
Abstract
α-Synuclein (SNCA) is a substantive component of Lewy bodies, the pathological hallmark of Parkinson’s disease (PD). The discovery and subsequent derivation of its role in PD has led to a suprising but fruitful convergence of the fields of biochemistry and molecular genetics. In particular, the manipulation of the cell lines of a number of forms of familial PD has implicated SNCA in distinct and diverse biochemical pathways related to its pathogenesis. This current and rapidly evolving concept indicates PD is a disease in which interacting pathways of oxidative stress, mitochondrial dysfunction and impaired regulation of protein turnover interact to cause dopaminergic cell dysfunction and death. SNCA has a central role in these processes and manipulation of its expression, degradation and aggregation appear to be promising neuroprotective therapeutic targets.
Similar content being viewed by others
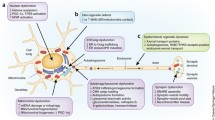


Parkinson’s Disease and α-Synuclein
PD is a clinical diagnosis characterised by an asymetrical resting tremor, increased tone and bradykinesia. Anosmia, sleep disturbance, constipation and other premotor symptoms may precede this presentation by some years. Cognitive impairment is an early sign of PD and advanced disease is frequently accompanied by a dementia with or without visual hallucinations which overlaps clinically with dementia with Lewy bodies. It has a lifetime risk of 4–5 %, making it the second most common neurodegenerative disease after Alzheimer’s disease. Current demographic trends predict a doubling in the number of cases by 2050. It is caused in part by progressive dopaminergic degeneration predominately in the substantia nigra. Traditionally, this was characterized histologically by the presence of Lewy bodies although in recent years a number of familial forms of PD have been identified without them.
The characterisation of α-synuclein’s (SNCA) role in the pathogenesis of PD began with the discovery of three familial forms of PD associated with mutations in the SNCA gene. Three autosomal dominant mutations which are associated with Lewy body formation—an alanine-to-threonine substitution at position 53 (A53T) with onset of PD in the fifth to sixth decade, an alanine to proline substitution at position 30 (A30P) with onset in the third to fifth decade and a glutamate to lysine substituion at position 46 (E46K) with onset in the fifth to seventh decade—were identified [1–3]. The SNCA locus was subsequently shown to be associated with sporadic PD [4–6] and SNCA was demonstrated within the Lewy bodies of sporadic PD patients in its fibrillar form [7–9]. Transgenic animal models carrying the A30P and A53T mutations have been produced; however, precise replication of the clinical and histological findings of PD patients have been inconsistent. In particular, only A30P and A53T Drosphilia and A53T rats mimic l-DOPA-responsive motor symptoms whilst Lewy body formation does not occur in A30P and A53T Thy1 mice or A53T rats [10]
α-Synuclein
α-synuclein (SNCA) is a small, highly acidic native unfolded protein molecule of unknown function. It is the principal component of Lewy bodies [7–9] and is localized in neuronal and glial cells of the substantia nigra, thalamus, neocortex and hippocampus. It is predominantly a cystolic protein which has been shown to disrupt directly mitochondrial morphology and to bind rapidly and specifically with mitochondrial membranes [11–13].
SNCA’s function is unclear. It appears to be particularly prone to rapid conformational change and has been implicated in a diverse range of roles including the ubiquitin–proteasome system, maintenance of synaptic functionality and regulation of oxidative stress [14–17]. Dopamine release has been shown to be reduced in the presence of overexpressed wild-type and mutant A53T SNCA. The protein’s ability to modulate the activity of tyrosine hydroxylase suggests it may have a role in dopamine metabolism [18, 19]. Dopamine has been shown to bond covalently with SNCA and this compound leads to dopamine dependent cell toxicity [20, 21]. Mutant SNCA appears to aggregate more readily with dopamine compared to wild-type [22]. Overexpression of mutant or wild-type SNCA leads to dose-dependent dopamine cell toxicity [23] Thus there is evidence to support an interaction direct or indirect, between SNCA and dopamine which enhances dopaminergic neuronal dysfunction.
All three pathological SNCA mutations have been shown to cause dopaminergic toxicity in vitro and in vivo [24]. Polymorphisms of SNCA have shown associations with sporadic PD [4–6] which have correlated with increased levels of plasma SNCA [25]. Moreover, duplications in the REP1 transcription promoter region of the SNCA represent a risk factor for idiopathic PD [26–28]. Expansion of this region upregulates production of SNCA in mice suggesting that SNCA overproduction mediates the associated parkinsonian features [29].
Whilst Lewy bodies seem to be indicative of sporadic PD, their appearance appears to be an early manifestation of pathogenesis which may occur in advance of the motor features of classical PD; although the extent of the correlation between Lewy bodies and preclinical PD is controversial [30–32]. Nitrated and phosphorylated forms of SNCA have been observed within Lewy bodies and SNCA can be made to undergo fibrillisation in vitro [33]. Lewy pathology appears to be capable of spreading from neuron to neuron and there is some evidence to suggest that SNCA may propagate aggregation in a prion-like manner [34–37]. Mutant SNCA appears to show either faster or slower aggregation dynamics than wild-type SNCA [38, 39] suggesting that the process of aggregation is related to SNCA-induced neurotoxicity. That said, some data suggest SNCA aggregates in a neuroprotective response to oxidative stress [40, 41] and the exact contribution of SNCA or indeed SNCA aggregation to dopaminergic cell toxicity in PD remains unclear.
