
Abstract
Harnessing the power of immune cells, especially T cells, to enhance anti-tumor activities has become a promising strategy in clinical management of hematologic malignancies. The emerging bispecific antibodies (BsAbs), which recruit T cells to tumor cells, exemplified by bispecific T cell engagers (BiTEs), have facilitated the development of tumor immunotherapy. Here we discussed the advances and challenges in BiTE therapy developed for the treatment of hematologic malignancies. Blinatumomab, the first BiTE approved for the treatment of acute lymphocytic leukemia (ALL), is appreciated for its high efficacy and safety. Recent studies have focused on improving the efficacy of BiTEs by optimizing treatment regimens and refining the molecular structures of BiTEs. A considerable number of bispecific T cell-recruiting antibodies which are potentially effective in hematologic malignancies have been derived from BiTEs. The elucidation of mechanisms of BiTE action and neonatal techniques used for the construction of BsAbs can improve the treatment of hematological malignancies. This review summarized the features of bispecific T cell-recruiting antibodies for the treatment of hematologic malignancies with special focus on preclinical experiments and clinical studies.
Similar content being viewed by others


Background
Over the past few decades, bispecific antibodies (BsAbs) have been developed rapidly for the treatment of hematologic malignancies. There are more than 100 formats for BsAbs, of which bispecific T cell engagers (BiTEs) are well-designed formats, and novel structures of BsAbs are emerging constantly [1]. The concept of BsAbs first appeared in the early 1960s, with the first example constructed in 1985 [2]. BiTE is the BsAb designed to target CD3 and tumor-specific antigens simultaneously and promote the cytotoxicity of T cells. Since Blinatumomab, a canonical CD3/CD19 BiTE, was approved by the United States Food and Drug Administration (FDA) in December 2014 for adult Philadelphia chromosome negative (Ph-) relapsed or refractory (R/R) B cell progenitor acute lymphoblastic leukemia (B-ALL), BiTEs for the management of hematologic malignancies have been developed rapidly [3]. This review summarized the current research status of BiTEs for the treatment of hematologic malignancies. Many bispecific T cell-recruiting antibodies with novel structures have been derived from BiTEs. Some bispecific T cell-recruiting antibodies have been approved for the treatment of hematologic malignancies and multiple promising drugs are currently in clinical trials. In order to maximize the therapeutic effects of bispecific T cell-recruiting antibodies, research issues including the response rates, the recommended doses and adverse events need to be discussed.
Structures of BsAbs
BsAbs are divided into three categories according to their targets: (i) antibodies targeting two different tumor antigens; (ii) antibodies targeting one tumor antigen and one immune-related molecule; (iii) antibodies targeting two immune-related molecules. BiTEs belong to the second category because one BiTE molecule usually targets one CD3 molecule and one tumor antigen simultaneously.
BsAbs are developed on the basis of monoclonal antibodies. In the early days of BsAb development, BsAbs were produced by the reduction and reoxidation of hinged cysteine in monoclonal antibodies [4]. At the present time, according to the structures of BsAbs, BsAbs are divided into two categories: the immunoglobulin G (IgG)-based antibodies and the variable fragment (Fv)-based antibodies [5].
BsAbs based on the IgG structure display a similar structure to native antibodies. The major method of producing IgG-based BsAbs is recombing half-molecules from heterogenous parental antibodies. New techniques of recombining functional half-molecules to produce IgG-based BsAbs include, but are not limited to orthogonal Fab interface, DuoBody, XmAb, CrossMab, and knobs-into-holes (KiH) [6,7,8,9,10]. Concerning the selection of IgG subclass, IgG2 and IgG4 are suitable options because IgG1-based antibodies can cause the elimination of activated T cells [11]. Duobody developed by Genmab is the platform which enables production of BsAbs by exchanging half-molecules from different parental IgGs. The mutation in the constant region of the heavy chain (CH) can recognize the heterologous half-molecule and promote the procedure of heterodimerization. KiH technology developed by Roche also enables production of antibodies through exchanging half-molecules. Knobs and holes mean mutations on CH3 domains which can promote heterodimerization between half-molecules. Based on KiH technology, Roche developed the CrossMab platform by exchanging the CH1 and the constant region of the light chain (CL) of one parental antibody. This technique can solve the problem of light chain mismatching. XmAb technology developed by Xencor also enables production of BsAbs nearly identical to natural antibodies. Compared with Fv-based BsAbs, IgG-based antibodies have longer half-lives in vivo because they are larger in size and are hard to be cleared by the kidney. The solubility and stability of BsAbs are also improved for the presence of the fragment crystallizable (Fc) domains [12]. Fc domains of BsAbs can recruit natural killer (NK) cells and macrophages to induce antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [13]. However, the disadvantages of IgG-based BsAbs are also notable. The permeability of IgG-based BsAbs to tumor tissues is lower than Fv-based BsAbs because of the increased molecular weight. And the production of IgG-based antibodies requires more complex techniques.
The Fv-based BsAbs usually consist of single-chain variable fragments (scFvs) simply. Due to their short half-lives, continuous infusion is required, which has restricted their promotion [12]. The BiTE technique is the prime platform to produce Fv-based BsAbs. Except for BiTE, single-chain diabody, dual-affinity retargeting antibody (DART), and tandem diabody (TandAb) are also platforms used for producing Fv-based BsAbs [14,15,16]. The BiTE molecule developed by Micromet is the antibody consisting of two scFvs connected by a short peptide linker. The scFv is an antibody fragment produced by fusing one variable region of the heavy chain (VH) and one variable region of the light chain (VL) artificially. The DART molecule developed by MacroGenics is the antibody consisting of two engineered heterogenous scFvs which have exchanged their VH regions. The TandAb molecule developed by Affimed is the antibody consisting of two single-chain diabodies which contain four variable domains, respectively. One TandAb molecule is constructed of two binding sites for CD3 and two binding sites for tumor antigens [17]. The BsAbs mentioned in this passage and their structures are illustrated in Table 1 and Fig. 1.

Structures of bispecific T cell-recruiting antibodies. A bispecific T cell engager (BiTE) consists of two single-chain variable fragments (scFvs); a dual-affinity retargeting antibody (DART) consists of two engineered scFvs whose VH exchanged with the other one; a TandAb consists of two single-chain diabodies with four variable domains; a XmAb consists of one scFv, one Fab fragment and one hetero-Fc domain; a 2:1 Crossmab contains two tumor antigen binders and one CD3 binder; “knob in hole” technique and duobody technique enable production of bispecific antibodies with similar structures to natural IgG
Mechanisms of BiTE action
Different from natural antibodies, BiTEs can redirect T cells to specific tumor antigens and activate T cells directly. Natural antibodies are unable to recruit T cells directly because T cells lack Fcγ receptors [18]. The BiTE molecule usually targets one tumor antigen and one CD3 molecule simultaneously. The CD3 molecule non-covalently associates with the T cell receptor (TCR) and participates in antigen-specific signals transduction which can induce the activation of T cells. Activated T cells express high levels of CD69 and CD25 which promote the proliferation of T cells [19]. BiTE therapy can be a strategy to activate exhausted T cells induced by long-term exposure to tumor antigens. Some features of T cell activation induced by BiTEs have been observed. Firstly, the tumor cell plays an indispensable role in the T cell activation induced by the BiTE. Secondly, T cells can be activated without costimulatory signals such as CD28 and interleukin (IL)-2 [20]. This feature is attributed to memory T cells which play an important role in the reaction to BiTEs [21]. Another explanation is that immunological synapses between T cells and tumor cells can assemble TCRs and amplify first signals [20].
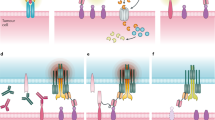
Immunological synapses between T cells and tumor cells are also essential to the BiTE-mediated tumor lysis [22,23,24,25]. Activated T cells secrete perforin and other granzymes through immunological synapses. These cytolytic proteins can form pores on cancer cell membrane. During the process of membrane self-repair, perforin, and other granzymes are endocytosed by cancer cells and then form endosomes. Perforin inside endosomes can form pores on the endosomal membrane and cause the release of granzymes inside targeted cells, then cancer cells are lysed (Fig. 2) [26, 27].

