
Background
SMA is a neuromuscular genetic disorder causing irreversible degeneration of the anterior horn cells of lower motor neurons. According to the age of onset and severity of the condition, it is classified into 5 subtypes. SMA carrier’s frequency worldwide is 1:40–80. We used quantitative real-time PCR to determine the copy number of the disease-determining SMN1 gene by rapid and reliable assays. We studied the SMN1 gene copy number in Egyptian sample of 115 individuals, as well as in 10 SMA families.
Results
Our results showed that 57.4% of the couples with the previous history of an affected family members were carriers. On the individual level, carriers of single SMN1 gene copy rate are much higher than the previously reported frequency rates. The effect of consanguineous marriages appears evident in SMA as an autosomal recessive disorder.
Conclusions
In conclusion, the carrier frequency detected in our cohort was high, which possibly corresponds with the worldwide report of SMA as a leading genetic cause of death among infants. Considering the high rate of consanguinity in developing countries confirms the importance of national SMA carrier screening in Egypt. The qPCR carrier screening test is a rapid-cost effective test that can detect approximately 90% of carriers. A population-based preconception prenatal screening for couples will also help reduce the disease burden.
Similar content being viewed by others

Background
Spinal muscular atrophy (SMA) is one of the most common autosomal recessive neuromuscular disorders affecting infants and children. SMA incidence was estimated to be 1 in 6000–10,000 live births worldwide with a carrier frequency of 1:40–80 among different ethnic groups [5, 7]. The highest reported prevalence of disease-prone mutation carrier is among European Caucasoids with 1/47 frequency, followed by 1/52 in Asian Indians, 1/59 in Asians, 1/68 in Hispanics, and 1/72 in African Americans [32, 33].
SMA is a neuromuscular disorder resulting from the irreversible degeneration of the anterior horn cells of the α-motor neurons of the spinal cord, producing a proximal progressive muscles atrophy, and may lead to paralysis. Respiratory muscle weakness together with the thoracic cage deformity frequently results in respiratory failure and death, particularly in severe cases or with early-onset patients [3].
Clinical phenotypes of SMA have a heterogeneous range from a severe to a mild phenotype. It is now classified into five subtypes (types 0 to IV) based on the age of onset and severity of the condition. SMA type 0 is the most severe form with uterine onset, and death usually occurs before six months of age. Type I (Werdnig-Hoffmann disease, OMIM# 253300) represented the most common subtype in over half of the reported SMA cases with muscle weakness persisting at birth or before six months of age and patients usually die of respiratory failure within two years. Type II (OMIM# 253550), has an onset usually 18 months after birth, with patients able to sit but never walk by themselves and can survive beyond four years of age. The late-onset types are type III (Wohlfart–Kugelberg–Welander disease, OMIM#253400), in which the onset is delayed to more than 18 months and patients are able to walk but often become wheelchair-bound during youth or adulthood), [5, 33], while SMA type IV is the mildest late-onset form with normal life expectancy [32].
The SMA determining gene is called the “survival motor neuron” gene (SMN, OMIM #600354, #601627) located on 5q13. The large inverted duplication consists of two homologous genes arranged in tandem on each chromosome; SMN1 (telomeric copy, the disease-causing gene) and SMN2 (paralog centromeric copy). Both genes consist of nine exons and share more than 99% nucleotide identity with exon 8 remaining untranslated. They differ only by five single nucleotide variants (SNVs) within their 3′ ends; two SNVs are located in the coding region of exons 7 & 8, one in intron 6 and two in intron 7. These five unique SNVs are used as a diagnostic tool that allows the distinction between SMN1 and SMN2 genes [1, 7, 10].
Although both genes produce equal transcript amounts, almost 70–85% of SMN2 derived transcripts are unstable, truncated and not fully functioning due to the exon 7 nucleotide exchange NM_000344.3:c.840C > T that interrupts a splicing enhancer and results in exon 7 skipping [33]. SMN2 gene partially compensates the SMN protein with small amount of functional protein, thus an inverse correlation arises between the number of SMN2 copies and the severity of the disease. SMA patients commonly have at least one SMN2 copy. Carriers on the other hand are asymptomatic because they retain one functioning copy of SMN1 gene but can pass their nonfunctioning copy to their children [3, 4, 16].