Oxidative Stress and α-Synuclein
The observation in the 1980s that 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) produced parkinsonism suggested that environmental agents might be involved in dopaminergic degeneration in PD [42, 43]. MPTP is a complex I inhibitor which may additionally generate free radicals including nitric oxide through its toxic metabolite 1-methyl 4-phenylpyridinium (MPP+) [44, 45]. Rotenone—a pesticide commonly used in the USA which has been shown to cause parkinsonism with SNCA aggregates in animal models—is also a potent complex I inhibitor. Exposure to both provides useful models for in vivo and in vitro study of parkinsonian degeneration [46–48]. Both agents induce cell apoptosis that can be blocked by ciclosporin [49]. There is evidence that MPTP-induced mitochondrial inhibition may be mediated by an alteration of endoplasmic reticulum and mitochondrial calcium homeostasis and that SNCA has a role in its regulation. Endoplasmic reticulum stress has also been implicated in the formation of toxic synuclein oligomers [50, 51]. Cigarette smoking, which has consistently been associated with a reduced risk of PD, has been shown to influence complex I activity [52]. Complex I inhibition is therefore a useful paradigm in dopaminergic degeneration [53].
The question of whether these or other environmental insults may act as trigger events in PD has been raised. Although a unifying theory of PD as an unfolding consequence of one or multiple environmental insults leading to oxidative stress and mitochondrial inhibition is attractive, there is little evidence to support it. Although statistically significant associations between PD and environmental toxins have been demonstrated, the odds ratios are at best modest and imply a weak influence only, compared to age which is epidemiologically by far the most significant risk factor for PD [54–56].
Complex I inhibitors have been shown to interact with SNCA. Normal expression but not overexpression of wild-type SNCA protects against oxidative stress [57]. In comparison to wild-type, mutant SNCA potentiates rotenone induced cell toxicity [58–60]. Hence environmental toxins can be linked to the pathogenesis of parkinsonism and extrinsic mediators of oxidative stress would seem likely to be one of many contributing factors in its pathogenesis.
Age and α-Synuclein
Age has consistently been found to be the greatest risk factor for the development of PD [54, 55]. Lewy body formation occurs in the absence of parkinsonian features in 8–17 % of over 60 year olds [61–63]; however, the extent of the correlation is controversial. Broadly, Lewy bodies are seen as a precursor marker of PD, although it has been suggested that some cases may instead be indicative of preclinical dementia with Lewy bodies [30–32]. A distinct pattern of dopaminergic cell loss has been demonstrated in the histology of PD brains compared to aged controls [64–67]. Moreover, expression of SNCA increases with age in normal primate brains. This occurs in parallel to decreased expression of tyrosine hydroxylase, a surrogate marker of dopaminergic neurons [68]. These findings and others have led parkinsonian degeneration to be hypothesised as an accelerated variant of the normal process of ageing, with inadequate degradation of mutant or overexpressed levels of wild-type SNCA playing a central role [69, 70].
Disposal of SNCA
SNCA is degraded predominantly by chaperone-mediated autophagy (CMA). There is evidence of less significant disposal of SNCA by proteasomal and alternative autophagal mechanisms intracellularly [71, 72]. CMA is a form of autophagy dependent on the presence of heat shock protein 70 (Hsp70) to chaperone protein transfer into the lysosome via lysosome-associated membrane protein 2 (LAMP2A). Here, it is engulfed into the matrix with the assistance of another molecular chaperone, lysosomal hsc73 (lys hsc73). Hsp70 is upregulated in response to ischaemia and other stressors [73] and has been shown to sequester SNCA in an adenosine triphosphate (ATP)-independent manner [74, 75], impair fibrillisation of SNCA [76], reduce SNCA aggregates in vivo and protect against SNCA toxicity in vitro [75, 77].
CMA differs from other forms of autophagy in that it is not dependent on vesicle formation or altered membrane potential [78]. Impaired CMA through downregulation of LAMP2A proteins slows down SNCA degradation in wild-type SNCA neuroblastoma cell lines and may induce enhanced macroautophagy. Markers of CMA are reduced in PD brains [73] whilst A53T mutant SNCA and dopamine-modified SNCA have been shown to inhibit CMA causing cell toxicity [79, 80]. Interestingly, CMA and other types autophagy have been identified as significant mediators of cell ageing and this resonates with the observation that age is a risk factor for sporadic PD. Cellular dependence on CMA as the preferred mechanism of SNCA degradation has aroused interest in CMA as a potential target susceptible to upregulation.