Mechanisms of tumor cell lysis mediated by the BiTEs. BiTEs can redirect T cells to tumor cells and active T cells. Activated T cells release perforin and other granzymes through immunological synapses. These cytolytic proteins can form endosomes in tumor cells and lyse tumor cells ultimately
Mechanisms of tumor escape
Although BiTEs have been proved to be efficient in many relapsed or refractory hematological malignancies, a subset of hematological malignancy patients still have no response to BiTEs. In order to improve the efficacy of BiTEs, further studies on tumor escape should be implemented. The suppression of immune system, especially suppression of T cells, is an important reason for tumor escape [28]. The relationship between BiTE resistance and programmed cell death protein 1/ programmed death-ligand 1 (PD-1/PD-L1) axis has been demonstrated [29]. The expression level of PD-L1 increased in blinatumomab-resistant patients, which indicated the potential efficacy of BiTE therapy in combination with PD-1/PD-L1 inhibitors [30]. AMG 330, a CD33/CD3 BiTE, caused elevated PD-L1 expression on AML cells. And PD-1/PD-L1 blockade therapy enhanced the anti-tumor efficacy of AMG 330 [31]. A large amount of clinical trials focusing on combination therapy with bispecific T cell-recruiting antibodies and PD-1/PD-L1 inhibitors are ongoing. An antibody which targets PD-1, CD3 and CD33 simultaneously has been developed and proved to be efficient in treating acute myeloid leukemia (AML) in preclinical experiments [32].
Loss of target antigen expression is another explanation for tumor escape. It was reported that 8% R/R acute lymphoblastic leukemia (ALL) cases after blinatumomab therapy were CD19 negative [33, 34]. It means other therapies targeting CD19 are potentially effective in most patients after blinatumomab therapy. However, understanding the mechanism of CD19 downregulation can improve the prognosis of CD19-relapsed patients. Antigenic shift is not the only reason for the downregulation of target antigens. It can also be attributed to the disrupted trafficking of the target antigens [35].
Acute lymphoblastic leukemia
ALL is characterized by the proliferation of a huge number of immature lymphocytes in different tissues. R/R ALL patients used to have poor clinical outcomes even after heavy salvage chemotherapy and hematopoietic stem cell transplantation (HSCT) [36, 37]. However, in recent years, new targeted drugs have been developed as remedies for ALL. These drugs include BsAbs, CAR-T cells, anti-CD20 monoclonal antibodies, and tyrosine kinase inhibitors [38,39,40]. BsAbs targeting CD19 and CD3 for the treatment of ALL have become a subject undergoing intense study recently because CD19 is overexpressed in ALL cells (Fig. 3) [5].

Targets of bispecific B cell-recruiting antibodies. CD19 and CD20 are targets for treatment of NHL; CD19 is the target for treatment of acute lymphocytic leukemia; CD123, CD33, CLEC12A, WT1, and FLT3 are targets for treatment of acute myeloid leukemia; BCMA, CD38, and GPRC5D are targets for treatment of multiple myeloma
Blinatumomab
Blinatumomab (MT103) is a BiTE that consists of two scFvs which can combine with CD19 and CD3 separately. The anti-CD19 domain and anti-CD3 domain of blinatumomab are connected with a short glycine–serine linker. The flexible structure of blinatumomab enables blinatumomab to bind T cells and tumor cells efficiently [41]. Blinatumomab has been proved to be an efficient drug in ALL and non-Hodgkin lymphoma (NHL) patients. Blinatumomab could activate cytotoxic T cells and induce regression of NHL cells at the concentration as low as 0.06 mg/m2/day [42, 43]. The FDA has approved blinatumomab for the treatment of R/R Philadelphia chromosome-negative (Ph-) B-ALL in 2014 and R/R Philadelphia chromosome-positive (Ph +) B-ALL in 2017. Blinatumomab is also conducive to deleting minimal residual disease (MRD) in AML [44]. MRD is the status of persistent detectable leukemia cells after complete remission assessed by traditional pathological examinations. MRD is predictive of poor patient prognosis and the deletion of MRD is beneficial to patients [45]. Blinatumomab was approved for the treatment of MRD in B-ALL patients in 2018 and it is the first FDA-approved treatment for MRD.
Efficacy of blinatumomab
Blinatumomab is an effective therapy for R/R Ph- B-ALL. In a phase II study which included primary R/R Ph- ALL patients, 43% of the patients achieved complete remission (CR) (33%) or complete remission with partial hematologic recovery in peripheral blood (CRh) (10%) after two cycles of blinatumomab treatment [46]. More detailed information about clinical trials of blinatumomab and some drugs mentioned in this passage are listed in Table 2. In patients who relapsed after allogeneic hematopoietic stem cell transplantation (allo-HSCT), the CR/CRh rate after two cycles of blinatumomab treatment was 45%. These data have corroborated that blinatumomab is significantly effective in aggressive ALL (NCT01466179) [47]. In a phase II study which included 108 Ph- B-ALL patients in CR with MRD, 85 patients reached MRD negativity after one cycle of blinatumomab treatment at the dose of 15 µg/m2/day. And blinatumomab administered in first CR is associated with the long-term survival of patients. Thus, early administration of blinatumomab is beneficial to B-ALL patients with MRD (NCT01207388) [48]. Compared with standard chemotherapy, blinatumomab was associated with a higher rate of CR and longer overall survival [49]. Moreover, administration of blinatumomab before allogeneic stem cell transplantation improved the rate of CR, which indicated that blinatumomab could be a bridge to allogeneic stem cell transplantation (NCT02013167) [50].
Blinatumomab is also a treatment option for Ph + ALL patients. The prognosis of Ph + ALL patients has been greatly improved since the appearance of tyrosine kinase inhibitors (TKIs) exemplified by imatinib. However, R/R Ph + ALL patients who are resistant to imatinib therapy still lack access to effective therapies. And blinatumomab is a feasible solution to this problem [51]. Blinatumomab has more advantages than chemotherapy, including the increased rate of CR/CRh and longer overall survival (OS) [52]. In a phase II study which included Ph + ALL patients intolerant or refractory to imatinib, 36% of study subjects reached CR/CRh after two cycles of blinatumomab monotherapy (NCT02000427) [53]. Another phase II trial aimed to evaluate the efficacy of dasatinib–blinatumomab therapy in newly diagnosed Ph + ALL patients. 98% of the patients included in this trial achieved CR after the dasatinib induction treatment and the following blinatumomab cycles at the dose of 28 μg/day. Fifteen patients were MRD-positive after the dasatinib induction treatment. ABL1 mutations were detected in 7 of 15 MRD-positive patients. However, blinatumomab eliminated all the mutations related to MRD in this trial, which proved that blinatumomab had an advantage in deleting MRD cells with ABL1 mutations [44].
Meanwhile, the efficacy of blinatumomab against NHL has been evaluated by a series of clinical trials. In 2011, Blinatumomab was proved to be efficient in R/R diffuse large B cell lymphoma (DLBCL) patients and the objective response rate (ORR) reached 56%. In a phase I clinical trial which included R/R NHL patients, the maximum tolerated dose (MTD) of blinatumomab was determined to be 60 μg/m2 per day. Among patients treated at the MTD, the ORR reached 69% (FL, 80%; MCL, 71%; DLBCL, 55%) and the CR/CRu rate reached 37% [54]. In another phase II study which included R/R DLBCL patients (n = 25), the ORR and CR rate were 43% (9/21) and 19% (4/21) respectively at the dose of 112 μg per day [55].
Administration of blinatumomab
The optimal dosing regimen of blinatumomab to maximize therapeutic effects has been determined. The molecular weight of blinatumomab is low (~ 55 kDa) because the BiTE molecule only consists of two scFvs. And the estimated mean (standard deviation) of elimination half-life of blinatumomab is 2.11 (1.42) h. Because of its short half-life, blinatumomab is usually administered by continuous intravenous infusion to maintain its therapeutic concentration [56]. The recommended dose of blinatumomab was determined in a phase II study (NCT01209286) and the regimen is described below. Each cycle lasts 6 weeks. During the first cycle, blinatumomab should be administered at the dose of 9 μg/day in the first week. Then the dose rises to 28 μg/day during the following 3 weeks. There is an interval for 2 weeks at the end of the first cycle. During the following cycles, each cycle includes continuous blinatumomab administration at the dose of 28 μg/day for 4 weeks and an interval for 2 weeks [57].
