
Abstract
In this study, the mitogenomes of two Paxillus species were assembled, annotated and compared. The two mitogenomes of Paxillus involutus and P. rubicundulus comprised circular DNA molecules, with the size of 39,109 bp and 41,061 bp, respectively. Evolutionary analysis revealed that the nad4L gene had undergone strong positive selection in the two Paxillus species. In addition, 10.64 and 36.50% of the repetitive sequences were detected in the mitogenomes of P. involutus and P. rubicundulus, respectively, which might transfer between mitochondrial and nuclear genomes. Large-scale gene rearrangements and frequent intron gain/loss events were detected in 61 basidiomycete species, which revealed large variations in mitochondrial organization and size in Basidiomycota. In addition, the insertion sites of the basidiomycete introns were found to have a base preference. Phylogenetic analysis of the combined mitochondrial gene set gave identical and well-supported tree topologies, indicating that mitochondrial genes were reliable molecular markers for analyzing the phylogenetic relationships of Basidiomycota. This study is the first report on the mitogenomes of Paxillus, which will promote a better understanding of their contrasted ecological strategies, molecular evolution and phylogeny of these important ectomycorrhizal fungi and related basidiomycete species.
Similar content being viewed by others

INTRODUCTION
Most boreal and north temperate forest tree species form mutualistic symbiotic relationships with ectomycorrhizal fungi (ECMF), which are of great significance to the balance and stability of forest ecosystems, carbon and nitrogen cycles, and the growth and reproduction of the fungi and host tree.
The genus Paxillus is an important ectomycorrhizal fungal group, widely distributed in the northern hemisphere. Paxillus species form ectomycorrhizal associations with various host plant species, including coniferous and hardwood trees (Hedh et al. 2009). It was reported that Paxillus can enhance the abiotic stress tolerance and promote the growth of host plants (Franco et al. 2014; Li et al. 2012; Ma et al. 2014). Paxillus involutus, the most studied species in the genus, has become one of the model organisms for revealing the evolution, life-style and physiology of ECMF (Rineau et al. 2012; Shah et al. 2013).
Studies of Paxillus nuclear genomes have found that species of this genus have a reduced complement of genes encoding plant cell wall-degrading enzymes (PCWDEs) and retain a unique array of PCWDEs to adapt to mutualistic symbiotic life-styles (Kohler et al. 2015). Some nuclear genes have also evolved to better adapt to such life-styles (Kohler et al. 2015; Rajashekar et al. 2007). However, up to now, their mitochondrial genetic information has not yet been elucidated, limiting our overall understanding of the genetics and evolution of Paxillus species.
The genus Paxillus was divided into two groups in Europe, the P. involutus complex and P. rubicundulus. Paxillus rubicundulus occurs mainly in wetland habitats and only forms a relationship with Alnus species (Jargeat et al. 2016), while the P. involutus complex occurs in more diverse habitats and forms mutualistic relationships with various tree species. Therefore, host specificity could be used as a useful feature to distinguish P. rubicundulus (Jargeat et al. 2016), but accurately identifying species in the P. involutus complex remains difficult (Jargeat et al. 2014). Phylogenetic studies have revealed four genetic lineages within the P. involutus complex (Jargeat et al. 2014), and in addition there are some species whose taxonomic status remains confusing. This presents difficulties for efficient utilization and research into those Paxillus species. Mitochondrial genes have been reported as a powerful tool for the study of phylogeny due to several advantages (Delsuc et al. 2003; Nie et al. 2019), but up to now no mitochondrial genome (mitogenome) from the genus Paxillus, or even the family Paxillaceae, has been available.
The mitogenome of eukaryotic organisms was reported to have been obtained from the common ancestral Alphaproteobacteria though endosymbiosis (Lang et al. 1999). In basidiomycete species, they generally contain 15 core protein coding genes (PCGs), including 14 genes for energy metabolism (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, and nad6) and 1 rps3 gene for transcriptional regulation. In addition, 20–36 tRNA genes and 2 rRNA genes were also detected in basidiomycete mitogenomes (Li et al. 2018a; Li et al. 2018d). The mitogenome has become a powerful tool for the study of phylogeny due to several available molecular markers and uniparental inheritance (Andersen and Balding 2018; Wang et al. 2018). With the rapid development of next-generation sequencing technology, more and more nuclear and mitochondrial genomes of eukaryotic organisms have been obtained (Tajima et al. 2016). However, compared with their animal counterparts, the mitogenomes of fungi have been less studied (Sandor et al. 2018), especially that of basidiomycetes in the basidiomycete class Agaricomycetes (the largest mushroom-forming fungal group). The limited studies that have been carried out on fungal mitogenomes have, however, shown that these vary greatly in genome size, gene content, gene arrangement, intron number, and content of repeat sequences, even between closely related species (Li et al. 2018c; Li et al. 2019b).
In this study, the mitogenomes of two Paxillus species, P. involutus and P. rubicundulus, were assembled and annotated in order to: (1) reveal features of Paxillus mitogenomes and the variations or similarities between those of the two species in genome size, gene content, gene arrangement, and repeat sequence; (2) demonstrate any dynamic changes of introns and gene rearrangement in the two Paxillus mitogenomes compared with other basidiomycete mitogenomes to contribute to a more comprehensive understanding of their size and organization variation in basidiomycetes; and (3) analyze the phylogenetic relationships of Basidiomycota based on the combined mitochondrial gene set. The mitogenomes of the two Paxillus species could further our understanding of the taxonomy, evolution, and potential molecular mechanisms driving host specificity in an important ectomycorrhizal.
MATERIALS AND METHODS
Mitochondrial genome assembly and annotations
The raw sequencing data of Paxillus involutus and P. rubicundulus were downloaded from the Joint Genome Institute (JGI) database (Kohler et al. 2015). A series of quality control steps were conducted to obtain clean reads from the raw sequencing data. First, we removed adapter reads using AdapterRemoval v2 (Schubert et al. 2016), and then ngsShoRT (Chen et al. 2014) to filter low-quality sequences. The Paxillus mitogenomes were assembled with the resulting clean reads using SPAdes 3.9.0 (Bankevich et al. 2012), and gaps between contigs were filled using MITObim V1.9 (Hahn et al. 2013). The complete mitogenomes obtained were first annotated according to our previously described methods (Li et al. 2018a; Li et al. 2018d). That is, the protein-coding genes (PCGs), rRNA genes and tRNA genes of the mitogenomes were initially annotated using MFannot (Valach et al. 2014) and MITOS (Bernt et al. 2013), based on the genetic code 4. PCGs were then predicted or modified with the NCBI Open Reading Frame Finder (Coordinators 2017), and further annotated by BLASTP searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton 1990). tRNA genes were also predicted and identified with tRNAscan-SE v1.3.1 (Lowe and Chan 2016). Graphical maps of the two complete mitogenomes were drawn with OGDraw v1.2 (Lohse et al. 2013).
