
Abstract
Background
Over the last decade, aggregating evidences suggested that there is a causative link between mutation in gene associated with mitochondrial dysfunction and development of several neurodegenerative disorders.
Main text
Recent structural and functional studies associated with mitochondrial genes have shown that mitochondrial abnormalities possibly lead to mitochondrial dysfunction. Several studies on animal models of neurodegenerative diseases and mitochondrial genes have provided compelling evidence that mitochondria is involved in the initiation as well as progression of diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and Friedreich ataxia (FA).
Conclusion
In this mini-review, we have discussed the different etiologic and pathogenesis connected with the mitochondrial dysfunction and relevant neurodegenerative diseases that underlie the dominant part of mitochondrial genes in the disease development and its progress.
Similar content being viewed by others

Background
Mitochondria basically allied with formation of adenosine triphosphate (ATP) through oxidative phosphorylation process. However, mitochondria also take part in a lots of necessary cellular functions like iron and calcium homeostasis, steroids, pyrimidines and heme biosynthesis [1, 2]. Mitochondria have both inner and outer membrane, which is impermeable to charged ions including all other molecules. The ATP generation process occurs at the inner membrane of mitochondria through electrons donation by nicotinamide adenine dinucleotide (NAD) or flavin adenine dinucleotide (FAD) equivalents formed by the tricarboxylic acid (TCA) cycle. This process is called as the electron transport system [3, 4]. Mitochondrial DNA (mtDNA) involves circular structure of 569, 16 base pairs which are active in the synthesis of proteins and mitochondrial ribonucleic acids (RNSs). This mtDNA encodes 13 polypeptides, 22 ribosomal RNAs and 22 transfer RNAs, all of which are important for ATP formation and electron transport, consequently for normal cellular physiology. There are many human diseases which are strongly related with mutated mitochondrial genes [5,6,7,8]. Oxidative damage and subsequent dysfunction are occurring to mitochondria which is known to be a major site of free radical generation in cells [9]. Mitochondrial genome (mtDNA) is more susceptible to oxidative damage as compared to the nuclear DNA [10]. Due to the decline of defense mechanism in the cell, oxidative stress occurs and damages the nucleic acids. If the damage DNA is not repaired then it is considered being highly mutagenic upon DNA replication [11].
Basic mitochondrial genetics
It is well known that mitochondria contribute to ageing and neurodegeneration through accumulation of mtDNA mutation and generation of reactive oxygen species (ROS) [12]. Excessive production of ROS stimulates several signaling molecule that governs the endogenous mitochondrial apoptotic pathway. Similarly, mtDNA dysfunction can be induced by many signaling molecule that are regulated by nuclear gene and intrinsic factors involved in mitochondrial metabolism. Thus, it is speculated that mtDNA is directly linked with nuclear signaling pathway and thus, can be influence ageing process and associated neurodegeneration [13, 14]. In view of this background, this review summarizes some possible pathophysiology of mitochondrial dysfunction associated with mutation in genes, as a cause of Parkinson’s disease (PD), Alzheimer’s disease (AD), Friedreich's ataxia (FRDA), and Huntington's disease (HD).
Main text
Mitochondrial genes in major neurodegenerative disorders
Parkinson’s disease (PD)
Parkinson’s disease is one of the progressive and most common neurodegenerative diseases characterized by some common clinical features such as bradykinesia, rigidity, tremor, and some non-motor symptoms such as depression, apathy, and sleep disorders [15]. Recently, several reports demonstrated the involvement of mitochondrial genes in the pathogenesis of PD.