Glucocerebrosidase and α-Synuclein
Deficiency of the glucocerebrosidase gene (GBA) leads to the lysosomal storage disease Gaucher’s disease. It has been observed that those with GBA mutations are predisposed to early onset PD [81–83]. Heterozygous mutations of GBA which do not cause glucosylceramide accumulation have been shown to be associated with an increased risk of PD [83–86] and glucocerebrosidase enzyme (GCase) deposits have been demonstrated within Lewy bodies [87]. Within a large cohort of PD patients the presence of Lewy bodies correlated with GBA mutations (in a subset where brain tissue was available) as did early onset symptoms, dementia and hallucinations [88]. Moreover, severity of parkinsonian features corresponds to that of the Gauchers phenotype encoded for by the respective GBA mutation [89] and non-motor features are more commonly seen in PD patients with GBA mutations [90]. A number of mechanisms for these observations have been proposed. One hypothesis is that defective GCase activity may disrupt lysosomal breakdown and disposal of SNCA, leading to an accumulation of intracellular SNCA [91]. GBA heterozytous mutations result in reduced activity and expression of GCase with evidence of endoplasmic reticulum stress in brains of patients with PD [92]. Recent data have also indicated that there may be a symbiotic relationship between SNCA and GBA mutations. Reduced GCase activity has been demonstrated in both GBA PD and sporadic PD brains compared to aged and Alzheimer’s disease control brains [92]. GCase activity appears to be reduced by overexpression of wild-type SNCA in SH-SY5Y cells, possibly by way of inhibition of intracellular GCase trafficking in the endoplasmic reticulum/Golgi apparatus [93]. GCase inhibition of SH-SY5Y cells leads to decreased mitochondrial membrane potential, reduced ADP phosphorylation and increased free radical production [94]. It has been suggested that SNCA disrupts GCase trafficking leading to altered GCase activity within the lysosome, causing impaired autophagy and, in turn, impaired disposal of defective mitochondria [93]
Mitochondrial Quality Control
Neuronal and glial cells contain a network of mitochondria propagated along neuronal axons with maximal density at the perinuclear region. The cellular function of eukaryotic cells is dependent on an abundant and dynamic supply of ATP supplied by a healthy and productive pool of mitochondria. Neurons are particularly dependent upon oxidative phosphorylation for their function including the dynamic process of synaptic remodeling. Defective mitochondria can result from defective mitochondrial DNA (mtDNA), nuclear mutations of mitochondrial proteins or following external metabolic insults. These factors in isolation or in combination will reduce mitochondrial membrane potential and (if unchecked) lead to cell apoptosis [95].
Quality control and exchange of mtDNA within the mitochondrial pool are regulated through a constant and dynamic process of fission, fusion and autophagic destruction (mitophagy) with subsequent lysosomal degradation. The importance of this mechanism of quality control is emphasised by the observation that fusion/fission-deficient mitochondria display a loss of directed movement and are swollen, aggregated and unable to migrate into neurites [96, 97]. Moreover, manipulation of fusion and fission machinery can lead to spontaneous generation of mtDNA mutations associated with hereditary mitochondrial disease, whilst impairment of proper fusion/fission dynamics is directly implicated in a diverse range of neurodegenerative diseases [96, 98].
Mitophagy is a selective process in which damaged or incompetent mitochondria are isolated and removed. This is independent of the process of bulk autophagy which occurs in response to a major cellular insult [99, 100]. In contrast, defective mitochondria in healthy cells are selectively removed. This process appears to be mediated by impaired mitochondrial fusion and can be halted by impairment of fission machinery [99, 101].
Mitochondrial Dysfunction and α-Synuclein
SNCA has been shown to cause direct mitochondrial toxicity. Overexpression of wild-type SNCA in GT-17 cells leads to giant mitochondria containing laminated bodies, autophagosomes and decreased MTT levels [102]. A53T transgenic mice develop dysmorphic mitochondria with inclusions and damaged mitochondrial DNA [103]. In vitro culture of Caenorhabditis elegans overexpressing SNCA leads to fragmented mitochondria with inhibition of mitochondrial membrane fusion. Conversely, siRNA knock down of SNCA results in elongated mitochondria [104]. In primary cell culture of rat cortical neurons overexpressing wild-type SNCA, increased expression of autophagial markers and co-localisation of mitochondria with autophagosomes was demonstrated. This effect was amplified in cells carrying the A53T mutation [105]. A53T has also been shown to localise to the mitochondrial membrane, result in complex I inhibition and cause an increase in mitochondrial autophagy [106]. Thus it is clear that SNCA not only interacts directly with and damages mitochondria, but also it appears to do so through interference with mitophagy and mitochondrial fusion.