Sequence analysis of the Paxillus mitogenomes
Base compositions of the two Paxillus mitogenomes were calculated using DNASTAR Lasergene v7.1 (http://www.dnastar.com/). We then used the following formulas to assess strand asymmetries of the two mitogenomes: AT skew = [A - T] / [A + T], and GC skew = [G - C] / [G + C] (Wang et al. 2017). The synonymous (Ks) and nonsynonymous (Ka) substitution rates for core PCGs in the two mitogenomes were calculated with DnaSP v6.10.01(Rozas et al. 2017). MEGA v6.06 (Caspermeyer 2016) was used to calculate the overall mean genetic distances between each pair of the 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3), using the Kimura-2-parameter (K2P) substitution model.
Repetitive element analysis
To identify if there were interspersed repeats or intra-genomic duplications of large fragments throughout the two mitogenomes, we conducted BLASTN searches (Chen et al. 2015) of each mitogenome against itself using an E-value of < 10− 10. Tandem repeats (> 10 bp in length) in the two mitogenomes were detected using the Tandem Repeats Finder (Benson 1999) with default parameters. Repeated sequences in the two mitogenomes were also searched by REPuter (Kurtz et al. 2001) to identify forward (direct), reverse, complemented, and palindromic (reverse complemented) repeats. To identify any gene fragments transferring between nuclear genomes and mitochondrial genomes of the two Paxillus species, we performed BLASTn searches of the two mitogenomes against their previously published nuclear genomes (JOMD00000000.1 and JMDR00000000.1).
Comparative mitogenomic analysis and intron analysis
Up to now, only two mitogenomes from Boletales have been published (Li et al. 2019a). We compared the genome sizes, base compositions, gene numbers, intron numbers, and gene arrangements among different Boletales species to assess variations and conservativeness among the mitogenomes. Group I introns in cox1 genes of the 61 published Basidiomycota mitogenomes were classified into different position classes (Pcls) according to the method of Férandon et al. (Ferandon et al. 2010). Each Pcl was constituted by introns inserted at the same position in the coding region of the cox1 gene. The same Pcl from different species usually has a high sequence similarity, and contains orthologous intronic ORF. The Pcls of cox1 gene were named by letters according to the similarity with the described Pcls (Ferandon et al. 2010). The conservativeness and variations of sequences around intron insertion sites (− 15 bp - 15 bp) were evaluated using WebLogo (Crooks et al. 2004).
Phylogenetic analysis
In order to investigate the phylogenetic status of the two Paxillus species within the Basidiomycota, we constructed a phylogenetic tree of 62 species based on the combined mitochondrial gene set (15 core PCGs + 2 rRNA genes) (Li et al. 2018d). Annulohypoxylon stygium (Ascomycota) was used as outgroup (Deng et al. 2018). Individual mitochondrial genes were first aligned using MAFFT v7.037 (Katoh et al. 2017), and these alignments were then concatenated in SequenceMatrix v1.7.8 (Vaidya et al. 2011) to obtain a combined mitochondrial gene set. Potential phylogenetic conflicts between different genes were detected by a partition homogeneity test; PartitionFinder 2.1.1 (Lanfear et al. 2017) was used to determine best-fit models of evolution and partitioning schemes. Both Bayesian Inference (BI) and Maximum Likelihood (ML) methods were used to construct phylogenetic trees. MrBayes v3.2.6 (Ronquist et al. 2012) was used to perform the BI analysis, and RAxML v 8.0.0 (Stamatakis 2014) the ML analysis.
Data availability
The complete mitogenomes of Paxillus involutus and P. rubicundulus were deposited in GenBank under accession numbers MK993563 and MK993564, respectively.
RESULTS
Genome features and PCGs of Paxillus mitogenomes
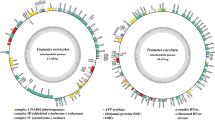
The mitogenomes of both species were both composed of circular nucleotide molecules, of 39,109 bp and 41,061 bp, respectively (Fig. 1). The average GC content of the two mitogenomes was 21.39%. Both species contained negative AT skews and positive GC skews (Table S1).

Circular maps of the mitogenomes of two Paxillus species. Genes are represented by different colored blocks. Coloured blocks outside each ring indicate that the genes are on the direct strand, while coloured blocks within the ring indicates that the genes are located on the reverse strand
There were 19 and 17 PCGs detected in the P. involutus and P. rubicundulus mitogenomes, respectively (Table S2), of which 78.95–82.35% were located in the direct strand. Both the mitogenomes contained 15 core PCGs, including 14 core PCGs for energy metabolism and one rps3 gene for transcriptional regulation. In addition, the P. involutus mitogenome contained a PCG located in the second intron of the cox1 gene, which encoded LAGLIDADG endonuclease. Paxillus rubicundulus contained a PCG encoding DNA polymerase. Three and one PCGs with unknown functions were detected in the P. involutus and P. rubicundulus mitogenomes, respectively. P. involutus and P. rubicundulus contained a homologous PCG showing no significant similarity with known protein sequences in the public databases checked; this encoded 732 amino acids in the P. involutus mitogenome and 792 amino acids in that of P. rubicundulus.
rRNA genes and tRNA genes
Both the Paxillus mitogenomes contained two rRNA genes, the large subunit ribosomal RNA (rnl) and small subunit ribosomal RNA (rns) genes (Table S2). The length of the rRNA genes varied between the two: there were five nucleotide length variations between the two rnl genes, and nine between the two rns genes.
The two mitogenomes both contained 25 tRNA genes, which encode 20 standard amino acids (Table S2). Both contained two tRNAs that code for arginine and serine with different anticodons, two coding for leucine, and three coding for methionine with the same anticodons. All tRNAs in the two mitogenomes were folded into a classical cloverleaf structures (Fig. S1), with each 71–86 bp long. Three tRNA genes, including trnY, trnS and trnL contained large extra arms, which resulted in their tRNA gene length exceeding 85 bp. Of the 25 tRNA genes shared by the two Paxillus mitogenomes, 10 contained sites that varied between the two. Thirteen variable sites were detected in the 25 tRNAs between the two species, of which 53.85% occurred on the acceptor arm, suggesting that the acceptor arm was highly variable in the two Paxillus mitogenomes.