Adverse events of blinatumomab
A number of adverse events have been observed during blinatumomab cycles. In a phase II study which included R/R B-ALL patients, the common adverse events during blinatumomab therapy included pyrexia (81%), fatigue (50%), headache (47%), tremor (36%), and leukopenia (19%). Most of the adverse events occurred during the first cycle of administration [57]. In a study including adult Ph- R/R ALL patients in Korean, the most common side effects included infection (50%), neurological adverse events (36%), and cytokine release syndrome (CRS) (20%; CRS of grade 3 or higher, 4%) [58]. In another trial including Ph + ALL patients, the most common adverse events of grade 3 or higher involved neutropenia (27%), thrombocytopenia (22%), and anemia (16%) [52]. Adverse effects were compared between the blinatumomab group and the chemotherapy group in a clinical trial which included advanced ALL patients. Although patients in the blinatumomab group suffered more adverse events, the rate of severe adverse effects in the blinatumomab group was lower than the rate in the chemotherapy group [68]. Compared with CAR-T therapy, blinatumomab is associated with a lower incidence of CRS, which is the advantage of blinatumomab [34].
Severe CRS and neurological adverse events are the main reasons for the interruption of blinatumomab therapy. CRS is caused by the release of a large number of cytokines and the following systemic inflammation. The clinical manifestations of CRS include high fever, skin rash, vomiting, and nausea [59]. This problem can be attributed to abnormal activation of effector T cells and macrophages induced by blinatumomab. Activated T cells release high levels of IFN-γ and other cytokines, which can induce macrophages to release high levels of IL-6 and IL-10 subsequently. Severe patients even suffer the problem of hemophagocytic lymphohistiocytosis (HLH) [60]. IL-6 is the center of this pathological process. Other cytokines including TNF, IL-2, GM-CSF, and IL-5 also participate in this process [61, 62]. Severe CRS can be prevented by the administration of dexamethasone beforehand and stepwise dosing [57, 58, 63]. Tocilizumab, an anti-IL-6 receptor monoclonal antibody, is effective in patients with HLH [60].
The neurological adverse events can be attributed to the redistribution of activated T cells. Activated T cells induced by blinatumomab attach to cerebral vessels and move to cerebrospinal fluid. Sedimentation of T cells causes microcirculation dysfunction and local ischemia which ultimately result in neurological symptoms. Patients with diminished B/T cell ratios are more likely to suffer neurological adverse events [64]. Although neurological adverse events can occur in patients with CRS, there is still no evidence that neurological adverse events are associated with the release of cytokines [65]. The neurological symptoms can be controlled after the withdrawal of blinatumomab treatment [57]. Severe neurological events can be prevented by administration of steroids in advance and close clinical monitoring [66].
AFM11
Although blinatumomab is a promising drug to defeat ALL, this antibody is still limited in clinical practice. Firstly, blinatumomab requires persistent administration due to its short half-life, which limits the generalizing of blinatumomab. Secondly, only less than half of the patients with R/R ALL had a partial or complete response to blinatumomab therapy whereas other R/R ALL patients lacked effective treatments. A new drug with higher efficacy and a longer half-life is in demand.
AFM11 is a TandAb with a longer half-life and better affinity to both CD3 and CD19. The efficacy of AFM11 has been demonstrated in preclinical studies. In vitro experiments have proved that AFM11 mediated the CD19 specific cytotoxic effect. AFM11 could activate T cells and promote apoptosis of leukemia cells in chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma. The cytotoxicity of AFM11 was stronger than blinatumomab in vitro [67]. AFM11 could also reinvigorate exhausted T cells induced by chemotherapy [68]. AFM11 was investigated in a phase I clinical trial which included patients with R/R B-ALL. However, the development of AFM11 was interrupted after one death caused by neurological adverse events.
Acute myeloid leukemia
Acute myeloid leukemia (AML) is the most frequent acute leukemia in adults and the incidence increases with age [69]. AML is a highly heterogeneous disease caused by mutations in hematopoietic stem/progenitor cells. Multiple mechanisms including upregulation of Wnt signaling pathway result in abnormal proliferation and differentiation of bone marrow stem cell clones [70]. The treatment of AML is still a gigantic challenge in the field of hematologic oncology. Cytosine arabinoside and anthracyclines are the basis of AML therapy. About 40%-45% of young patients and 10%-20% of elderly patients can be cured in this way [71]. However, the cure rate for patients with R/R AML is lower than 10% [72]. Allo-HSCT used to be the single choice for R/R AML patients, but only a fraction of patients received this treatment [73]. Elderly patients rarely received this treatment because allo-HSCT was associated with poor prognosis and a high recurrence rate among them [74]. Immunotherapy which stimulates the immune system by targeting immune pathways has revolutionized the field of AML therapy. Gemtuzumab ozogamicin was approved as an antibody–drug conjugate for the treatment of AML in 2000, which has significantly promoted the development of other immunotherapies such as BsAbs [75]. Currently, a considerable number of BsAbs for the treatment of AML are in clinical trials and the members include BiTEs, DARTs, and TandAbs. Many tumor surface antigens are potential targets of BsAbs, such as CD123, CD33, FMS-like tyrosine kinase 3 (FLT3), C-type lectin domain family 12 member A (CLEC12A), and Wilms' tumor gene 1 (WT1) [76].
CD123/CD3
CD123, also known as the interleukin 3 receptor alpha chain (IL-3Rα), is overexpressed in many hematological malignances, including AML, Hodgkin lymphoma (HL), and blastic plasmacytoid dendritic cell neoplasm [77]. CD123 is mainly overexpressed in CD34 + /CD38- AML cells [78]. And the overexpression of CD123 is predictive of inferior prognosis [79]. Several BsAbs targeting CD123 and CD3 simultaneously such as MGD006, XmAb14045, and JNJ-63709178 are currently in clinical trials for the treatment of AML. And the preliminary results of clinical trials focusing on MGD006 and XmAb14045 have been published.
MGD006
MGD006 is a DART which targets CD123 and CD3 simultaneously [80]. Compared with BiTEs, it has better stability and manufacturability. It recruits T lymphocytes to CD123 + tumor cells, which induces T cell activation, T cell proliferation and CD123 + tumor cell lysis [81]. Currently, the safety and efficacy of MGD006 in AML patients has been demonstrated in a phase I/II clinical trial (NCT02152956). The CR/CRh rate was 26.7% and the total effective rate (CR/CRh/ complete remission with incomplete count recovery) was 30.0% among primary induction failure/early relapsed patients treated at the recommended phase II dose of 500 ng/kg/day. The median overall survival was 10.2 months and the 6- and 12-month survival rates were 75% and 50%, respectively, among patients who achieved CR/CRh [82]. MGD006 needs continuous infusion because the half-life of MGD006 is short [83]. Like other BsAbs, the most common adverse reaction to MGD006 is CRS. The trial is ongoing and the focus of the research is laid on primary induction failure/early relapsed AML.
XmAb14045 (Vibecotamab)
XmAb14045 is an anti-CD123/CD3 BsAb produced by XmAb technique. Different from MGD006, XmAb14045 is intermittently administered because the half-life of XmAb14045 is extended. XmAb14045 has been investigated in a phase I clinical study since 2016. In part A of the study, 23% of the 64 R/R AML patients had complete responses to XmAb14045 monotherapy. One of them remained in remission 14 weeks after the initial treatment without stem cell transplantation. CRS was present in 77% of the R/R AML patients treated with XmAb1404. The optimal dosing regimen is being studied in part B of this clinical trial [77].
CD33/CD3
CD33, a member of sialic acid-binding sialoadhesin receptors, is selectively expressed on AML cells [84]. It was reported that CD33 was expressed in 87.8% of 319 AML patients, thus CD33 could be an ideal therapeutic target for AML [85]. Since gemtuzumab ozogamicin was approved for treating AML, BsAbs targeting CD33 and CD3 have been developed rapidly. BsAbs including AMG330 (NCT02520427), AMG673 (NCT03224819), AMV564 (NCT03144245), and GEM333 (NCTT03516760) are currently in clinical trials.