Synuclein alpha (SNCA)
SNCA encodes for α-Synuclein (α-Syn), a small polypeptide consisting of 140 amino acids. Even though its role is not discovered, it has been observed that it mediates the release of neurotransmitter at the presynaptic terminals and interact with membranes of organelles, including mitochondria. Interestingly, α-Syn has shown influence on structure of mitochondria and its function [16]. Initially, α-Syn was associated with PD as important component of Lewy bodies [17]. Elevated levels of wild-type (WT) α-Syn to a larger extent, leads to PD-associated mutations such as E46K, H50Q, and A53T which induces in vivo and in vitro mitochondrial fragmentation and reactive oxygen species (ROS) formation. Further, α-Syn is confined to mitochondria-associated membranes (MAM), a special structure which forms an interface between the mitochondria-endoplasmic reticulum (ER) is important for the regulation of apoptosis and Ca2+ signaling. Mutant α-Syn was found to decrease binding to MAM and elevation in mitochondrial fragmentation, suggesting a role for α-Syn in mitochondrial morphology regulation [18, 19]. Mutant α-Syn was reported as reason of separation of mitochondria and ER at MAM, which impairs Ca2+ exchange and reduces ATP formation in mitochondria [20]. Additionally, a recent study has been reported that α-Syn also affects mitochondrial biogenesis through peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) [21]. Accordingly, treatment of dopaminergic neurons containing A53T with mitochondrial toxins such as S-nitrosylation of transcription factor myocyte-specific enhancer factor 2C (MEF2C), leads to reduced mitochondrial biogenesis through down regulation of PGC1α [22]. α-synuclein aggregation is a pathological characteristic common to PD, as well as other neurodegenerative diseases, such as dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) which are collectively called “α-synucleinopathies” [22].
Leucine rich repeat kinase 2 (LRRK2)
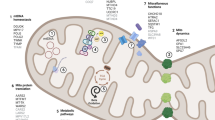
In humans, LRRK2 gene encodes leucine rich repeat kinase 2 (LRRK2) which is also called as dardarin and PARK8 is a kinase enzyme [23, 24]. Mutation in LRRK2 causes penetrant autosomal dominant type of PD and it is the most common cause of familial PD. Basically, LRRK2 is a type of protein kinase that is multifunctional and LRRK2 mutants exerts their pathogenic action by elevating kinase activity. The mutant LRRK2 may contribute in elevating the mitochondrial toxins, ROS production and defects in mitochondrial dynamics as shown in Fig. 1 [22, 25]. In addition to this, a common heterozygous mutation, 2877510G→A leads to idiopathic PD. This heterozygous mutation leads to formation of glycine to serine amino acid substitution at codon 2019 (Gly2019Ser) [22].

Representative pathways of mitochondrial dysfunctions involved in Parkinson’s disease pathophysiology. SNCA: synuclein alpha; LRRK2: leucine rich repeat kinase 2; VPS35: vacuolar protein sorting-associated protein 35; CHCHD2: coil-helix-coiled-coil-helix domain 2; PARK2: parkin 2; PINK1: PTEN-induced kinase 1; ATP13A2: ATPase 13A2. ↑increase, ↓decrease,  increase or decrease
increase or decrease
Vacuolar protein sorting-associated protein 35 (VPS35)
The relation between VPS35 and PD was observed first in European PD cohorts with history of an autosomal dominant inheritance [26,27,28]. The key role of VPS35 in mitochondria is the mitochondrial dynamics regulation through interaction with mitochondrial fusion/fission proteins. Multiple studies have been reported that mutation in VPS35 can trigger mitochondrial fragmentation, which leads to neurodegeneration [29, 30]. Apart from this, it was reported that increased interaction of dynamin-like protein (DLP) 1 with VPS35 mutant, supports change of the mitochondrial DLP1 complexes through the mitochondria-derived vesicle–dependent trafficking of various complexes to lysosomes for degradation. Interestingly, oxidative stress elevates the VPS35-DLP1 interaction, which also observed to be raises in the sporadic PD patients [30].
