
Abstract
Background
Findings of new targeted treatments with adequate safety evaluations are essential for better cancer cures and mortality rates. Immunotherapy holds promise for patients with relapsed disease, with the ability to elicit long-term remissions. Emerging promising clinical results in B-cell malignancy using gene-altered T-lymphocytes uttering chimeric antigen receptors have sparked a lot of interest. This treatment could open the path for a major difference in the way we treat tumors that are resistant or recurring.
Main body
Genetically altered T cells used to produce tumor-specific chimeric antigen receptors are resurrected fields of adoptive cell therapy by demonstrating remarkable success in the treatment of malignant tumors. Because of the molecular complexity of chimeric antigen receptors-T cells, a variety of engineering approaches to improve safety and effectiveness are necessary to realize larger therapeutic uses. In this study, we investigate new strategies for enhancing chimeric antigen receptors-T cell therapy by altering chimeric antigen receptors proteins, T lymphocytes, and their relations with another solid tumor microenvironment (TME) aspects. Furthermore, examine the potential region of chimeric antigen receptors-T cells therapy to become a most effective treatment modality, taking into account the basic and clinical and practical aspect.
Short conclusions
Chimeric antigen receptors-T cells have shown promise in the therapy of hematological cancers. Recent advancements in protein and cell editing, as well as genome-editing technologies, have paved the way for multilayered T cell therapy techniques that can address numerous important demands. At around the same time, there is crosstalk between various intended aspects within the chimeric antigen receptors-T cell diverse biological complexity and possibilities. These breakthroughs substantially improve the ability to comprehend these complex interactions in future solid tumor chimeric antigen receptor-T cell treatment and open up new treatment options for patients that are currently incurable.
Similar content being viewed by others

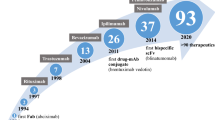
1 Background
Cancer immunotherapy (CI) is fast progressing, and it is currently regarded as the "5th pillar" of cancer treatment, alongside surgeries, toxic chemotherapy, irradiation, and various targeted therapies. Antibodies that target suppressive immune checkpoint molecules have generated the most research in cancer immunotherapy. Combining treatment with another immunotherapeutic medicine, as suggested by the 50% of responses in patients with carcinoma cells, has even greater potential for patients with this type of cancer [67]. Cancer immunotherapy (CI) has also been demonstrated to be useful in different types of cancer, and its use in association with other therapeutic methods like immuno-oncology is quite beneficial. There are two arms to the immune system: innate and adaptive. The initial defense mechanism toward foreign substances is innate immune cells, which do not involve antigen activation. B- and T cell cells are produced by adaptive immunity [4, 87]. Immunotherapy is a type of cancer treatment that targets the immune system instead of tumor cells. In the fight against cancer, tumor vaccines, cytokine killers, monoclonal and bispecific antibodies, immune blockade checkpoint, tumor-infiltrating lymphocytes, and chimeric antigen receptors have all been employed.
Whereas a tumor cell may have over 11,000 genetic variants, several unique tumor-associated antigens (TAAs) may be presented. Tumor-associated antigens and major histocompatibility complex molecules can be observed on the cell surface. The identification of an antigen-MHC complex by a T-lymphocyte receptor activates the body's immune system [28]. To become "effective," some malignant tumors can modify their qualities as well as the properties of the cells in their environment. Interleukin-2 and other immunostimulatory cytokines are conventional and non-specific immunotherapies [32]. The BCG vaccine was the first to be used in the treatment of bladder cancer. BCG promotes the expression of tumor antigens indirectly. As a result, several cytokines, including those produced by T helper 1 cells, are released in a sophisticated manner [116]. Oncolytic viruses are a sort of cancer treatment that falls between biologics and immunotherapy. These viruses have been genetically altered to be non-virulent to healthy cells. They can enter cancerous cells that have dropped many of their antiviral mechanisms and lyse them [118]. Lysis is one of the numerous ways that cause malignant cells to die when they are infected with a virus. Different viral vectors are now being investigated in the clinical creation of numerous malignancies. Tumor antigens have elicited a response, with some trials combining many cancers treatment [42]. Antigens in tumors have elicited a reaction, although there are various "checkpoints" in place [119]. T cell receptors interact with molecules on other cells in the micro-environment. Some of the co-stimulatory and co-inhibitory checkpoint component combinations include TIM3/GAL-9 and LAG-3/MHCII [6]. LAG-3 has a structure comparable to CD4 but has a higher affinity for MHC class II antigens than CD4 [65, 70]. Antibody-like molecules (MAbs) could be known to cure many types of cancer [43]. MAbs that are currently being used in clinical trials target CTLA-4, PD-L1, etc., which "minimize unfavorable T-cell blocking," hence improving anti-tumor immunity [44]. The wide range of reported adverse effect percentages reflects the various drug regimens, concentration levels, and types of cancers treated [1].
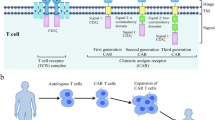
Adoptive cell therapy (ACT) is immunotherapy in which cancer T cells are isolated and grown in a laboratory before even being injected directly into the person. ACT responds quickly to tumoral metastasis and hematologic malignancies. Natural killer cells were also used in these tests, which respond quickly and effectively to these conditions [75]. ACT therapies include several that require growing malignant cells lymphocytes gathered from the tumor. Chimeric antigen receptor T lymphocytes, which produced and activated in vitro to recognize and destroy tumors, are used in some treatments [101].
T cells can recognize tumor-associated antigens without using the major histocompatibility complex if they have a chimeric antigen receptor made artificially [57]. In the therapy of resistant B cell malignancies, chimeric antigen receptor-T cells that targeted the pan–B-cell marker CD19 showed an excellent reaction [126]. To overcome tumor-defensive pathways such as antigen resistance, physiological hurdles, and immunosuppressive entrance into solid tumors, creating appropriate chimeric antigen receptor-T immunotherapy for non-B-cell cancers has necessitated more technically complicated techniques [97]. We look at current and future chimeric antigen receptors design methods, tumor-specific T lymphocytes receptors, and how designed T-lymphocytes interface with the tumor microenvironment shown in Fig. 1. The goal of this research is to improve T-lymphocyte immunotherapy for tumor types related to effectiveness and safety.

Approaches for enhancing the activity of chimeric antigen receptor-T cells in solid tumors
2 Main text
2.1 Development in chimeric antigen receptor designs
The first brilliant idea for combining antibody-type specificity with T-lymphocyte activation was to combine the constant area of a T cell receptor to the variable regions of a bacterial antigen-recognizing antibody [66]. Single-chain variable fragments are still widely seen as the exogenous antigen-sensing domain in chimeric antigen receptors. They are made up of a flexible binder that joins a monoclonal antibody's variable heavy and light chains [95].
Antigens identified by single-chain variable fragments linked to the CD3z activation domain may produce tumor-specific toxicity, according to the first tests using tumor-targeting chimeric antigen receptors. In the great majority of instances, T cells produce these “1st generation” chimeric antigen receptors, which only would include the CD3z chain for T-lymphocytes signaling, unsuccessful to provoke substantial antitumor reactions [22]. Second- and third-generation chimeric antigen receptors with one or two costimula were produced after it was recognized that the endogenous T cell receptor required connotation with the other costimulatory or adjunct molecules for vigorous signaling [53, 54].
These costimulatory domains, which are primarily derivative from CD28, improved overall cytotoxicity, cytokine secretion, and chimeric antigen receptors-T cell tumor development and persistence [89]. The costimulatory domain employed affects a lot of things, namely metabolic activities, T cell memory development, and antigen-independent regulatory stimulation, all of which are essential driving variables [123].
The release of the anti-inflammatory cytokine interleukin (IL)-10 was inhibited by third-generation chimeric antigen receptors containing OX40 and CD28 costimulatory domains. Chimeric antigen receptors of the fourth generation, named "armored" chimeric antigen receptors because of their extra stimulation domains, have recently been developed. Chimeric antigen receptors T cells that generate pro-inflammatory cytokines are known as T cells redirected for ubiquitous cytokine-mediated killing (TRUCK) [7].
The ability of soluble agents like CCL-7 and CCL-19 to increase T-lymphocytes activation, as well as a combination of IL-15 and IL18 to recruit endogenous immune system has been studied [82]. Advances in chimeric antigen receptors design have permitted much more optimization of each of the four primary components of a chimeric antigen receptor hinged domains or extracellular spacer, extracellular antigen-sensing domains, intracellular signaling domains, and the single transmembranes, enabling chimeric antigen receptors therapy to progress [112].