In this highly homologous region, a gene conversion between SMN1 and SMN2 can occur producing a hybrid SMN gene. Gene conversion mainly takes place by fusion of SMN1 exon 8 with SMN2 exon 7 converting SMN1 into SMN2 or vice versa. This results in variable copy numbers (CNs) of SMN1 and SMN2 [5, 27].
Approximately 95% of SMA patients are due to homozygous deletion of exon 7 of SMN1 gene. The remaining 5% shows other pathogenic point mutations in compound either in homozygous form or in compound heterozygosity with SMN1 deletion. Since the disease is autosomal recessive; de-novo variants are causative reasons in only 2% of the affected patients [21].
Three early treatments, Spinraza®(nusinersen), Zolgensma® (onasemnogene abeparvovec-xioi, OA), and Evrysdi® (risdiplam) received FDA approval for the amelioration of SMA symptoms and enhancing of long-term quality of life and survival [5, 11].
Spinraza® (Biogen, Cambridge, MA, USA) is the first SMA effective treatment and is a modified antisense oligonucleotide-based therapy that enhances the production of SMN protein by increasing the production of full-length SMN proteins [11, 21] .
Zolgensma® (onasemnogene abeparvovec “OA”) is a gene replacement therapy that delivers a cDNA coding for the SMN protein using Adeno-associated virus 9 (AAV9) as a vector. It is systemically applied to children less than 2 years employed at two different doses [14].
Evrysdi® is an oral SMN2 pre-mRNA splicing modifier recently approved for the treatment of SMA patients aged 2 months and older. Risdiplam directly promotes the generation of full-length SMN2 mRNA—which increases the production of functional SMN protein [23].
SMA treatment in pre-symptomatic infants increased the likelihood of survival and improved motor function. For an early diagnosis and the initiation of treatment, newborn SMA screening, carrier frequency of SMA, and SMN2 copy number are important [21, 34].
The comparative Ct (threshold cycle) method can be used to determine the copy number of SMN1 gene by simple quantitative real-time PCR assays. It detects the most common mutation in SMA and approximately 90% of carriers. This test can be used for Genetic counseling in such patients to reduce the likelihood of having an affected child in the future [1, 10].
This study is a pilot study aiming at throwing some light on the importance of SMA carrier detection in Egypt. Herein, we report the results of qPCR quantification of the SMN1 gene in geographically heterogeneous Egyptian samples, as well as in SMA families seeking genetic counseling.
Subjects and methods
Subjects
The study is a case-series study conducted between 2018 and 2020 and included 115 adults, 10 SMA patients with homozygous deletion of SMN1 gene, and their parents (20 obligate heterozygotes of SMA parents), and 50 normal controls (for normalization of the results). All the subjects were seeking genetic counseling for a family history of a dead sibling with a provisional diagnosis of SMA or with reported SMA in another family member.
Six different families in which carrier status has been confirmed came later for fetal diagnosis. Amniotic fluid (AF) samples were withdrawn from the pregnant mothers. Herein, we offered SMA screening tests with adequate counseling about the disease, for all subjects included in the study (couples, pregnant women, or women seeking pregnancy).
The Medical Research Ethics Committee of the host research centre approved the study protocol and results. Written informed consents were obtained from all participants according to the guidelines of the Medical Research Ethics Committee. All participants were seeking genetic counseling either premarital or before pregnancy.
Methods
For each participant, samples of 3–5 ml venous blood were withdrawn and transferred immediately to polypropylene tubes containing 0.5 M EDTA (pH 8.0, violet cap) and mixed thoroughly to prevent clotting and to stop nuclease activity.
Genomic DNA was extracted from peripheral blood leukocytes using GeneJET Whole Blood Genomic DNA Purification Mini Kit (Thermo Scientific, EU). DNA concentration and purity were determined using NanoDrop® 2000 (Thermo Scientific, EU).