Coiled-coil-helix-coiled-coil-helix domain 2 (CHCHD2)
Recently, it has been reported that CHCHD2 mutation causes late-onset, autosomal dominant PD in three Japanese families [31]. Mitochondrial inter-membrane space protein CHCHD2 shows functions in the nucleus and mitochondria. In normal conditions, CHCHD2 is bound to mitochondrial complex IV and mutation of CHCHD2 has shown decreased mitochondrial complex IV activity, which results into mitochondrial fragmentation and increases in ROS production [32, 33]. Interestingly, CHCHD2 was found that it translocates in the nucleus and act as a transcription factor under stress conditions. Furthermore, several models expressing mutants associated PD also shown biochemical and structural mitochondrial abnormalities leading to motor dysfunction and dopaminergic neurodegeneration. The mutant CHCHD2 leads to impairment of mitochondrial function which results into the progression of PD [34].
Parkin protein gene (PARK2)
Parkin protein in humans is encoded by the PARK2 gene. Mutations in PARK2 gene cause Parkinson’s disease, especially autosomal recessive juvenile Parkinson’s disease. Parkin is a cytosolic E3 ubiquitin ligase. Target proteins for proteasomal degradation are ubiquitinated by Parkin [35, 36]. Parkin plays key role in maintaining healthy mitochondria by regulating their biogenesis and degradation through mitophagy [36]. The removal of damaged mitochondria from the healthy mitochondrial pool and allows their degradation through the autophagy-lysosomal pathway by the process of mitophagy. Parkin is also known to regulate the functional mitochondrial pol by mitochondrial biogenesis regulation [37]. In normal condition, it interposes the degradation of parkin interaction substrate (PARIS), leading to transcriptional activation of nuclear translocation of PGC1α and mitochondria-associated genes [38]. Accordingly, loss in Parkin function facilitates PARIS to accumulate and supress mitochondrial biogenesis, which results in mitochondrial functional defects.
PTEN-induced kinase 1 (PINK1)
Mutations in PINK1 is very known common causes of the autosomal recessive early-onset PD. PINK1, which plays an important role in balancing mitochondrial homeostasis, impairs several aspects of mitochondrial biology, including morphology, degradation, and trafficking [39,40,41]. Most widely reported role of PINK1 is in the mitochondria mitophagy, promoting removal of damaged mitochondria by activating and recruiting Parkin [42]. PINK1 activates Parkin by two mechanisms: (1) trans-activation by phosphorylation of ubiquitin at S65 and further binding to Parkin and (2) direct Parkin phosphorylation at S65. Loss of PINK1 leads to wide range of mitochondrial dysfunction in mice, cell models, and Drosophila [41]. These mainly result into loss of PINK1 mitophagy. Mutant PINK1 protein leads to development of improperly folded proteins in the mitochondria. Mutations in the threonine/serine kinase domain of PINK1 have been found Parkinson’s patients [39, 43]. There are several studies reported that PINK1 is basically mitochondrial site located and this may show a protective effect on cell that is affected by the mutations, and that leads to increased susceptibility to cellular stress. This suggests a direct relation between the pathogenesis of PD and mitochondria [39].
ATPase cation transporting 13A2 (ATP13A2)
An enzyme found in humans, probable cation-transporting ATPase 13A2, mainly involved in the transport of divalent transition metal cations [44]. Mutant ATP13A2 causes Kufor-Rakeb syndrome (KRS). It is an autosomal recessive juvenile-onset PD [45]. ATP13A2 mitochondrial function was firstly recognized in mitochondrial dysfunction in KRS patient-derived skin fibroblast [46]. Several studies have been also reported that ATP13A2-deficient models demonstrates mitochondrial dysfunction, decreased ATP production, elevated ROS production, and increased mitochondrial fragmentation as shown in Fig. 1. Additionally, deregulation of Zn2+ metabolism leads to lysosomal dysfunction, which further lead to defective mitophagy. This shows that associated pathways is involved in the pathogenesis of PD [47, 48].