2.2 Logic-gated T cell activation through combinatorial antigen sensing
Chimeric antigen receptors-T cells stability and anticancer efficiency, Boolean logic gates were used to identify many antigens in the combination shown in Fig. 2(1). To trigger a chimeric antigen receptors-T cell, AND-gate logic necessitates the coexistence of several antigens, decreasing the threat of off-target recognition or toxicities that kill healthy cells [51]. The synthetic Notch (synNotch) receptor was intended to measure a TAA and trigger the production of chimeric antigen receptors, which can then induce T-lymphocytes response when secondary TAA is detected [124, 127]. When the targeted non-cancer is not close to the cancer cells, this method has been demonstrated to reduce inflammatory reactions [20]. Because synNotch recognizes TAA #1 before chimeric antigen receptors recognize TAA #2, TAA #1 from a malignant cell could trigger a T cell's synNotch receptor, which then targets a normal cell producing TAA #2. In the alternate AND-gate technique, the CD3z chain and costimulatory domain split into binary distinct receptors, to each one recognizes the other [90].

Protein designing methodologies to enhance the safety, program, and effectiveness of chimeric antigen receptor 1. Antigen recognition via AND and AND-NOT logic, respectively, can improve binding ability and efficiency. 2. Chimeric antigen receptor-T cell activity may be rapidly and effectively changed with ON/OFF switches. 3. Chimeric antigen receptors to activate only when an adapter is assembled can provide chimeric antigen receptor-T cells more control over their activity
This technique, though, is susceptible to “leakiness” because first-generation chimeric antigen receptors that just incorporate the CD3z chain already are proficient in signaling. Another way instructs T cells to release a specifically active cytotoxic protein in response to TAA #1 on the surface of the cell getting identified by chimeric antigen receptors or TCR; the designed protein gets cytotoxic only if TAA #2 within the cell is identified [72, 73]. OR-gate logic was used to improve therapeutic impact by preventing antigen spillage and carcinoma cells' loss of the target epitope [94].
Chimeric antigen receptors immunotherapy designed to use AND–NOT logic can also assist protect normal tissue from toxicity. This method combines inhibiting chimeric antigen receptors that target a TAA with activation chimeric antigen receptors that target an antigen present on normal tissue [100]. A prostate-specific membrane antigen binds to chimeric antigen receptors, triggering the production of apoptosis protein. Although AND and AND–NOT logic can enhance chimeric antigen receptors-T cell protection by enhancing specificity, OR-gate logic is used to increase the antitumor effect to avoid leakage of antigen, or tumor cells losing targeting epitope [56]. OR-gate chimeric antigen receptors can identify two distinct TAAs, and attaching to one of them activates T cells. One OR-gate technique makes use of a pooled combination of two chimeric antigen receptors-T cell populations (chimeric antigen receptors pool), each of which expresses monospecific chimeric antigen receptors. A variation on this topic is to give two separate chimeric antigen receptors-T cell products at the same time [114]. Signaling domain 1 PD1inhibitory was coexpressed with CD19 inhibiting chimeric antigen receptors in solid evidence research, and the inhibiting chimeric antigen receptors suppressed chimeric antigen receptors-T cell stimulation in the vicinity of prostate-specific membrane antigen (PSMA) [8]. Coexpression of two distinct chimeric antigen receptors in each T cell (dual chimeric antigen receptors) is another technique [37]. Another option employs tandem bispecific chimeric antigen receptors (Tan-chimeric antigen receptors), which consist of split single receptor chain on 2 scFv domains, and has been proven to be substantially better both to the chimeric antigen receptors pool and dual-chimeric antigen receptors strategies [101]. CD19-CD20 and CD19-CD22 bispecific chimeric antigen receptors, for example, have been studied in the treatment of B-cell malignancies and are currently being evaluated in clinical trials for myeloma and lymphocytic leukemia, respectively [40].
2.3 Controllability and safety using ON/OFF switches
Self-regulating on/off switches have also been utilized to increase the security and adaptability of chimeric antigen receptor modifications. Chimeric antigen receptors-T cells that have previously been employed are “always on.” When it comes to chimeric antigen receptors derived cytotoxicity, however, this is not necessarily the greatest solution. Patients treated with chimeric antigen receptors-T cells have reported a variety of systemic adverse effects in adding to off-target damage. A common occurrence is cytokine release syndrome (CRS). However, when it comes to chimeric antigen receptors derived cytotoxicity, this is not always the best option [19, 20].
Changing the structure of the chimeric antigen receptors protein can access the availability of functional chimeric antigen receptors, allowing the receptor to be signaling-competent only under specified conditions, rather than modifying protein half-life. For example, chimeric antigen receptors have an antigen-binding region that can be masked by a tailored inhibiting peptide, allowing them to function only when the inhibiting peptide is removed [49]. Modifying the chimeric antigen receptors protein illustrated in Fig. 2(2) can affect the function of functional chimeric antigen receptors.
2.4 Chimeric antigen receptors that are adapter-dependent
Genetically manipulated T lymphocytes in order to generate receptor that requires the addition of a second protein component before it can translate antigen detection into T cell activation shown in Fig. 2(3). A biotin-binding region is coupled to an intracellular T cell triggering region in a "ubiquitous" receptor. T cells that express this biotin-specific receptor may theoretically be employed to attack antibody along with biotinylated which is a target [14]. Scientists can use biotin-specific receptors to assault a wide range of TAAs and manage the ON/off condition of T cells by providing or withholding biotinylated antibodies. To tackle tumor variants, researchers used a wide chimeric antigen receptor in combination with one or more antigen-targeting adaptor proteins [21]. The discovery of a new form of receptor that binds to FITC and folate that alleviates the negative effects of CRS-like harmful effects in lab rats made possible by adapter-dependent chimeric antigen receptor designs [16]. The SUPRA chimeric antigen receptors system is based on the idea of overexpressing a ubiquitous receptor on T cells surfaces in combination with an extrinsically administered adaptor protein, by exhibiting multiple receptors, each holding various adaptor protein associations recreated by the leucine-zipper dime, which can produce diverse Boolean logic gates in designed T lymphocytes [110].
Using AND-NOT or AND gates, researchers can increase sensitivity and cure tumor diversity by treating numerous antigens with different adaptor proteins [21]. Multi-component processes, on the other hand, have a larger range of factors to optimize, such as the adapter protein's half-life, bioavailability, and interface kinetics, as well as the base receptors, adaptor proteins, altered T cell's half-life, bioavailability, and interfaces kinetics [122]. As a result, it is unclear whether adaptor-dependent chimeric antigen receptors designs' sophisticated signal processing will convert into effective medicinal possibilities [24].
3 Designing a car expressing cell
3.1 Designing allogeneic compatibility and site-specific chimeric antigen receptors transgene insertion
Synthesized inducible promoters are utilized in the aforementioned cases of regulatory expression of genes, T cell genomes frequently contain a gene-expression cassette, employing lentiviral and retroviral vectors, resulting in a range of integration sites and copy counts [77]. Integrating the transgenic into specific genomic regions is another way for establishing dynamic chimeric antigen receptors levels of expression [77]. CRISPR and CRISPR-associated protein 9 are two gene-editing techniques (Cas9). T cell designing has become possible mainly to the use of zinc finger nucleases (ZNFs) and activator-like effector nucleases (TALENs) [37]. Chimeric antigen receptor-T cells wipe out retrovirus-mediated random chimeric antigen receptor-transgene integration after CRISPR-Cas9-mediated incorporation of the CD19 chimeric antigen receptor transgenic into the TRAC loci [61, 62]. The CD19 chimeric antigen receptor transgene was introduced into the TRAC region using CRISPR-Cas9, resulting in chimeric antigen receptor-T cells with enhanced in vivo studies [78]. However, new information suggests that whether site-specific chimeric antigen receptor insertion into TRAC loci improves T cell activation is dependent on the chimeric antigen receptor design employed. Allogeneic T cell therapy has also been made easier due to gene-editing techniques [81].
T cell products obtained from better and healthier people can help resolve some of the challenges that arise with producing autologous cell therapy, such as getting enough high-quality T cells from individuals who have been heavily pretreated and have a chronic illness [12]. Allogeneic T cell transplant, on either hand, is a fairly new concept. Allogeneic T cell transfer has been proposed to limit allograft resistance by including the deletion class-I major histocompatibility complex and TCR to avoid host disease-versus-graft [12]. ZFN-mediated suppression of HLA-A or TCRab expression in CD19 chimeric antigen receptor-T cells was used to achieve this [93]. Gene editing has been utilized to defend against GvHD in addition to avoiding the disease [115]. Before injecting chimeric antigen receptor-T cells, lymphodepletion is a typical preconditioning method boost the efficiency of the transplanted cells endogenous gene deletion and chimeric antigen receptors transgenic injection was conducted separately in the work mentioned above, resulting in a wide range of chimeric antigen receptor-T cells [92].