Intergenic sequence and mitogenome composition
Both the mitogenomes contained two overlapping regions (Table S2), one of which was located between the nad2 and nad3 genes (13 bp). Another overlapping region in P. involutus was located between cox3 and orf732 (46 bp), and a further one in P. rubicundulus was across the neighboring cox3 and orf792 genes (88 bp). The two Paxillus mitogenomes lacked the common overlapping region found in some other Basidiomycota mitogenomes located between the nad4L and nad5 genes. A total of 11,295 bp and 13,790 bp intergenic sequences were detected in the P. involutus and P. rubicundulus mitogenomes, respectively, with each intergenic sequence ranging from 9 bp to 3537 bp. The longest intergenic sequence detected was between cob and cox3 in the P. rubicundulus mitogenome.
Protein coding regions accounted for the largest proportion (40.92–43.60%) of the two Paxillus mitogenomes, followed by the intergenic region, which accounted for 20.88–33.58% of the whole mitogenomes (Fig. 2). RNA regions, including rRNA and tRNA, accounted for 19.41–20.38% of the two mitogenomes. The intronic region accounted for the smallest proportion of the two mitogenomes, with only 6.09–7.13%. Compared with the P. involutus mitogenome, the size of that of P. rubicundulus was expanded by 1952 bp, to which the intergenic region contributed 127.82% of the size expansion, while the other regions contracted in the P. rubicundulus mitogenome relative to rgose in that of P. involutus, indicating that the intergenic region was the most important factor contributing to the dynamic changes of mitogenomes in the two Paxillus species.

The protein-coding, intronic, intergenic, and RNA gene region proportions of the entire mitogenomes of the two Paxillus species. The bottom panel shows the contribution of different gene regions to the expansion of the P. rubicundulus mitogenome
Repetitive elements in the two Paxillus mitogenomes
A total of 53 and 47 tandem repeats were detected in the P. involutus and P. rubicundulus mitogenomes, respectively (Table S3). The longest tandem repeat sequence was located in the P. rubicundulus mitogenome, with a length of 49 bp. Most of the tandem repeats contained 2–4 copies in the two mitogenomes. Using REPuter, we detected 1 complemented, 24 forward, 4 palindromic, and 21 reverse repeats in the P. involutus mitogenome, accounting for 3.13% of the whole mitogenome (Table S4). Repeated sequences identified by REPuter accounted for 3.25% of the P. rubicundulus mitogenome.
In order to detect whether there were gene fragments that naturally transferred between the mitochondrial and nuclear genomes of the two Paxillus species, we compared the two newly sequenced mitogenomes with their published nuclear genomes (Kohler et al. 2015) by BLASTN. Eleven repetitive fragments were identified between the mitochondrial and nuclear genomes of P. involutus, ranging from 33 bp to 3202 bp in length (Table S5). The pair-wise nucleotide similarities of these repetitive fragments ranged from 85.31–100%. The repetitive sequences detected in the P. involutus mitogenome accounted for 10.64% of the entire mitogenome. A total of 62 repetitive gene fragments were detected between the P. rubicundulus mitochondrial and nuclear genomes, with pair-wise nucleotide similarities ranging from 87.93–100%. The largest repeat fragment in the P. rubicundulus mitogenome reached 2367 bp, which contained a partial sequence of the rnl gene, the whole sequence of the cox2 gene, and the intergenic sequences around them. The repetitive gene fragments between P. rubicundulus mitochondrial and nuclear genomes accounted for 36.50% of the entire mitogenome.
Mitochondrial gene arrangement in Basidiomycota species
The mitochondrial gene arrangement in Basidiomycota varied greatly at the order level (Fig. 3). Of the 13 orders in Basidiomycota with data, no identical arrangement of mitochondrial genes was found between any two orders, suggesting that large-scale rearrangements of Basidiomycota mitochondrial genes had occurred in the course of evolution. At the level of family, only some species from Paxillaceae and Rhizopogonaceae were found to have the same gene order. Gene orders in other families were highly variable. At the generic level, we found large-scale gene rearrangement events in aevearl genera, including Cantharellus, Ganoderma, Lyophyllum, Pleurotus, and Russula. However, the gene arrangement of the two species of Paxillus studied was found to be conservative.

Mitochondrial gene arrangement analyses of 61 species from Basidiomycota. Genes are represented with different colour blocks. All genes are shown in order of occurrence in the mitochondrial genome, starting from cox1. Fourteen core protein coding genes, one rps3 gene, and two rRNA genes were included in the gene arrangement analysis. Species and NCBI accession number used for gene arrangement analysis in the present study are listed in Supplementary Table S6
Variation, genetic distance, and evolutionary rates of core PCGs
Among the 15 core PCGs detected, three genes showed length variations between the two Paxillus species, including cox3, nad2 and rps3 (Fig. 4). Among them, cox3 and nad2 genes showed two amino acid length variations between the two, while rps3 had one amino acid length variation between them. The GC content of 13 of the 15 core PCGs differed between the two Paxillus species, except for that of the atp6 and nad1 genes. Among the 15 core PCGs tested, atp9 had the highest GC content and atp8 the lowest. Most AT skews of core PCGs were negative, except for the rps3 gene. Eleven of the 15 core PCGs showed positive GC skews, while atp6, atp8, nad2 and nad6 had negative GC skews.

Variation in the length and base composition of each of 15 protein-coding genes (PCGs) between two Paxillus mitogenomes. a PCG length variation; b GC content of the PCGs; c AT skew; d GC skew
Among the 15 core PCGs detected, rps3 contained the largest K2P genetic distance between the two species (Fig. 5), followed by cox3 and nad2, which suggested that these genes were largely differentiated in the Paxillus species. The K2P genetic distance of atp8 was the lowest among the 15 core PCGs between the two mitogenomes, indicating that this gene was highly conserved in the two species. The cox3 gene exhibited the highest synonymous substitution (Ks) rate, while nad4L had the lowest substitution rate among the 15 core PCGs detected. The highest non-synonymous substitution rate (Ka) was in cox3, while atp9, cob and nad3 exhibited the lowest Ka values among the 15 core PCGs. The Ka/Ks values for most core PCGs were less than 1, indicating that these genes were subjected to purifying selection. However, the Ka/Ks value of nad4L gene was far greater than 1, indicating that this gene has been subjected to strong positive selection in the two Paxillus species. A non-synonymous mutation was detected in the nad4L gene, of which the 46th nucleotide ‘G’ in P. involutus mutated into nucleotide ‘A’ in P. rubicundulus.