Alzheimer’s disease (AD)
Alzheimer disease (AD) is a very common and disabling neurodegenerative disorder which is a form of dementia of the aged [49]. Its incidence increases along with age and thereby it is a significant public-health concern. In the late stages of AD, severe memory loss is observed and serious neurodegeneration is obvious [50].
Amyloid protein precursor (APP)
Highly conserved and an ancient protein APP is a precursor, produces amyloid beta (Aβ), a polypeptide which contains 37 to 49 amino acid residues. In the brain of Alzheimer’s patient the amyloid fibrillar form of Aβ that is amyloid plaques was observed [51]. Mutations in APP gene, causes familial susceptibility to Alzheimer’s disease. It has been reported that APP duplications or APP mutations located around the β cleavage site leads to the overall increase in production of Aβ species which causes early-onset Alzheimer disease (EOAD) [51].
Presenilin
Presenilin, a sub-component of γ-secretase, is basically responsible for the APP cutting. A γ-secretase can intercept APP at multiple points within protein, which leads to the formation of Aβ of different lengths in relation with Alzheimer’s disease; 40 and 42 amino acids long [52, 53]. It has been reported that Aβ 42 have more chances to form plaques in the brain than Aβ 40. Mutant presenilin leads to an elevation in the ratio of Aβ 42 production as compared to Aβ 40. This mutation also results into a decrease in amyloid precursor protein-derived amyloid β-peptide generation [54]. The loss in presenilin function causes an incomplete degradation of the amyloid β-peptide which contributes to an increased vulnerability of the brain, and therefore became a cause EOAD [55].
AvaII16390
The human mitochondrial genome contains AvaII in a non-coding region. The frequency of AvaII16390 in the Alzheimer’s brain was investigated [50]. The DNA sequence analysis of the AvaII16390 has been shown that the major change in the sequence was a C to T transition mutation at position 5 of the AvaII site [56]. Increase frequency of AvaII16390 in the Alzheimer’s brain may contribute to the formation of oxidative radicals. Some studies found that no notable relation in between the person age and AvaII16390 frequency [57].
Cytochrome c oxidase CO1 and CO2
Cytochrome c oxidase (CO) encoded exclusively by two mitochondrial genes, CO1 (subunits I) and CO2 (subunits II). It was found that cytochrome c oxidase activity decline in peripheral tissue and brain especially in late-onset AD patients [58]. Higher frequency of specific missense mutations in the mitochondrial CO1 and CO2 genes were AD showed the strong association in between the genes and AD. A mutant mitochondrial DNA molecule revealed the decrease in CO activity and elevated production of ROS [59]. It has been reported that a CO defect may directly participate in a cascade of events that result in AD. They again identified that AD mother’s asymptomatic child had more number of these mutations than child of AD fathers. These mutations are maternally inherited [60].
16S rRNA
Mitochondrial 16S rRNA alteration in most of the AD patients is considering the possible reason for its involvement in AD but additional studies are required to clarify the possible role of rRNA mutations in pathogenesis. One of the possible roles of genetic mutation is to interfere with the normal tRNA protection activity [61].
Apolipoprotein E (APO E)
APO E consists of three different isoforms such as apo e2, e3, and e4 which are different at 299 amino acid chain [62]. Presence of APO E e4 allele confers considerable risk for late-onset AD, which may be sporadic or familial [63]. The brain APO E is the principal cholesterol carrier involve in the disease by cholesterol dyshomeostasis [64]. Existing studies suggested that rise in the cholesterol content, increased the risk of developing AD [65].
Huntington’s disease (HD)
Huntington’s disease (HD) is an untreatable, late-onset, slowly progressive, neurodegenerative disease caused by genetic mutation which leading to an expanded polyglutamine (polyQ) for which no suitable therapy is currently available [66, 67]. HD is identified by ataxia, chorea and dementia [68]. Another type of peptide having 23 aa known as P42 shows protective action by preventing the polyQ-hHtt aggregation [69]. The neuropathological classification of HD disease involved 5 classes (0–IV). Among all the classes, class-IV is considered to be more severe which shows increase loss of neurons [70].