Quantitative real-time PCR of SMN1 gene
Real-time PCR analysis of SMN1 gene copy number was performed in a total volume of 15 μl in each reaction using 0.5 μM of specific primers of SMN1 forward primer is 5’-CCTTTTATTTTCCTTACAGGGTTTC-3’; reverse primer is 5’-GATTGTTTTACATTAACCTTTCAACTTTT-3’, 1 μM of GAPDH gene primers as a reference gene; GAPDH forward is 5’-CTACTGGCGCTGCCAAGGCTGT-3’; and reverse is 5’- GCCATGAGGTCCACCACCCTGT-3’, 10 ng/μl DNA and 7.5 μl of SYBR Green I PCR Master Mix (Qiagen, Germany). All samples were analyzed in duplicate using StepOne real-time PCR system (Applied Biosystems, USA).
Multiplex ligation-dependent probe amplification (MLPA) analysis
MLPA analysis was done by SLSA MLPA P21-B1 (MRC-Holland, Amsterdam, Netherlands) according to manufacturer protocol (https://www.mlpa.com/WebForms). It was used for the analysis of amniotic fluid samples and one family.
Data analysis
Data were calculated by the comparative Ct method to detect the relative gene copy numbers. The copy number of the sample was determined by the following formula:
ΔΔCt = [(ΔCt GAPDH – ΔCt SMN1) in calibrator sample]–[(ΔCt GAPDH – ΔCt SMN1) of unknown sample].
The relative gene copy numbers (RQ—Relative Quantification) were calculated by the expression: 2−ΔΔCt (it is expected to be ≥ 1 in normal controls, about 0.5–1 in carriers ,and 0 in patients with SMA) [15].
Results
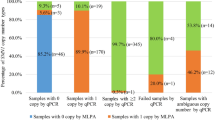
The study was conducted on 115 apparently normal individuals including 53 married couples. From the 53 families, both parents were carriers in 33 families (66 individuals) with RQ values from 0.326 to 0.93 (mean 0.65, SD 0.15, SE 0.019, CV% 23.41). These families were advised to consider prenatal diagnosis during any subsequent pregnancy. Prenatal diagnosis for 6 of these families was carried out using MLPA technique. Both exons 7 and 8 of SMN1 gene were deleted in 2 samples confirming an SMA diagnosis. Carrier status was confirmed in 2 other samples with only one copy of SMN1 gene exons 7 and 8; and 4 copies of exon 7 of SMN2 gene. The other 2 samples showed normal copy numbers (Table 1).
The remaining 19 families were; 16 families with one carrier parent and the other partner showed normal copy number, and only 3 families both parents had normal copy number. The RQ values for the 16 families ranged from 0.48 and 0.93 (mean 0.7, SD 0.14, SE 0.035, CV% 19.85) for the carrier partner, and the other parent was non-carrier with RQ values between 1.0 and 4.0 (mean 1.57, SD 0.68, SE 0.17, CV% 43.39). For the 3 couples with both parents within the normal RQ values ranged from (1.03–2.4) (mean 1.5, SD 0.5, SE 0.2, CV% 31.9) (Table 1).
The last family (Family 53, not shown in Table1) was seeking genetic counseling due to a previously dead child, and a newborn with hypotonia and difficulty in breathing, suckling and swallowing. Real-time PCR has shown that the mother (19 years old) was the carrier with heterozygous deletion (1 copy) of exon 7 of SMN1 gene, whereas the father had homozygous deletion of exon 7. The daughter was affected with homozygous deletion of exon 7 with type I SMA (confirmed by MLPA). The patient died at the age of 5 months. The real-time PCR results for the father were confirmed by MLPA showing homozygous deletion of exons 7 and 8 of SMN1 gene, 5 copies of exon 7 of SMN2 gene, 4 copies of exon 8 of SMN2 gene and heterozygous deletion (1 copy) of exon 5 of NAIP gene. The rest of the cases were 4 males and 5 females referred to our lab individually. One male and 4 females were carriers and the rest had normal copy number.
MLPA was performed for 6 families for detection of SMA in amniotic fluid samples in pregnant mothers (Fig. 1). Results is shown in Table 2.


