
Abstract
In the present study, we assembled and analyzed the mitogenomes of two Ramaria species. The assembled mitogenomes of Ramaria cfr. rubripermanens and R. rubella were circularized, with sizes of 126,497 bp and 143,271 bp, respectively. Comparative mitogenome analysis showed that intron region contributed the most (contribution rate, 43.74%) to the size variations of Ramaria mitogenomes. The genetic contents, gene length, tRNAs, and codon usages of the two Ramaria mitogenomes varied greatly. In addition, the evolutionary rates of different core protein coding genes (PCGs) in Phallomycetidae mitogenomes varied. We detected large-scale gene rearrangements between Phallomycetidae mitogenomes, including gene displacement and tRNA doubling. A total of 4499 bp and 7746 bp aligned fragments were detected between the mitochondrial and nuclear genomes of R. cfr. rubripermanens and R. rubella, respectively, indicating possible gene transferring events. We further found frequent intron loss/gain and potential intron transfer events in Phallomycetidae mitogenomes during the evolution, and the mitogenomes of R. rubella contained a novel intron P44. Phylogenetic analyses using both Bayesian inference (BI) and Maximum Likelihood (ML) methods based on a combined mitochondrial gene dataset obtained an identical and well-supported phylogenetic tree for Basidiomycota, wherein R. cfr. rubripermanens and Turbinellus floccosus are sister species. This study served as the first report on mitogenomes from the genus Ramaria, which provides a basis for understanding the evolution, genetics, and taxonomy of this important fungal group.
Similar content being viewed by others

INTRODUCTION
Species from the genus Ramaria are usually known as coral fungi because of their colorful and finely branched sporocarps. Ramaria species are conspicuous ectomycorrhizal fungi associated with conifers, which are widely distributed in the northern hemisphere (Nouhra et al. 2005). The micro and macro morphological features of Ramaria are quite diverse. Some Ramaria fruiting bodies are considered to be poisonous, including Ramaria flavo-brunnescens and Ramaria rufescens (Huang et al. 2009; Perez-Moreno and Ferrera-Cerrato 1995; Scheid et al. 2022), while other Ramaria species are edible, such as Ramaria botrytis, Ramaria madagascariensis, Ramaria largentii, Ramaria flava, and Ramaria formosa (Aprotosoaie et al. 2017; Khaund and Joshi 2014; Liu et al. 2014, 2015). Polysaccharide, sesquiterpenes, and glucan extracted from the basidiocarps of Ramaria exhibited antioxidant, antigenotoxic, antitumor, antimicrobial, and immunoenhancing activities (Barros et al. 2008; Bhanja et al. 2013; Centko et al. 2012; Dong et al. 2020; Fu et al. 2022; Li 2017; Liu et al. 2013; Zou et al. 2021). All in all, the accurate identification and classification of Ramaria species is very important for the development and utilization of Ramaria species.
Ramaria species belong to the order Gomphales and the subclass Phallomycetidae, which occupy a unique phylogenetic position in the higher Basidiomycetes (Nouhra et al. 2005). Taxonomy of the Gomphales has traditionally relied upon morphological features now known to be subject to parallel evolution and phenotypic plasticity. Inaccurate classification limits the discovery and utilization of Ramaria species. Subsequently, morphological and molecular data (mit-atp6-DNA, mit-12S-rDNA, and nuc-25S-rDNA) have been used to infer inter- and intra-specific relationships among Ramaria and closely related genera, which reveal that Gomphales species are closely related to taxa in the Geastrales, Phallales, and Hysterangiales (Giachini et al. 2010). Mitochondrial genomes (mitogenomes) have been reported to be an effective tool for analyzing the phylogeny of basidiomycetes. Up to now, we have reported four complete mitogenomes from Phallomycetidae, including Turbinellus floccosus (Cheng et al. 2021), Sphaerobolus stellatus (Ye et al. 2020), Dictyophora indusiate, and Phallus echinovolvatus (Chen et al. 2020a). However, the characteristics of mitogenomes from the genus Ramaria are still unknown, which limits our comprehensive understanding of mitochondrial evolutionary pattern and genetics of Ramaria species.
Mitochondrial genome is called the ‘second genome’ of eukaryotes, which is considered to be obtained from bacteria through endosymbiosis (Gray et al. 2001). Independent origin, single parent inheritance and several available molecular markers make the mitogenome an effective tool to infer the phylogenetic relationship of species (Abuduaini et al. 2021; Basse 2010). In addition, mitochondrial gene arrangement, variation of conserved genes, dynamic changes of introns, structural variation of tRNA, codon usage and so on also provide rich genetic information for the analysis of species variation and evolution (Fonseca et al. 2021; Ren et al. 2021; Wu et al. 2021a; Zhang et al. 2021a). The mitogenome of fungi has been less studied than that of animals. Mitogenome mutations in animals are considered to be closely related to the occurrence of diseases (Du et al. 2022; Pickett et al. 2022; Zhong et al. 2022). In addition, mitogenomes have also become a powerful tool for analyzing animal systematics and population genetics (Garcia-Souto et al. 2022; Wolfsberger et al. 2022; Zhang et al. 2021b). However, the relationship between fungal mitogenome variation and fungal growth, development and stress response are still unknown. The purpose of obtaining the complete sequence of fungal mitogenome is to further analyze the relationship between fungal mitochondrial sequence mutation and fungal traits. It is reported that the mitogenome of fungi varies greatly in gene arrangement, repeat sequence content, gene number and intron species, even among closely related species (Araujo et al. 2021, Fonseca et al. 2021; Li et al. 2020b; Li et al. 2021b). While most basidiomycetes have been reported to contain a series of core protein coding genes, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3 (Li et al. 2021a), the large variations in the content and structure of fungal mitogenomes make it very difficult to obtain a complete fungal mitogenome. Up to now, less than 150 basidiomycete mitogenomes have been published (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/), while hundreds of thousands of basidiomycetes have been described in nature. The limited number of available mitogenomes limits our understanding of the genetics and evolution of this diverse fungal group (Basidiomycota).
