
Abstract
Background
Spinal muscular atrophy (SMA) is the most common autosomal recessive disorder in humans after cystic fibrosis. It is classified into five clinical grades based on age of onset and severity of the disease. Although SMN1 was identified as the SMA disease-determining gene, modifier genes mapped to 5q13 were affirmed to play a crucial role in determination of disease severity and used as a target for SMA therapy. In this study, we determined SMN2 copy number and NAIP deletion status in SMA Egyptian patients with different clinical phenotypes and had homozygous deletion of SMN1. We aimed at finding a prognostic genetic pattern including SMN1, SMN2, and NAIP gene genotypes to determine the clinical SMA type of the patient to help in genetic counseling and prenatal diagnosis.
Results
Copy number variations (CNVs) of exon 7 of SMN2 gene were significantly decreased with the increase in disease severity. Homozygous deletion of exon 5 of NAIP was detected in 60% (12/20) of type I SMA and in 73% (8/11) of type III SMA cases. Combining the data of the SMN2 and NAIP genes showed 8 genotypes. Patients with D2 genotype (0 copies of NAIP and 2 copies of SMN2) were likely to have type I SMA. Type II SMA patients mostly had no homozygous deletion of NAIP and 2 copies of SMN2. However, patients with N3 genotype (> 1 copy of NAIP and 3 copies of SMN2) and patients with D3 genotype (0 copies of NAIP and > 3 copies of SMN2) had type III SMA.
Conclusion
SMN2 and NAIP are the most important modifier genes whose copy numbers can affect the severity of SMA. We concluded that the combination of modifier genes to provide prognostic genetic pattern for phenotype determination is preferable than using CNVs of exon 7 of SMN2 gene only. CNVs of exon 7 of SMN2 are of high importance to predict patients’ response to genetic therapy. On the other hand, deletion of exon5 of NAIP gene alone is not a sufficient predictor of SMA severity.
Similar content being viewed by others

Background
Spinal muscular atrophy (SMA) is one of the most common autosomal recessive diseases. SMA is characterized by degeneration of alpha motor neurons in the spinal cord and the medulla oblongata, causing symmetrical proximal muscular atrophy. The incidence of SMA is approximately 1/6000 to 1/10,000 live births, and heterozygous healthy carriers show a very high frequency in general population (1 in 35) [1]. In the Middle East countries, the carrier frequencies are higher. In Morocco, it is 1/25 individuals, as well as in Iran, and Saudi Arabia, the carrier frequency is 1 in 20 [2,3,4].
SMA can be classified into five clinical grades based on age of onset and severity of the disease [5, 6]. Type 0 SMA patients present with very severe hypotonia and respiratory distress at birth. These SMA infants do not survive beyond 6 months. Type I SMA patients (Werdnig-Hoffmann disease) have an age of onset before 6 months at which hypotonia can be observed followed by progressive weakness which is worse proximally than distally and is initially more obvious in the legs making it difficult for them to sit up. They do not have the ability to sit without support and their life expectancy is less than 2 years; because muscle weakness affects the chest wall and diaphragm causing breathing difficulty and respiratory failure. Type II SMA patients (chronic spinal muscular atrophy) have an age of onset before 18 months. These patients ultimately attain the ability to sit independently when placed. However, most of these patients can hardly stand without support. Their life expectancy is until early adulthood. Type III SMA patients (juvenile spinal muscular atrophy or Kugelberg-Welander disease) have an age of onset > 18 months. These patients are able to walk independently and their legs are weaker than their arms. Type III patients usually have a normal lifespan. Type IV SMA patients (adult onset) have an age of onset after the age of 10 years. They exhibit slowly progressive limb weakness and associated with a normal life expectancy.