Genetic analysis of 15 protein coding genes conserved in two Paxillus mitogenomes. K2P, the Kimura-2-parameter distance; Ka, the mean number of nonsynonymous substitutions per nonsynonymous site; Ks, the mean number of synonymous substitutions per synonymous site. The asterisk indicates that the gene is under strong pressure of positive selection
Intron dynamics of cox1 gene in Basidiomycota
According to previous studies (Ferandon et al. 2013), introns can be divided into different Pcls according to their precise insertion sites in the protein coding region of PCGs. Introns belonging to the same Pcls were considered as homologous and often contained homologous intronic open reading frames (ORFs). Introns belonging to different Pcls were considered as non-homologous introns, and showed low sequence similarities. In the present study, 367 introns were detected in cox1 genes of 61 Basidiomycota species, including 359 group I introns and 8 group II introns. The number of introns in the cox1 gene of individual species ranged from 0 to 19. A total of 32 position classes (Pcls) of group I introns were detected in the 61 species (Fig. 6), including five novel Pcls that had not been identified in previous studies, and 27 known Pcls. Pcls K was the most common intron class, which occurred in 36 of the species. Pcls P, AD, AI, N, and S were present in more than 30% of the species tested, and considered as the common Pcls in Basidiomycota. Pcls E was detected only in the cox1 gene of Ganoderma leucocontextum (MH252534) (Li et al. 2019e). However, it was present in the cox1 gene of the distantly related aquatic species Blastocladiella emersonii (Tambor et al. 2008) which belongs to the phylum Blastocladiomycota, a situation not readily explained as horizontal gene transfer between fungi which grow in such different environments seems improbable. In the two Paxillus species, we detected three group I introns, which belonged to different Pcls, indicating great variation of introns between them.

Position class (Pcl) information of cox1 genes of 61 Basidiomycota species. The same Pcl (orthologous intron) is represented by the same letter. The phylogenetic positions of the species were established using the Bayesian inference (BI) method and Maximum Likelihood (ML) method based on 15 concatenated mitochondrial core proteins and 2 rRNA genes. N1 - N5 represent the newly detected Pcl in this study; The II in the figure shows the intron belongs to the group II intron. Species ID are shown in Supplementary Table S6
The conservativeness and variation of nucleic acid sequences (30 bp) around insertion sites of different Pcls was also assessed (Fig. 7). We found that the insertion site preferences of different Pcls were quite different, while the nucleic acid sequences around the same Pcls were relatively conservative. Over 13 of the 30 nucleic acid loci detected were highly consistent around the same Pcls. Of the 32 group I intron Pcls we detected, 28 were inserted downstream of base T, while 11 Pcls were inserted behind GGT. In addition, 14 Pcls of 32 group I introns were located before base A, indicating a preference of intron insertion sites. These results could be useful for accurately identifying intron classes and annotating mitochondrial genes with multiple introns.

The insertion sites of different Pcls in the coding region of 61 Basidiomycete cox1 genes and the conservative analysis of the sequence around the insertion sites (− 15 bp – 15 bp). The symbols “+ 1” and “+ 2” indicate that the insertion of the intron occurs inside the indicated codon: between the nt 1 and nt 2 of this codon for “+ 1” and between the nt 2 and nt 3 for “+ 2”
Comparative mitogenome analysis and phylogenetic analysis
Compared with previously published Rhizopogon mitogenomes, the two Paxillus species contained smaller mitogenomes (Table S1). That of P. involutus was 97.16% smaller than the largest mitogenome, R. vinicolor. The two Paxillus species contained a higher proportion of protein coding regions, RNA regions, and intergenic regions, but a lower proportion of intronic regions relative to the Rhizopogon species. Amongst the four Boletales species we tested, they all contained negative AT skews and positive GC skews. The two Paxillus species contained less PCGs, intron and intronic ORFs than Rhizopogon species. However, the four species from Boletales had the same number of rRNA genes and tRNA genes.
Phylogenetic analysis using Maximum likelihood (ML) and Bayesian Inference (BI) methods based on the combined mitochondrial gene set (14 core PCGs + rps3 + 2 rRNA genes) yielded identical and well-supported tree topologies (Fig. 8). All major clades within the trees were well supported (BS ≥ 97; BPP ≥0.98). According to the phylogenetic tree, the 61 Basidiomycota species included could be divided into 13 major clades, corresponding to the orders Agaricales, Boletales, Cantharellales, Microbotryales, Microstromatales, Polyporales, Pucciniales, Russulales, Sporidiobolales, Tilletiales, Tremellales, Trichosporonales, and Ustilaginales (Table S6). Boletales had a close phylogenetic relationship with Agaricales and Russulales, and the four Boletales species could be divided into two groups, with P. involutus as a sister species to P. rubicundulus.

Molecular phylogeny of 61 Basidiomycete species based on Bayesian inference (BI) and Maximum likelihood (ML) analysis of 15 protein coding genes and two rRNA genes. Support values are Bayesian posterior probabilities (before slash) and bootstrap (BS) values (after slash). The asterisk indicates that the BPP value is 1 and the BS value is 100 of the branch. Species and NCBI accession numbers for genomes used in the phylogenetic analysis are provided in Supplementary Table S6
DISCUSSION
Genome size variation in Boletales
The mitochondrial genome size of fungi is highly variable, the largest being in Rhizoctonia solani (235.85 kb; (Losada et al. 2014) and the smallest in Spizellomyces punctatus (1.14 kb; (Forget et al. 2002). Intronic and intergenic regions, the content of repeat sequence and horizontally transferred genes have been found to be the main factors contributing to size dynamic changes in fungal mitogenomes (Losada et al. 2014; Salavirta et al. 2014). So far, only four complete mitogenomes from Boletales have been published, and these also varied in size. The two Rhizopogon species contained larger mitogenomes than those from Paxillus, was mainly due to their larger intronic regions. In Paxillus, the intergenic region was the main factor contributing to the size variation of the mitogenomes. Intronic and intergenic regions together contribute most to variations in mitogenome size in Boletales, as well as in other Basidiomycota so far examined (Ferandon et al. 2010).
Gene content variation in Paxillus species
Since eukaryotic organisms first acquired a mitogenome from endosymbiotic bacteria, most mitochondrial genes have been transferred to the nuclear genome during evolution, which is considered to have many advantages (Adams and Palmer 2003). However, most fungal mitochondria have so far been found to retain 14 core PCGs for energy metabolism and one rps3 gene for transcriptional regulation (Allen 2015; Bjorkholm et al. 2015). We found that the core PCGs in the two Paxillus species varied in base composition and length. In addition, the two core PCGs had different genetic distances between the species, indicating that different genes evolve at different rates. The nad4L gene is an important functional protein for ATP synthesis in mitochondria, which plays an important role in maintaining cell homeostasis and regulating celluar responses to the environment (Kazama et al. 2001). Interestingly, we found that the nad4L gene had experienced a strong positive selection between the two Paxillus species, which might be related to their host specificity. In addition, several PCGs coding homing endonucleases, DNA polymerase, and several unknown ORFs, were found in the species, indicating that some PCGs remain to be revealed. The tRNA and rRNA genes shared by the two Paxillus species also underwent base and length variations. Base variation in tRNA has been thought to affect the efficiency of amino acid transport and protein synthesis in animals (Ding et al. 2019; Lin et al. 2019). Further studies are needed to reveal the effect of tRNA variation on the growth and development of basidiomycete species.