In the present study, the mitogenomes of two Ramaria species, including R. cfr. rubripermanens and R. rubella, were assembled and annotated. We also compared with the two Ramaria mitogenomes with other mitogenomes from subclass Phallomycetidae. The aims of this study are: 1) to reveal the characterizations of the two Ramaria mitogenomes; 2) to reveal the variations or conservativeness between Ramaria and other Phallomycetidae mitogenomes by comparative mitogenomic analysis; 3) to reveal the intron dynamics of Phallomycetidae mitogenomes; 4) to understand the phylogenetic status of Ramaria in the phylum Basidiomycota based on the combined mitochondrial gene set. This study served as the first report on the mitogenome from the genus Ramaria, which will promote our understanding of the origin, evolution, and genetics of Ramaria species and closely related genera.
MATERIALS AND METHODS
Mitogenome assembly and annotation
We assembled the complete mitogenomes of R. cfr. rubripermanens and R. rubella using the raw sequencing data from the Sequence Read Archive (SRA) database under the accession numbers of SRR5801920 and SRR3747475, respectively, which are publically available (Li et al. 2018a, Miyauchi et al. 2020). The specimen of R. cfr. rubripermanens was that collected in Yunnan used in our previous study (Li et al. 2019a), deposited in the Yunnan Edible Mushroom Research Initiative, Yunnan Agricultural University (number MG17).That of R. rubella was collected by the US Joint Genome Institute (JGI; https://mycocosm.jgi.doe.gov/Ramac1/Ramac1.home.html) for the 1000 Fungal Genome project, and is stored in the USDA Forest Products Laboratory collection, Madison, WI (number UT-36052-T). Quality control steps were conducted to obtained clean reads from the raw sequencing data, which included removing adapter reads using AdapterRemoval v2 (Schubert et al. 2016) and filtering low-quality sequences using ngsShoRT 2.2 (Chen et al. 2014). The two mitogenomes of Ramaria were assembled using NOVOPlasty v4.2.1 based on the K-mer size of 29 (Dierckxsens et al. 2017). Gaps between contigs were complemented based on polymerase chain reaction and pyrosequencing. We further obtained the two circularized assembled Ramaria mitogenomes. The two complete Ramaria mitogenomes were annotated according to previously described methods (Li et al. 2018b). First, the protein-coding genes (PCGs), open reading frames (ORFs), rRNAs, tRNAs, and introns of the two Ramaria mitogenomes were annotated according to the results of MFannot (Valach et al. 2014) and MITOS (Bernt et al. 2013) based on the mitochondrial genetic code 4. ORFs longer than 100 aa were selected for further modification or annotation based on the NCBI Open Reading Frame Finder (Coordinators 2017) and BLASTP searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton 1990). Intron and exon boundaries were detected by using exonerate v2.2 (Slater and Birney 2005). The previously annotated tRNA genes in the Ramaria mitogenomes were further verified by tRNAscan-SE v1.3.1 software (Lowe and Chan 2016). We drew the physical maps of the two Ramaria mitogenomes using OGDraw v1.2 (Lohse et al. 2013).
Sequence analysis
We calculated base compositions of the two Ramaria mitogenomes and other mitogenomes from subclass Phallomycetidae using DNASTAR Lasergene v7.1 software (http://www.dnastar.com/). Strand asymmetries of the tested Phallomycetidae mitogenomes were calculated according to the following formulas: AT skew = [A − T]/[A + T], and GC skew = [G − C]/[G + C]. We determined codon usages of the two Ramaria mitogenomes based on Sequence Manipulation Suite (Stothard 2000). The nonsynonymous (Ka) and synonymous (Ks) substitution rates for the 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3) in the 6 Phallomycetidae mitogenomes were calculated using the DnaSP v6.10.01 (Rozas et al. 2017). The genetic distances between each pair of the 15 core PCGs were calculated using MEGA v6.06 based on the Kimura-2-parameter (K2P) substitution model (Caspermeyer 2016). In addition, we conducted BlastN searches of the two Ramaria mitogenomes against themselves (Chen et al. 2015) to find intra-genomic duplications or interspersed repeats in the two Ramaria mitogenomes (> 50 bp). Tandem repeats in the two Ramaria mitogenomes were determined by using Tandem Repeats Finder (Benson 1999) with default parameters. We also conducted BlastN searches of the two Ramaria mitogenomes against their nuclear genomes to identify any gene fragments that transferred between their nuclear and mitochondrial genomes.