All five clinical subtypes were mapped to chromosome 5q13.2 region with a large inverted duplication with several repeated genes. In 1995, the survival motor neuron 1 (SMN1) was identified as the SMA disease-determining gene [7]. The SMN gene exists in 2 highly homologous copies (SMN1 and SMN2) that have been mapped to chromosome region 5q13 [8]. Approximately 94% of clinically typical SMA patients have a homozygous deletion of exon 7 of the telemetric copy of the gene (SMN1) [9], and most carriers have only one copy of SMN1 exon 7, as determined by SMN gene dosage analysis [1]. The centeromeric copy of SMN gene (SMN2) is almost identical to SMN1. The only critical difference is (840C > T) transition in exon 7 which is translationally silent; however, it affects the splicing pattern [8]. Both SMN genes code for full-length RNA with nine exons, but SMN2 mainly produces a transcript without exon 7 that encodes a truncated, non-functioning SMN protein (SMNΔ7). As a result, SMNΔ7 is the major product of SMN2 and the number of copies of SMN2 is linked to the severity of the disease. Approximately 20% of mRNA generated from SMN2 locus encodes SMN by virtue of alternative splicing; thus, a larger number of SMN2 copies correlate with a milder form of the disease [10]. Another gene in the region (5q13) is neuronal apoptosis inhibitory protein (NAIP). The functional role of NAIP in the pathogenesis of SMA has not been fully elucidated. However, some reports have demonstrated a correlation between deletion of the NAIP gene and severity of SMA [11]. This gene was also found to be frequently deleted in 45% of the Egyptian patients [12]. Each patient affected with SMA retains at least one SMN2 copy. An inverse correlation between SMN2 protein level and SMA severity has been reported in humans and transgenic SMA mice [13, 14]. The quantitative analysis of SMN2 copies in patients with type I, type II, or type III SMA showed a significant correlation between SMN2 copy number and type of SMA as well as duration of survival [15, 16]. Combined SMN1-SMN2-NAIP genotypes with fewer copies were associated with earlier onset age and poorer survival probability [17]. So, a child with homozygous absence of SMN1 to develop type I, type II, or type III SMA can be calculated on basis of SMN2 copy number. Moreover, it was found that better survival for type I SMA might due to higher SMN2 copies and homozygous deletion of NAIP [17].
Management of SMA is multidisciplinary supporting respiratory, gastrointestinal systems, nutritional, orthopedic care, and rehabilitation [18]. Advancement in gene therapy allows introduction of the first tailored molecular therapy for SMA. After FDA (U.S. Food and Drug Administration) and EMA (European Medicines Agency) approvals, nusinersen (Spinraza®) is currently the genuine SMA treatment. It is an antisense oligonucleotide that binds to SMN2 pre-mRNA and prevents exon 7 from being removed by alternative splicing. This mechanism increases the expressed full length SMN2 gene resulting into larger amount of functional SMN protein [19].
In the current study, we determined copy number variations (CNVs) of exon7 of SMN2 gene and the homozygous deletion of exon 5 of NAIP gene in Egyptian SMA patients with homozygous deletion of exon 7 of SMN1 gene in spite of having different clinical phenotypes. We aimed at reaching a correlation between CNVs of SMN1, SMN2, and NAIP genes to determine the clinical SMA type of each patient in an attempt to allow better disease prognosis, which would help in genetic counseling and prenatal diagnosis.
Methods
Forty-four patients were referred from Clinical Genetics out-patient clinics. SMA diagnosis was according to the criteria defined by the International SMA Consortium. Ten healthy individuals were used as a control group, for normalization of the real-time quantification data results within each run. Written informed consents were taken from all subjects or from their guardians.
Genomic DNA was extracted from the peripheral blood of SMA patients and controls according to standard methods. All patients had homozygous deletion of exon 7 of SMN1 gene, which was determined by PCR-RFLP. Both exon 5 deletion of NAIP gene and copy number variations of exon 7 of SMN2 gene were also estimated.
Deletions within SMN1 and NAIP genes
Homozygous absence of exon 7 of SMN1 gene was detected using restriction fragment length polymorphism (PCR-RFLP) using DraI restriction enzyme [18]. Deletion of the Neuronal Apoptosis Inhibitory Protein (NAIP) gene was analyzed using multiplex PCR amplification of exon 5 and 13 of the gene [12].