Huntington (Htt) gene
Htt is a 3144 amino acids containing protein having molecular weight about 350 kDa, ever present in the brain as well as peripheral tissues [71]. Htt mostly found in the cell organelles like cytoplasm, along with the mitochondria [72]. The role of Htt protein is not well established but various studies show that it may play a vital role in the development of neurons [66, 73]. Huntington protein is an essential for regulating axonal transport of vesicles including brain-derived neurotrophic factor (BDNF) [74].
Mitochondrial structural genes (Drp1, Fis1, Mfn1, Mfn2 and Opa1)
Different forces like fission and fusion are responsible for changing morphology of mitochondria [75]. Various mitochondrial structural genes are also known as shaping proteins because which are responsible for maintaining the proper morphology of mitochondria [76]. Free radical in mitochondria activates the Dynamin-related protein 1 (Drp1) and mitochondrial fission 1 (Fis1) protein which are responsible for the mitochondrial fission. Mitochondrial fusion proteins are Mfn1 (mitofusin 1), Mfn2 (mitofusin 2), and Opa1 (optric atrophy 1) which are the GTPase proteins [77]. Increase expression of fission proteins as well as decreased expression of fusion proteins may be the reason for change in mitochondrial dynamics which leads to neuronal damage in HD brain [78].
Friedreich’s ataxia (FA)
The neurodegenerative disorder Friedreich’s ataxia (FRDA) is an adolescent autosomal recessive disorder caused by mutations in frataxin, a mitochondrial protein whose function remains controversial [79]. It is a prevalence of approximately 1 in 50,000. Dysarthria, progressive ataxia, skeletal deformities, pyramidal features, hypertrophic cardiomyopathy, and hyporefexia are the major clinical sign of the FA [80]. Some established reports shows that mitochondrial enzymes such as pyruvate glutamate dehydrogenase, α-ketoglutarate dehydrogenase, and dehydrogenase activities are decreased in FA cells [81,82,83].
Frataxin
Friedreich’s ataxia is caused due the mutation of a 210 amino acid protein called frataxin [84]. Although the exact role is not fully understood, it may be vital for the proper functioning of mitochondria. The main cause involves the tri-nucleotide GAA repeat expansion within the intron of the frataxin gene [85]. The increase iron content has been reported in mitochondria suggesting that frataxin plays a major role in transportation of the iron [86]. Mutated frataxin result into the aconatse and mitochondrial Fe-S (iron-sulfur) respiratory enzyme deficiency in FA [87].
Conclusion
There are several reported factors which cause neurodegenerative disorders such as PD, AD, HD, and FRDA. Among all these, mitochondrial dysfunction plays an important role in the etiology and pathogenesis of these disorders. Hence, this article is mainly focusing on the causality relationship between gene-associated mitochondrial dysfunction which leads to development of neurodegenerative disorders as shown in Table 1. The additional studies are needed to clarify the possible pathogenic role of mtDNA mutations. Rapid advances in these types of knowledge have created an unmatched and great opportunity towards the study of mitochondrial dysfunction in neurodegenerative disorders. This also creates an opportunity for the research and development of drugs or therapies which targets mitochondrial genes, whose mutation leads to the generation of neurodegenerative disorders.