Comparative mitogenomic analysis and intron analysis
We conducted comparative mitogenomic analysis of the 6 Phallomycetidae mitogenomes reported to assess variations and conservations between different mitogenomes, which included genome sizes, GC contents, base compositions, codon usage, gene, and intron numbers. We calculated the contribution rate of different regions to the size variation of the two Ramaria mitogenomes, which is calculated by the following formula: size difference of region / size different of the entire mitogenome *100%. Introns in cox1 genes of the 6 Phallomycetidae mitogenomes were classified into different position classes (Pcls) according to previously described methods (Ferandon et al. 2010). Cox1 genes of the 6 Phallomycetidae mitogenomes were first aligned with the cox1 gene of Ganoderma calidophilum (Li et al. 2019b), which was used as reference in previous studies (Ye et al. 2020), by using Clustal W (Thompson et al. 1994). The same Pcl was judged by the same insert site in the corresponding reference gene and conservation of sequences, which was named according to their insert site (nt) in the reference gene. Introns belonging to the same Pcl were considered as orthologous intron and usually had a high sequence similarity (Ferandon et al. 2010).
Phylogenetic analysis
We constructed a phylogenetic tree of 84 Basidiomycota mitogenomes using two phylogenetic inference methods based on the combined mitochondrial gene set (14 core PCGs), which included Bayesian inference (BI) method and Maximum Likelihood (ML) methods. The phylogenetic status of Ramaria species in the phylum Basidiomycota was investigated according to the phylogenetic tree. The phylogenetic tree was constructed based on previously described methods (Huang et al. 2021). Tuber calosporum from the phylum Ascomycota was used as the outgroup (Li et al. 2020d). Individual mitochondrial genes were first aligned using MAFFT v7.037 (Katoh et al. 2019), and then concatenated into a combined mitochondrial gene dataset using the SequenceMatrix v1.7.8 (Vaidya et al. 2011). Ambiguous regions of these sequences were trimmed. In this dataset, the alignment sequence length of each species used to construct the evolutionary tree reached 21,990 bp. We conducted preliminary partition homogeneity test to detect if there were potential phylogenetic conflicts between different mitochondrial genes. Best-fit models of partitioning schemes and evolution for the combined mitochondrial gene dataset were detected by using PartitionFinder 2.1.1 software (Lanfear et al. 2017). MrBayes v3.2.6 (Ronquist et al. 2012) was used to conduct the BI analysis. Two independent runs with four chains (three heated and one cold) each were conducted simultaneously for 1.0 × 107 generations. Each run was sampled every 1000 generations. We assumed that stationarity had been reached when the estimated sample size (ESS) was greater than 100, and the potential scale reduction factor (PSRF) approached 1.0. The first 50% samples were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) in a 50% majority-rule consensus tree (Wang et al. 2020b). RAxML v 8.0.0 (Stamatakis 2014) was used to conduct the ML analysis. We assessed Bootstrap values (BS) through an ultrafast bootstrap approach with 1,000 replicates.
Data availability
The complete mitogenomes of R. cfr. rubripermanens and R. rubella were deposited in the GenBank database (Benson et al. 2018) under the accession numbers OM272988and OM272989, respectively.
RESULTS
PCGs in the two Ramaria mitogenomes
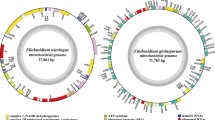
In the present study, we assembled the two Ramaria mitogenomes. The assembled mitogenomes of R. cfr. rubripermanens and R. rubella were circularized, with sizes of 126,497 bp and 143,271 bp, respectively (Fig. 1). The GC content of the R. cfr. rubripermanens and R. rubella mitogenomes were 28.92% and 31.69%, respectively (Additional file 1: Table S1). Both the Ramaria mitogenomes had negative AT skews and positive GC skews. A total of 18 and 20 free-stranding PCGs were detected in the mitogenomes of R. cfr. rubripermanens and R. rubella, respectively. Both the Ramaria mitogenomes contained a whole set of core PCGs, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3 (Additional file 1: Table S2). In addition, the mitogenome of R. cfr. rubripermanens contained 2 non-conserved PCGs encoding proteins with unknown functions and 1 non-conserved PCG encoding a putative GIY-YIG endonuclease. The R. rubella mitogenome contained 2 non-conserved PCGs encoding proteins with unknown functions, 1 non-conserved PCG encoding an RNA polymerase and 2 non-conserved PCGs encoding DNA polymerases. The mitogenomes of R. cfr. rubripermanens and R. rubella contained 38 and 37 introns, respectively, which were distributed in atp6, cob, cox1, cox2, cox3, nad1, nad2, nad4, nad4L, nad5, rns, and rnl genes. Two of the 38 introns from R. cfr. rubripermanens mitogenome belonged to the group II, 32 belonged to the group I, and 4 were of unknown types. The mitogenome of R. rubella contained 3 group II introns, 30 group I introns, and 4 unknown introns. The mitogenomes of R. cfr. rubripermanens and R. rubella also contained 40 and 38 ORFs in introns, which encoded putative LAGLIDADG homing endonucleases, GIY-YIG homing endonucleases, reverse transcriptase/maturase, and proteins with unknown functions.