Potential gene transfer between nuclear genome and mitogenome
The synergistic effects of nuclear and mitochondrial genes maintain the growth and development of eukaryotes (Baris et al. 2017; Latorre-Pellicer et al. 2016). During the evolution of eukaryotes, most mitochondrial genes have been transferred to the nucleus (Adams and Palmer 2003). However, some nuclear genes have also been found transferred to the mitogenome, and this has affected the evolution of eukaryotes (Li et al. 2019f; Srirattana and St John 2018). In our study, 10.64 and 36.50% of gene fragments were found potentially transferred between nuclear and mitochondrial genomes of the two Paxillus, respectively, which is a higher proportion than reported from Ganoderma lucidum (Li et al. 2013) and Pleurotus eryngii (Li et al. 2018a). Our result indicates that frequent natural gene transfer has occurred in the two Paxillus species. The impact of this transfer on the evolution and function of the Paxillus mitogenomes, however, remains unknown.
Gene rearrangement of basidiomycete species
The evolutionary rate of mitochondrial gene in fungi has been reported as intermediate between that of animals and plants (Aguileta et al. 2014; Barr et al. 2005). As more mitochondrial genomes have been sequenced, gene rearrangements found to be widespread in animals, plants, and fungi (Park et al. 2016; Sankoff et al. 1992). Various gene rearrangement models have been proposed to reveal gene rearrangement events in animals (Lavrov et al. 2002; Xia et al. 2016), but mitochondrial arrangements in fungi have been much less studied. We found that the mitochondrial gene arrangement in Basidiomycota was highly variable. At the order level, no two orders had the same gene order. In addition, we also observed large-scale gene rearrangements at the family or even generic level (Li et al. 2018a; Li et al. 2018b; Li et al. 2019b; Li et al. 2019c). The Basidiomycota mitogenome contains more repetitive sequences than animals, which we consider to be one of the main factors resulting in the high variation seen in the mitochondrial genome; this result is consistent with the findings of previous studies (Aguileta et al. 2014).
Dynamic changes of introns in cox1 gene of Basidiomycota
As a mobile genetic element (Burke 1988; Repar and Warnecke 2017), introns have been considered one of the main factors contributing to the variations of mitogenome organization and size (Hamari et al. 2002; Kanzi et al. 2016; Repar and Warnecke 2017). In Basidiomycota, the cox1 gene was found to harbour the most introns, with the largest proportion belonging to group I (Li et al. 2020). We detected 32 Pcls of the group I introns and 3 Pcls of the group II introns amongst the 61 species for which data were available. Among these intron Pcls, five were newly identified here, indicating that the phylum contains diverse intron types. Pcl K was the most widely distributed Pcl in Basidiomycota, and so was presumed to be derived from the phylum’s ancestors. Pcl E was only detected in Ganoderma leucocontextum (Li et al. 2019e), but this has also been reported in the distantly related Blastocladiella emersonii (Tambor et al. 2008), something suggestive of horizontal gene transfer, but difficult to imagine in view of the different ecologies of the two phyla. Different Basidiomycota species, even from within the same genera, could contain different numbers and Pcls of introns, indicating frequent intron loss or acquisition events during the evolution of the phylum. Introns contained homing endonucleases that can insert into specific sites in the mitogenome could potentially be used as gene editing tools (Chi et al. 2018; Zubaer et al. 2018). We assessed the conservativeness and variability of nucleic acid sequences around insertion sites of introns from basidiomycetes where information was available which showed that the insertion sites had certain base preferences. Our results provide a reference source for the accurate annotation of basidiomycete mitogenomes and also indicate a potential application of homing endonucleases in gene editing.
Phylogenetic relationships of Basidiomycota based on mitochondrial genes
Because of their independence from the nuclear genome, their rapid evolutionary rate, and the presence of many molecular markers, the mitogenome has become a powerful tool for studying phylogeny, population genetics, and taxonomy (Cameron 2014; Li et al. 2015; Li et al. 2019d). With the rapid development of next generation sequencing technology, more and more mitogenomes have been acquired, which has promoted the development of eukaryotic systems biology (Johri et al. 2019; Wang et al. 2018). However, the mitogenomes of fungi have been less studied than those of animals. Up to now, few mitochondrial molecular markers have been employed in studying their population genetics or phylogeny (Li et al. 2020). We obtained identical and well-supported tree topologies by BI and ML revealing phylogenetic relationships amongst 61species of Basidiomycota, based on the combined mitochondrial gene set. This demonstrates that the mitochondrial gene is a reliable molecular marker for the analysis of phylogenetic relationships of basidiomycetes and perhaps other fungal groups More mitogenomes are, however, needed to comprehensively assess the origin and evolution of the Basidiomycota.
CONCLUSION
In this study, the mitogenomes of two Paxillus species were assembled, annotated, and analyzed. Comparative mitogenome analysis indicated that the intronic and intergenic regions were the main factors contributing to mitogenome size variations in Boletales. The genome content, gene length, base composition, tRNAs, and rRNAs also varied greatly between the two Paxillus species studied. In addition, the nad4L gene was found to have been subjected to strong positive selection in the two species, which we suggest may be related to their contrasting ecological strategies. Potential gene fragment transferance between mitochondrial and nuclear genomes was also detected in the two Paxillus species.