MLPA analysis by Coffalyser® software showing the tested probes (32 probes for SMN1, SMN2, NAIP and reference genes) against the median values ratios detected in three amniotic fluid samples from different three families (Normal range: 0.7—1.3). a Heterozygous deletion of exons 7 and 8 of SMN1 in amniotic fluid sample AF5. b Homozygous deletion of exons 7 & 8 of SMN1 in asymptomatic parent of special case family (Family 53). c Normal copies of SMN1 gene in amniotic fluid sample AF4
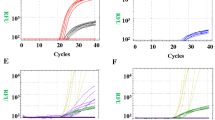
The amplification plots of the 50 normal controls showed almost identical Ct values (threshold cycle) of SMN1 and GAPDH genes. In the 10 SMA patients, only GAPDH gene was amplified showing Ct values near to that of normal controls. Whereas in obligate carriers (SMA parents), the Ct values of SMN1 increased by 2.2 (range 1.2–2.5) compared with Ct values of GAPDH gene.
The RQ (2−ΔΔCt) for normal controls ranged from 1.0 to 2.4, in SMA patients the RQ were 0.0 indicating homozygous absence of SMN1 gene and RQ values of the 20 SMA parents (obligate carriers) were between 0.4 and 0.6 (mean 0.5, SD 0.07, SE 0.02, CV% 13.3).
Discussion
SMA is a severe neurogenetic disease with high mortality rate among infants and young children. It is a widely spread autosomal recessive disease. Reports of carrier frequencies were 1:40–80 among different ethnic groups worldwide, while the prevalence of disease-prone mutation carriers ranged from 1/47 for European Caucasoids, to 1/72 for African Americans [7, 33]. Due to the high frequency of SMA and the severe clinical outcomes of the disease in general population, the American College of Obstetricians and Gynecologists (ACOG) and American College of Medical Genetics and Genomics (ACMG) recommend universal SMA carrier screening and prenatal screening of SMA in couples regardless of race or ethnicity [6, 9].
Since SMA is an autosomal recessive disorder, consanguinity is expected to play a major role in its prevalence and incidence. Egypt is a home to many different ethnic groups. Consanguineous marriages rate reached 39% [26, 31].
We established quantitative real-time PCR to determine the carrier status of SMA in families where a history of SMA-like neurologically affected previously deceased family member exists. Neurologic disorders have the highest frequency among all genetic disorders in Egypt with a ratio of 31.38%. In a retrospective study conducted in Egypt from January 1966 to December 2009, SMA constituted 18.66% of neuromuscular disorders and 0.02% of all patients attending the Pediatric Hospital (17.7/ 100,000). Comparing this frequency with published data, Egypt showed higher SMA frequency than USA, Germany, Italy, Poland, England, Saudi Arabia, and Libya. Consanguinity among the studied group (47.8%) was higher than the reported rate among the general population in Egypt (38.9%) [12, 25].
Our results showed that 57.4% of the couples seeking genetic counseling were carriers.
On the individual level, SMA carriers represented 75.7% of the studied individuals. That is 87 of 115 apparently healthy individuals included in this study were carriers with only one copy of SMN1 gene. This rate is much higher than the previously reported frequency rates [24], which could be explained by the selection criteria of the current study. As to validate the analysis, 50 healthy controls with no familial history of any neurogenetic disorders were tested showing normal copies of SMN1 gene.
The effect of the habit of consanguineous marriages appears evident in Arab populations. It raised the frequency of SMA carriers to reach 1 in 25 for morocco and 1 in 20 for both Iran and Saudi Arabia. Since India also practices consanguinity, in a group of 606 Indians, the carrier frequency was 1 in 38. SMA carrier frequency is heterogeneous among different ethnic populations. It was found to be higher among Caucasians and Ashkenazi Jews than Hispanics, Asians, and African American populations [20].