AMG330
AMG330 is a BiTE binding with CD33 and CD3 simultaneously. AMG330 can promote the proliferation and activation of T cells, which result in the lysis of CD33 + human AML cells [86]. AMG330 has been demonstrated to attack CD33 + myeloid derived suppressor cells (MDSCs) through T cell-mediated cytotoxicity [87]. An ongoing phase I clinical trial of AMG330 is investigating the safety and tolerated dose of this antibody (NCT02520427).
AMV564
AMV564 is a TandAb containing two binding sites for CD3 and two binding sites for CD33 separately. This structure improves not only its binding affinity for target cells, but also its molecular weight. Because of the increased molecular weight, AMV564 is more difficult to be cleared by the kidney compared with AMG330, which gives it a longer half-life [88]. Phase I clinical trials of AMV564 in patients with AML, myelodysplastic syndromes (MDS), and solid tumors are currently ongoing. In a phase I clinical trial, R/R AML patients received increasing doses of intravenous injection of AMV564 for 14 days. It has been demonstrated that AMV564 was well tolerated, safe and selective. The annual meeting of the Cancer Immunotherapy Society (SITC) also reported that AMV564 could bolster anticancer immunity by depleting immunosuppressive MDSCs and promoting T cell activation.
FLT3/CD3
FLT3 is a class III receptor tyrosine kinase that plays an important role in the proliferation of hematopoietic cells and lymphocytes. It regulates the survival and growth of hematopoietic stem cells, maturation of dendritic cells, and maintenance of regulatory T cell homeostasis [89]. Aberrant expression of FLT3 is closely related to the occurrence of AML and other malignant tumors. FLT3 is overexpressed in more than 70% of AML cases, so FTL3 is an effective target for AML treatment [90]. 7370 is an anti-FLT3/CD3 IgG-based BsAb with advantages of a long half-life and high affinity. 7370 could potently activate human T cells against FLT3 + AML cells in vivo. And 7370 was well tolerated in cynomolgus monkeys. Moreover, 7370 has shown potential clinical values in AML patients regardless of the FLT3 mutation status [91].
CLEC12A/CD3
CLEC12A, also known as human C-type lectin like molecule-1 (CLL-1) or myeloid inhibitory C‐type lectin‐like receptor (MICL), is specifically expressed in AML progenitor cells and leukemia stem cells [92]. CLEC12A is overexpressed in 90–95% of new or recurrent AML cases, but it is rare in normal tissues. Thus, CLEC12A is a potentially effective target for AML therapy [93]. Supported by Biclonics platform, MCLA-117 is a full-length human bispecific IgG that specifically binds CLEC12A + AML cells and CD3 + T cells. Pieter Fokko Van Loo et al. demonstrated that MCLA-117 effectively recruited cytolytic T cells to attack tumor cells in 10 of 11 primary AML samples. MCLA-117 strongly induced killing of AML cells (23%-98%) at low effector-to-target ratios (1:3–1:97) through activating autologous bone marrow T cells in primary AML patient samples. Because MCLA-117 has the potential to selectively target CLEC12A + myeloid cells without affecting normal hematopoietic stem cells, it has the ability to restore normal hematopoietic function and prevent the hematotoxicity induced by MCLA-117 [94]. MCLA-117 is currently under a phase I clinical study (NCT03038230) which focuses on evaluating the efficacy and safety of MCLA-117 in adult patients with AML.
WT1/CD3
WT1 is a tumor-associated antigen located on chromosome 11p13. WT1 plays an important role in the control of cell growth and differentiation [95]. WT1 is overexpressed in leukemia and many solid tumors, especially in AML samples. Therefore, WT1 is an ideal target for AML therapy [96]. A BiTE molecule derived from ESK1, a TCR-mimic monoclonal antibody, can specifically bind WT1 + cells from HLA-A*02:01 + AML cell lines and CD3 + T cells. Its efficacy of killing WT1 + AML cells has been proved by experiments in vitro and in mouse models [97].
Multiple myeloma
Multiple myeloma (MM) is characterized by the proliferation of malignant plasma cells [98]. The median overall survival of MM patients has been improved to 5 years because of the introduction of new drugs [99]. Although the survival of patients is increasing with the continuous progress of therapeutic regimens, MM is still an incurable disease and almost all MM patients eventually relapsed [100]. Many bispecific T cell-recruiting antibodies for the treatment of MM have been developed rapidly in recent years. The ideal targets of BsAbs for treating MM include B cell maturation antigen (BCMA), G protein-coupled receptor 5D (GPRC5D), CD38, and Fc receptor-like 5 (FCRL5).
BCMA/CD3
BCMA, a tumor necrosis factor receptor, is generally expressed on mature B cells. It plays a key role in the survival and proliferation of B cells, especially the long term survival of plasma cells [101]. The ligand of BCMA, a proliferation-inducing ligand, also participates in the progression of MM [102]. BCMA is expressed specifically on MM cells, which makes it an ideal target for the therapy of MM. Serum B cell maturation antigen (sBCMA) is produced by BCMA cleavage and is related to poor clinical outcomes [103]. The poor prognosis can be attributed to its interaction with BCMA signaling pathways in normal cells [104]. There have been many BCMA targeted therapies for the treatment of MM patients, such as BsAbs, CAR-T cells, and antibody–drug conjugates [102, 105, 106]. BiTEs targeting BCMA have been investigated in clinical trials and the preliminary results are promising.
AMG 420
AMG 420 is a BiTE composed of two scFvs derived from one anti-BCMA antibody and one anti-CD3 antibody respectively. Preclinical studies have proved that AMG420 could induce BCMA-dependent T cell activation and MM cell apoptosis. In vivo experiments also proved that AMG420 could consume BCMA + MM cells in cynomolgus monkey models [107]. The first in-human dose-escalation trial has been executed to evaluate the efficacy and safety of AMG420 in R/R MM patients. The dose ranged from 0.2 µg/ d to 800 µg/ d and the drug was administered by continuous intravenous infusion. This study demonstrated that the dose of 800 µg/d could not be tolerated by patients. But the dose of 400 µg/d was appropriate. At the dose of 400 µg/d, the response rate and CR rate reached 70% (7/10) and 50% (5/10), respectively. In this trial, the overall response rate was 31% (13/42).
The adverse effects of AMG 420 in this clinical trial were acceptable. 38% of 42 R/R MM patients suffered CRS and one of them reached grade 3. Severe central nervous system adverse events were not observed. Common severe adverse events included infections (14/42) and polyneuropathy (2/42). Compared with blinatumomab, the adverse event rates of AMG 420 were lower, which might be attributed to the distinctions between CD19 and BCMA and the differences between diseases [108].
AMG 701
AMG 420 should be administered by persistent intravenous infusion to maintain its blood concentration. This character has restricted the application of AMG 420. Another BCMA/CD3 BiTE with a longer half-life is required. And AMG 701 meets the demand because the Fc domain enables it to have a longer half-life. An experiment in vitro demonstrated that AMG 701 enabled T cells to attack tumor cells more efficiently. Animal experiments demonstrated that AMG 701 restricted MM tumor growth in mouse models and resulted in the consumption of plasma cells in cynomolgus monkey models. The combination therapy with PD-1 blockers resulted in synthetic effects [109]. Immunomodulatory imide drugs including lenalidomide and pomalidomide could make AMG 701 more potent in xenograft models [110]. Phase I clinical trials of AMG 701 in patients with R/R MM are being conducted.
GPRC5D/CD3
GPRC5D is a seven transmembrane protein expressed on MM cells specifically [111]. It is predictive of the poor outcomes of MM patients. High expression of GPRC5D is associated with translocation t (4;14) (p16; q32), which is also a poor predictive factor [112, 113]. Compared with BCMA, GPRC5D is fixed more firmly to the membrane because of its seven transmembrane structure. So the protein is less likely to shed from the membrane and T cells can also be bound to tumor cells in a tighter way, which promotes the cytotoxicity of BsAbs [114]. GPRC5D is a promising target for treating MM and anti-GPRC5D/CD3 BsAbs are being developed.