Mitochondrial Trafficking of SNCA Causing Complex I Inhibition
Mitochondrial DNA codes for 13 proteins, seven of which are essential components of the network of protein complexes which are responsible for the production of ATP. Substrate proteins for the other cellular functions performed by mitochondria must therefore be imported—recent proteomic studies suggest these number some 1,500 nuclear-encoded proteins [107, 108]. SNCA has been identified predominately in the mitochondrial outer membrane although under favourable energy and pH conditions it appears to migrate to the inner membrane [109–112]. The interaction between SNCA and mitochondrial membranes appears to be rapid, specific and independent of changes in membrane potential. There is some evidence that membrane contact causes a change of the SNCA conformation from a closed to an open state [13]. Once incorporated into the mitochondria, SNCA leads to fragmented, aggregated mitochondria. Interestingly, this finding is duplicated in the other SNCA mutations with the exception of A30P SNCA which does not cause such morphological abnormalities and does not interact with the mitochondrial membrane [13, 113].
Imported SNCA has been shown to cause mitochondrial complex I inhibition at low concentrations in a dose-dependent fashion [107, 109]. Import of extracellular proteins is mediated by a series of mitochondrial protein complexes which recognise targeting signals [114–116]. The location of the potential targeting sequence of SNCA remains unclear although the N terminus of the SNCA molecule appears to be a likely candidate [107]. Migration into the mitochondria is energy dependent and appears to be mediated through translocase of the outer membrane (TOM) 40 protein located in the mitochondrial extracellular membrane [117, 118].
Mutations in Mitochondrial Proteins in Parkinson’s Disease
Parkin
Parkin encodes E3 ubiquitin–protein ligase of unknown function associated with the endoplasmic reticulum, golgi apparatus, synaptic vesicles and mitochondria [109, 113, 116]. The autosomal recessive forms of familial juvenile onset PD caused by Parkin mutations lead to PD without Lewy body formation [119, 120]. Parkin also localizes to Lewy bodies in sporadic PD and dementia with Lewy bodies [121]. Homozygous alleles of Parkin cause early onset familial PD and heterozygous alleles may increase the risk of development of late-onset sporadic PD [122–125]. The lack of SNCA pathology in Parkin-mediated mitochondrial dysfunction and Parkin’s presence in (SNCA-mediated) sporadic PD implicates it in the dynamic processes of mitochondrial regulation and selection. This is confirmed by the observation that mutations of Parkin and PINK1 result in accumulation of defective mitochondria [126, 127].
Although the exact mechanism of its involvement in the mitochondrial regulation remains unclear, Parkin has been shown to arrest cell apoptosis through cytochrome C inhibition [128]. Other mechanisms have also been proposed including a number that are independent of E3 ligase activity (reviewed in further detail in [129]). Parkin knockout mice exhibit abnormal motor function but do not display dopaminergic neuronal degeneration in the substantia nigra [130, 131]; however, reduced respiratory capacity and increased susceptability to oxidative damage which results in dopaminergic neuronal damage have been demonstrated [132]. Functional interactions between mutant SNCA and Parkin have been observed in vitro—of particular interest is the fact that toxicity of mutant SNCA can be arrested by the E3 ligase activity of Parkin [133]. SNCA aggregates causing oxidative stress could represent the neuronal insult needed to overcome impaired Parkin-mediated mitochondrial quality control, leading to DA cell death. Accordingly, Parkin may not only represent a potential genetic marker of predisposition to PD but a therapeutic target susceptible to upregulation.
PINK1
PTEN-induced putative kinase 1 mutations (PINK1) encode a widely expressed 63 kDa relatively insoluble protein which incorporates an 8-kDa amino terminal mitochondrial targeting sequence located primarily near the mitochondrial inner membrane. Mutations of PINK1 cause an autosomal recessive form of PD. A number of pathological homozygous and heterozygous variants have been identified, leading to Parkinsonian features of varying type and severity with histological evidence of nigrostriatal cell loss [134, 135]. Overexpression of wild-type PINK1 protects against proteasome-induced cell damage and there is evidence that PINK1 overexpression may mediate protection against some features of SNCA-mediated cell damage [136].
Recent studies indicate that PINK1 and Parkin interact in a common pathway implicit in mitochondrial autophagy. Parkin overexpression can rescue mutant PINK1 phenotypes but not the other way round suggesting that Parkin functions downstream of PINK1 [137]. Assuming that Parkin overexpression replaces the deficit induced by PINK1 deficiency, it would seem logical that Parkin is a mediator of PINK1 mitoprotection [98]. This is borne out with the finding that co-expression of PINK1, Parkin or DJ1 rescues mitochondrial damage caused by overexpressed A53T SNCA in vitro, an effect which is not duplicated by the 9309D mutation in PINK1 [104].
DJ1
DJ1 is a cystolic and mitochondrial protein expressed in the peripheral tissues and brain. It is primarily located inner membrane space and matrix although DJ1 has been shown to translocate to the mitochondrial outer membrane in response to oxidative stress [138]. Mutations in DJ1 are a rare cause of autosomal recessive PD [139]. Downregulation of DJ1 may influence dopaminergic neuronal activity in the substantia nigra in a specific manner through modulation of uncoupling protein function and calcium signaling [140].