For the parent with SMN1 exons 7 and 8 deletion and 5 copies of SMN2 exon 7; two hypotheses arise. The first suggests that the father is an asymptomatic SMA type 4. Studies have shown that increasing copies of SMN2 provide a protective effect against severe forms of SMA in a direct relationship manner. Also, the presence of one copy of NAIP gene was accompanied by an increased copy number of SMN2 gene. Thus, individuals with homozygous SMN1 deletion and five or more copies of the SMN2 produce full-length SMN protein enough to compensate for the loss of the non-functioning SMN1 protein till adulthood [18, 30]. Both SMN2 and the NAIP are SMA-modifying genes and can ameliorate the disease severity while only SMN1 is SMA causing gene [13, 19].
The second possible explanation for our family concerns with the limitations of MLPA technique. Although MLPA is accurate and reliable in detecting SMN1 gene deletion, it cannot detect both pathogenic point mutations and certain paralogous sequence variants between SMN1 and SMN2 genes e.g. c.859G > C and c.835-44A > G), these SMN2 positive modifying variants are associated with better than expected phenotypes [2, 22].
Carrier detection in our study was carried out by two techniques. Real-time qPCR represents the simple, rapid, cheap, screening tool. While MLPA is considered leveling up for more detailed insight into the genotype. Yet, their limitations in detecting rare genotypes (“2 + 0” or pathogenic point mutations) must be convoyed to families in whom non-carrier by MLPA testing would be reported [17, 28. 35].
Multiple treatment options become available. As a national response to the high number of both SMA cases and carriers in Egypt, the Egyptian National Drug Authority has approved in June 2021 the first spinal muscular atrophy (SMA) therapy, Risdiplam.
Studies in Egypt have shown that a great ratio of neurologically affected dead children does not have a definite diagnosis. Thus, a considerable ratio of SMA patients is missed undiagnosed. Lack of proper awareness of SMA carriers in Egypt is one of the main reasons behind that [8, 24].
Because of the cost of the treatment, the importance of carrier detection or preconception carrier screening and prenatal diagnosis grows up, as a countermeasure to tackle the burden of a disease with low to moderate outcome country.
Conclusions
In conclusion, the carrier frequency detected in our cohort was high, which signifies SMA as a leading genetic cause of death among the infants in our study. This corresponds with the literature reports of SMA as a leading inherited cause of infant death [29]. Conjoining our results with the high prevalence of SMA worldwide and the high rate of consanguinity in developing countries confirms the importance of national SMA carrier screening in Egypt. Proper genetic counseling, carrier testing, and prenatal diagnosis all along with lowering the consanguinity rate are the SMA preventing scheme. This study highlights the importance of SMA carrier testing in Egypt. Thus, a large-scale population study to determine the SMA carrier frequency in Egypt is highly needed. As Egypt has a unique genetic pool, more population-specific studies on different geographical regions of the country with detailed demographical data would provide a more accurate insights about SMA carrier status. A population-based preconception carrier screening for couples will also help reduce the disease burden.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AAV9:
-
Adeno-associated virus 9.
- ACMG:
-
American College of Medical Genetics and Genomics.
- ACOG:
-
American College of Obstetricians and Gynecologists.
- AF:
-
Amniotic fluid
- CNs:
-
Copy numbers
- DNA:
-
Deoxyribonucleic acid
- FDA:
-
Food and drug administration
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase.