Circular maps of the two Ramaria mitochondrial genomes. Genes are represented by different colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, while colored blocks within the ring indicates that the genes are located on the reverse strand. The inner grayscale bar graph shows the GC content of the mitochondrial sequences. The circle inside the GC content graph marks the 50% threshold
RNA genes in the Ramaria mitogenomes
We detected two rRNA genes in both the Ramaria mitogenomes, which included the small subunit ribosomal RNA (rns) and the large subunit ribosomal RNA (rnl) (Additional file 1: Table S2). The average lengths of rns and rnl genes were 1687 bp and 3778 bp, respectively. The rns and rnl genes of R. rubella were 29 bp and 117 bp longer than that of R. cfr. rubripermanens. Both the Ramaria mitogenomes contained 25 tRNA genes, which were folded into classical cloverleaf structures (Fig. 2). The two mitogenomes contained 2 tRNAs with different anticodons coding for arginine, serine, and leucine, and 3 tRNAs with the same anticodons coding for methionine. The size of individual tRNAs ranged from 71 to 88 bp. tRNA genes of sizes > 85 bp, including trnS(tga) and trnL(tag), contained larger extra arms than other tRNAs, indicating the size variations of tRNAs were mainly due to size variations of extra arms in the Ramaria mitogenomes. Of the 25 tRNA genes shared by the two Ramaria mitogenomes, 23 contained mutational sites that varied between the two mitogenomes. A total of 83 mutational sites were detected in the 25 tRNA genes between the two Ramaria mitogenomes, of which 24.10% variable sites were located on the extra arm, followed by the D arm.

Putative secondary structures of tRNA genes identified in the two Ramaria mitochondrial genomes. Residues conserved across the two mitochondrial genomes are shown in green, while variable sites are shown in red. All genes are shown in order of occurrence in the mitochondrial genome of Ramaria cfr. rubripermanens, starting from trnW
Mitogenome composition analysis
The mitogenomes of R. cfr. rubripermanens and R. rubella both contained one overlapping nucleotide (Additional file 1: Table S2), which located across the neighboring genes nad4L and nad5 (− 1 bp). The length of intergenic sequences in the two Ramaria mitogenomes ranged from 1 to 4480 bp, and the longest intergenic sequence was located between cox3 and atp8 in the R. cfr. rubripermanens mitogenome.
The intronic regions accounted for the largest proportions of the R. cfr. rubripermanens and R. rubella mitogenomes, which accounted for 47.26% and 46.85% of their mitogenomes, respectively (Fig. 3). Intergenic regions accounted for the second largest proportions of the R. cfr. rubripermanens and R. rubella mitogenomes, accounting for 33.80% and 34.31% of the entire mitogenomes, respectively, which indicated that the two Ramaria mitogenomes have relatively relaxed structures. Protein coding regions occupied 13.20–13.67% of the two mitogenomes. RNA coding regions accounted for the smallest proportion of the two mitogenomes (5.17–5.74%). The R. rubella mitogenome was 16,774 bp larger than that of R. cfr. rubripermanens. Intronic regions contributed the most to the size expansion of the R. rubella mitogenome, with the contributing rate of 43.74%. Intergenic regions contributed to 38.17% of the size variation, and protein coding regions contributed to 17.23% of the R. rubella mitogenome expansion.

Mitogenome composition of the two Ramaria mitochondrial genomes. The bottom panel shows the contribution of different gene regions to the expansion of the Ramaria rubella mitogenome. The y-axis represents the contribution rate of different regions to the size variation of the whole mitogenome, which is calculated by the following formula: size difference of region/size different of the entire mitogenome *100%
Codon usage analysis
Most of the core PCGs in the 6 Phallomycetidae mitogenomes used ATG as start codons, except that the cox2 gene of Sphaerobolus stellatus used GTG, the nad3 gene of R. cfr. rubripermanens used TTG, and the nad6 gene of S. stellatus used TTG as start codons (Additional file 1: Table S3). The TAA was the most common used stop codon in the core PCGs of the 6 Phallomycetidae mitogenomes, followed by the TAG. The start and stop codons varied greatly between the two Ramaria species. The atp8 and cob genes of R. cfr. rubripermanens used TAG as stop codons, while R. rubella used TAA as stop codons. The nad4 gene of R. rubella used TAG as stop codon, while R. cfr. rubripermanens used TAA as stop codon.
Codon usage analysis indicated that the most frequently used codons in the two Ramaria mitogenomes were AAA (for lysine; Lys), TTT (for phenylalanine; Phe), TTA (for leucin; Leu), AAT (for asparagine; Asn), ATT (for isoleucine; Ile), and TAT (for tyrosine; Tyr) (Fig. 4)). The frequent use of A and T in codons contributed to a relative high AT content in the two Ramaria mitogenomes (average: 69.70%).

Codon usage of the two Ramaria mitochondrial genomes. Frequency of codon usage is plotted on the y-axis. a, Ramaria cfr. rubripermanens; b, Ramaria rubella
Repetitive sequences analysis
We conducted BlastN searches of the two Ramaria mitogenomes against themselves, and identified 142 and 74 repeat elements in the mitogenomes of R. cfr. rubripermanens and R. rubella, respectively (Additional file 1: Table S4). The length of these repeat sequences ranged from 50 to 413 bp, with pair-wise nucleotide similarities ranging from 76.47 to 100%. The largest repeats were located in the intergenic region between atp9 and cox1 gene in the R. cfr. rubripermanens mitogenome. The largest repeats in the R. rubella mitogenome were located in the intergenic region between trnM and trnE, as well as in intergenic region between trnE and trnT genes, with each of 100 bp long. Repeat sequences accounted for 9.28% and 3.25% of the R. cfr. rubripermanens and R. rubella mitogenomes, respectively. A total of 37 and 58 tandem repeats were detected in the mitogenomes of R. cfr. rubripermanens and R. rubella, respectively (Additional file 1: Table S5). The longest tandem repeat sequence was detected in the mitogenome of R. cfr. rubripermanens, comprising 48 bp, which was located in the intergenic region between cox3 and atp8. Tandem repeat sequences accounted for 1.49% and 1.98% of the R. cfr. rubripermanens and R. rubella mitogenomes, respectively.