Screening for SNCA
The preclinical course of PD may last several years and interruption of dopaminergic cell damage by SNCA at an early stage as possible would likely offer the best chance of preventing the development of PD. SNCA and other markers of PD pathology have been proposed as targets for screening in premotor PD. SNCA and DJ1 have been detected in human saliva although significant differences in levels were not found in PD patients compared with controls [141]. DJ-1 appears to be present in the serum although correlation with parkinsonian features appear to be poor [142, 143]. Although early data correlating levels of cerebrospinal spinal fluid (CSF) SNCA and DJ-1 with PD were conflicting, recent studies have demonstrated promising results which indicate that CSF SNCA and DJ-1 may predict PD with a reasonable degree of sensitivity and specificity [144–147]. There is also interest in colonic and olfactory bulb biopsies as a means of` early histological diagnosis of premotor PD, although interpretation of the finding of Lewy pathology in these sites is controversial [148–150]. It seems likely that the practical use of any screening technology would be most useful in those with mild premotor symptoms or even asymptomatic PD: whether observed correlations are preserved in these groups is unclear. Similarly, the practicality, safety and cost-effectiveness of using invasive procedures to screen for PD remain to be seen.
Therapeutics
Whilst the prospect of neuroprotective therapeutics in PD is some years away the rapidly evolving understanding of the mechanisms underlying dopaminergic degeneration in PD has focused attention on a number of therapeutic targets [151, 152]. Downregulation of SNCA either by modifying its transcription or reducing expression post translationally would seem a promising area for future development [153–156]. However, this approach may be something of a blunt instrument as SNCA’s role in the dynamics of normal neuronal function has not been fully established. SNCA may to some extent actually mediate a dose-dependent neuroprotective effect, and some data suggest that post translational SNCA depletion may actually cause neurotoxicity [157]. That said, the observation that SNCA knockout mice appear not to display hallmarks of neuronal damage and have a reduced susceptibility to neurotoxicity is reassuring [158]. A more nuanced approach may be to target the chaperone mediated autophagy system that is primarily responsible for the disposal of intracellular SNCA. SNCA’s prion like characteristics have also aroused interest in SNCA as a potential therapeutic target. If the observation that Lewy pathology is propagated from PD brains to fetal stem cell implants is mediated by the transfer of SNCA from cell to cell, sequestering SNCA may halt its progression. Immunization with the SNCA antigen of transgenic PD mice expressing SNCA appears to show slowed PD development which may be due to antibody binding intracellularly inhibiting transcellular spread [159].
Concluding Remarks
The convergence of the fields of molecular genetics and biochemistry in PD have allowed progressive elucidation of the mechanisms of familial PD phenotypes from protein translation, through impaired intracellular function to their histological sequelae. The model that is evolving is complex and multifaceted and it is clear that a range of mechanisms are implicated in its pathogenesis [160]. Although its precise role in the routine maintenance of neuronal cell function is unclear, SNCA appears to be a principal (but not the sole) ingredient of the complex biochemical map of PD that is developing. Overexpression, mutation and impaired degradation of SNCA all give rise to parkinsonism or an increased predisposition to sporadic PD. It has been shown to be imported directly into the mitochondrial outer membrane and to interact with complex I, deficiency of which in PD brain provided the first link to mitochondrial dysfunction [161, 162]. Manipulation of neuronal SNCA levels presents a promising target for therapeutic interventions mediating neuroprotection against PD progression.
References
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez TE, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173
Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH (2009) PSG-PROGENI and GenePD investigators, coordinators and molecular genetic laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124:593–605
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron 66:646–661
Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF (2007) Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18:1543–1546
Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH (2011) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 286:20710–20726
Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH (2008) Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J Neurosci 28:12305–12317
Clayton DF, George JM (1998) The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 21:249–254
Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS (2010) Alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci USA 107:19573–19578
Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79
Tompa P (2005) The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett 579:3346–3354
Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ (2002) A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci 22:3090–3099
Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, Schapira AH (2000) Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum Mol Genet 9:2683–2689
Orth M, Tabrizi SJ, Tomlinson C, Messmer K, Korlipara LV, Schapira AH, Cooper JM (2004) G209A mutant alpha synuclein expression specifically enhances dopamine induced oxidative damage. Neurochem Int 45:669–676
Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA (2002) Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med 8:600–606
Moussa CE, Mahmoodian F, Tomita Y, Sidhu A (2008) Dopamine differentially induces aggregation of A53T mutant and wild type alpha-synuclein: insights into the protein chemistry of Parkinson’s disease. Biochem Biophys Res Commun 365:833–839
Bisaglia M, Greggio E, Maric D, Miller DW, Cookson MR, Bubacco L (2010) Alpha-synuclein overexpression increases dopamine toxicity in BE2-M17 cells. BMC Neurosci 11:41
Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci USA 108:4194–4199
Mata IF, Shi M, Agarwal P, Chung KA, Edwards KL, Factor SA, Galasko DR, Ginghina C, Griffith A, Higgins DS, Kay DM, Kim H, Leverenz JB, Quinn JF, Roberts JW, Samii A, Snapinn KW, Tsuang DW, Yearout D, Zhang J, Payami H, Zabetian CP (2010) SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch Neurol 67:1350–1356
Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C (2006) Genetic epidemiology of Parkinson’s disease (GEO-PD) consortium. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296:661–670
Mellick GD, Maraganore DM, Silburn PA (2005) Australian data and meta-analysis lend support for alpha-synuclein (NACP-Rep1) as a risk factor for Parkinson’s disease. Neurosci Lett 375:112–116
Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T (2006) Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum Mol Genet 15:1151–1158
Cronin KD, Ge D, Manninger P, Linnertz C, Rossoshek A, Orrison BM, Bernard DJ, El-Agnaf OM, Schlossmacher MG, Nussbaum RL, Chiba-Falek O (2009) Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum Mol Genet 18:3274–3285
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AH et al (1994) Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 35(1):38–44
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157
Frigerio R, Fujishiro H, Ahn TB, Josephs KA, Maraganore DM, DelleDonne A, Parisi JE, Klos KJ, Boeve BF, Dickson DW, Ahlskog JE (2011) Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies. Neurobiol Aging 32:857–863
Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA (2000) Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci USA 97:4897–4902
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106:13010–13015
Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) Alpha-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506
Schapira AH, Tolosa E (2010) Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol 6:309–317
Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 576:363–368
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA 97:571–576
Bayir H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, Zhao Q, Belikova NA, Vlasova II, Maeda A, Zhu J, Na HM, Mastroberardino PG, Sparvero LJ, Amoscato AA, Chu CT, Greenamyre JT, Kagan VE (2009) Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem 284:15951–15969
Orth M, Tabrizi SJ, Tomlinson C, Messmer K, Korlipara LV, Schapira AH, Cooper JM (2004) G209A mutant alpha synuclein expression specifically enhances dopamine induced oxidative damage. Neurochem Int 45:669–676
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980
Schapira AH (2006) Etiology of Parkinson’s disease. Neurology 66:S10–S23
Cleeter MW, Cooper JM, Schapira AH (1992) Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement. J Neurochem 58:786–789
Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF (1996) Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med 2:1017–1021
Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH (1994) Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett 345:50–54
Schober A (2004) Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res 318:215–224
Xiong N, Huang J, Zhang Z, Zhang Z, Xiong J, Liu X, Jia M, Wang F, Chen C, Cao X, Liang Z, Sun S, Lin Z, Wang T (2009) Stereotaxical infusion of rotenone: a reliable rodent model for Parkinson’s disease. PLoS One 4:e7878
Seaton TA, Cooper JM, Schapira AH (1998) Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res 809:12–17
Arduino DM, Esteves AR, Cardoso SM, Oliveira CR (2009) Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: relevance to Parkinson’s disease. Neurochem Int 55:341–348
Cali T, Ottolini D, Brini M (2011) Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 37:228–240
Smith PR, Cooper JM, Govan GG, Harding AE, Schapira AH (1993) Smoking and mitochondrial function: a model for environmental toxins. Q J Med 86:657–660
Schapira AH (2010) Complex I: inhibitors, inhibition and neurodegeneration. Exp Neurol 224:331–335
Bennett DA, Beckett LA, Murray AM, Shannon KM, Goetz CG, Pilgrim DM, Evans DA (1996) Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med 334:71–76
Morens DM, Davis JW, Grandinetti A, Ross GW, Popper JS, White LR (1996) Epidemiologic observations on Parkinson’s disease: incidence and mortality in a prospective study of middle-aged men. Neurology 46:1044–1050
Warner TT, Schapira AH (2003) Genetic and environmental factors in the cause of Parkinson’s disease. Ann Neurol 53(Suppl 3):S16–S23, discussion S23-5
Byers B, Cord B, Nguyen HN, Schule B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD (2011) SNCA triplication Parkinson’s Patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One 6:e26159
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306
Choong CJ, Say YH (2011) Neuroprotection of alpha-synuclein under acute and chronic rotenone and maneb treatment is abolished by its familial Parkinson’s disease mutations A30P, A53T and E46K. Neurotoxicology 32:857–863
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, Palmer TD, Pera RR (2011) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8:267–280
Bloch A, Probst A, Bissig H, Adams H, Tolnay M (2006) Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol 32:284–295
Frigerio R, Fujishiro H, Maraganore DM, Klos KJ, DelleDonne A, Heckman MG, Crook JE, Josephs KA, Parisi JE, Boeve BF, Dickson DW, Ahlskog JE (2009) Comparison of risk factor profiles in incidental Lewy body disease and Parkinson disease. Arch Neurol 66(9):1114–1119
Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF, DeLucia MW, Dickson DW (2006) Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology 66:1100–1102
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301
Irwin I, DeLanney LE, McNeill T, Chan P, Forno LS, Murphy GM Jr, Di Monte DA, Sandy MS, Langston JW (1994) Aging and the nigrostriatal dopamine system: a non-human primate study. Neurodegeneration 3:251–265
McCormack AL, Di Monte DA, Delfani K, Irwin I, DeLanney LE, Langston WJ, Janson AM (2004) Aging of the nigrostriatal system in the squirrel monkey. J Comp Neurol 471:387–395
Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson’s disease? Neurobiol Dis 25:134–149