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MREC:
-
Medical Research Ethics Committee
- NAIP:
-
Neuronal apoptosis inhibitory protein
- OA:
-
Onasemnogene Abeparvovec-Xioi
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Quantitative real-time polymerase chain reaction
- RNA:
-
Ribonucleic acid
- RQ:
-
Relative quantification
- SMA:
-
Spinal muscular atrophy
- SMN:
-
Survival motor neuron
- SNVs:
-
Single nucleotide variants
References
Alías L, Barceló MJ, Bernal S, Martínez-Hernández R, Also-Rallo E, Vázquez C et al (2014) Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin Genet 85(5):470–475
Blasco-Pérez L, Paramonov I, Leno J, Bernal S, Alias L, Fuentes-Prior P et al (2021) Beyond copy number: a new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum Mutat 42(6):787–795
Burns JK, Kothary R, Parks RJ (2016) Opening the window: the case for carrier and perinatal screening for spinal muscular atrophy. Neuromuscul Disord 26(9):551–559. https://doi.org/10.1016/j.nmd.2016.06.459
Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, Aller E et al (2018) Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. J Neuromuscul Dis 28:208–215. https://doi.org/10.1016/j.nmd.2018.01.003
Chen X, Sanchis-Juan A, French CE, Connell AJ, Delon I, Kingsbury Z et al (2020) Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med 22(5):945–953. https://doi.org/10.1038/s41436-020-0754-0
Dejsuphong D, Taweewongsounton A, Khemthong P, Chitphuk S, Stitchantrakul W, Sritara P et al (2019) Carrier frequency of spinal muscular atrophy in Thailand. Neurol Sci 40(8):1729–1732
Eggermann K, Gläser D, Abicht A, Wirth B (2020) Spinal muscular atrophy (5qSMA): best practice of diagnostics, newborn screening and therapy. Medizinische Genet 32(3):263–272
Essawi M, Effat L, Shanab G, Al-Ettribi G, El-Haronui A, Karim A (2007) Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratislava Med J 108(3):133–137
Essawi M, Al-Attribi G, Gaber K, El-Harouni A (2012) Molecular prenatal diagnosis of autosomal recessive childhood spinal muscular atrophies (SMAs). Gene 509(1):120–123. https://doi.org/10.1016/j.gene.2012.07.085
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightcycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70(2):358–368
Finkel RS, Fischbeck KH (2021) Maybe too much of a good thing in gene therapy. Nat Neurosci. https://doi.org/10.1038/s41593-021-00882-w
Hasanzad M, Azad M, Kahrizi K, Saffar BS, Nafisi S, Keyhanidoust Z et al (2010) Carrier frequency of SMA by quantitative analysis of the SMN1 deletion in the Iranian population. Eur J Neurol 17(1):160–162
Hassan HA, Zaki MS, Issa MY, El-Bagoury NM, Essawi ML (2020) Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt J Med Hum Genet 21(1):6
Hensel N, Kubinski S, Claus P (2020) The need for SMN-independent treatments of spinal muscular atrophy (SMA) to complement SMN-enhancing drugs. Front Neurol 11(February):1–10
Lee TM, Kim SW, Lee KS, Jin HS, Koo SK, Jo I et al (2004) Quantitative analysis of SMN1 gene and estimation of SMN1 deletion carrier frequency in korean population based on real-time PCR. J Korean Med Sci 19(6):870–873
Li C, Geng Y, Zhu X, Zhang L, Hong Z, Guo X, et al (2020) The prevalence of spinal muscular atrophy carrier in China: evidences from epidemiological surveys. Medicine (Baltimore) 99(5):e18975
Li L, Zhou W, Fang P, Zhong Z, Xie J, Yan T, Zeng J, Tan X, Xu X (2017) Evaluation and comparison of three assays for molecular detection of spinal muscular atrophy. Clin Chem Lab Med 55(3):358–367. https://doi.org/10.1515/cclm-2016-0275
Macdonald WK, Hamilton D, Kuhle S (2014) SMA carrier testing: a meta-analysis of differences in test performance by ethnic group. Prenat Diagn 34(12):1219–1226
Nicolau S, Waldrop MA, Connolly AM, Mendell JR (2021) Spinal muscular atrophy. Semin Pediatr Neurol 37(00168):1–10
Nilay M, Moirangthem A, Saxena D, Mandal K, Phadke SR (2021) Carrier frequency of SMN1-related spinal muscular atrophy in north Indian population: the need for population based screening program. Am J Med Genet Part A 185(1):274–277
Park JE, Yun SA, Roh EY, Yoon JH, Shin S, Ki CS (2020) Carrier frequency of spinal muscular atrophy in a large-scale Korean population. Ann Lab Med 40(4):326–330