We further conducted BLAST searches of the two Ramaria mitogenomes against their nuclear genomes to identify duplication events between nuclear and mitochondrial genomes. A total of 18 and 22 aligned fragments were detected in the mitogenome of R. cfr. rubripermanens and R. rubella, respectively (Additional file 1: Table S6). The length of these aligned fragments ranged from 100 to 1702 bp, with sequence identities ranging from 76.82 to 100%. The largest aligned fragment was found located in the third intron of rnl gene in the R. rubella mitogenome. The largest aligned fragment in the R. cfr. rubripermanens mitogenome (972 bp) was found located in the first exon of nad2. A total of 4499 bp and 7746 bp aligned fragment were detected in the mitogenomes of R. cfr. rubripermanens and R. rubella, respectively.
Evolution and variation of core PCGs
Twelve of the 15 core PCGs showed sequence length variations, with the exceptions of atp9, cox3 and nad4L, which had identical gene length among the 6 Phallomycetidae species (Fig. 5). The length variation of rps3 gene was the largest among the 15 core PCGs detected, up to 1545 bp. Among the 15 core PCGs detected, the average GC content of atp9 was the highest, reaching 39.04%, followed by cox1 gene, reaching 35.57%. The GC content of atp8 gene was the lowest, with an average of 22.29%. The GC contents of core PCGs varied among the 6 Phallomycetidae species, indicating that there are frequent sequence mutations in core PCGs of Phallomycetidae species. Fourteen of the 15 core PCGs contained negative AT skews, and only rps3 gene contained positive AT skew in the six Phallomycetidae species, which showed that most core PCGs tend to evolve in the direction of T-rich rather than A-rich in the leading strand of core PCGs. The GC skews of core PCGs among different Phallomycetidae species varied, indicating frequent G/C mutations in the core PCGs.

Sequence information of core protein coding genes in the 6 Phallomycetidae species
The rps3 gene was found had the largest average Kimura-2-parameter (K2P) genetic distance between the 6 Phallomycetidae species among the 15 core PCGs detected, followed by the nad3 gene, which showed that these genes differentiated greatly in evolution (Fig. 6). The nad4L gene had the smallest average K2P genetic distance between the 6 Phallomycetidae species, indicating this gene was highly conserved. The rps3 gene had the largest non-synonymous substitutions rate (Ka) among the 15 core PCGs, followed by the nad2 and nad3 genes. While the nad4L gene had the smallest Ka value. The average synonymous substitution rate (Ks) of nad3 gene was the largest, while that of nad2 gene was the smallest among the 15 core PCGs. The Ka/Ks values for all the 15 core PCGs were < 1, indicating that these genes were subjected to purifying selection.

Genetic analysis of the 15 core protein coding genes in the 6 Phallomycetidae species. K2P, the Kimura-2-parameter distance; Ka, the number of nonsynonymous substitutions per nonsynonymous site; Ks, the number of synonymous substitutions per synonymous site
Intron dynamics in cox1 genes of Phallomycetidae
A total of 156 introns were detected in the 6 Phallomycetidae mitogenomes, which were distributed in atp6, cob, cox1, cox2, cox3, nad1, nad2, nad4, nad4L, nad5, rns, and rnl genes (Fig. 7). Six of the 156 introns belonged to the group II (Lambowitz and Zimmerly 2004, 2011), 10 were twintrons, and the rest belonged to the group I. We did not detect any putative ORF in 27 of the 156 introns, and about 82.69% introns contained putative ORFs, which encoded putative homing endonuclease, maturases, or reverse transcriptases (Michel et al. 1989). These introns are unevenly distributed in the host gene. Cox1 gene harbored the largest number of introns, accounting for 31.41% of the total introns, followed by cob and rnl genes, which harbored 16.03% and 13.46% of the total introns, respectively. We found that introns have a preference for particular host genes, for example, cox1 gene tends to be rich in introns, which is considered to be independent of the length of host genes (Megarioti and Kouvelis 2020; Michel and Ferat 1995). The atp8, atp9, nad3, and nad6 did not contain any intron in the 6 Phallomycetidae mitogenomes. Intron dynamics in cox1 gene could significantly affect organization and size of Phallomycetidae mitogenomes. We further classified introns in cox1 genes of the 6 Phallomycetidae mitogenomes into different position classes (Pcls) using the cox1 gene of medical fungus Ganoderma calidophilum (Li et al. 2019b) as the reference. Introns belonging to the same Pcl were considered as orthologous introns and had similar intron sequence or structures. The 49 introns in cox1 genes of the 6 Phallomycetidae mitogenomes could be classified into 27 Pcls, which showed the rich diversity of Phallomycetidae intron types. The class and number of introns in different Phallomycetidae species varied, indicating potential intron loss/gain events in Phallomycetidae evolution. The P612, P706, and P821 were the most widely distributed introns (belonging to group I) in Phallomycetidae, which were distributed in 4 of the 6 Phallomycetidae species. Intron P209, P273, P717, P731, and P1057 were the second common introns (belonging to group I), which could be detected in 3 of the 6 Phallomycetidae mitogenomes. However, some rare Pcls could only be detected in one of the 6 Phallomycetidae species, such as P237, P369, P807, and P971 (belonging to group I). These rare introns in Phallomycetidae could be detected in distantly related species, such as Pleurotus eryngii (Li et al. 2018b) and Laccaria bicolor (Li et al. 2020c) from the order Agaricales, Ganoderma meredithae (Li et al. 2019b) from order Polyporales, and Rhizopogon vinicolor from order Boletales (Li et al. 2019a), which indicated possible intron transfer events may have occurred in Phallomycetidae evolution. A novel intron P44 was detected in R. rubella, which belonged to the group II and contained a putative intronic ORF encoding reverse transcriptase. No homologous intron of P44 was detected in other Basidiomycota species.