Collier TJ, Kanaan NM, Kordower JH (2011) Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci 12:359–366
Obeso JA (2010) Modeling clinical features of neurodegeneration. Nat Med 16:1372
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283:23542–23556
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67:1464–1472
Falsone SF, Kungl AJ, Rek A, Cappai R, Zangger K (2009) The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J Biol Chem 284:31190–31199
Pemberton S, Madiona K, Pieri L, Kabani M, Bousset L, Melki R (2011) Hsc70 protein interaction with soluble and fibrillar alpha-synuclein. J Biol Chem 286:34690–34699
Dedmon MM, Christodoulou J, Wilson MR, Dobson CM (2005) Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem 280:14733–14740
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem 279:25497–25502
Koga H, Cuervo AM (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 43:29–37
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118:777–788
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L (2009) Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 4:e5515
Bras J, Paisan-Ruiz C, Guerreiro R, Ribeiro MH, Morgadinho A, Januario C, Sidransky E, Oliveira C, Singleton A (2009) Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging 30:1515–1517
De Marco EV, Annesi G, Tarantino P, Rocca FE, Provenzano G, Civitelli D, Ciro Candiano IC, Annesi F, Carrideo S, Condino F, Nicoletti G, Messina D, Novellino F, Morelli M, Quattrone A (2008) Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov Disord 23:460–463
Socal MP, Bock H, Michelin-Tirelli K, Hilbig A, Saraiva-Pereira ML, Rieder CR, Jardim LB (2009) Parkinson’s disease and the heterozygous state for glucocerebrosidase mutations among Brazilians. Parkinsonism Relat Disord 15:76–78
Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, Bar-Shira A, Orr-Urtreger A (2008) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70:2277–2283
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Tan EK, Tong J, Fook-Chong S, Yih Y, Wong MC, Pavanni R, Zhao Y (2007) Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 64:1056–1058
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E (2010) Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol 120:641–649
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, Li A, Holton J, Guerreiro R, Paudel R, Segarane B, Singleton A, Lees A, Hardy J, Houlden H, Revesz T, Wood NW (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132:1783–1794
Gan-Or Z, Giladi N, Orr-Urtreger A (2009) Differential phenotype in Parkinson’s disease patients with severe versus mild GBA mutations. Brain 132:e125
McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A, Hardy J, Wood NW, Schapira AH (2012) Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord 27:526–532
Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, Rochet JC, Glicksman MA, Cheng SH, Grabowski GA, Shihabuddin LS, Schlossmacher MG (2011) Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol 69:940–953
Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy, Wood NW, Schapira AHV (2012) Glucocerebrosidase deficiency in substantia nigra of Parkinson’s disease brains. Ann. Neurol. 72(3):455-63. doi:10.1002/ana.23614
Schapira AHV, Gegg ME (2013) Glucocerebrosidase in the pathogenesis and treatment of Parkinson’s disease. PNAS (in press)
Cleeter MW, Chau KY, Gluck C, Mehta A, Hughes DA, Duchen M, Wood NW, Hardy J, Mark CJ, Schapira AH (2012) Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int 62:1–7
Gorman AM (2008) Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med 12:2263–2280
Chen H, Chan DC (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 18:R169–R176
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Gegg ME, Schapira AH (2011) PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: implications for Parkinson disease pathogenesis. Autophagy 7:243–245
Mijaljica D, Prescott M, Devenish RJ (2007) Different fates of mitochondria: alternative ways for degradation. Autophagy 3:4–9
Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC (2005) Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 12:1613–1621
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E (2000) Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 157:401–410
Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK (2006) Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci 26:41–50
Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K, Eimer S, Winklhofer KF, Haass C (2010) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 29:3571–3589
Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286:10814–10824
Chinta SJ, Mallajosyula JK, Rane A, Andersen JK (2010) Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett 486:235–239
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283:9089–9100
Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M (2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115:629–640
Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL (2008) Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res 314:2076–2089
Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, Yang H, Ueda K, Chan P, Yu S (2009) Alpha-synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett 454:187–192
Shavali S, Brown-Borg HM, Ebadi M, Porter J (2008) Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 439:125–128
Zhang L, Zhang C, Zhu Y, Cai Q, Chan P, Ueda K, Yu S, Yang H (2008) Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res 1244:40–52
Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH (2011) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem 286:20710–20726
Becker T, Gebert M, Pfanner N, van der Laan M (2009) Biogenesis of mitochondrial membrane proteins. Curr Opin Cell Biol 21:484–493
Dolezal P, Likic V, Tachezy J, Lithgow T (2006) Evolution of the molecular machines for protein import into mitochondria. Science 313:314–318
Neupert W, Herrmann JM (2007) Translocation of proteins into mitochondria. Annu Rev Biochem 76:723–749
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283:9089–9100
McFarland MA, Ellis CE, Markey SP, Nussbaum RL (2008) Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics 7:2123–2137
Mori H, Kondo T, Yokochi M, Matsumine H, Nakagawa-Hattori Y, Miyake T, Suda K, Mizuno Y (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892