Ruhno C, McGovern VL, Avenarius MR, Snyder PJ, Prior TW, Nery FC et al (2019) Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet 138(3):241–256. https://doi.org/10.1007/s00439-019-01983-0
Sergott RC, Amorelli GM, Baranello G, Barreau E, Beres S, Kane S et al (2021) Risdiplam treatment has not led to retinal toxicity in patients with spinal muscular atrophy. Ann Clin Transl Neurol 8(1):54–65
Shawky RM, El-Sayed NS (2011) Clinico-epidemiologic characteristics of spinal muscular atrophy among Egyptians. Egypt J Med Hum Genet 12(1):25–30. https://doi.org/10.1016/j.ejmhg.2011.02.015
Shawky RM, Elsayed NS, Ibrahim DS, Seifeldin NS (2012) Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egypt J Med Hum Genet. 13(1):45–62. https://doi.org/10.1016/j.ejmhg.2011.10.002
Shawky RM, Elsayed SM, Zaki ME, Nour El-Din SM, Kamal FM (2013) Consanguinity and its relevance to clinical genetics. Egypt J Med Hum Genet 14(2):157–164. https://doi.org/10.1016/j.ejmhg.2013.01.002
Stabley DL, Holbrook J, Scavina M, Crawford TO, Swoboda KJ, Robbins KM et al (2021) Detection of SMN1 to SMN2 gene conversion events and partial SMN1 gene deletions using array digital PCR. Neurogenetics 22(1):53–64
Strom C, Anderson B, Peng M, Patel U, Braastad C, Sun W (2013) 1000 sample comparison of MLPA and RT-PCR for carrier detection and diagnostic testing for Spinal Muscular Atrophy Type 1. Open J Genet 3:111–114. https://doi.org/10.4236/ojgen.2013.32014
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, Flynn K, Hendrickson BC, Scholl T, Sirko-Osadsa DA, Allitto BA, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Euro J Human Genet. 2012;20(1):27–32. https://doi.org/10.1038/ejhg.2011.134.
Szunyogova E, Zhou H, Maxwell GK, Powis RA, Francesco M, Gillingwater TH et al (2016) Survival motor neuron (SMN) protein is required for normal mouse liver development. Sci Rep 6(October):1–15
Temtamy S, Aglan M (2012) Consanguinity and genetic disorders in Egypt. Middle East J Med Genet 1(1):12–17
Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC et al (2017) Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy—a literature review. Orphanet J Rare Dis 12(1):1–15
Yépez Y, Paradisi I, Arias S (2020) Spinal muscular atrophy in Venezuela: quantitative analysis of SMN1 and SMN2 genes. Egypt J Med Hum Genet 21(1)
Wijaya YO, Purevsuren J, Fatimah Harahap NI, Eko Niba ET, Bouike Y, Nurputra DK et al (2020) Assessment of spinal muscular atrophy carrier status by determining SMN1 copy number using dried blood spots. Int J Neonatal Screen 6(2):1–12. https://doi.org/10.3390/ijns6020043
Zhao S, Wang W, Wang Y, Han R, Fan C, Ni P et al (2021) NGS-based spinal muscular atrophy carrier screening of 10,585 diverse couples in China: a pan-ethnic study. Eur J Hum Genet 29(1):194–204. https://doi.org/10.1038/s41431-020-00714-8
Acknowledgments
The authors thank the patients and their families for accepting to participate in this study.
Funding
No Funding was received.
Author information
Authors and Affiliations
Contributions
NE contributed to the molecular studies, statistical analysis, and writing and revising the article. HH contributed to molecular studies and writing the article. SS contributed to prenatal diagnosis and AF sample collection from pregnant mothers. HS contributed to data analysis and revising the article. ME contributed to the study design, molecular studies, and revising of the article. All authors have read and approved the manuscript, and all authors equally contributed to the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Medical Research Ethics Committee of the host research centre approved the study protocol and results. Written informed consents were obtained from all participants according to the guidelines of the Medical Research Ethics Committee. Reference number is not applicable.
Consent for publication
Written informed consents for publication were obtained from all participants according to the guidelines of the medical research ethics committee (MREC).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eissa, N.R., Hassan, H.A., Senousy, S.M. et al. SMA carrier testing using Real-time PCR as a potential preconception screening tool. Egypt J Med Hum Genet 23, 24 (2022). https://doi.org/10.1186/s43042-022-00233-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00233-9