Position class (Pcl) information of cox1 genes in the 6 Phallomycetidae species. Introns in cox1 genes of the 6 Phallomycetidae mitogenomes were classified into different position classes (Pcls) using the cox1 gene of Ganoderma calidophilum as the reference. Each Pcl was constituted by introns inserted at the same position of corresponding cox1 gene and named according to its insertion site in the aligned corresponding reference sequence (nt). Phylogenetic positions of the 6 Phallomycetidae species were established using the Bayesian inference (BI) method and Maximum Likelihood (ML) method based on concatenated mitochondrial genes
Gene arrangement and comparative mitogenomic analysis
We further compared mitochondrial gene arrangements of the 6 Phallomycetidae species, including PCGs, rRNA genes, and tRNA genes. The results showed that five of the six Phallomycetidae species have identical gene arrangement of PCGs and rRNA genes (Fig. 8). Only Sphaerobolus stellatus had gene displacement, including nad2, nad3, rps3 and atp9 genes. As far as tRNA genes are concerned, S. stellatus had the location transfer of four tRNA genes, including trnD, trnR, trnW, and trnQ. In addition, four tRNA doubling events have occurred in the S. stellatus mitogenome, including trnM, trnE, trnT, and trnS. The mitogenome of R. cfr. rubripermanens and R. rubella each had one tRNA gene doubling event involving trnM gene, respectively.

Mitochondrial gene arrangement analyses of the 6 Phallomycetidae species. Genes in the green color block indicate that genes from all the six species have identical arrangements; genes in the yellow color block indicate that genes from five of the six species have identical arrangements; genes in the red color block indicate that the gene has been transferred; and genes in the purple color block indicate that the gene has doubled
The size of the 6 Phallomycetidae mitogenomes varied, ranging from 50,098 to 152,722 bp, with an average size of 112,345 bp (Additional file 1: Table S1). The two Ramaria mitogenomes are larger than the average size of Phallomycetidae mitogenomes. The mitogenome of R. rubella was the second largest among the 6 Phallomycetidae mitogenomes detected, which was only smaller than Sphaerobolus stellatus (152,722 bp) from the order Geastrales. The GC content of the 6 Phallomycetidae mitogenomes ranged from 24.3 to 31.69%, with an average GC content of 27.46%. The two Ramaria mitogenomes had a higher GC content than the average value. In addition, there is a large gap (2.77%) in GC content between the two Ramaria mitogenomes. Four mitogenomes from Gomphales and Geastrales had negative AT skews, while the other two mitogenomes from Phallales had positive AT skews. Four of the six mitogenomes had positive GC skews, including R. cfr. rubripermanens, R. rubella, Dictyophora indusiata, and Phallus echinovolvatus. Each mitogenomes contained 16–58 PCGs, and the two Ramaria mitogenomes contained the greatest number of PCGs. There were 4–38 introns harboured in the mitogenomes of the 6 Phallomycetidae species, and the two Ramaria mitogenomes contained the greatest number of introns. All the 6 Phallomycetidae mitogenomes contained two rRNA genes. In addition, 24–26 tRNA genes were detected in the 6 Phallomycetidae mitogenomes.
Phylogenetic analysis
In the present study, the phylogenetic status of 84 Basidiomycete species was assessed based on combined mitochondrial gene dataset. An identical and well-supported phylogenetic tree was obtained by using both Bayesian inference (BI) and Maximum Likelihood (ML) methods (Fig. 9). All major clades within the phylogenetic tree had a high support value (BPP ≥ 0.96; BS ≥ 98). According to the phylogenetic tree, the 84 Basidiomycota species could be divided into 18 major clades, corresponding to the orders Agaricales, Boletales, Cantharellales, Filobasidiales, Geastrales, Gomphales, Hymenochaetales, Microbotryales, Microstromatales, Phallales, Polyporales, Pucciniales, Russulales, Sporidiobolales, Tilletiales, Tremellales, Trichosporonales, and Ustilaginales (Additional file 1: Table S7). Phylogenetic analysis indicated that R. cfr. rubripermanens and T. floccosus are sister species, which had a close relationship. The result is consistent with previous studies, indicating the mitogenome was an effective molecular marker for analyze the phylogenetic relationship of basidiomycetes.