By inserting a single-guide RNA into chimeric antigen receptors U3 domain encoding lentiviral vector's 30 long terminal repeat sequence, the scientists were able to combine genome—editing and chimeric antigen receptors integration. Following Cas9 mRNA was electroporated, magnetic resonance imaging was employed [34].
3.2 Negative regulators are eliminated
Negative T cell regulators' expression can also be suppressed via gene-editing approaches. Immune-checkpoint ligands are generally upregulated in tumor cells [13]. T cell action is evaluated in the tumor microenvironment when cytotoxic T-cells-associated antigen (CTLAA) receptors PD-1 AND CTLA-4 are activated on T lymphocytes, which inhibits the activity. In recent times, antibodies have been utilized to impair immunological checkpoints [27]. On modified T cells, checkpoint suppression can also be substituted by deletion of checkpoint receptor expression [61, 62]. Several knockouts employing CRISPR-Cas9 in chimeric antigen receptor-T cells have been shown in numerous studies to reduce alloreactivity while also improving T cell functionality [88]. Cas9:sgRNA ribonucleoprotein (RNP) complexed with mRNA expressing was electroporated into chimeric antigen receptor-T cells [124, 127].
3.3 Switching from an inhibited to a stimulatory mode of receptors
Despite having shown tremendous therapeutic effectiveness against hematologic malignancies, chimeric antigen receptor-T cells have been limited in their ability to treat specific solid tumors due to a variety of obstacles. Furthermore, tumor cells induce tumorigenic and antigen heterogeneity TME immunosuppressive factors, and thus inhibiting environment is exacerbated by immunosuppressive cells MDSCs and Tregs are examples of these forms of cells [65, 70]. Among the transforming growth factor-b is a predominantly immunosuppressive soluble factor identified in the TME. T cell differentiation into Tregs is triggered by this powerful cytokine as well as the immunosuppressive polarization of macrophages phenotype M2 [59, 99]. The development of a chimeric antigen receptor that responds to transforming growth factor-b showed that chimeric antigen receptors usage to identify a soluble feature and T cells are rearranged to transform inhibitory signals into antitumor activity signals [25]. Transforming growth factor-b responsive chimeric antigen receptor-T cells multiply in the presence of soluble transforming growth factor-b and generate cytokines related with t helper cells type 1 (Th1). A combination of transforming growth factor-b internalization and paracrine Th1 cell stimulation protects neighboring cells against transforming growth factor-inhibitory effects. [48].
Switch receptors, which are chimeras made up of an extracellular domain that attaches to a repressive protein and an endodomain that controls the modulatory path, can be utilized to rewire signals, for example, IL-4 is a kind of cytokine performing numerous functions inside the body. [55]. Tumor microenvironment functions include promoting tumor development and producing M2 polarization, suppressing the development of malignant cells effector T lymphocytes [109]. IL-4Ra in IL-4 receptors ectodomain is linked to either the IL-4 receptor or the IL-4Rb receptor ectodomain in IL-4 switching receptor [121]. The bc receptor subunits or IL-7Ra endodomain similar to IL2 and IL-15 transmission has been reported to increase T lymphocytes and IL-4 multiplication [35]. As a consequence, chimeric antigen receptor with coexpression of the IL4Ra: bC switching receptor had the better cytotoxic capability [50].
3.4 Chimeric antigen receptor-T cells and cancer cells in tumor microenvironment
Chimeric antigen receptor-T cells' limited therapeutic efficiency in solid tumors is due to the TME's immunosuppression properties [76]. The solid tumor microenvironment is characterized by physical barriers to immune cell invasion by tumors, elevated checkpoint receptors, and a pro-tumor stromal microenvironment, all of which have been thoroughly discussed elsewhere, an excess of immunosuppression and pro-metastatic soluble molecules, a pro-tumor stromal microenvironment, an overabundance of immunosuppression, pro-metastatic soluble chemicals, and increased production of chemokines that primarily recruit immunosuppression leukocytes [96]. Researchers are striving to improve the development of tumor microenvironment-responsive chimeric antigen receptor-T cells resulting from these methods shown in Fig. 3 [96].These methods are discussed below.

Approaches for enhancing chimeric antigen receptor with cancer interactions. Through such a range of strategies, chimeric antigen receptor were designed to be used, counter, or avoid tumor-driven immunosuppressive mediators and pathways
3.5 Penetration and tumor homing
The effectiveness of chimeric antigen receptor-T cell treatment in solid tumors is dramatically reduced due to decreased immune activation [52]. The chemokine axis regulates T cell migration. Tumor cells may influence tumor-associated cell chemokine expression and upregulation or downregulation of inflammatory cytokines, leading to poor chimeric antigen receptor-T cell attraction [79]. Chimeric antigen receptor-T cells that are designed to regulate the chemokine receptors expression that is highly activated in the tumor microenvironment can turn a cancer defense pathway against it [107].
CCR2b, the main subtype of the chemokine receptor CCL2, has been transformed into chimeric antigen receptor-T cells that target GD2 and mesothelin, subsequent in improved T lymphocytes adhesion to CCL2-expressing malignant glioblastoma and mesothelial [83]. The better structured extracellular matrix associated with solid-tumors nodules prevents chimeric antigen receptor-T cells from accessing the cancer site. As a result, transgenic chimeric antigen receptor-T cells that produce heparinase, an anticoagulant, have been developed [53, 54].
Chimeric antigen receptor-T cell activation can be aided or hindered by cytokines, which are signaling substances. Co-expression of chimeric antigen receptors with immunomodulatory cytokines may have a big impact. It is possible to improve chimeric antigen receptor-T cell growth, survivability, and mediator activity [15]. The immune system is inhibited by the microenvironment of solid tumors. For example, T cells coexpress a CD19-targeting chimeric antigen receptor-T cells that generate CD19-targeting chimeric antigen receptors plus IL-IL-21, IL-7, IL-15 and IL-2 inhibit tumors more efficiently in vivo than T lymphocytes the chimeric antigen receptors individually. Surprisingly, the gamma subunit is found in all four cytokine receptor complexes. On the proliferative chain, each cytokine had a distinct effect (GC) [125]. T cells that generate IL-21, IL-18, in association with chimeric antigen receptors that target several antigens have also been reported to be more active, multiply, and/or persist in vivo. But on the other hand, persistent overexpression of immunomodulatory cytokines can worsen damage [23].
3.6 Disruption of immune-suppressive axes
TME is rich in immune-checkpoint receptors and mediators including PD-L1 and PD-1, which reduce chimeric antigen receptor-T cell cytotoxic effects and cause anergy [91]. Immune checkpoint suppression has been shown in various studies to have a substantial synergistic impact with chimeric antigen receptor-T cell treatment [44]. A combined therapy combining chimeric antigen receptor-T cells and externally given checkpoint inhibitors are now being investigated for the treatment. Chimeric antigen receptor-T cells are sometimes used to develop immune-checkpoint blockers such PD-1 scFvs and PD-L1 antibody, and the ability to express PD-1 receptors that are notably negative (DNRs) [45].
Tumor development and chimeric antigen receptor-T cell therapy resistance are connected to aberrant cytokine expression in the TME. Transforming growth factor b interacts with cancer cells, stroma, and both to contribute a complex role in cancer development [91]. Adaptive and innate immune cells secrete chemokines, cytokines, and growth factors that weaken the immune system; (2) ECM remodeling (3) macrophage, neutrophil, and monocyte immunosuppressive T cells; and (4) prevent T cells from multiplying. Chimeric antigen receptor-T cells have been developed to produce a transforming growth factor b receptor to counteract these exceptional effects [124, 127]. DNR suppresses endogenous transforming growth factor b signaling in a prostate cancer model, resulting in enhanced T cell proliferation and antitumor effectiveness. These findings prompted the activation of T cells in a new therapy trial for the prevention of chronic and resistant metastatic prostate cancer that coexpressed PSMA chimeric antigen receptors and the DNR [118]. Unlike transforming growth factor b-targeting chimeric antigen receptors and transforming growth factor b switch receptors, the DNR does not send a signal to the transformed T cell. It has been shown that the anticancer potential of CD19-targeting chimeric antigen receptor-T cells that additionally expressed mbaIL6, membrane-bound IL-6 receptor, is maintained in vivo. However, it's unclear whether this chimeric antigen receptor-T cells can prevent CRS [36]. Cancer penetration and death were accelerated in chimeric antigen receptor cells that generated ezrin-mediated which is a peptide inhibition of PKA transport to immunological synapse. Increased levels of relevant entities molecules in the TME, such as reactive oxygen species, play a key role in carcinogenesis [98]. Increased intracellular catalase levels have been shown to boost the cytolytic activity of chimeric antigen receptor cells. The ability of chimeric antigen receptor cells to metabolise hydrogen peroxide has been studied [63].