SMN2 genes dosage analysis
Copy number variations (CNVs) of exon 7 of SMN2 gene were determined by quantitative real-time PCR on StepOne Real-Time PCR System (Applied Biosystems, USA) using primers designed to distinguish between SMN1 and SMN2 according to Feldkötter and Schwarzer [15]. The PCR reaction was carried out in a final volume of 15 ul in duplicates containing 7 ul SYBR Green I master mix (Qiagen, Germany), 1 pmol of each primer, and 10 ng of DNA. Melting curve was performed after each reaction to confirm appropriate amplification. Calibration curve was constructed using Human Genomic DNA (Promega, Germany) by 2 fold dilutions (1.25, 2.5, 5, and 10 ng) representing 1, 2, 4, and 8 copies of SMN2 gene, respectively.
Determination of CNVs of exon 7 of SMN2 gene was performed through plotting the Ct (cycle threshold) value of each sample on the calibration curve to calculate CNVs of exon 7 of SMN2 gene.
Statistical analysis
Kruskal-Wallis test was performed to detect the relationship between CNV of exon 7 of SMN2, the deletion in exon 5 of NAIP gene, and patients’ phenotype. Statistical analysis was performed using SPSS software.
Results
The 44 patients were classified according to the age of onset and the severity of the disease into three types: SMA type I, 20 patients (45%); SMA type II, 13 patients (30%); and SMA type III, 11 patients (25%). PCR-RFLP analysis showed homozygous deletion of exon 7 of SMN1 gene in all the 44 patients.
Detection of homozygous deletion of exon 5 of NAIP gene: multiplex PCR analysis of exons 5 and 13 of NAIP gene revealed homozygous deletion of exon 5 in 26 patients (45%, 26/44), 12 patients with SMA type I (60%, 12/20), 6 patients with SMA type II (46%, 6/13), and 11 patients with SMA type III (73%, 8/11) (Fig. 1).

2% Agarose gel visualized by ethidium bromide showing, amplification of exon 5 of NAIP gene by multiplex PCR using exon 13 as an internal control. Lanes 1 and 4 showed no homozygous deletion. Lanes 2, 3, 5, and 6 showed homozygous deletion of exon 5
SMN2 gene copy number analysis
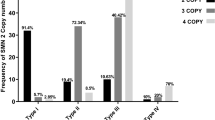
Real-time PCR was performed to determine the SMN2 gene dosage, through detection of CNVs of exon 7 of SMN2 gene using specific primers and SYBR Green master mix (Qiagen) (Fig. 2). Cycle threshold (Ct) values of all patients with the 3 clinical SMA types were normalized according to the constructed standard curve. The copy number variations estimated were 1 copy, 2 copies, 3 copies, or more than 3 copies. Figure 3 shows the number of patients with different SMA types and the corresponding CNVs of exon 7 of SMN2 gene. It was found that there were significant difference between the three clinical groups (SMA type I, II, and III); p value < 0.00001. In addition, 70% of type I-SMA patients had 1 copy of exon 7 of SMN2 gene. In type III-SMA patients, 82% of patients had more than 3 copies of exon 7 of SMN2 gene. In type I SMA, no patient had more than 2 copies of exon 7 of SMN2 gene. Patients with one copy of exon 7 of SMN2 gene were only of type I-SMA. Patients with two copies of exon 7 of SMN2 gene were either type I or II SMA, whereas patients with 3 copies or > 3 copies were of either type II or III SMA.