Large-scale gene rearrangements were also found to have occurred in basidiomycete species for which data were avaliable, with introns experiencing frequent loss or gain events, resulting in large variations of mitogenome organization and size in Basidiomycota. In addition, the insertion sites of introns in basidiomycete mitogenomes were found to have a certain base preference. Phylogenetic analysis revealed that mitochondrial genes were reliable tools for analyzing phylogenetic relationships within Basidiomycota. As the first report on the mitogenome from the genus Paxillus or even the suborder Paxilineae of Boletales, this study contributes to our understanding of the evolution, genetics and phylogeny of both Paxillus species and the phylum Basiiomycota as a whole.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- mitogenome :
-
Mitochondrial genome
- BI:
-
Bayesian inference
- ML:
-
Maximum likelihood
- ECMF:
-
Ectomycorrhizal fungi
- PCG:
-
Protein-coding gene
- PCWDEs:
-
Plant cell wall–degrading enzymes
- Pcls:
-
Position classes
- Ks:
-
Synonymous substitution rates
- Ka:
-
Nonsynonymous substitution rates
References
Adams KL, Palmer JD (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Molecular Phylogenetics and Evolution 29(3):380–395
Aguileta G, de Vienne DM, Ross ON, Hood ME, Giraud T, Petit E, Gabaldon T (2014) High variability of mitochondrial gene order among fungi. Genome Biology and Evolution 6(2):451–465. https://doi.org/10.1093/gbe/evu028
Allen JF (2015) Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proceedings of the National Academy of Sciences of the United States of America 112(33):10231–10238. https://doi.org/10.1073/pnas.1500012112
Andersen MM, Balding DJ (2018) How many individuals share a mitochondrial genome? PLoS Genetics 14(11):ARTN e1007774. https://doi.org/10.1371/journal.pgen.1007774
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19(5):455–477. https://doi.org/10.1089/cmb.2012.0021
Baris TZ, Wagner DN, Dayan DI, Du X, Blier PU, Pichaud N, Oleksiak MF, Crawford DL (2017) Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genetics 13(3):ARTN e1006517. https://doi.org/10.1371/journal.pgen.1006517
Barr CM, Neiman M, Taylor DR (2005) Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. The New Phytologist 168(1):39–50. https://doi.org/10.1111/j.1469-8137.2005.01492.x
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27(2):573–580
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution 69(2):313–319. https://doi.org/10.1016/j.ympev.2012.08.023
Bjorkholm P, Harish A, Hagstrom E, Ernst AM, Andersson SG (2015) Mitochondrial genomes are retained by selective constraints on protein targeting. Proceedings of the National Academy of Sciences of the United States of America 112(33):10154–10161. https://doi.org/10.1073/pnas.1421372112
Bleasby AJ, Wootton JC (1990) Construction of validated, non-redundant composite protein sequence databases. Protein Engineering 3(3):153–159
Burke JM (1988) Molecular genetics of group I introns: RNA structures and protein factors required for splicing--a review. Gene 73(2):273–294
Cameron SL (2014) Insect mitochondrial genomics: implications for evolution and phylogeny. Annual Review of Entomology 59:95–117. https://doi.org/10.1146/annurev-ento-011613-162007
Caspermeyer J (2016) MEGA evolutionary software re-engineered to handle Today's big data demands. Molecular Biology and Evolution 33(7):1887. https://doi.org/10.1093/molbev/msw074
Chen C, Khaleel SS, Huang H, Wu CH (2014) Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code for Biology and Medicine 9:8. https://doi.org/10.1186/1751-0473-9-8
Chen Y, Ye W, Zhang Y, Xu Y (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Research 43(16):7762–7768. https://doi.org/10.1093/nar/gkv784
Chi SI, Urbarova I, Johansen SD (2018) Expression of homing endonuclease gene and insertion-like element in sea anemone mitochondrial genomes: lesson learned from Anemonia viridis. Gene 652:78–86. https://doi.org/10.1016/j.gene.2018.01.067
Coordinators NR (2017) Database resources of the national center for biotechnology information. Nucleic Acids Research 46(D1): D8-D13.https://doi.org/10.1093/nar/gkx1095
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Research 14(6):1188–1190. https://doi.org/10.1101/gr.849004
Delsuc F, Stanhope MJ, Douzery EJ (2003) Molecular systematics of armadillos (Xenarthra, Dasypodidae): contribution of maximum likelihood and Bayesian analyses of mitochondrial and nuclear genes. Molecular Phylogenetics and Evolution 28(2):261–275
Deng Y, Hsiang T, Li S, Lin L, Wang Q, Chen Q, Xie B, Ming R (2018) Comparison of the mitochondrial genome sequences of six Annulohypoxylon stygium isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Frontiers in Microbiology 9:2079. https://doi.org/10.3389/fmicb.2018.02079
Ding Y, Teng YS, Zhuo GC, Xia BH, Leng JH (2019) The mitochondrial tRNAHis G12192A mutation may modulate the clinical expression of deafness-associated tRNAThr G15927A mutation in a Chinese pedigree. Current Molecular Medicine 19(2):136-146. https://doi.org/10.2174/1566524019666190308121552
Ferandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, Barroso G (2010) The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group i introns. PLoS One 5(11):e14048. https://doi.org/10.1371/journal.pone.0014048
Ferandon C, Xu J, Barroso G (2013) The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal genetics and biology : FG & B 55:85–91. https://doi.org/10.1016/j.fgb.2013.01.009
Forget L, Ustinova J, Wang Z, Huss VA, Lang BF (2002) Hyaloraphidium curvatum: a linear mitochondrial genome, tRNA editing, and an evolutionary link to lower fungi. Molecular Biology and Evolution 19(3):310–319. https://doi.org/10.1093/oxfordjournals.molbev.a004084
Franco AR, Sousa NR, Ramos MA, Oliveira RS, Castro PM (2014) Diversity and persistence of ectomycorrhizal fungi and their effect on nursery-inoculated Pinus pinaster in a post-fire plantation in northern Portugal. Microbial Ecology 68(4):761–772. https://doi.org/10.1007/s00248-014-0447-9
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads--a baiting and iterative mapping approach. Nucleic Acids Research 41(13):e129. https://doi.org/10.1093/nar/gkt371
Hamari Z, Juhasz A, Kevei F (2002) Role of mobile introns in mitochondrial genome diversity of fungi (a mini review). Acta Microbiologica et Immunologica Hungarica 49(2–3):331–335. https://doi.org/10.1556/AMicr.49.2002.2-3.22
Hedh J, Johansson T, Tunlid A (2009) Variation in host specificity and gene content in strains from genetically isolated lineages of the ectomycorrhizal fungus Paxillus involutus s. lat. Mycorrhiza 19(8):549–558. https://doi.org/10.1007/s00572-009-0252-3