3.7 Promoting endogenous immunoresponses by restructuring TMEs
To promote immune regulation or repression, tumors have acquired the ability to recruit or reject particular types of leukocytes, such as chimeric antigen receptor-T cells. Additionally, cancer is on the rise it can cause a pro-metastatic phenotypic or immunosuppression in the localized stroma, in addition to anti-inflammatory or phenotypic defects in localized lymphocytes [11].
Redesigning the tumor-cellular composition and reversing the immunosuppressive cell niche phenotype are two further ways to enhance the chimeric antigen receptor-T cell treatment efficiency [64]. To do this, designed chimeric antigen receptor-T cells to release cytokines or other soluble chemicals that promote paracrine or endocrine TME modification [113].
In syngeneic animal models, designed chimeric antigen receptor-T cells that generate IL-12 were able to redesign the tumor microenvironment by modifying M1 phenotype (tumor associated macrophages) and lowering Tregs and myeloid derived suppressor cells variety [96]. Chimeric antigen receptor-T cells that release IL-18 regularly can change the organization of the tumor microenvironment by boosting M1 macrophages, activating DC, and provoking NK cells [26]. The number of M2 macrophages and Treg cells is decreasing, whereas the number of cells is increasing [23].
In a study, IL-12-producing chimeric antigen receptor-T cells were compared to IL-18-producing chimeric antigen receptor-T cells. In a syngeneic murine model, IL-18 is more efficient at changing the immunosuppression tumor microenvironment [1]. The tumor microenvironment can be regulated using chimeric antigen receptor-T cells, which also create proinflammatory ligands that bind to the cell's surface [60]. CD40L, for example, is frequently expressed transiently on T cells. After activation of the TCR and contact with the CD40 receptor, a variety of types of immune cells can trigger APCs [9].
BiTEs, or bispecific T cell engagers, are formed by two scFvs linked together, can be generated by chimeric antigen receptor-T cells to induce non-engineered T cells to encounter cancer cells, as well as CD40 + cancer cell apoptosis and DC licensing [47]. The researchers created bispecific T cell engagers, one with CD3 activation on T cells and another scFv EGFR targeting, which is found in glioma cells, according to the researchers [47]. In several ways, chimeric antigen receptor-T cells targeted EGFRvIII that create EGFR/CD3 bispecific T cell engagers have been shown to eradicate orthotopic cancer xenograft models [46].
4 Clinical experience with chimeric antigen receptor-T cells on cancers
Malignant tumors can block T cells in an infinite variety of ways. Detailed preclinical models for testing potential chimeric antigen receptor immunotherapy combinations are very inspiring in this sector [128]. We analyzed and summarized new research on tumor chimeric antigen receptor-T cell treatment and clinical trials.
To explore if chimeric antigen receptor-T immunotherapy can be utilized to treat malignancy, researchers are doing basic and clinical studies. This strategy in pancreatic cancer targets antigens such as MUC1, mesothelin, CD133, HER2, prostate stem cell antigen, and CEA [3]. Chimeric antigen receptor immunotherapy are used in PDAC and other solid cancers treatments. Early experiments are being conducted on chimeric antigen receptor cells designed to identify mesothelin and create antibodies against PD-1 and CTLA-4 (NCT03182803, NCT03030001) [40]. Patients with non-hematologic malignancies are being enrolled in a clinical study to test a potential MUC1-redirected chimeric antigen receptor cell fusion. [72]. Non-self-recognition chimeric antigen receptor cell techniques are being developed using CRISPR/Cas9. Other genome-editing options are being investigated, such as combining suicide genes to improve safety [73].
Patients diagnosed with chronic HER2-positive sarcoma and osteosarcoma, chimeric antigen receptor-T cell that displays the 2nd-genera on HER2– chimeric antigen receptor-T cell (CD28) were estimated. They were well tolerated with no dose-limiting effects and survived for at least 6 weeks after treatment [68]. In patients with recurrent rhabdomyosarcoma who only had chronic bone marrow illness, a clinical trial of the HER2–chimeric antigen receptor therapy showed effectiveness. Patients were given lymphodepleting chemotherapy before receiving the injection and showed significant improvements in their quality of life [2]. A clinical trial (NCT02932956) to examine the effectiveness and safety of GPC3-chimeric antigen receptor treatment in children with solid tumors, such as rhabdomyosarcoma, has been approved by the FDA [74].
Chimeric antigen receptors that target EGFRvIII, and interleukin 13 receptor 2 and HER2 have been studied in children with glioblastoma. In two investigations, only adults were infused, but in the HER2–chimeric antigen receptor treatment study, 10 of 17 participants were children. [85]. T—Lymphocytes were capable of traveling to glioblastoma spots after becoming infused intravenously with EGFRvIII– chimeric antigen receptor. Glioblastoma activity of a transgene was suppressed, suggesting the chimeric antigen receptor-T cell response [108]. Immunotherapeutic substances such as indoleamine 2,3 dioxygenase and IL10 were shown to be upregulated in glioblastoma, indicating that they can neutralize pro-inflammatory chimeric antigen receptor lymphocytes. Individuals with glioblastoma, particularly young people, are increasingly being aggressively courted for clinical trials [71]. In their initial clinical trial, chimeric antigen receptors were found to be safe for brain and pediatric solid cancer [58].
ROR1 is an oncofetal protein that can sustain pro-apoptotic and survivor signaling in lung cancer cells. Overexpression of the protein has been discovered in a range of tumors, notably lung cancer. It has been proposed as a target molecule for chimeric antigen receptor T cell immunotherapy [104]. Carcinoembryonic antigen is an oncofetal protein that is usually expressed throughout pregnancy but decreases after birth. CEA levels rise dramatically during lung cancer carcinogenesis and progression. Advanced lung carcinomas have been reported to be eradicated by chimeric antigen receptor-T cell [120]. Experts claim that using the PD-L1-chimeric antigen receptor in the treatment of small-cell lung cancer could have antitumor cytotoxic efficacy. Delta-like 3 has been proposed as a possible strategy for the therapy of lung cancer [18].
The use of chimeric antigen receptor immunotherapy to combat liver cancer is still in its early stages, and thus further studies need to be done. It has been verified that chimeric antigen receptor-based epithelial cell adhesion antigen, CEA, glypican-3, and mucin-1 can be used to cure liver cancer. In vitro, the 32A9 antibody fused chimeric antigen receptor kills (GPC3+) HCC cells, and liver xenograft tumors are reduced in vivo. The use of Glypican-3 chimeric antigen receptor immunotherapy treatment of liver cancer could be effective [29, 86]. Another study found that GPC3/chimeric antigen receptor-T cells that expressed IL15/21 enhanced T cell anticancer responses against HCC [108].
TAG-72, GUCY2C, CD133, and NKG2D are the most appropriate target antigens in colorectal tumors. The cytotoxic potential of chimeric antigen receptor cells are increased when mesenchymal stem cells are designed to produce IL-7/12 cytokines [5, 105]. Primary and recurrent colon cancer cells are successfully eradicated by DCLK1's/chimeric antigen receptor immunotherapy [72, 105].
Novel therapeutic approaches for the fight against ovarian cancer are urgently needed due to the high likelihood of recurrence following surgery and chemotherapy. TAG72, protein that is exceedingly expressed on ovarian cancerous cells surface, has been used as a chimeric antigen receptor treatment target [41, 80]. Recent research has created CD19- and Mesothelin-chimeric antigen receptor NK-92 cells for CD19 and mesothelin targeting [106]. Furthermore, 5T4 and FR specific/chimeric antigen receptor t lymphocytes suppressed the proliferation and growth of ovarian cancer cells considerably [38]. OVCAR3 and SK-OV-3 ovarian cancerous cells were killed in vitro by MSLN/NK chimeric antigen receptor cells. The human ovarian SKOV3 cell line's proliferative capability was limited by Her2/ chimeric antigen receptor [17, 117].
Mesothelin, MUC1, chondroitin sulfate proteoglycan 4, and receptor tyrosine kinase EGFR are among the antigens that clinical trials for a chimeric antigen receptor therapy for triple-negative breast cancer are ongoing. C-Met-chimeric antigen receptor was injected into triple-negative patients with breast cancer and generated with little drug-related adverse effects, according to the results of clinical trial [111]. Chimeric antigen receptor cells were previously used to target MUC-1 for triple-negative breast malignance cure, suggesting that this antigen could be regarded as a therapeutic method in the future [31].