Availability of data and materials
Not applicable
Abbreviations
- ATP:
-
Adenosine triphosphate
- NAD:
-
Nicotinamide adenine dinucleotide
- FAD:
-
Flavin adenine dinucleotide
- TCA:
-
Tricarboxylic acid
- RNAs:
-
Ribonucleic acids
- PD:
-
Parkinson’s disease
- AD:
-
Alzheimer’s disease
- HD:
-
Huntington’s disease
- FRDA:
-
Friedreich’s ataxia
- α-Syn:
-
α-synuclein
- WT:
-
Wild type
- ROS:
-
Reactive oxygen species
- MAM:
-
Mitochondria-associated membranes
- ER:
-
Endoplasmic reticulum
- MSA :
-
Multiple system atrophy
- DLB :
-
Dementia with Lewy bodies
- DLP:
-
Dynamin-like protein
- PGC1α:
-
Proliferator-activated receptor gamma coactivator 1-α
- MEF2C:
-
Myocyte-specific enhancer factor 2C
- LRRK2:
-
Leucine rich repeat kinase 2
- VPS35:
-
Vacuolar protein sorting-associated protein 35
- CHCHD2:
-
Coil-helix-coiled-coil-helix domain 2
- PARK2:
-
Parkin 2
- PARIS:
-
Parkin interaction substrate
- PINK1:
-
PTEN-induced kinase 1
- ATP13A2:
-
ATPase 13A2
- APP:
-
Amyloid protein precursor
- Aβ:
-
Amyloid beta
- CO:
-
Cytochrome c oxidase
- rRNA:
-
Ribosomal ribonucleic acid
- APOE:
-
Apolipoprotein E
- PolyO:
-
Polyglutamine
- Htt :
-
Huntington
- Drp1:
-
Dynamin-related protein 1
- Fis1:
-
Fission 1
- Mfn1:
-
Mitofusin 1
- Opa1:
-
Optric atrophy 1
- Tomm40 :
-
Translocase of outer membrane 40
- CypD:
-
Cyclophilin D
- GAA:
-
Guanine-adenine-adenine
References
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Beal MF (1995) Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 38(3):357–366
DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348(26):2656–2668
Mao P, Reddy PH (2010) Is multiple sclerosis a mitochondrial disease. Biochim Biophys Acta 1802(1):66–79
Chomyn AA (2003) G MtDNA mutations in aging and apoptosis. Biochem Biophys Res Commun 304(3):519–529
Wallace DC, Ruiz-Pesini E, Mishmar D (2003) mtDNA variation, climatic adaptation, degenerative diseases, and longevity. Cold Spring Harb Symp Quant Biol 68:471–478
McKenzie M, Liolitsa D, Hanna MG (2004) Mitochondrial disease: mutations and mechanisms. Neurochem Res 29(3):589–600
Aruoma OI, Kaur H, Halliwell B (1997) Oxygen free radicals and human diseases. J R Soc Health 111:172–177
Sullivan PG, Brown MR (2005) Mitochondrial aging and dysfunction in Alzheimer's disease. Prog Neuropsychopharmacol 29(3):407–410
Grazina M, Pratas J, Silva F, Oliveira S, Santana I, Oliveira C (2006) Genetic basis of Alzheimer's dementia: role of mtDNA mutations. Genes Brain Behav 5:92–107
Zhu X, Smith MA, Perry G, Aliev G (2004) Mitochondrial failures in Alzheimer's disease. Am J Alzheimers Dis Other Demen 19(6):345–352
Cha MY, Kim DK, Mook-Jung I (2015) The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med 47(3):e150–e150
Keogh MJ, Chinnery PF (2015) Mitochondrial DNA mutations in neurodegeneration. Biochimica et Biophysica Acta (BBA)-Bioenerg 1847(11):1401–1411
Zhunina OA, Yabbarov NG, Grechko AV, Yet SF, Sobenin IA, Orekhov AN (2020) Neurodegenerative diseases associated with mitochondrial DNA mutations. Curr Pharm Des 26(1):103–109
Chaudhuri KR, Schapira AH (2009) Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8(5):464–474
Mullin S, Schapira A (2003) Alpha-Synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol Neurobiol 47(2):587–597
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276(5321):2045–2047
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T (2013) α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 125(5):753–769
Ryan BJ, Hoek S, Fon EA (2015) Wade-Martins R mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 40(4):200–210
Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon EA, Przedborski S (2014) α-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34(1):249–259
Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ, Noble W, Hanger DP, Miller CC (2017) α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol 134(1):129–149
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44(4):601–607
Lill CM (2016) Genetics of Parkinson’s disease. Mol Cell Probes 30(6):386–396
Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 40(4):200–210
Santos D, Esteves AR, Silva DF, Januario C, Cardoso SM (2015) The impact of mitochondrial fusion and fission modulation in sporadic Parkinson’s disease. Mol Neurobiol 52(1):573–586
Papkovskaia TD, Chau KY, Inesta-Vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AH, Cooper JM (2012) G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum Mol Genet 21(19):4201–4213
Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89(1):168–175
Small SA, Petsko GA (2015) Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci 16(3):126–132
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, Xiong WC (2015) VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep 12(10):1631–1643
Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X (2016) Parkinson's disease–associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22(1):54–63
Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H, Tomiyama H (2015) CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol 14(3):274–282
Aras S, Bai M, Lee I, Springett R, Huttemann M, Grossman LI (2015) MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 20:43–51
Meng H, Yamashita C, Shiba-Fukushima K, Inoshita T, Funayama M, Sato S, Hatta T, Natsume T, Umitsu M, Takagi J, Imai Y (2017) Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat Commun 8(1):1–18
Tio M, Wen R, Lim YL, Zukifli ZHB, Xie S, Ho P, Zhou Z, Koh TW, Zhao Y, Tan EK (2017) Varied pathological and therapeutic response effects associated with CHCHD2 mutant and risk variants. Hum Mutat 38(8):978–987
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608
Von Coelln R, Dawson VL, Dawson TM (2004) Parkin-associated Parkinson’s disease. Cell Tissue Res 318(1):175–184
Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37(6):315–324
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206(5):655–670
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304(5674):1158–1160
Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W (2010) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6(7):871–878
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460(1):127–141
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510(7503):162–166
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205(2):143–153
Park JS, Blair NF, Sue CM (2015) The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mo Disord 30(6):770–779
Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, Sue CM, Klein C (2012) ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging 33(8):1843–18e1
Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ (2012) PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet 21(8):1725–1743
Park JS, Koentjoro B, Davis RL, Sue CM (2016) Loss of ATP13A2 impairs glycolytic function in Kufor-Rakeb syndrome patientderived cell models. Parkinsonism Relat Disord 27:67–73
Tsunemi T, Krainc D (2014) Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alphasynuclein accumulation. Hum Mol Genet 23(11):2791–2801
Swomley AM, Förster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA (2014) Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim Biophys Acta Mol Basis Dis 1842(8):1248–1257
Markesbery WR (1999) The role of oxidative stress in Alzheimer disease. Arch Neurol 56(12):1449–1452
Wallon D, Rousseau S, Rovelet-Lecrux A, Quillard-Muraine M, Guyant-Maréchal L, Martinaud O, Pariente J, Puel M, Rollin-Sillaire A, Pasquier F, Le Ber I (2012) The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J Alzheimers Dis 30(4):847–856
Annaert W, De Strooper B (2002) A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol 18(1):25–51
McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K (2005) Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47(2):191–199
Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, Haass C (2002) Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J 21(20):5408–5416
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45:358–368
Chang SW, Zhang D, Chung HD, Zassenhaus HP (2000) The frequency of point mutations in mitochondrial DNA is elevated in the Alzheimer’s brain. Biochem Biophys Res Commun 273(1):203–208
Kadenbach B, Münscher C, Frank V, Müller-Höcker J, Napiwotzki J (1995) Human aging is associated with stochastic somatic mutations of mitochondrial DNA. Mutat Res 338(1-6):161–172
Parker WD, Parks JK (1995) Cytochrome c oxidase in Alzheimer's disease brain: purification and characterization. Neurology 45(3):482–486
Wei YH, Lu CY, Lee HC, Pang CY, Ma YS (1998) Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function a. Ann N Y Acad Sci 854(1):155–170
Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD (1997) Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci 94(9):4526–4531
Noller HF (1991) Ribosomal RNA and translation. Annu Rev Biochem 60(1):191–227
Davignon J, Bouthillier D, Nestruck AC, Sing CF (1988) Apolipoprotein E polymorphism and atherosclerosis: insight from a study in octogenarians. Trans Am Climatol Clin Assoc 99:100
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PS, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C (1993) Association of apolipoprotein E allele ϵ4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43(8):1467
Mori T, Paris D, Town T, Rojiani AM, Sparks DL, Delledonne A, Crawford F, Abdullah LI, Humphrey JA, Dickson DW, Mullan MJ (2001) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APPsw mice. J Neuropathol Exp Neurol 60(8):778–785
Vance JE, Hayashi H, Karten B (2005) Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol 16(2):193–212
MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72(6):971–983
Zhang Q, Lei YH, Zhou JP, Hou YY, Wan Z, Wang HL, Meng H (2019) Role of PGC-1α in mitochondrial quality control in neurodegenerative diseases. Neurochem Res 44(9):2031–443.