a. Amplification plot of SMN2 in SMA patients showing cycle threshold (CT) versus fluorescence. b. Calibration curve showing log quantity of serial dilutions of Human Genomic DNA (Promega) versus cycle threshold (CT)

The chart shows number of patients with different number of copies of SMN2 gene in each clinical phenotype (SMAI, II, or III)
Combining the genotypes of SMN2 and NAIP genes, 8 genotypes were determined (Table 1). No significant difference was detected between D1 and N1 genotypes. Significant difference was found in patients with type II SMA (p = 0.018) for the genotype N2. Patients carrying two SMN2 copies of exon 7 and with no deletion in exon 5 of NAIP gene (N2 genotype) were significantly related to type II SMA, whereas patients with the D2 genotype were likely to have type I SMA. Additionally, all patients with the D3 genotype had type II SMA; however, patients with the N3 genotype had type III SMA. It was found that the D4 genotype is significantly related to type III SMA rather than the N4 genotype (p = 0.01).
Discussion
Loss of both copies of exon 7 of the survival of motor neuron (SMN1) gene is the most common causative mutation of SMA. However, the phenotype and the clinical severity differ greatly. Modifier genes mapped to 5q13 chromosomal region were affirmed to play a crucial role in determination of disease severity. Being the major modifier gene of spinal muscular atrophy (SMA) disease, the higher the SMN2 copy number, the larger the amount of full-length SMN protein produced, and the milder the associated SMA phenotype [15, 19]. Also, NAIP gene is another disease-modifying gene, correlating with disease severity and duration of survival [20].
In this study, we determined the copy number variations (CNVs) of SMN2 and NAIP genes in 44 SMA Egyptian patients with homozygous deletion of exon 7 of SMN1 gene. Genotype-phenotype correlation revealed that CNVs of exon 7 of SMN2 significantly decreased with the increase in disease severity. The copy number variations of exon 7 of SMN2 were 1 or 2 in type I SMA, whereas in type II SMA, CNVs of exon 7 of SMN2 gene were 2, 3, or > 3. Finally, 3 copies or more than 3 copies were detected in SMA III patients. The revealed data could not discriminate clearly between the clinical types specifically in patients with 3 or more than 3 copies. Although SMN2 is considered as a positive modifier, it could not be a clear-cut phenotype predictor. This finding was also concluded by a Cypriotic study where phenotype could not be predicted by the SMN2 copy number in Cypriot SMA patients [21]. On the other hand, increasing the number of patients might give better discrimination due to the increase of the statistical values. Calucho and Bernal [22] assumed that the prediction based on SMN2 CNVs could be better attempted on a large Spanish cohort.
Our results showed that NAIP deletion alone also was not a good predictor of SMA severity as homozygous deletion was detected in 60% (12/20) of type I SMA and in 73% (8/11) of type III SMA, which was much lower than the published ratios in other populations. In Tunisian, Iranian, Kuwaiti, Malaysian, and Cypriot patients, the frequency of deletion of both SMN1 and NAIP genes in type I SMA patients ranged from 80 to 100% of the patients [21, 23,24,25,26]. However, type I SMA patients with homozygous deletion of exon 5 of NAIP gene have shown lower ratios of between 26% and 46% of German, Vietnamese, Chinese, and Serbian patients [17, 27,28,29,30]. It was noted that populations with higher parental consanguinity rates showed higher frequency of NAIP gene deletion. Therefore, deletion of NAIP gene might be related more to the mechanism of SMN1 gene deletion rather than the phenotype.
Combination of CNVs of exon 7 of SMN2 and deletion of exon 5 of NAIP genes revealed that they are closely related to the occurrence and development of spinal muscular atrophy in patients. In the current study, patients with homozygous deletion of exon 5 of NAIP gene and two copies of SMN2 gene were likely to have type I SMA, whereas type II SMA patients mostly had no homozygous deletion of exon 5 of NAIP and 2 copies of exon 7 of SMN2. Finally, patients with 3 copies of exon 7 of SMN2 and no homozygous deletion of exon 5 of NAIP in addition to patients with homozygous deletion of exon 5 of NAIP and more than 3 copies of exon 7 of SMN2 had type III SMA. He and Zhang [31] reported that 50% of SMA type I patients had homozygous deletion of exon 7 of SMN1 and exon 5 of NAIP genes and with either one or two copies of exon 7 of SMN2 gene, which is compatible with our results where 60% (12/20) of type I SMA patients had homozygous deletion of exon 5 of NAIP and 1 or 2 copies of exon 7 of SMN2 gene. Theodorou and Nicolaou [21] showed that patients with 2 copies of exon 7 of SMN2 and homozygous deletion of exon 5 of NAIP were type I SMA patients. In the current study, the presence or absence of exon 5 of NAIP gene was of less value in determining the type of SMA when CNVs of exon 7 of SMN2 were 1 or 2 copies. On the other hand, presence of exon 5 of NAIP was more likely to be associated with milder types of SMA, when CNVs of exon 7 of SMN2 were 2 or more copies and vice versa.