The Food and Drug Administration has approved tisagenlecleclecleucel, the first chimeric antigen receptor immunotherapy for acute lymphoblastic leukemia. Axicabtagtagene ciloleucel, a medication, is a second treatment option for B-cell non-lymphoma Hodgkin’s. There are concerns about the long-term negative impact of gene mutations [10]. Other antigens, such as CD30 in refractory Hodgkin's lymphoma and FLT3, and CD123 in acute myeloid leukemia are among the antigens targeted by chimeric antigen receptor-T lymphocytes. Commercial chimeric antigen receptor therapy must be used with caution in cancer patient’s cure [69].
4.1 Difficulties facing chimeric antigen receptor-T cell therapy
The most important targets for chimeric antigen receptor design are tumor-linked antigens. Tumor-associated antigens expression by many types of tumor cells poses a substantial problem. Antigen expression levels at distinct tumor tissues may hinder the efficacy of chimeric antigen receptor treatment in the targeted site [102]. Co-expression of various chimeric antigen receptors and programmed chimeric antigen expression levels have both been employed to facilitate the targeting of multiple tumor-associated antigens by identified chimeric antigen receptors [39]. On the other side, targeting cancer stem cells, which are strongly related to tumor heterogeneity, is one method for eradicating tumor heterogeneity. Cancer stem cell marker CD133 is overexpressed in a lot of solid cancers and is linked to a worse prognosis [40].
In solid tumor cells, the chimeric antigen receptor would be unable to reach tumor tissue through to the vascular endothelial cells. The existence of some means in cancerous tissue reduces the release of vascular-related factors. Endothelin B receptor overexpression in tumor tissue lowers ICAM-1 expression, inhibiting T lymphocytes from exiting blood vessels [30, 98]. The absence of expression of chemokines crucial for T cell penetration into tumor tissues, as well as the presence of substantial fibrotic substrate in tumors, limit the ability of chimeric antigen receptors to move and infiltrate tumor cells. The levels of these chemokines are lower in tumor tissue [124, 127]. In regions at which the tumor area is confined, localized injection of chimeric antigen receptors seems to be more effective versus systemic therapy. In glioblastoma, intracranial transportation is secure and has an adequate anti-cancer impact, while intra-pleural transport has also been shown to be safe and efficacious [107]. CCR2 and CCR4 are chemokine-specific ligands that can be genetically developed to produce chimeric antigen receptors. In cancers, these ligands are highly expressed, allowing them to interact with cancerous cells [98]. Increasing knowledge of what facilitates or hinders T cell accessibility to malignancies may pave the way for new methods to fix chimeric antigen receptor cell trafficking [33].
Chimeric antigen receptors have been shown to express a variety of receptors. Several kinds of cells can infiltrate cancer cells significantly, allowing tumor development, angiogenesis, and proliferation to occur. The suppressive tumor microenvironment is a huge obstacle to successful chimeric antigen receptor treatment for cancers. In clinical studies designed chimeric antigen receptors, in combination with RANTES receptors such as CCR5, CCR3, and CCR1, as well as CCL5-expressing oncolytic virus, significantly boosted treatment methods and eradication [73].
T-lymphocytes are the most prominent immunological suppressor cells in the tumor site. In solid tumors, these cells help cancer progression and multiplication by producing signaling molecules, cytokines, and chemokine [59, 99]. CTLA-4, as well as PD-1, are immune checkpoint molecules that block the immune system's capacity to combat cancer [11]. Chimeric antigen receptor therapy is less effective in a tumor microenvironment containing a lot of cells and inhibitory substances. T cell responses in malignancy are hampered by high adenosine and reactive oxygen species levels. Cancer patients have greater extracellular potassium expression that prevents TCR-mediated Akt-mTOR activation and subsequent lethal activity [84]. The purpose of T cell design is to increase potassium channel activity in terms of improving potassium flow. Several scientists have sought to increase chimeric antigen receptor-T cell performance by combining ACT and TME immunomodulatory [59, 99]. Checkpoint inhibitors that target the PD-1/PD-L1 mechanisms are effective therapy [103]. Chimeric antigen receptor responses to PD-1 and LAG3 suppression were created using CRISPR. Antibodies such as Anti-CTLA-4 shown to increase immune activity against tumors, but the pathway is unknown [9].
5 Conclusions
Hematologic malignancies have shown considerable potential with chimeric antigen receptor-T cell treatment. Solid tumors, on the other hand, present distinct obstacles that necessitate more advanced technologies to treat these refractory cancers magnificently. Recent developments in protein and cell editing have produced some remarkable discoveries. T cells' intrinsic fitness has been improved, and their anticancer function is on the rise. Even though the majority of engineering solutions published to date have centered on providing individual results. Advances in genome-editing technologies and genomic circuitry development have paved the way for multilayered strategies to T cell therapeutic strategies that can simultaneously address multiple critical demands. Simultaneously, the biological complexity and potential within the T cell, there is crosstalk between several designed features. Cancer cells and other malignant cells components, as well as synthetic and endogenous immune cells, must all be examined carefully. When it comes to chimeric antigen receptor-T cell clinical translation, to be employed in the treatment of solid tumors, it is critical to achieve a balance. These achievements vastly improve the capability to understand and rationally plan these complicated interactions in future solid tumor chimeric antigen receptor-T cell treatment. T cell innovations that can be combined and tweaked for optimal efficacy and safety will continue to advance human health and provide new treatment options for diseases that are presently incurable.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author.
Abbreviations
- TME:
-
Tumor microenvironment
- PSMA:
-
Prostate-specific membrane antigen
- CRS:
-
Cytokine release syndrome
- ZNFs:
-
Zinc finger nucleases
- TALENS:
-
Transcription activator-like effector nucleases
References
Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Núñez N, Stowe CL, Friebel E, Nannini F, Rindlisbacher L, Roberts TA, Ramasawmy R, Williams IP, Siow BM, Lythgoe MF, Kalber TL, Quezada SA, Pule MA, Tugues S, Straathof K, Becher B (2021) Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun 12:1–11. https://doi.org/10.1038/s41467-020-20599-x
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, Gray T, Wu M, Liu H, Hicks J, Rainusso N, Dotti G, Mei Z, Grilley B, Gee A, Rooney CM, Brenner MK, Heslop HE, Wels WS, Wang LL, Anderson P, Gottschalk S (2015) Human epidermal growth factor receptor 2 (HER2)—Specific chimeric antigen receptor—modified T cells for the immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33:1688–1696. https://doi.org/10.1200/JCO.2014.58.0225
Akce M, Zaidi MY, Waller EK, El-rayes BF (2018) The potential of CAR T cell therapy in pancreatic. Cancer 9:1–11. https://doi.org/10.3389/fimmu.2018.02166
Akkın S, Varan G, Bilensoy E (2021) A review on cancer immunotherapy and applications of nanotechnology to chemoimmunotherapy of different cancers. Molecules. https://doi.org/10.3390/molecules26113382
Aparicio C, Belver M, Espeso F, Ana S, Miguel Á, Fuente D, Gonz M (2021) Cell therapy for colorectal cancer: the promise of chimeric antigen receptor (CAR) -T Cells. Int J Mol Sci 22:11781