Talquetamab, also known as GPRC5D T cell–redirecting antibody, recruits T cells to tumor cells and activate T cells. It is produced on the base of DuoBody technology. This drug has shown anti-tumor activity in xenograft mouse models. However, we still lack information about the safety of this drug [111]. A clinical trial focusing on the safety and recommended phase II dose of talquetamab in R/R MM patients is ongoing (NCT03399799).
CD38/CD3
CD38 is a transmembrane protein which consists of an intracellular domain, a transmembrane helix, and a larger extracellular domain [115]. CD38 is selectively expressed on MM cells, thus this molecule enables researchers to distinguish MM cells from normal cells [116]. Daratumumab, a CD38 monoclonal antibody, has been demonstrated to be efficient and safe in MM patients [117]. Researchers have developed several anti-CD38/CD3 BsAbs including AMG 424 and Bi38-3.
AMG 424, a bispecific T cell-recruiting antibody based on XmAb platform, was selected from a group of anti-CD38/CD3 bispecific drugs because it could delete target cells efficiently without causing severe CRS. AMG 424 can delete CD38 + tumor cells at low concentration and it is expected to be potent in relapsed patients after daratumumab therapy. Although T cells expressing CD38 might be attacked by AMG 424, this side effect is acceptable because the advantages outweigh the disadvantages [118]. Another anti-CD38/CD3 BiTE, Bi38-3, has also shown its efficacy. Bi38-3 could restrict the growth of xenograft MM tumor in mouse models. Compared with AMG 424, Bi38-3 has fewer adverse effects on normal cells [119].
FCRL5/CD3
FCRL5 is expressed on the sub-groups of B cells. Anti-FcRH5/CD3 BsAbs can recruit T cells to attack plasma cells and MM cells [120]. The extracellular domain of FcRH5 is large, which interferes with the crosstalks between T cells and tumor cells. The anti-FcRH5/CD3 T cell-dependent bispecific antibody (TDB) developed on the base of KiH technique has shown efficacy in attacking FCRL5 + MM cells. In mouse models and cynomolgus monkey models, anti-FcRH5/CD3 TDB could restrict the growth of xenograft MM cells. The combination therapy with PD-1/PD-L1 blockers could improve the potency of anti-FcRH5/CD3 TDB. The half-life of anti-FcRH5/CD3 TDB is long, thus it should be administered intermittently. Release of cytokines usually happened immediately after administration, but it was not severe nor persistent [121].
Non-Hodgkin lymphoma
Non-Hodgkin lymphoma (NHL) includes all lymphomas except for Hodgkin lymphomas. There are huge differences between aggressive lymphomas and indolent lymphomas. It has been proved that the interaction between lymphomas and the immune system plays a key role in the development of lymphomas [122,123,124,125]. Antibodies which regulate the immune system have been proved to be efficient in deleting NHL cells [126]. BsAbs which target NHL cells and T cells simultaneously can form synapses between targeted cells and induce cytotoxicity of T cells [127]. Ideal targets of BsAbs include CD19, CD20, and CD47. The Efficacy of blinatumomab in NHL patients has been illustrated in the efficacy of blinatumomab part. Although BsAbs have shown impressive efficacy in NHL therapy, they are not satisfying in CLL therapy. CLL is a highly heterogeneous disease with acquired immune dysfunction. Prognostic biomarkers and risk scoring systems play important roles in guiding CLL treatment decisions [128]. The strong immunomodulatory effect of CLL causes low response rates to immunotherapy strategies. The emerging antibody products with experimental evidence, such as OTSSP167, mosunetuzumab, and blinatumomab, have offered hope to CLL patients[129, 130].
CD20/CD3
CD20 is a kind of unglycosylated phosphoprotein. It is specifically expressed on B cells and is an ideal target for the treatment of B cell malignancies. Rituximab, an anti-CD20 monoclonal antibody, has been widely used in the clinical treatment of NHL [131]. However, there are a fraction of patients who have no response to rituximab therapy [132]. Anti-CD20/CD3 BsAbs which can mediate the interaction between T cells and CD20 + tumor cells are a viable option for the treatment of R/R NHL.
REGN1979
REGN1979, an anti-CD20/CD3 BsAb with the natural IgG-like structure, is being evaluated among R/R B-NHL patients in a phase I clinical trial. At the doses of 5-27 mg, the ORR in R/R FL (Gr 1–3a) patients reached 100% and the ORR in R/R DLBCL patients was 40%. In R/R DLBCL patients treated with REGN1979 and REGN2810, the response rate rose with the dose of REGN1979. At doses of 5-12 mg, the response rate in R/R DLBCL patients was 18% (2/11). At doses of 18-40 mg, the rate was 55% (6/11). When the dose increased to 80 mg, the response rate was 100% (2/2). But the higher dose was related to the higher rate of CRS.
CD20-TCB
CD20-TCB (RG6026) is an anti-CD20/CD3 BsAb with two CD20 binding sites and one CD3 binding site. This unique 2:1 Crossmab format promotes the function of T cells efficiently. Compared with other BsAbs, CD20-TCB has a longer half-life and higher potency. A phase I study has proved that the ORR in R/R NHL patients was 38% when the dose reached 300 µg or higher. The common adverse drug reactions included neutropenia and CRS. In order to improve the safety of CD20-TCB, researchers have combined CD20-TCB with obinutuzumab pretreatment and this therapeutic regimen is being investigated [133].
Mosunetuzumab
Mosunetuzumab is an anti-CD20/CD3 BsAb with a full-length humanized structure and the production of this antibody is supported by KiH technology [134]. This drug, also known as CD20-TDB, can recruit T cells to attack xenograft CLL cells in mouse models [135]. In a phase I study which included R/R NHL patients, mosunetuzumab has shown promising potency. The ORR in indolent NHL patients and aggressive NHL patients were 64.1% and 34.7%, respectively.
CD47
CD47 is composed of an extracellular V-set IgSF domain, a transmembrane-spanning domain and a selectively spliced cytoplasmic tail [136]. CD47 is widely expressed in normal human cells, but it is specifically overexpressed in NHL cells [137]. The interaction between CD47 and signal regulatory protein alpha (SIRPα) plays an important role in the progression of NHL. CD47 interacts with SIRPα and sends a "don't eat me" signal to macrophages, thus inhibiting the macrophage phagocytosis of tumor cells [123]. Therefore, several therapeutic strategies have been developed to block the CD47-SIRPα signaling pathway. These drugs include TTI-621, HU5F9-G4, ALX-148, and CC-90002 [138]. CD20-CD47SL, a BsAb targeting CD47 and CD20, was investigated by Piccione et al. for the treatment of NHL. CD20-CD47SL was demonstrated to eliminate detectable NHL cells in mouse models. Compared with anti-CD20 monotherapy and anti-CD47 monotherapy, CD20-CD47SL significantly prolonged the survival of mice [139]. BsAbs targeting CD47 belong to the rapidly advancing frontier and more novel antibodies are being developed.
Hodgkin lymphoma
Hodgkin lymphoma (HL) is a sort of B cell lymphoid hematopoietic malignancy. HL is characterized by CD30 + Reed-Sternberg cells surrounded by the immunoinhibitory microenvironment consisting of lymphocytes, eosinophils, plasma cells, and neutrophils [140]. Therefore, there are immune barriers to the success of immunotherapy for HL. To optimize the treatment of HL, one anti-CD30 antibody–drug conjugate (brentuximab vedotin) and two immune checkpoint inhibitors (nivolumab and pembrolizumab) have been developed and these drugs can significantly improve the prognosis of R/R HL [141]. BsAbs targeting CD30 are also promising for the treatment of HL. AFM13 is a bispecific NK-cell engager that specifically targets CD30 + HL cells and CD16A + NK cells. It can activate NK cells and induce tumor cell apoptosis effectively [142]. The safety and tolerability of AFM13 in HL treatment has been demonstrated in a phase I trial implemented by Achim Rothe et al. The overall effective rate of AFM13 in R/R HL patients was as high as 61.5% [143]. More studies focusing on AFM13 in HL patients are being conducted to improve the efficacy of the treatment.