Conclusions
We implemented PCR-RFLP, multiplex PCR, and real-time PCR to analyze the correlation between CNVs of SMN1, NAIP, and SMN2 genes and the SMA phenotype in Egyptian patients. Although we used three different molecular techniques to generate the genotype of SMA patients, the study showed the preference of the combination of modifier genes as a prognostic genetic pattern for phenotype determination rather than using SMN2 CNVs only. The genetic pattern including SMN1, SMN2, and NAIP genes can help more in the prognosis of the more severe SMA type rather than the less severe types. Moreover, determination of CNVs of exon 7 of SMN2 is of great importance in placement of the effectiveness of the new therapy (nusinersen), as it depends on having at least one copy of exon 7 of SMN2 gene.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- CNV:
-
Copy number variation
- Ct:
-
Cycle threshold
- EMA:
-
European Medicines Agency
- FDA:
-
U.S. Food and Drug Administration
- NAIP:
-
Neuronal Apoptosis Inhibitory Protein gene
- PCR-RFLP:
-
Polymerase Chain Reaction - Restriction Fragment Length Polymorphism
- SMA:
-
Spinal muscular atrophy
- SMN1:
-
Survival motor neuron 1 gene
- SMN2:
-
Survival motor neuron 2 gene
References
Ogino S, Wilson RB (2002) Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 111(6):477–500
Al Jumah M, Majumdar R, Rehana Z, Al Rajeh S, Eyaid W (2007) A pilot study of spinal muscular atrophy carrier screening in Saudi Arabia. J Pediatr Neurol 5(03):221–224
Hasanzad M, Azad M, Kahrizi K, Saffar BS, Nafisi S, Keyhanidoust Z et al (2010) Carrier frequency of SMA by quantitative analysis of the SMN1 deletion in the Iranian population. Eur J Neurol 17(1):160–162
Lyahyai J, Sbiti A, Barkat A, Ratbi I, Sefiani A (2012) Spinal muscular atrophy carrier frequency and estimated prevalence of the disease in Moroccan newborns. Genet Test Mol Biomarkers 16(3):215–218
Munsat TL, Davies KE (1992) International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul Disord 2(5–6):423–428
Russman BS (2007) Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol 22(8):946–951
Roy N, McLean MD, Besner-Johnston A, Lefebvre C, Salih M, Carpten JD et al (1995) Refined physical map of the spinal muscular atrophy gene (SMA) region at 5q13 based on YAC and cosmid contiguous arrays. Genomics 26(3):451–460
Bürglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C et al (1996) Structure and organization of the human survival motor neurone (SMN) gene. Genomics 32(3):479–482
Wirth B (2000) An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 15(3):228–237
Cho S, Dreyfuss G (2010) A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev 24(5):438–442
Anderson K, Talbot K (2003) Spinal muscular atrophies reveal motor neuron vulnerability to defects in ribonucleoprotein handling. Curr Opin Neurol 16(5):595–599
Essawi ML, Effat LK, Shanab GM, Al-Ettribi GM, El-Haronui AA, Karim AM (2007) Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratisl Lek Listy 108(3):133–137
Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR et al (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6(8):1205–1214
Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in SMN(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9(3):333–339
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70(2):358–368
Zheleznyakova GY, Kiselev AV, Vakharlovsky VG, Rask-Andersen M, Chavan R, Egorova AA et al (2011) Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med Genet 12:96
Y-j Q, X-s G, J-l B, Wang L-w, Y-y C, Lu Y-y et al (2015) Association of copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein gene with the natural history in a Chinese spinal muscular atrophy cohort. J Child Neurol 30(4):429–436