Appleton E, Hassan J, Chan C, Hak W, Sivamanoharan N, Wilkins A, Samson A, Ono M, Harrington KJ, Melcher A, Wennerberg E (2021) Kickstarting immunity in cold tumours: localised tumour therapy combinations with immune checkpoint blockade. Front Immunol 12:1–21. https://doi.org/10.3389/fimmu.2021.754436
Ajina A, Maher J (2019) Europe PMC Funders Group strategies to address chimeric antigen receptor tonic signalling. https://doi.org/10.1158/1535-7163.MCT-17-1097
Andrea AE, Chiron A, Bessoles S, Hacein-Bey-abina S (2020) Engineering next-generation car-t cells for better toxicity management. Int J Mol Sci 21:1–25. https://doi.org/10.3390/ijms21228620
Bedoya DM, Dutoit V, Migliorini D (2021) Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol. https://doi.org/10.3389/fimmu.2021.640082
Bracaglia (2017) 乳鼠心肌提取 HHS public access. Physiol Behav 176:139–148. https://doi.org/10.1016/j.jcyt.2019.12.004
Bruno A, Salemme V, Centonze G, Cavallo F, De P (2021) The crosstalk between tumor cells and the immune microenvironment in breast cancer: implications for Immunotherapy. Front Oncol 11:1–20. https://doi.org/10.3389/fonc.2021.610303
Bair SM, Porter DL (2019) Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: the development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am J Hematol 94:10–17. https://doi.org/10.1002/ajh.25457
Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR (2020) Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol 13:100738. https://doi.org/10.1016/j.tranon.2019.12.010
Bedard PL, Hansen AR, Ratain MJ, Siu LL, Centre MC (2017) HHS Public Access 501:355–364. https://doi.org/10.1038/nature12627.Tumour
Bell M, Gottschalk S (2021) Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol 12:1–16. https://doi.org/10.3389/fimmu.2021.684642
Beyes S, Bediaga NG, Zippo A (2021) An epigenetic perspective on intra-tumour heterogeneity : novel insights and new challenges from multiple fields. Cancers 13:4969
Boissel L, Betancur M, Lu W, Krause D, Van R, Wels W, Klingemann H, Boissel L, Betancur M, Lu W, Krause D, Van R, Boissel L, Betancur-boissel M, Lu W, Krause DS (2013) Retargeting NK-92 cells utilizing CD19- compares favorably with antibody-dependent cellular cytotoxicity retargeting NK-92 cells using CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody- dependent cellular cytotoxi. OncoImmunology 2:e26527. https://doi.org/10.4161/onci.26527
Chen X, Amar N, Zhu Y, Wang C, Xia C, Yang X, Wu D, Feng M (2020) Combined DLL3- targeted bispecific antibody with PD-1 inhibition is efficient to suppress small cell lung cancer growth. J Immunother Cancer 8:e000785. https://doi.org/10.1136/jitc-2020-000785
Caliendo F, Dukhinova M, Siciliano V (2019) Engineered cell-based therapeutics: synthetic biology meets immunology. Front Bioeng Biotechnol 7:1–8. https://doi.org/10.3389/fbioe.2019.00043
Caruso HG, Heimberger AB, Cooper LJN (2019) Steering CAR T cells to distinguish friend from foe. OncoImmunology. https://doi.org/10.1080/2162402X.2016.1271857
Caswell DR, Swanton C (2017) The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 15:1–9. https://doi.org/10.1186/s12916-017-0900-y
Cells CART, Benmebarek M, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S (2019) Killing mechanisms of chimeric antigen receptor. Int J Mol Sci. https://doi.org/10.3390/ijms20061283
Chmielewski M (2020) TRUCKS, the fourth-generation CAR T cells: current developments and clinical translation. Adv Cell Gene Ther 3:1–9. https://doi.org/10.1002/acg2.84
Cooperation W, Guo F, Cui J (2021) CAR-T in cancer treatment : develop in self-optimization
Dexter A Jr, August B (2018) CAR T cells in solid tumors: blueprints for building effective therapies 9:1–20. https://doi.org/10.3389/fimmu.2018.01740
Dong Y, Wan Z, Gao X, Yang G, Liu L (2021) Reprogramming immune cells for enhanced cancer immunotherapy: targets and strategies. Front Immunol 12:1–14. https://doi.org/10.3389/fimmu.2021.609762
Dummer RG (2018) Anti-PD-1 and anti-CTLA-4 therapies in cancer : mechanisms of action, efficacy, and limitations. Front Oncol 8:1–14. https://doi.org/10.3389/fonc.2018.00086
Dhatchinamoorthy K, Colbert JD, Rock KL (2021) Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. https://doi.org/10.3389/fimmu.2021.636568
Duel C, Spear C, Shield T, Jo Y, Ali LA, Shim JA, Lee BH, Hong C (2020) Innovative CAR-T cell therapy for solid tumor
Dana H, Mahmoodi G, Amir S, Reza H, Grupp SA, Webster TJ, Rabello E, Rapo C (2021) CAR-T cells: early successes in blood cancer and challenges in solid tumors. Acta Pharmaceutica Sinica B 11:1129–1147. https://doi.org/10.1016/j.apsb.2020.10.020
Dees S, Ganesan R, Singh S, Grewal IS (2020) Emerging CAR-T cell therapy for the treatment of triple-negative breast cancer. Mol Cancer Ther 19:2409–2421. https://doi.org/10.1158/1535-7163.MCT-20-0385
Ehx G, Perreault C (2019) Discovery and characterization of actionable tumor antigens. Genome Med 11:10–12
Evgin L, Vile RG (2021) Parking CAR T cells in tumours: oncolytic viruses as valets or vandals ? Cancers 13:1106
Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, Danyi A, Humphryes-kirilov N, Ajetunmobi A, Kim D, Kim J, Qasim W (2018) Long Terminal repeat CRISPR-CAR-coupled “Universal” T cells mediate potent anti-leukemic effects. Mol Ther 26:1215–1227. https://doi.org/10.1016/j.ymthe.2018.02.025
Gill N, Paltser G, Ashkar AA (2009) Interleukin-15 expression affects homeostasis and function of B cells through NK cell-derived interferon- c. Cell Immunol 15:1–6. https://doi.org/10.1016/j.cellimm.2009.03.010
Gowrishankar K, Birtwistle L, Micklethwaite K (2018) Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells. Mamm Genome. https://doi.org/10.1007/s00335-018-9756-5
Guedan S, Calderon H, Posey AD, Maus MV (2019) Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev 12:145–156. https://doi.org/10.1016/j.omtm.2018.12.009
Guo C, Wu M, Lu Y (2020) Effective antitumor activity of 5T4-specific CAR-T cells against ovarian cancer cells in vitro and xenotransplanted tumors in vivo. MedComm 1:338–350. https://doi.org/10.1002/mco2.34
Han D, Xu Z, Zhuang Y, Ye Z, Qian Q (2021) Current progress in CAR-T cell therapy for hematological malignancies. J Cancer 12:326–334. https://doi.org/10.7150/JCA.48976
Han X, Wang Y, Wei J, Han W (2019) Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol 12:1–10. https://doi.org/10.1186/s13045-019-0813-7
Hartnett EG, Knight J, Radolec M, Buckanovich RJ, Edwards RP, Vlad AM (2020) Immunotherapy advances for epithelial ovarian cancer. Cancers 12:1–27. https://doi.org/10.3390/cancers12123733
Hemminki O, Manuel J, Hemminki A (2020) Oncolytic viruses for cancer immunotherapy. J Hematol Oncol 1:1–15
Hoteit M, Oneissi Z, Reda R, Wakim F, Zaidan A, Farran M, Abi-Khalil E, El-Sibai M (2021) Cancer immunotherapy: a comprehensive appraisal of its modes of application (review). Oncol Lett. https://doi.org/10.3892/ol.2021.12916
Huemer F, Leisch M, Geisberger R, Melchardt T, Rinnerthaler G, Zaborsky N, Greil R (2017) Combination strategies for immune-checkpoint blockade and response prediction by Artificial Intelligence. Int J Mol Sci 4:1–35
Hassanpour SH, Dehghani M (2017) Review of cancer from perspective of molecular. J Cancer Res Pract 4:127–129. https://doi.org/10.1016/j.jcrpr.2017.07.001
Hematol J, Edeline J, Houot R, Marabelle A, Alcantara M (2021) CAR-T cells and BiTEs in solid tumors: challenges and perspectives. J Hematol Oncol. https://doi.org/10.1186/s13045-021-01067-5
Hematol J, Tian Z, Liu M, Zhang Y, Wang X (2021) Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol. https://doi.org/10.1186/s13045-021-01084-4
Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY (2018) TGF- b – responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med 3:75–86. https://doi.org/10.1002/btm2.10097
Hughes-parry HE, Cross RS, Jenkins MR (2019) The evolving protein engineering in the design of chimeric antigen receptor T cells. Int J Mol Sci 21:204