Myelodysplastic syndromes
Myelodysplastic syndromes (MDS) are a group of highly heterogeneous hematologic malignancies originating from hematopoietic stem cells. They are characterized by ineffective intramedullary hematopoiesis which results in myeloid hyperplasia and peripheral blood cytopenia [144]. The biological and clinical manifestations of MDS vary widely but the treatment options are limited, including follow-up observation, erythropoietic stimulants therapy, immunosuppressive therapy, demethylation therapy, and HSCT [145]. The survival rate of patients with MDS is low. About 25% of high-risk and very high-risk MDS develop to AML within one year [146]. Therefore, it is necessary to develop novel and effective therapies to treat patients with MDS.
It has been demonstrated that CD123 is overexpressed in the bone marrow of MDS patients, which indicates that CD123 is an ideal biological marker and therapeutic target of MDS [147]. Several antibodies targeting CD123, such as KHK2823 and IMGN632, are in clinical trials (NCT02181699 and NCT03386513). MGD006, a DART that targets CD123 and CD3, is currently under a phase I clinical trial.
There is evidence that the proportion of CD33 + cells increases in the bone marrow of MDS patients and bispecific T cell-recruiting antibodies targeting CD33 are being developed. AMV564, a tetravalent anti-CD33/CD3 BsAb, has been proved to be efficient in treating MDS. It could delete MDS cells dose-dependently and restore immune homeostasis in vitro. The phase I clinical trial of AMV564 in MDS patients is underway [148, 149].
Conclusions
BiTEs have shown great potency in treating patients with hematologic malignancies. By binding T cells and tumor cells through the specific structure, bispecific T cell engagers enhance tumor lysis effectually and provide relapse/refractory patients with feasible options regardless of mutations or T cell dysfunction. Further advances in molecular structures, dosing regimens and combination therapies can help to improve the efficacy and safety of BiTEs. Innovative platforms enable the production of novel bispecific T cell-recruiting antibodies with higher affinity, greater flexibility and longer half-lives. The efficacy and toxicity of emerging new drugs are evaluated in clinical trials. Deeper investigation of combination therapy with PD-1/PD-L1 blockers is expected to prevent tumor escape efficiently. We are optimistic that such knowledge will facilitate the evolution of anti-tumor strategies focusing on BiTEs.
Availability of data and materials
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ALL:
-
Acute lymphocytic leukemia
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- BCMA:
-
B cell maturation antigen
- BiTE:
-
Bispecific T cell engager
- B-ALL:
-
B cell progenitor acute lymphoblastic leukemia
- CAR-T:
-
Chimeric antigen receptor T cell
- CD:
-
Cluster of differentiation
- CDC:
-
Complement-dependent cytotoxicity
- CH :
-
Constant region of the heavy chain
- CL :
-
Constant region of the light chain
- CLEC12A:
-
C-type lectin domain family 12 member A
- CLL:
-
Chronic lymphocytic leukemia
- CLL-1:
-
C-type lectin like molecule-1
- CR:
-
Complete remission
- CRh:
-
Complete remission with partial hematologic recovery in peripheral blood
- CRP:
-
C-reactive protein
- CRS:
-
Cytokine release syndrome
- DART:
-
Dual-affinity retargeting antibody
- DLT:
-
Dose-limiting toxicities
- DLBCL:
-
Diffuse large B cell lymphoma
- FCRL5:
-
Fc receptor-like 5
- FDA:
-
United States Food and Drug Administration
- FISH:
-
Fluorescence in situ hybridization
- FLT3:
-
FMS-like tyrosine kinase 3
- Fv:
-
Variable fragment
- Fc:
-
Fragment crystallizable
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- GPRC5D:
-
G protein-coupled receptor 5D
- HSCT:
-
Hematopoietic stem-cell transplantation
- HL:
-
Hodgkin lymphoma
- HLH:
-
Hemophagocytic lymphohistiocytosis
- IFN:
-
Interferon
- IgG:
-
Immunoglobulin G
- IGHV:
-
Immunoglobulin heavy chain variable region
- IL-3Rα:
-
Interleukin 3 receptor alpha chain
- IMiD:
-
Immunomodulatory imide drugs
- IL:
-
Interleukin
- KiH:
-
Knobs-into-holes
- MDS:
-
Myelodysplastic syndromes
- MDSC:
-
Myeloid-derived suppressor cell
- MICL:
-
Myeloid inhibitory C‐type lectin‐like receptor
- MM:
-
Multiple myeloma
- MP:
-
Melphalan plus prednisone
- MRD:
-
Minimal residual disease
- MTD:
-
Maximum tolerated dose
- NHL:
-
Non-Hodgkin lymphoma
- NK:
-
Natural killer
- OS:
-
Overall survival
- ORR:
-
Objective response rate
- PBMC:
-
Peripheral blood mononuclear cells
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PFS:
-
Progression-free survival
- Ph:
-
Philadelphia chromosome
- R/R:
-
Refractory or relapsed
- s-BCMA:
-
Serum B cell maturation antigen
- scFv:
-
Single-chain variable fragment
- SIRPα:
-
Signal regulatory protein alpha
- TandAb:
-
Tandem Diabody
- TCB:
-
T cell bispecific antibody
- TCR:
-
T cell receptor
- TDB:
-
T cell-dependent bispecific antibody
- TFS:
-
Treatment-free survival
- TKIs:
-
Tyrosine kinase inhibitors
- VH :
-
Variable region of the heavy chain
- VL :
-
Variable region of the light chain
- WT1:
-
Wilms' tumor gene 1
References
Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212.
Perez P, Hoffman RW, Shaw S, Bluestone JA, Segal DM. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature. 1985;316(6026):354–6.
Löffler A, Gruen M, Wuchter C, Schriever F, Kufer P, Dreier T, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia. 2003;17(5):900–9.
Brennan M, Davison PF, Paulus H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science. 1985;229(4708):81–3.
Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: a review. Pharmacol Ther. 2018;185:122–34.
Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. 2014;32(2):191–8.
Engelberts PJ, Hiemstra IH, de Jong B, Schuurhuis DH, Meesters J, Beltran Hernandez I, et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EBioMedicine. 2020;52:102625.
Kumar A, Blum KA, Fung HC, Smith MR, Foster PA, Younes A. A phase 1 dose-escalation study of XmAb® 2513 in patients with relapsed or refractory Hodgkin lymphoma. Br J Haematol. 2015;168(6):902–4.
Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, et al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019;154:21–31.
Xu Y, Lee J, Tran C, Heibeck TH, Wang WD, Yang J, et al. Production of bispecific antibodies in “knobs-into-holes” using a cell-free expression system. MAbs. 2015;7(1):231–42.
Yu J, Song Y, Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J Hematol Oncol. 2020;13(1):45.
Thakur A, Huang M, Lum LG. Bispecific antibody based therapeutics: strengths and challenges. Blood Rev. 2018;32(4):339–47.
Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A. 2019;116(2):609–18.
Lu L, Liu N, Fan K, Zhang G, Li C, Yan Y, et al. A tetravalent single chain diabody (CD40/HER2) efficiently inhibits tumor proliferation through recruitment of T cells and anti-HER2 functions. Mol Immunol. 2019;109:149–56.
Circosta P, Elia AR, Landra I, Machiorlatti R, Todaro M, Aliberti S, et al. Tailoring CD19xCD3-DART exposure enhances T-cells to eradication of B-cell neoplasms. Oncoimmunology. 2018;7(4):e1341032.
Azhar A, Ahmad E, Zia Q, Rauf MA, Owais M, Ashraf GM. Recent advances in the development of novel protein scaffolds based therapeutics. Int J Biol Macromol. 2017;102:630–41.
McAleese F, Eser M. RECRUIT-TandAbs: harnessing the immune system to kill cancer cells. Future Oncol. 2012;8(6):687–95.
Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res. 2011;317(9):1255–60.
Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother. 2007;30(8):798–807.
Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290–6.
Godbersen C, Coupet TA, Huehls AM, Zhang T, Battles MB, Fisher JL, et al. NKG2D ligand-targeted bispecific T-Cell engagers lead to robust antitumor activity against diverse human tumors. Mol Cancer Ther. 2017;16(7):1335–46.
Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285(5425):221–7.