Jargeat P, Chaumeton JP, Navaud O, Vizzini A, Gryta H (2014) The Paxillus involutus (Boletales, Paxillaceae) complex in Europe: genetic diversity and morphological description of the new species Paxillus cuprinus, typification of P. involutus s.s., and synthesis of species boundaries. Fungal Biology 118(1):12–31. https://doi.org/10.1016/j.funbio.2013.10.008
Jargeat P, Moreau PA, Gryta H, Chaumeton JP, Gardes M (2016) Paxillus rubicundulus (Boletales, Paxillaceae) and two new alder-specific ectomycorrhizal species, Paxillus olivellus and Paxillus adelphus, from Europe and North Africa. Fungal Biology 120(5):711–728. https://doi.org/10.1016/j.funbio.2016.02.008
Johri P, Marinov GK, Doak TG, Lynch M (2019) Population genetics of paramecium mitochondrial genomes: recombination, mutation spectrum, and efficacy of selection. Genome Biology and Evolution. https://doi.org/10.1093/gbe/evz081
Kanzi AM, Wingfield BD, Steenkamp ET, Naidoo S, van der Merwe NA (2016) Intron derived size polymorphism in the mitochondrial genomes of closely related Chrysoporthe species. PLoS One 11(6):e0156104. https://doi.org/10.1371/journal.pone.0156104
Katoh K, Rozewicki J, Yamada KD (2017) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics 20(4):1160-1166.https://doi.org/10.1093/bib/bbx108
Kazama F, Yatomi Y, Ohmori T, Hosogaya S, Ozaki Y (2001) Elevated expression of mitochondrial NADH dehydrogenase subunit 4/4L genes in vascular endothelial cells undergoing sphingosine-induced apoptosis. Thrombosis and Haemostasis 85(3):572–573
Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, Canback B, Choi C, Cichocki N, Clum A, Colpaert J, Copeland A, Costa MD, Dore J, Floudas D, Gay G, Girlanda M, Henrissat B, Herrmann S, Hess J, Hogberg N, Johansson T, Khouja HR, LaButti K, Lahrmann U, Levasseur A, Lindquist EA, Lipzen A, Marmeisse R, Martino E, Murat C, Ngan CY, Nehls U, Plett JM, Pringle A, Ohm RA, Perotto S, Peter M, Riley R, Rineau F, Ruytinx J, Salamov A, Shah F, Sun H, Tarkka M, Tritt A, Veneault-Fourrey C, Zuccaro A, Mycorrhizal Genomics Initiative C, Tunlid A, Grigoriev IV, Hibbett DS, Martin F (2015) Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nature Genetics 47(4):410–415. https://doi.org/10.1038/ng.3223
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research 29(22):4633–4642
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution 34(3):772–773. https://doi.org/10.1093/molbev/msw260
Lang BF, Gray MW, Burger G (1999) Mitochondrial genome evolution and the origin of eukaryotes. Annual Review of Genetics 33:351–397. https://doi.org/10.1146/annurev.genet.33.1.351
Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sanchez-Cabo F, Torroja C, Acin-Perez R, Calvo E, Aix E, Gonzalez-Guerra A, Logan A, Bernad-Miana ML, Romanos E, Cruz R, Cogliati S, Sobrino B, Carracedo A, Perez-Martos A, Fernandez-Silva P, Ruiz-Cabello J, Murphy MP, Flores I, Vazquez J, Enriquez JA (2016) Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535(7613):561–565. https://doi.org/10.1038/nature18618
Lavrov DV, Boore JL, Brown WM (2002) Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Molecular Biology and Evolution 19(2):163–169. https://doi.org/10.1093/oxfordjournals.molbev.a004068
Li H, Shao R, Song N, Song F, Jiang P, Li Z, Cai W (2015) Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Scientific Reports 5:8527. https://doi.org/10.1038/srep08527
Li J, Bao S, Zhang Y, Ma X, Mishra-Knyrim M, Sun J, Sa G, Shen X, Polle A, Chen S (2012) Paxillus involutus strains MAJ and NAU mediate K(+)/Na(+) homeostasis in ectomycorrhizal Populus x canescens under sodium chloride stress. Plant Physiology 159(4):1771–1786. https://doi.org/10.1104/pp.112.195370
Li J, Zhang J, Chen H, Chen X, Lan J, Liu C (2013) Complete mitochondrial genome of the medicinal mushroom Ganoderma lucidum. PLoS One 8(8):e72038. https://doi.org/10.1371/journal.pone.0072038
Li Q, Chen C, Xiong C, Jin X, Chen Z, Huang W (2018a) Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Applied Microbiology and Biotechnology 102(14):6143-6153.https://doi.org/10.1007/s00253-018-9082-6
Li Q, He X, Ren Y, Xiong C, Jin X, Peng L, Huang W (2020) Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and asymbiotic Amanita species. Frontiers in Microbiology 11: 1382.
Li Q, Liao M, Yang M, Xiong C, Jin X, Chen Z, Huang W (2018b) Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of Agaricomycetes. International Journal of Biological Macromolecules 118(Pt a):756-769. https://doi.org/10.1016/j.ijbiomac.2018.06.129
Li Q, Ren Y, Shi X, Peng L, Zhao J, Song Y, Zhao G (2019a) Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum Agaricomycotina. International Journal of Molecular Sciences 20(20):5167. https://doi.org/10.3390/ijms20205167
Li Q, Wang Q, Chen C, Jin X, Chen Z, Xiong C, Li P, Zhao J, Huang W (2018c) Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. International Journal of Biological Macromolecules 119:792–802. https://doi.org/10.1016/j.ijbiomac.2018.07.197
Li Q, Wang Q, Jin X, Chen Z, Xiong C, Li P, Liu Q, Huang W (2019b) Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. International Journal of Biological Macromolecules 121:249–260. https://doi.org/10.1016/j.ijbiomac.2018.10.029
Li Q, Wang Q, Jin X, Chen Z, Xiong C, Li P, Zhao J, Huang W (2019c) Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes. International Journal of Biological Macromolecules 121:364–372. https://doi.org/10.1016/j.ijbiomac.2018.10.037
Li Q, Wang Q, Jin X, Chen Z, Xiong C, Li P, Zhao J, Huang W (2019d) The first complete mitochondrial genome from the family Hygrophoraceae (Hygrophorus russula) by next-generation sequencing and phylogenetic implications. International Journal of Biological Macromolecules 122:1313–1320. https://doi.org/10.1016/j.ijbiomac.2018.09.091
Li Q, Xiang D, Wan Y, Wu Q, Wu X, Ma C, Song Y, Zhao G, Huang W (2019e) The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. International Journal of Biological Macromolecules 139:397–408. https://doi.org/10.1016/j.ijbiomac.2019.08.003
Li Q, Yang L, Xiang D, Wan Y, Wu Q, Huang W, Zhao G (2020) The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. International Journal of Biological Macromolecules 145:974–984. https://doi.org/10.1016/j.ijbiomac.2019.09.188
Li Q, Yang M, Chen C, Xiong C, Jin X, Pu Z, Huang W (2018d) Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Scientific Reports 8(1):9104. https://doi.org/10.1038/s41598-018-27489-9
Li W, Freudenberg J, Freudenberg J (2019f) Alignment-free approaches for predicting novel nuclear mitochondrial segments (NUMTs) in the human genome. Gene 691:141–152. https://doi.org/10.1016/j.gene.2018.12.040
Lin L, Cui P, Qiu Z, Wang M, Yu Y, Wang J, Sun Q, Zhao H (2019) The mitochondrial tRNA (ala) 5587T>C and tRNA (Leu (CUN)) 12280A>G mutations may be associated with hypertension in a Chinese family. Experimental and Therapeutic Medicine 17(3):1855–1862. https://doi.org/10.3892/etm.2018.7143