Isidoro-García M, Dávila I, Laffond E, Moreno E, Lorente F, González-Sarmiento R (2005) Polymorphisms in asthma: a case control study. Clin Mol Allergy 4:1–7. https://doi.org/10.1186/1476-7961-3-15
Jaspers JE, Brentjens RJ, Sloan M, Cancer K, Sloan M, Cancer K, Program C, Sloan M, Cancer K (2017) HHS Public Access 28:83–91. https://doi.org/10.1016/j.pharmthera.2017.03.012
Ji F, Zhang F, Zhang M, Long K, Xia M, Lu F, Li E, Chen J, Li J, Chen Z, Jing L, Jia S, Yang R, Hu Z, Guo Z (2021) Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J Hematol Oncol 14:1–5. https://doi.org/10.1186/s13045-021-01168-1
Jin KT, Chen B, Liu YY, Lan HR, Yan JP (2021) Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer. Cancer Cell Int. https://doi.org/10.1186/s12935-021-01763-9
Jin L, Cao L, Zhu Y, Cao J, Li X, Zhou J, Liu B (2021) Enhance anti-lung tumor efficacy of chimeric antigen receptor-T cells by ectopic expression of C–C motif chemokine receptor 6. Sci Bull 66:803–812. https://doi.org/10.1016/j.scib.2020.12.027
Junttila IS (2018) Tuning the Cytokine Responses : An Update on interleukin (iL)-4 and iL-13 receptor complexes. Front Immunol. https://doi.org/10.3389/fimmu.2018.00888
Koristka S, Ziller-Walter P, Bergmann R, Arndt C, Feldmann A, Kegler A, Cartellieri M, Ehninger A, Ehninger G, Bornhäuser M, Bachmann MP (2019) Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol Immunother 68:1401–1415. https://doi.org/10.1007/s00262-019-02376-y
Kowalski J, Rzyman W, Biernat W, Montesano C, Bernardini R, Marek-Trzonkowska N (2020) Adoptive cell therapy—harnessing antigen-specific T cells to target solid tumours 1–30
Kim K, Gwak H, Han N, Hong EK, Choi BK, Cho H, Lee S, Lee Y, Nam KT, Song S (2021) Chimeric antigen receptor T cells with modified interleukin-13 preferentially recognize IL13R a 2 and suppress malignant glioma: a preclinical study. Front Immunol 12:1–13. https://doi.org/10.3389/fimmu.2021.715000
Kozani PS, Kozani PS, Rahbarizadeh F, Olive D (2021) Strategies for dodging the obstacles in CAR T cell therapy. Front Oncol 11:1–20. https://doi.org/10.3389/fonc.2021.627549
Lanitis E, Kosti P, Ronet C, Cribioli E, Rota G, Spill A, Reichenbach P, Zoete V, Laniti DD, Coukos G, Irving M, Car V (2021) VEGFR-2 redirected CAR-T cells are functionally impaired by soluble A competition for receptor binding. J Immunother Cancer 8:1–13. https://doi.org/10.1136/jitc-2020-002151
Li C, Jiang P, Wei S, Xu X, Wang J (2020) Regulatory T cells in tumor microenvironment : new mechanisms , potential therapeutic strategies and future prospects. Mol Cancer 1–23
Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X (2020) Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. https://doi.org/10.1038/s41392-019-0089-y
Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, Kiessling R (2016) Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol 196:759–766. https://doi.org/10.4049/jimmunol.1401710
Liu B, Yan L, Zhou M (2019) Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res 9:228–241
Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, Degaramo DA, Levine BL, Fraietta JA (2018) CAR T cell therapy of non-hematopoietic malignancies: detours on the road to clinical success. Front Immunol 9:15. https://doi.org/10.3389/fimmu.2018.02740
Lu R, Hwang Y, Liu I, Lee C, Tsai H, Li H, Wu H (2020) Development of therapeutic antibodies for the treatment of diseases 1–30
Lee Ventola C (2017) Cancer immunotherapy, part 1: current strategies and agents. Pharm Ther 42:375–383
Li T, Zhang F, Cao Y, Ning S, Bi Y, Xue W, Ren L (2017) Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the ileum: case report of a 16-year-old Chinese female and literature review. Diagn Pathol 12:1–9. https://doi.org/10.1186/s13000-017-0626-3
Lin WY, Wang HH, Chen YW, Lin CF, Fan HC, Lee YY (2020) Gene modified car-t cellular therapy for hematologic malignancies. Int J Mol Sci 21:1–21. https://doi.org/10.3390/ijms21228655
Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P (2018) The promising immune checkpoint LAG-3: microenvironment to cancer immunotherapy from tumor. Genes Cancer 9:176–189
Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S (2021) CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci 15:17. https://doi.org/10.3389/fnins.2021.662064
Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, Shomali N, Chartrand MS, Pathak Y, Jarahian M (2021) CAR T cells in solid tumors : challenges and opportunities. Stem Cell Res Ther 1:1–16
Marofi F, Rahman HS, Al-Obaidi ZMJ, Jalil AT, Abdelbasset WK, Suksatan W, Dorofeev AE, Shomali N, Chartrand MS, Pathak Y, Hassanzadeh A, Baradaran B, Ahmadi M, Saeedi H, Tahmasebi S, Jarahian M (2021) Novel CAR T therapy is a ray of hope in the treatment of seriously ill AML patients. Stem Cell Res Ther 12:1–23. https://doi.org/10.1186/s13287-021-02420-8
Muralidhar D, Vasugi GA, Sundaram S (2021) Incidence and demographic profile of Ewings sarcoma: experience from a tertiary care Hospital. Cureus 2021:1–12. https://doi.org/10.7759/cureus.18339
Morotti M, Albukhari A, Alsaadi A, Artibani M, Brenton JD, Curbishley SM, Dong T, Dustin ML, Hu Z, Mcgranahan N, Miller ML, Santana-gonzalez L, Seymour LW, Shi T, Loo PV, Yau C, White H, Wietek N, Church DN (2021) Promises and challenges of adoptive T-cell therapies for solid tumours. Br J Cancer. https://doi.org/10.1038/s41416-021-01353-6
Martinez M, Moon EK (2019) CAR T cells for solid tumors: new Strategies for finding infiltrating, and surviving in the tumor microenvironment. Front Immunol 10:1–21. https://doi.org/10.3389/fimmu.2019.00128
Mavilio, F., 2021. Designing lentiviral vectors for gene therapy of genetic diseases 1–14
Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, Carlos CJJ, Liu A, Yin S, Wang W, Yao H, Yu Z, Tang Z, Xie S, Ding Z, Zhao X, Hammock BD, Lu X (2021) Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduct Target Ther. https://doi.org/10.1038/s41392-021-00462-1
Morgan MA, Schambach A (2018) Engineering CAR-T cells for improved function against solid tumors. Front Immunol 9:2493. https://doi.org/10.3389/fimmu.2018.02493
Nimmagadda S, Penet MF (2020) Ovarian cancer targeted theranostics. Front Oncol 9:1–13. https://doi.org/10.3389/fonc.2019.01537
Od Z, Condori J, Peterson N, Zhou S, Krenciute G (2020) Integration and expression in T cells: protocol and application for T-cell therapy
Pinel S, Thomas N, Boura C, Barberi-heyob M, Pinel S, Thomas N, Boura C, Approaches MB, Pinel S, Thomas N, Boura C, Barberi-heyob M (2019) Approaches to physical stimulation of metallic nanoparticles for glioblastoma treatment To cite this version : HAL Id : hal-01935483 Approaches to physical stimulation of metallic nanoparticles for glioblastoma treatment. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2018.10.013
Poorebrahim M, Melief J, Coaña YPD, Wickström SL, Cid-arregui A, Kiessling R (2021) Counteracting CAR T cell dysfunction. Oncogene. https://doi.org/10.1038/s41388-020-01501-x
Pan Y, Yu Y, Wang X, Zhang T (2020) Tumor-associated macrophages in tumor immunity. Front Immunol. https://doi.org/10.3389/fimmu.2020.583084
Patterson JD, Henson JC, Breese RO, Bielamowicz KJ (2020) CAR T cell therapy for pediatric brain tumors. Front Oncol 10:1–14. https://doi.org/10.3389/fonc.2020.01582
Pharmd UP, Ms JA, Savani BN (2022) Review CAR T cell therapy in solid tumors : a review of current clinical trials. EJHaem 3:24–31. https://doi.org/10.1002/jha2.356
Pucci C, Martinelli C, Ciofani G (2019) Innovative approaches for cancer treatment: current perspectives and new challenges. Ecancermedicalscience 13:1–26. https://doi.org/10.3332/ecancer.2019.961
Qin S, Xu L, Yi M, Yu S, Wu K (2019) Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4 1–14
Quintarelli C, Orlando D, Boffa I, Guercio M, Assunta V (2018) Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. OncoImmunology 7:1–16. https://doi.org/10.1080/2162402X.2018.1433518