Scheffer H, Cobben JM, Matthijs G, Wirth B (2001) Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur J Hum Genet 9(7):484–491
Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A et al (2006) Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 119(4):422–428
Scharf JM, Endrizzi MG, Wetter A, Huang S, Thompson TG, Zerres K et al (1998) Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat Genet 20(1):83
Theodorou L, Nicolaou P, Koutsou P, Georghiou A, Anastasiadou V, Tanteles G et al (2015) Genetic findings of Cypriot spinal muscular atrophy patients. Neurol Sci 36(10):1829–1834
Calucho M, Bernal S, Alias L, March F, Vencesla A, Rodriguez-Alvarez FJ et al (2018) Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord 28(3):208–215
Amara A, Adala L, Ben Charfeddine I, Mamai O, Mili A, Lazreg TB et al (2012) Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur J Paediatr Neurol 16(2):167–174
Omrani O, Bonyadi M, Barzgar M (2009) Molecular analysis of the SMN and NAIP genes in Iranian spinal muscular atrophy patients. Pediatr Int 51(2):193–196
Samilchuk E, D'Souza B, Bastaki L, Al-Awadi S. Deletion analysis of the SMN and NAIP genes in Kuwaiti patients with spinal muscular atrophy. Hum Genet 1996;98(5):524–527
Watihayati MS, Zabidi-Hussin AM, Tang TH, Matsuo M, Nishio H, Zilfalil BA (2007) Deletion analyses of SMN1 and NAIP genes in Malaysian spinal muscular atrophy patients. Pediatr Int 49(1):11–14
Hahnen E, Forkert R, Marke C, Rudnik-Schoneborn S, Schonling J, Zerres K et al (1995) Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet 4(10):1927–1933
Tran VK, Sasongko TH, Hong DD, Hoan NT, Dung VC, Lee MJ et al (2008) SMN2 and NAIP gene dosages in Vietnamese patients with spinal muscular atrophy. Pediatr Int 50(3):346–351
Liang YH, Chen XL, Yu ZS, Chen CY, Bi S, Mao LG et al (2009) Deletion analysis of SMN1 and NAIP genes in southern Chinese children with spinal muscular atrophy. J Zhejiang Univ Sci B 10(1):29–34
Miskovic M, Lalic T, Radivojevic D, Cirkovic S, Vlahovic G, Zamurovic D et al (2011) Lower incidence of deletions in the survival of motor neuron gene and the neuronal apoptosis inhibitory protein gene in children with spinal muscular atrophy from Serbia. Tohoku J Exp Med 225(3):153–159
He J, Zhang Q-J, Lin Q-F, Chen Y-F, Lin X-Z, Lin M-T et al (2013) Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene. 518(2):325–329
Acknowledgements
The authors thank the patients and their families for accepting to participate in this study.
Funding
The research was funded by NRC (National Research Centre in Egypt) through the project entitled 'Assessment the role of SMN & 1 SMN2 Genes copy numbers and NAIP Gene deletion in prediction of severity of Spinal Muscular Atrophy' Grant number: P101308.
Author information
Authors and Affiliations
Contributions
HH contributed to the molecular studies, statistical analysis, and writing and revising the article. MZ contributed in clinical diagnosis and assessment of patients. MI contributed to the clinical diagnosis and assessment of patients. NE contributed to molecular studies and writing the article. ME contributed to the contributed in the study design, molecular studies, and revising the article. All authors have read and approved the manuscript, and all authors equally contributed to the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consents were taken from all subjects or from their guardians according to Medical Research Ethics Committee (MREC) of the NRC [registration number: 15-219].
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hassan, H.A., Zaki, M.S., Issa, M.Y. et al. Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt J Med Hum Genet 21, 4 (2020). https://doi.org/10.1186/s43042-019-0044-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-019-0044-z