Molecular phylogeny of 84 Basidiomycota species based on Bayesian inference (BI) and Maximum Likelihood (ML) analysis of 14 protein coding genes. Support values are Bayesian posterior probabilities (before slash) and bootstrap (BS) values (after slash). The asterisk on the evolutionary clades indicates that the BPP value is 1 and the BS value is 100. Species and NCBI accession numbers for mitogenomes used in the phylogenetic analysis are provided in Additional file 1: Table S7
DISCUSSION
Mitogenomes have been reported to play an important role in eukaryotic growth and development, oxidative stress, and environmental response (Cai et al. 2021; Du et al. 2022; Guan et al. 2021). The mitogenome mutation of animals may lead to animal diseases and affect the metabolic level of animals (Chen et al. 2020b). However, as one of the most diverse eukaryotes on earth, the mitogenome structure and variation of basidiomycetes have been less studied (Li et al. 2022; Xu and Wang 2015). Up to now, the complete mitogenome of basidiomycetes available in public databases is less than 0.05% of the described basidiomycete species in nature. It is reported that the mitogenome of basidiomycetes varies greatly in gene content, repeat sequence, genome structure and genome size, which makes it difficult to obtain the complete mitogenome of fungi (Basse 2010; Fonseca et al. 2021; Mendoza et al. 2020). In the family Phallomycetidae, the largest mitogenome (152,722 bp) (Ye et al. 2020) is three times larger than the smallest one (50,098 bp) (Chen et al. 2020a). Great genome size variation has also occurred in Ramaria species. Previous studies have shown that the size variation of fungal mitogenome is closely related to plasmid-derived genes, repeat accumulation, dynamic changes of introns and variation of intergenic sequence (Boussau et al. 2011; Chen et al. 2021; Wang et al. 2020a; Zubaer et al. 2018). In the present study, we found that introns contribute the most to the mitogenome variation of Ramaria species, which is consistent with previous studies (Ye et al. 2020), which showed that introns play an important role in Phallomycetidae size variation.
It is reported that the ancestors of eukaryotes obtained mitochondria from bacteria through endosymbiosis (Archibald 2015; Martin et al. 2015; Zimorski et al. 2014). In the long-term evolution process, most mitochondrial genes have been transferred to the nuclear genome, which is convenient for the overall regulation of cells (Adams and Palmer 2003; Barton and Jones 1983). However, some PCGs, tRNA genes and rRNA genes are still retained in the mitogenome (Burki 2016). Among them, a set of core PCGs play an important role in the energy metabolism of eukaryotes. We found that the length, codon usage and base composition of these core PCGs varied greatly between different Phallomycetidae species, even among closely related species. The effect of core PCG mutation on fungal growth needs to be further revealed. In addition, different core PCGs have different evolutionary rates and may undergo purifying selection. We also found that 23 of the 25 tRNAs in Ramaria species had site variation, and the frequency of mutation may have a certain impact on the growth or stress response of fungi. The mutation of tRNA was considered to be closely related to the efficiency of protein synthesis and may eventually affect the phenotype of eukaryotes (Hayashi et al. 2016; Lin et al. 2021; Povea-Cabello et al. 2020). In the present study, a number of non-conserved PCGs have been detected in Ramaria species, which encoded RNA polymerase, DNA polymerase, and proteins with unknown functions. RNA and DNA polymerase are plasmid-derived genes. In some species, they are integrated into the mitogenome, while in others, they exist independently (Wu et al. 2021a, 2021b). The dynamic change of plasmid-derived regions promoted the organization and size variations of fungal mitogenome, and finally makes the fungal mitogenome more diverse and complex (Himmelstrand et al. 2014). In addition, some PCGs with unknown functions exist in Ramaria mitogenomes, indicating that the functions of Ramaria mitogenomes need to be further analyzed, so as to promote a comprehensive understanding of the function and origin of fungal mitogenome.
Introns are considered as potentially mobile genetic elements in fungal mitogenome, and can affectthe organization and size of fungal mitogenomes (Mukhopadhyay and Hausner 2021). In the present study, we found that introns are closely related to the size variation of mitogenome in subclass Phallomycetidae. In basidiomycetes, most mitochondrial introns belong to the group I and only a few introns belong to the group II (Mullineux et al. 2010). Some introns in fungal mitogenome contain intronic ORFs, which can encode putative homing endonuclease, maturases, or reverse transcriptases (Belfort and Lambowitz 2019). Introns can be classified into different position classes according to their insertion sites on the mitochondrial PCGs. Introns belonging to the same Pcls are considered to be orthologous and have high sequence similarities (Cheng et al. 2021; Li et al. 2020a). In this study, we found that the number and types of introns in different Phallomycetidae species varied, even among closely related species, indicating that intron loss or gain events have occurred in the evolution of Phallomycetidae species. In addition, some rare introns in subclass Phallomycetidae were found to exist in a large number in distant species, indicating that there may be potential intron transfer events between them. We also detected a novel intron P44 in R. rubella species, which have not been detected in any basidiomycete species reported (Cheng et al. 2021). The origin and evolution of the novel intron P44 need to be further analyzed.