Weiner GJ, De Gast GC. Bispecific monoclonal antibody therapy of B-cell malignancy. Leuk Lymphoma. 1995;16(3–4):199–207.
Dustin ML. The immunological synapse. Cancer Immunol Res. 2014;2(11):1023–33.
Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15(5):751–61.
Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol. 2011;12(8):770–7.
Trapani JA. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust N Z J Med. 1995;25(6):793–9.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8.
Madorsky Rowdo FP, Baron A, Urrutia M, Mordoh J. Immunotherapy in cancer: a combat between tumors and the immune system; you win some. You Lose Some Front Immunol. 2015;6:127.
Köhnke T, Krupka C, Tischer J, Knösel T, Subklewe M. Increase of PD-L1 expressing B-precursor ALL cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J Hematol Oncol. 2015;8:111.
Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Köhnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia. 2016;30(2):484–91.
Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A, et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood. 2018;132(23):2484–94.
Jabbour E, Düll J, Yilmaz M, Khoury JD, Ravandi F, Jain N, et al. Outcome of patients with relapsed/refractory acute lymphoblastic leukemia after blinatumomab failure: No change in the level of CD19 expression. Am J Hematol. 2018;93(3):371–4.
Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–34.
Braig F, Brandt A, Goebeler M, Tony HP, Kurze AK, Nollau P, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. 2017;129(1):100–4.
Samra B, Jabbour E, Ravandi F, Kantarjian H, Short NJ. Evolving therapy of adult acute lymphoblastic leukemia: state-of-the-art treatment and future directions. J Hematol Oncol. 2020;13(1):70.
Gökbuget N, Dombret H, Ribera JM, Fielding AK, Advani A, Bassan R, et al. International reference analysis of outcomes in adults with B-precursor Ph-negative relapsed/refractory acute lymphoblastic leukemia. Haematologica. 2016;101(12):1524–33.
Ayyappan S, Maddocks K. Novel and emerging therapies for B cell lymphoma. J Hematol Oncol. 2019;12(1):82.
Zhao J, Song Y, Liu D. Recent advances on blinatumomab for acute lymphoblastic leukemia. Exp Hematol Oncol. 2019;8:28.
Timmerman J, Herbaux C, Ribrag V, Zelenetz AD, Houot R, Neelapu SS, et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am J Hematol. 2020;95(5):510–20.
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–42.
Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100(6):690–7.
Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–7.
Foà R, Bassan R, Vitale A, Elia L, Piciocchi A, Puzzolo MC, et al. Dasatinib-blinatumomab for ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383(17):1613–23.
Berry DA, Zhou S, Higley H, Mukundan L, Fu S, Reaman GH, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: a meta-analysis. JAMA Oncol. 2017;3(7):e170580.
Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66.
Stein AS, Kantarjian H, Gökbuget N, Bargou R, Litzow MR, Rambaldi A, et al. Blinatumomab for acute lymphoblastic leukemia relapse after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2019;25(8):1498–504.
Gökbuget N, Dombret H, Bonifacio M, Reichle A, Graux C, Faul C, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522–31.
Gökbuget N, Dombret H, Giebel S, Brüggemann M, Doubek M, Foa R, et al. Blinatumomab vs historic standard-of-care treatment for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukaemia. Eur J Haematol. 2020;104(4):299–309.
Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera J-M, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Chalandon Y, Thomas X, Hayette S, Cayuela JM, Abbal C, Huguet F, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. 2015;125(24):3711–9.
Rambaldi A, Ribera JM, Kantarjian HM, Dombret H, Ottmann OG, Stein AS, et al. Blinatumomab compared with standard of care for the treatment of adult patients with relapsed/refractory Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia. Cancer. 2020;126(2):304–10.
Martinelli G, Boissel N, Chevallier P, Ottmann O, Gökbuget N, Topp MS, et al. Complete hematologic and molecular response in adult patients with relapsed/refractory philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm. Multicenter Study J Clin Oncol. 2017;35(16):1795–802.
Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-hodgkin lymphoma: final results from a phase I Study. J Clin Oncol. 2016;34(10):1104–11.
Viardot A, Goebeler ME, Hess G, Neumann S, Pfreundschuh M, Adrian N, et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016;127(11):1410–6.
Zhu M, Wu B, Brandl C, Johnson J, Wolf A, Chow A, et al. Blinatumomab, a bispecific T-cell engager (BiTE(®)) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271–88.
Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–40.
Jung SH, Lee SR, Yang DH, Lee S, Yoon JH, Lee H, et al. Efficacy and safety of blinatumomab treatment in adult Korean patients with relapsed/refractory acute lymphoblastic leukemia on behalf of the Korean Society of Hematology ALL Working Party. Ann Hematol. 2019;98(1):151–8.
Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. 2016;2016(1):567–72.
Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–7.
Aldoss I, Khaled SK, Budde E, Stein AS. Cytokine release syndrome with the novel treatments of acute lymphoblastic leukemia: pathophysiology, prevention, and treatment. Curr Oncol Rep. 2019;21(1):4.
Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56.
Nägele V, Kratzer A, Zugmaier G, Holland C, Hijazi Y, Topp MS, et al. Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp Hematol Oncol. 2017;6(1):1–14.
Klinger M, Zugmaier G, Nägele V, Goebeler ME, Brandl C, Stelljes M, et al. Adhesion of T cells to endothelial cells facilitates blinatumomab-associated neurologic adverse events. Cancer Res. 2020;80(1):91–101.
Stein AS, Schiller G, Benjamin R, Jia C, Zhang A, Zhu M, et al. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: management and mitigating factors. Ann Hematol. 2019;98(1):159–67.
Magge RS, DeAngelis LM. The double-edged sword: neurotoxicity of chemotherapy. Blood Rev. 2015;29(2):93–100.
Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs. 2015;7(3):584–604.
Duell J, Lukic DS, Karg M, Reusch U, Koch J, Zhukovsky EA, et al. Functionally defective T cells after chemotherapy of B-cell malignancies can be activated by the tetravalent bispecific CD19/CD3 antibody AFM11. J Immunother. 2019;42(5):180–8.
Wang ES. Treating acute myeloid leukemia in older adults. Hematology. 2014;2014(1):14–20.
Zhang Y, Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol. 2020;13(1):165.
Zhang J, Gu Y, Chen B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019;12:1937–45.
Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. 2017;18(3):17.
Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301(22):2349–61.
Molica M, Breccia M, Foa R, Jabbour E, Kadia TM. Maintenance therapy in AML: the past, the present and the future. Am J Hematol. 2019;94(11):1254–65.
Wei AH, Tiong IS. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood. 2017;130(23):2469–74.
Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627–35.
Testa U, Pelosi E, Castelli G. CD123 as a therapeutic target in the treatment of hematological malignancies. Cancers. 2019;11(9):1358.
Braciak TA, Roskopf CC, Wildenhain S, Fenn NC, Schiller CB, Schubert IA, et al. Dual-targeting triplebody 33–16-123 (SPM-2) mediates effective redirected lysis of primary blasts from patients with a broad range of AML subtypes in combination with natural killer cells. Oncoimmunology. 2018;7(9):e1472195.
Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42.
Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399(3):436–49.
Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, et al. Targeting CD123 in acute myeloid leukemia using a T-cell–directed dual-affinity retargeting platform. Blood. 2016;127(1):122–31.
Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137(6):751–62.
Campagne O, Delmas A, Fouliard S, Chenel M, Chichili GR, Li H, et al. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. 2018;24(11):2631–41.
Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73–82.
Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–208.
Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123(3):356–65.
Jitschin R, Saul D, Braun M, Tohumeken S, Völkl S, Kischel R, et al. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6(1):116.
Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. 2016;22(23):5829–38.
Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312.
Poubel CP, Mansur MB, Boroni M, Emerenciano M. FLT3 overexpression in acute leukaemias: new insights into the search for molecular mechanisms. Biochim Biophys Acta Rev Cancer. 2019;1872(1):80–8.
Yeung YA, Krishnamoorthy V, Dettling D, Sommer C, Poulsen K, Ni I, et al. An optimized full-length FLT3/CD3 bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Mol Ther. 2020;28(3):889–900.
Zhao X, Singh S, Pardoux C, Zhao J, Hsi ED, Abo A, et al. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95(1):71–8.