Lohse M, Drechsel O, Kahlau S, Bock R (2013) OrganellarGenomeDRAW--a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Research 41(web server issue):W575–W581. https://doi.org/10.1093/nar/gkt289
Losada L, Pakala SB, Fedorova ND, Joardar V, Shabalina SA, Hostetler J, Pakala SM, Zafar N, Thomas E, Rodriguez-Carres M, Dean R, Vilgalys R, Nierman WC, Cubeta MA (2014) Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiology Letters 352(2):165–173. https://doi.org/10.1111/1574-6968.12387
Lowe TM, Chan PP (2016) tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Research 44(W1):W54–W57. https://doi.org/10.1093/nar/gkw413
Ma Y, He J, Ma C, Luo J, Li H, Liu T, Polle A, Peng C, Luo ZB (2014) Ectomycorrhizas with Paxillus involutus enhance cadmium uptake and tolerance in Populus x canescens. Plant, Cell & Environment 37(3):627–642. https://doi.org/10.1111/pce.12183
Nie Y, Wang L, Cai Y, Tao W, Zhang YJ, Huang B (2019) Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Applied Microbiology and Biotechnology 103(3):1379–1391. https://doi.org/10.1007/s00253-018-9549-5
Park JS, Kim MJ, Jeong SY, Kim SS, Kim I (2016) Complete mitochondrial genomes of two gelechioids, Mesophleps albilinella and Dichomeris ustalella (Lepidoptera: Gelechiidae), with a description of gene rearrangement in Lepidoptera. Current Genetics 62(4):809–826. https://doi.org/10.1007/s00294-016-0585-3
Rajashekar B, Samson P, Johansson T, Tunlid A (2007) Evolution of nucleotide sequences and expression patterns of hydrophobin genes in the ectomycorrhizal fungus Paxillus involutus. The New Phytologist 174(2):399–411. https://doi.org/10.1111/j.1469-8137.2007.02022.x
Repar J, Warnecke T (2017) Mobile introns shape the genetic diversity of their host genes. Genetics 205(4):1641–1648. https://doi.org/10.1534/genetics.116.199059
Rineau F, Roth D, Shah F, Smits M, Johansson T, Canback B, Olsen PB, Persson P, Grell MN, Lindquist E, Grigoriev IV, Lange L, Tunlid A (2012) The ectomycorrhizal fungus Paxillus involutus converts organic matter in plant litter using a trimmed brown-rot mechanism involving Fenton chemistry. Environmental Microbiology 14(6):1477–1487. https://doi.org/10.1111/j.1462-2920.2012.02736.x
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61(3):539–542. https://doi.org/10.1093/sysbio/sys029
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution 34(12):3299–3302. https://doi.org/10.1093/molbev/msx248
Salavirta H, Oksanen I, Kuuskeri J, Makela M, Laine P, Paulin L, Lundell T (2014) Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS One 9(5):e97141. https://doi.org/10.1371/journal.pone.0097141
Sandor S, Zhang Y, Xu J (2018) Fungal mitochondrial genomes and genetic polymorphisms. Applied Microbiology and Biotechnology 102(22):9433–9448. https://doi.org/10.1007/s00253-018-9350-5
Sankoff D, Leduc G, Antoine N, Paquin B, Lang BF, Cedergren R (1992) Gene order comparisons for phylogenetic inference: evolution of the mitochondrial genome. Proceedings of the National Academy of Sciences of the United States of America 89(14):6575–6579
Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Research Notes 9:88. https://doi.org/10.1186/s13104-016-1900-2
Shah F, Rineau F, Canback B, Johansson T, Tunlid A (2013) The molecular components of the extracellular protein-degradation pathways of the ectomycorrhizal fungus Paxillus involutus. The New Phytologist 200(3):875–887. https://doi.org/10.1111/nph.12425
Srirattana K, St John JC (2018) Additional mitochondrial DNA influences the interactions between the nuclear and mitochondrial genomes in a bovine embryo model of nuclear transfer. Scientific Reports 8(1):7246. https://doi.org/10.1038/s41598-018-25516-3
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Tajima N, Saitoh K, Sato S, Maruyama F, Ichinomiya M, Yoshikawa S, Kurokawa K, Ohta H, Tabata S, Kuwata A, Sato N (2016) Sequencing and analysis of the complete organellar genomes of Parmales, a closely related group to Bacillariophyta (diatoms). Current Genetics 62(4):887–896. https://doi.org/10.1007/s00294-016-0598-y
Tambor JH, Ribichich KF, Gomes SL (2008) The mitochondrial view of Blastocladiella emersonii. Gene 424(1–2):33–39. https://doi.org/10.1016/j.gene.2008.07.031
Vaidya G, Lohman DL, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27(2):171–180
Valach M, Burger G, Gray MW, Lang BF (2014) Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Research 42(22):13764–13777. https://doi.org/10.1093/nar/gku1266
Wang J, Zhang L, Zhang QL, Zhou MQ, Wang XT, Yang XZ, Yuan ML (2017) Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 5:e3661. https://doi.org/10.7717/peerj.3661
Wang L, Zhang S, Li JH, Zhang YJ (2018) Mitochondrial genome, comparative analysis and evolutionary insights into the entomopathogenic fungus Hirsutella thompsonii. Environmental Microbiology 20(9):3393–3405. https://doi.org/10.1111/1462-2920.14379
Xia Y, Zheng Y, Murphy RW, Zeng X (2016) Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genomics 17(1):965. https://doi.org/10.1186/s12864-016-3309-7
Zubaer A, Wai A, Hausner G (2018) The mitochondrial genome of Endoconidiophora resinifera is intron rich. Scientific Reports 8(1):17591. https://doi.org/10.1038/s41598-018-35926-y
Acknowledgements
Not applicable.
Adherence to national and international regulations
Not applicable.
Funding
This study was supported by the start up funds for talent introduction of Chengdu University.
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: QL, YR, and GZ. Analyzed the data: QL, XS, JZ, DX and LP. Wrote the Acknowledgements paper: QL. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Fig. S1.
Putative secondary structures of the 25 tRNA genes identified in the mitogenomes of two Paxillus species. Residues conserved across the two mitogenomes are shown in green, while variable sites are shown in red. All genes are shown in order of occurrence in the mitogenome of P. involutus, starting from trnM.
Additional file 2 Table S1.
Comparison on mitogenomes between four Boletales species. Table S2. Gene features and organization of the Paxillus mitogenomes. Table S3. Tandem repeats detected in the mitogenomes of two Paxillus species using the Tandem Repeats Finder. Table S4. Distribution of repeat loci in the two Paxillus mitogenomes searched by REPuter. Table S5. Local BLAST searches between the mitochondrial and the nuclear genomes of two Paxillus species. Table S6. Species and GenBank accession number used for phylogenetic analysis in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Ren, Y., Xiang, D. et al. Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11, 12 (2020). https://doi.org/10.1186/s43008-020-00038-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43008-020-00038-8