Rafiq S, Hackett CS, Brentjens RJ (2020) Therapy. Nat Rev Clin Oncol. https://doi.org/10.1038/s41571-019-0297-y
Ramachandran S, Verma AK, Dev K, Goyal Y, Bhatt D, Alsahli MA, Rahmani AH, Almatroudi A, Almatroodi SA, Alrumaihi F, Khan NA (2021) Review article role of cytokines and chemokines in NSCLC immune navigation and proliferation. Oxid Med Cell Longev 2021:1–20
Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR (2021) A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther 7:1–17
Redeker A, Arens R (2016) Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post-transfer vaccination. Front Immunol 7:1–17. https://doi.org/10.3389/fimmu.2016.00345
Richards DM, Sefrin JP, Gieffers C, Hill O, Merz C (2020) Concepts for agonistic targeting of CD40 in immuno-oncology. Hum Vaccin Immunother 16:377–387. https://doi.org/10.1080/21645515.2019.1653744
Roberto RBD, Castellanos-rueda R, Frey S, Egli D, Vazquez-lombardi R, Kapetanovic E, Kucharczyk J, Reddy ST (2020) A functional screening strategy for engineering chimeric antigen receptors with reduced on-target, off-tumor activation. Mol Ther 28:2564–2576. https://doi.org/10.1016/j.ymthe.2020.08.003
Rodriguez-garcia A, Palazon A, Noguera-ortega E Jr, D.J.P., Martin, F., (2020) CAR-T cells hit the tumor microenvironment : strategies to overcome tumor escape. Front Immunol 11:1–17. https://doi.org/10.3389/fimmu.2020.01109
Rosenblatt E, Zubizarreta E (n.d.) Radiotherapy in cancer care : facing the
Srivastava S, Riddell SR, Hutchinson F (2019) HHS Public Access 200:459–468. https://doi.org/10.4049/jimmunol.1701155.CAR
Kozani S, Pouya SK, Pooria R, F., (2021) CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res Ther 12:1–17. https://doi.org/10.1186/s13287-021-02595-0
Stoiber S, Cadilha BL, Benmebarek M-R, Lesch S, Endres S, Kobold S (2019) Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 8:472. https://doi.org/10.3390/cells8050472
Strohl N (2019) Bispecific T-cell redirection versus chimeric antigen receptor (CAR)-T cells as approaches to kill cancer cells. Antibodies 8:41. https://doi.org/10.3390/antib8030041
Schmitt M, Greten FR (2021) The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol 21:653–667. https://doi.org/10.1038/s41577-021-00534-x
Souza RRD (2021) Biologics: targets and therapy armored CAR T-cells : the next chapter in T-cell cancer immunotherapy. Biolog Targets Ther 15:95–105
Schiavone G, Epistolio S, Martin V, Molinari F, Barizzi J, Mazzucchelli L, Frattini M, Wannesson L (2020) Correction to: Functional and clinical significance of ROR1 in lung adenocarcinoma (BMC Cancer, (2020), 20, 1, (1085), DOI: 10.1186/s12885-020-07587-6). BMC Cancer 20:1–7. https://doi.org/10.1186/s12885-020-07704-5
Sur D, Havasi A, Cainap C, Samasca G, Burz C (2020) Chimeric antigen receptor T-cell therapy for colorectal cancer
Terlikowska KM, Terlikowski SJ (2021) Chimeric antigen receptor design and efficacy in ovarian cancer treatment
Tian Y, Li Y, Shao Y, Zhang Y (2020) Gene modification strategies for next- generation CAR T cells against solid cancers. J Hematol Oncol 6:1–16
Transl J, Land CA, Musich PR, Haydar D, Krenciute G, Xie Q (2020) Chimeric antigen receptor T - cell therapy in glioblastoma : charging the T cells to fight. J Transl Med. https://doi.org/10.1186/s12967-020-02598-0
Tavernier SJ, Athanasopoulos V, Verloo P, Behrens G, Staal J, Bogaert DJ, Naesens L, De Bruyne M, Van Gassen S, Parthoens E, Ellyard J, Cappello J, Morris LX, Van Gorp H, Van Isterdael G, Saeys Y, Lamkanfi M, Schelstraete P, Dehoorne J, Bordon V, Van Coster R, Lambrecht BN, Menten B, Beyaert R, Vinuesa CG, Heissmeyer V, Dullaers M, Haerynck F (2019) A human immune dysregulation syndrome characterized by severe hyperinflammation with a homozygous nonsense Roquin-1 mutation. Nat Commun. https://doi.org/10.1038/s41467-019-12704-6
Tellez-gabriel M, Ory B, Lamoureux F, Heymann M, Heymann D (2012) Tumour heterogeneity: the key advantages of single-cell analysis. Int J Mol Sci. https://doi.org/10.3390/ijms17122142
Toulouie S, Johanning G, Shi Y (2021) Chimeric antigen receptor T-cell immunotherapy in breast cancer: development and challenges. J Cancer 12:1212–1219. https://doi.org/10.7150/JCA.54095
Waldman AD, Fritz JM, Lenardo MJ (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. https://doi.org/10.1038/s41577-020-0306-5
Ware MB, El- BF, Lesinski GB (2020) Mirage or long–awaited oasis: cell responses in pancreatic cancer. J Immunother Cancer 8:e001100. https://doi.org/10.1136/jitc-2020-001100
Wen H, Qu Z, Yan Y, Pu C, Wang C, Jiang H, Hou T, Huo Y (2019) Preclinical safety evaluation of chimeric antigen receptor-modified T cells against CD19 in NSG mice. Ann Transl Med 7:735–735. https://doi.org/10.21037/atm.2019.12.03
Wiebking V, Lee CM, Mostrel N, Lahiri P, Bak R, Bao G, Roncarolo MG, Bertaina A, Porteus MH (2021) Genome editing of donor-derived T cells to generate allogeneic chimeric antigen receptor-modified T cells: optimizing aβ T-cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica 106:847–858. https://doi.org/10.3324/haematol.2019.233882
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z (2017) Role of tumor microenvironment in tumorigenesis. J Cancer 8:761–773. https://doi.org/10.7150/jca.17648
Wrona E, Borowiec M, Potemski P (2021) CAR-NK cells in the treatment of solid tumors. Int J Mol Sci 22:5899
Wu P, Gao W, Su M, Nice EC, Zhang W, Lin J (2021) Adaptive mechanisms of tumor therapy resistance driven by tumor microenvironment. Front Cell Dev Biol 9:1–23. https://doi.org/10.3389/fcell.2021.641469
Xie Q, Ding J, Chen Y (2021) Role of CD8 D T lymphocyte cells: Interplay with stromal cells in tumor microenvironment. Acta Pharm Sin B 11:1365–1378. https://doi.org/10.1016/j.apsb.2021.03.027
Xiao BF, Zhang JT, Zhu YG, Cui XR, Lu ZM, Yu BT, Wu N (2021) Chimeric antigen receptor T-cell therapy in lung cancer: potential and challenges. Front Immunol 12:1–16. https://doi.org/10.3389/fimmu.2021.782775
Yanagihara Y, Ikizawa K, Kajiwara K (1995) Functional significance of IL-4 receptor on B cells in IL-4-induced human IgE production. J Allergy Clin Immunol 96:1145–1151
Zaman R, Islam RA, Ibnat N, Othman I, Zaini A, Lee CY, Chowdhury EH (2019) Current strategies in extending half-lives of therapeutic proteins. J Control Release 301:176–189. https://doi.org/10.1016/j.jconrel.2019.02.016
Zambrano-Zaragoza ML, González-Reza R, Mendoza-Muñoz N, Miranda-Linares V, Bernal-Couoh TF, Mendoza-Elvira S, Quintanar-Guerrero D (2018) Nanosystems in edible coatings: a novel strategy for food preservation. Int J Mol Sci. https://doi.org/10.3390/ijms19030705
Zhang Q, Ping J, Huang Z, Zhang X, Zhou J, Wang G, Liu S, Ma J (2020) Review article CAR-T cell therapy in cancer: tribulations and road ahead 2020
Zhang Z, Miao L, Ren Z, Tang F, Li Y, Fantini M, Golubovskaya V, Biotechnologies P (2021) Gene-edited interleukin CAR-T cells therapy in the treatment of malignancies : present and future. Front Immunol 12:1–13. https://doi.org/10.3389/fimmu.2021.718686
Zhylko A, Winiarska M, Graczyk-Jarzynka A (2020) The great war of today: modifications of CAR-T cells to effectively combat malignancies. Cancers 12:2030
Zhang Y, Guan X, Jiang P (2020) Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol 11:1–20. https://doi.org/10.3389/fimmu.2020.594609
Zhao L, Cao YJ (2019) Engineered T cell therapy for cancer in the clinic. Front Immunol. https://doi.org/10.3389/fimmu.2019.02250
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AH contributed to concept and design, searching, data entry, and writing. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hussain, A. Therapeutic applications of engineered chimeric antigen receptors-T cell for cancer therapy. Beni-Suef Univ J Basic Appl Sci 11, 49 (2022). https://doi.org/10.1186/s43088-022-00238-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-022-00238-8