Larsen H, Roug AS, Just T, Brown GD, Hokland P. Expression of the hMICL in acute myeloid leukemia-a highly reliable disease marker at diagnosis and during follow-up. Cytometry B Clin Cytom. 2012;82(1):3–8.
van Loo PF, Hangalapura BN, Thordardottir S, Gibbins JD, Veninga H, Hendriks LJA, et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin Biol Ther. 2019;19(7):721–33.
Scharnhorst V, van der Eb AJ, Jochemsen AG. WT1 proteins: functions in growth and differentiation. Gene. 2001;273(2):141–61.
Lapillonne H, Renneville A, Auvrignon A, Flamant C, Blaise A, Perot C, et al. High WT1 expression after induction therapy predicts high risk of relapse and death in pediatric acute myeloid leukemia. J Clin Oncol. 2006;24(10):1507–15.
Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33(10):1079–86.
Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet. 2009;374(9686):324–39.
Landgren O, Rajkumar SV. New developments in diagnosis, prognosis, and assessment of response in multiple myeloma. Clin Cancer Res. 2016;22(22):5428–33.
Yang Y, Li Y, Gu H, Dong M, Cai Z. Emerging agents and regimens for multiple myeloma. J Hematol Oncol. 2020;13(1):150.
Coquery CM, Erickson LD. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit Rev Immunol. 2012;32(4):287–305.
Yu B, Jiang T, Liu D. BCMA-targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13(1):125.
Sanchez E, Li M, Kitto A, Li J, Wang CS, Kirk DT, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727–38.
Sanchez E, Gillespie A, Tang G, Ferros M, Harutyunyan NM, Vardanyan S, et al. Soluble B-cell maturation antigen mediates tumor-induced immune deficiency in multiple myeloma. Clin Cancer Res. 2016;22(13):3383–97.
Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141.
Tai YT, Anderson KC. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin Biol Ther. 2019;19(11):1143–56.
Hipp S, Tai Y, Blanset D, Deegen P, Wahl J, Thomas O, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31(8):1743–51.
Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol. 2020;38(8):775–83.
Goldstein RL, Goyos A, Li CM, Deegen P, Bogner P, Sternjak A, et al. AMG 701 induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv. 2020;4(17):4180–94.
Cho SF, Lin L, Xing L, Li Y, Wen K, Yu T, et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020;4(17):4195–207.
Kodama T, Kochi Y, Nakai W, Mizuno H, Baba T, Habu K, et al. Anti-GPRC5D/CD3 bispecific T-cell-redirecting antibody for the treatment of multiple myeloma. Mol Cancer Ther. 2019;18(9):1555–64.
Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585–98.
Atamaniuk J, Gleiss A, Porpaczy E, Kainz B, Grunt TW, Raderer M, et al. Overexpression of G protein-coupled receptor 5D in the bone marrow is associated with poor prognosis in patients with multiple myeloma. Eur J Clin Invest. 2012;42(9):953–60.
Pillarisetti K, Edavettal S, Mendonça M, Li Y, Tornetta M, Babich A, et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood. 2020;135(15):1232–43.
Deaglio S, Aydin S, Vaisitti T, Bergui L, Malavasi F. CD38 at the junction between prognostic marker and therapeutic target. Trends Mol Med. 2008;14(5):210–8.
Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841–86.
Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S, et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood. 2016;128(1):37–44.
Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell-recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin Cancer Res. 2019;25(13):3921–33.
Fayon M, Martinez-Cingolani C, Abecassis A, Roders N, Nelson E, Choisy C, et al. Bi38-3 is a novel CD38/CD3 bispecific T-cell engager with low toxicity for the treatment of multiple myeloma. Haematologica. 2021;106(4):1193–7.
Polson AG, Zheng B, Elkins K, Chang W, Du C, Dowd P, et al. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int Immunol. 2006;18(9):1363–73.
Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59(8):1197–209.
Maruhashi T, Okazaki IM, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol. 2018;19(12):1415–26.
Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Ann Rev Immunol. 2014;32:25–50.
Yang ZZ, Liang AB, Ansell SM. T-cell-mediated antitumor immunity in B-cell non-Hodgkin lymphoma: activation, suppression and exhaustion. Leuk Lymphoma. 2015;56(9):2498–504.
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69(12):4941–4.
Yun X, Zhang Y, Wang X. Recent progress of prognostic biomarkers and risk scoring systems in chronic lymphocytic leukemia. Biomark Res. 2020;8:40.
Zhang Y, Zhou X, Li Y, Xu Y, Lu K, Li P, et al. Inhibition of maternal embryonic leucine zipper kinase with OTSSP167 displays potent anti-leukemic effects in chronic lymphocytic leukemia. Oncogene. 2018;37(41):5520–33.
Wong R, Pepper C, Brennan P, Nagorsen D, Man S, Fegan C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica. 2013;98(12):1930–8.
Bauer K, Rancea M, Roloff V, Elter T, Hallek M, Engert A, et al. Rituximab, ofatumumab and other monoclonal anti-CD20 antibodies for chronic lymphocytic leukaemia. Cochrane Database Syst Rev. 2012;11(11):Cd008079.
Ghielmini M, Schmitz SF, Cogliatti S, Bertoni F, Waltzer U, Fey MF, et al. Effect of single-agent rituximab given at the standard schedule or as prolonged treatment in patients with mantle cell lymphoma: a study of the Swiss Group for Clinical Cancer Research (SAKK). J Clin Oncol. 2005;23(4):705–11.
Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S, et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res. 2018;24(19):4785–97.
Spiess C, Merchant M, Huang A, Zheng Z, Yang NY, Peng J, et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat Biotechnol. 2013;31(8):753–8.
Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015;7(287):287ra70.
Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009;19(2):72–80.
Seiffert M, Cant C, Chen Z, Rappold I, Brugger W, Kanz L, et al. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood. 1999;94(11):3633–43.
Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking “don’t eat me” signal of CD47-SIRPα in hematological malignancies, an in-depth review. Blood Rev. 2018;32(6):480–9.
Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. 2015;7(5):946–56.
Shanbhag S, Ambinder RF. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J Clin. 2018;68(2):116–32.
Tun AM, Ansell SM. Immunotherapy in Hodgkin and non-Hodgkin lymphoma: Innate, adaptive and targeted immunological strategies. Cancer Treat Rev. 2020;88:102042.
Bartlett NL, Herrera AF, Domingo-Domenech E, Mehta A, Forero-Torres A, Garcia-Sanz R, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2020;136(21):2401–9.
Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125(26):4024–31.
Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358–74.
Steensma DP. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. 2018;8(5):47.
Germing U, Hildebrandt B, Pfeilstöcker M, Nösslinger T, Valent P, Fonatsch C, et al. Refinement of the international prognostic scoring system (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia. 2005;19(12):2223–31.
Stevens BM, Zhang W, Pollyea DA, Winters A, Gutman J, Smith C, et al. CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp Hematol. 2019;74(52–63):e3.
Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127(1):122–31.
Liu Y, Bewersdorf JP, Stahl M, Zeidan AM. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019;34:67–83.
Acknowledgements
Not applicable.
Funding
This study was funded by National Natural Science Foundation (No.82000195, No.82070203, No.81770210, No.81473486 and No.81270598); Key Research and Development Program of Shandong Province (No.2018CXGC1213); Technology Development Projects of Shandong Province (No.2017GSF18189); Translational Research Grant of NCRCH (No.2021WWB02, No.2020ZKMB01); Shandong Provincial Natural Science Foundation (No.ZR2020QH094); Taishan Scholars Program of Shandong Province; Shandong Provincial Engineering Research Center of Lymphoma; Academic Promotion Programme of Shandong First Medical University (No. 2019QL018; No.2020RC007); Technology Development Project of Jinan City (No.202019182); Shandong Provincial Hospital Youth Talent Plan; Shandong Provincial Hospital Research Incubation Fund.
Author information
Authors and Affiliations
Contributions
Zheng Tian and Ming Liu wrote the initial manuscript. Xin Wang and Ya Zhang revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tian, Z., Liu, M., Zhang, Y. et al. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol 14, 75 (2021). https://doi.org/10.1186/s13045-021-01084-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-021-01084-4