Roos RA, Bots GT (1983) Nuclear membrane indentations in Huntington's chorea. J Neurol Sci 61(1):37–47
Arribat Y, Bonneaud N, Talmat-Amar Y, Layalle S, Parmentier ML, Maschat F (2013) A huntingtin peptide inhibits polyQ-huntingtin associated defects. PLoS One 8(7):68775
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44(6):559–577
Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621
Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR (2002) Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem 277(9):7466–7476
Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, Goldowitz D (2011) Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci Res 31(41):14794–14799
DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM (1995) Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14(5):1075–1081
Reddy PH (2009) Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol 218(2):286–292
Dhingra R, Kirshenbaum LA (2014) Regulation of mitochondrial dynamics and cell fate. Circulation 78(4):803–810
Reddy PH (2007) Mitochondrial dysfunction in aging and Alzheimer's disease: strategies to protect neurons. Antioxid Redox Signal 9(10):1647–1658
Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Hemachandra Reddy P (2011) Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum Mol Genet 20(7):1438–1455
Wilson RB, Roof DM (1997) Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet 16(4):352–357
Geoffroy G, Barbeau A, Breton G, Lemieux B, Aube M, Leger C, Bouchard JP (1976) Clinical description and roentgenologic evaluation of patients with Friedreich's ataxia. Can J Neurol Sci 3(4):279–286
Kark RP, Blass JP, Engel WK (1974) Pyruvate oxidation in neuromuscular diseases: evidence of a genetic defect in two families with the clinical syndrome of Friedreich's ataxia. Neurology 24(10):964
Blass JP, Kark RP, Menon NK, Harris SE (1976) Low activities of the pyruvate and oxoglutarate dehydrogenase complexes in five patients with Friedreich's ataxia. N Engl J Med 295(2):62–67
Barbeau A (1984) The Quebec cooperative study of Friedreich's ataxia: 1974-1984—10 years of research. Can J Neurol Sci 11(S4):646–660
Shan Y, Napoli E, Cortopassi G (2007) Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum Mol Genet 16(8):929–941
Campuzano V, Montermini L, Molto MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial iron–Sulphur protein deficiency in Friedreich ataxia. Nat Genet 17(2):215–217
Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276(5319):1709–1712
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
VM contributed in preparation primary content. He performed extensive literature survey and compile the content. NW contributed in preparation of figures and table. PT contributed in checking of manuscript and correction of grammatical mistake. MU contributed in preparation of figure. AU contributed in finalization of manuscript and in its correction. MK contributed in finalization of content, preparation of concrete manuscript, and in schematic presentation of content. BT contributed in checking of manuscript and correction of grammatical mistake. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marde, V.S., Tiwari, P.L., Wankhede, N.L. et al. Neurodegenerative disorders associated with genes of mitochondria. Futur J Pharm Sci 7, 66 (2021). https://doi.org/10.1186/s43094-021-00215-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00215-5