van de Warrenburg BP, Lammens M, Lucking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW (2001) Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56:555–557
Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS (2002) Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol 160:1655–1667
Kim S., Seong M., Jeon B., Kim S., Ko H., Kim J. and Park S., (2011) Phase analysis identifies compound heterozygous deletions of the PARK2 gene in patients with early-onset Parkinson disease. Clin. Genet
Kitada T, Asakawa S, Minoshima S, Mizuno Y, Shimizu N (2000) Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm Genome 11:417–421
Lucking CB, Brice A (2000) Alpha-synuclein and Parkinson’s disease. Cell Mol Life Sci 57:1894–1908
Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Ondo WG, Allen FH Jr, Scott BL, Goetz CG, Small GW, Mastaglia F, Stajich JM, Zhang F, Booze MW, Winn MP, Middleton LT, Haines JL, Pericak-Vance MA, Vance JM (2003) Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol 53:624–629
Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA 105:14503–14508
Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 103:10793–10798
Berger AK, Cortese GP, Amodeo KD, Weihofen A, Letai A, LaVoie MJ (2009) Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum Mol Genet 18:4317–4328
Yasuda T, Mochizuki H (2010) The regulatory role of alpha-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis 15:1312–1321
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100:4078–4083
Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B (2003) Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron 37:911–924
Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019
Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N (2004) Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol 56:424–427
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Todd AM, Staveley BE (2008) Pink1 suppresses alpha-synuclein-induced phenotypes in a Drosophila model of Parkinson’s disease. Genome 51:1040–1046
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441:1162–1166
Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 5:213–218
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468(7324):696–700
Devic I, Hwang H, Edgar JS, Izutsu K, Presland R, Pan C, Goodlett DR, Wang Y, Armaly J, Tumas V, Zabetian CP, Leverenz JB, Shi M, Zhang J (2011) Salivary alpha-synuclein and DJ-1: potential biomarkers for Parkinson’s disease. Brain 134:e178
Maita C, Tsuji S, Yabe I, Hamada S, Ogata A, Maita H, Iguchi-Ariga SM, Sasaki H, Ariga H (2008) Secretion of DJ-1 into the serum of patients with Parkinson’s disease. Neurosci Lett 431:86–89
Shi M, Zabetian CP, Hancock AM, Ginghina C, Hong Z, Yearout D, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Leverenz JB, Zhang J (2010) Significance and confounders of peripheral DJ-1 and alpha-synuclein in Parkinson’s disease. Neurosci Lett 480:78–82
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Leverenz JB, Baird G, Montine TJ, Hancock AM, Hwang H, Pan C, Bradner J, Kang UJ, Jensen PH, Zhang J (2010) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133:713–726
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG (2011) Alpha-synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10:230–240
Ohrfelt A, Grognet P, Andreasen N, Wallin A, Vanmechelen E, Blennow K, Zetterberg H (2009) Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett 450:332–335
Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T, Ishigami N, Tamaoka A, Nakagawa M, El-Agnaf OM (2010) Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75:1766–1772
Beach TG, 3rd White CL, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J, Bachalakuri J, Henry-Watson J, Akiyama H, Adler CH, Arizona Parkinson’s Disease Consortium (2009) Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174
Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N’Guyen JM, Chaumette T, Tasselli M, Paillusson S, Flamand M, Galmiche JP, Damier P and Derkinderen P (2010) Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One. 5, e12728
Parkkinen L, Silveira-Moriyama L, Holton JL, Lees AJ, Revesz T (2009) Can olfactory bulb biopsy be justified for the diagnosis of Parkinson’s disease? Comments on “olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders”. Acta Neuropathol 117:213–214, author reply 217–8
Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G (2010) Missing pieces in the Parkinson’s disease puzzle. Nat Med 16:653–661
Schapira AH (2011) Challenges to the development of disease-modifying therapies in Parkinson’s disease. Eur J Neurol 18(Suppl 1):16–21
Fountaine TM, Venda LL, Warrick N, Christian HC, Brundin P, Channon KM, Wade-Martins R (2008) The effect of alpha-synuclein knockdown on MPP + toxicity in models of human neurons. Eur J Neurosci 28:2459–2473
McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA (2010) Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS One 5:e12122
Sapru MK, Yates JW, Hogan S, Jiang L, Halter J, Bohn MC (2006) Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp Neurol 198:382–390
Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG (2008) GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci USA 105:10907–10912
Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP, Muzyczka N (2010) In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18:1450–1457
Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Sudhof TC (2004) Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA 101:14966–14971
Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46:857–868
Schapira AH. Mitochondrial diseases. Lancet. 2012 Apr 4. [Epub ahead of print]
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1(8649):1269
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54(3):823–827
Acknowledgement
The authors’ work described in this manuscript has been supported in part by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698), Parkinson’s UK and the Kattan Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mullin, S., Schapira, A. α-Synuclein and Mitochondrial Dysfunction in Parkinson’s Disease. Mol Neurobiol 47, 587–597 (2013). https://doi.org/10.1007/s12035-013-8394-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8394-x