors confirmed informed consent was obtained from all subjects participating in the experiments.
Consent of participate
The authors confirmed informed consent was obtained from all subjects participating in the experiments.
Consent for publication
Not applicable.
Competing of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Afify, K.K., Ali, R., El-Dosoky, M.A. et al. HPLC/UV approach method for the first simultaneous estimation of molnupiravir and ertapenem as a binary mixture in human plasma and dosage form as a regimen for COVID-19 treatments. BMC Chemistry 17, 121 (2023). https://doi.org/10.1186/s13065-023-01024-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-023-01024-y