In this study, we also compared the mitochondrial gene arrangement of the 6 Phallomycetidae species. Five of the six Phallomycetidae species had identical arrangement of PCGs and rRNA genes, which may inherit the gene arrangement from the ancestors of Phallomycetidae species. Compared with the gene arrangement of the ancestors of Phallomycetidae species, there was a large-scale gene rearrangement in PCGs and tRNA genes of Sphaerobolus stellatus (Ye et al. 2020). We also detected the doubling event of tRNAs in S. stellatus and the two Ramaria species. The mechanism of fungal mitochondrial gene rearrangement has not been fully analyzed. Previous studies have shown that fungal mitochondrial genome rearrangement may be related to the accumulation of repeat sequences (Aguileta et al. 2014). However, we only detected 2.50% repeat elements in the mitogenome of S. stellatus, which is lower than the repeat content of the two Ramaria species, indicating that the mitochondrial gene rearrangement of Phallomycetidae species may be affected by other factors other than repeats. In addition, we detected 4499 bp and 7746 bp aligned fragments between the mitochondrial and nuclear genomes of R. cfr. rubripermanens and R. rubella, respectively, indicating the potential gene fragment transfer events between mitochondrial and nuclear genomes. The coevolution of mitochondrial genome and nuclear genome affects a series of ecological adaptation processes of animals and plants (Bar-Yaacov et al. 2012), which may also be happening in Ramaria.
Ramaria species is a diverse fungal group. Some Ramaria species are edible while others are considered poisonous (Barros et al. 2008; Liu et al. 2014). The misidentification of Ramaria species may lead to serious poisoning events. However, some morphological features of Ramaria species are easy to overlap, so it is not feasible to classify Ramaria species only by morphological features. Mitochondrial genome is considered to be a powerful tool to analyze the phylogenetic relationship of eukaryotes (Luchetti and Plazzi 2021). Compared with traditional multiple molecular markers, which requires multiple PCR and pyrosequencing, mitogenome can provide more genetic information and operate more conveniently. Usually, 15 core PCGs and 2 rRNA genes can be used as molecular markers for phylogeny in mitogenome, resulting in reliable and high support rate of eukaryotic phylogeny (Li et al. 2018b, 2021a; Nie et al. 2019). Although the phylogenetic analysis based on nuclear genome provides more abundant genetic information, the acquisition cost of nuclear genome is high and the amount of data is too large, which limits large-scale access to fungal nuclear genomes. Therefore, phylogeny based on mitochondrial genome is an important choice. Ramaria species occupies a unique phylogenetic position in basidiomycetes. While no mitogenome of Ramaria species has been reported. In this study, we obtained a highly supported phylogenetic tree based on the combined mitochondrial gene set through two phylogenetic inference methods. Phylogenetic analysis shows that the phylogenetic relationships of R. cfr. rubripermanensand T. floccosus species is closer than that between the two Ramaria species, which is consistent with the previous studies based on multiple molecular markers (Giachini et al. 2010), indicating that the mitogenome is a reliable tool for analyzing the phylogenetic relationship of Phallomycetidae species. More mitogenomes of Phallomycetidae species need to be obtained to understand the population genetics and phylogeny of Ramaria.
CONCLUSIONS
In this study, we obtained two novel mitogenomes from the Ramaria genus. The complete mitogenomes of R. cfr. rubripermanens and R. rubella showed large size variations, and the intron region contributed the most to the size variation of two mitogenomes (contribution rate, 43.74%). We further detected large variations in genetic contents, gene length, tRNAs, and codon usages of the two Ramaria mitogenomes. Comparative genomic analysis detected large-scale gene rearrangements between Phallomycetidae mitogenomes, including gene displacement and tRNA doubling. We first found possible gene transferring events between the mitochondrial and nuclear genomes of the two Ramaria species. The Phallomycetidae mitogenomes have been experienced frequent intron loss/gain and potential intron transfer events during the evolution, and a novel intron P44 was found in the R. rubella mitogenome. Phylogenetic analyses showed that phylogenetic relationships of R. cfr. rubripermanens and T. floccosus species is closer than that between the two Ramaria species. The present study provides the first record of mitogenomes from this genus Ramaria, thus providing a foundation for studying the evolution, genetics, and taxonomy of this important genus and related fungal group.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its Additional files].
Abbreviations
- Mitogenome:
-
Mitochondrial genome
- PCG:
-
Protein-coding gene
- Pcls:
-
Position classes
- Ks :
-
Synonymous substitution rates
- Ka :
-
Nonsynonymous substitution rates
- BI:
-
Bayesian inference
- ML:
-
Maximum Likelihood
References
Abuduaini A, Wang YB, Zhou HY, Kang RP, Ding ML, Jiang Y, Suo FY, Huang LD (2021) The complete mitochondrial genome of Ophiocordyceps gracilis and its comparison with related species. IMA Fungus 12(1):31
Adams KL, Palmer JD (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol 29(3):380–395
Aguileta G, de Vienne DM, Ross ON, Hood ME, Giraud T, Petit E, Gabaldon T (2014) High variability of mitochondrial gene order among fungi. Genome Biol Evol 6(2):451–465
Aprotosoaie AC, Zavastin DE, Mihai CT, Voichita G, Gherghel D, Silion M, Trifan A, Miron A (2017) Antioxidant and antigenotoxic potential of Ramaria largentii Marr & D. E. Stuntz, a wild edible mushroom collected from Northeast Romania. Food Chem Toxicol 108(3):429–437
Araujo DS, De-Paula RB, Tome LMR, Quintanilha-Peixoto G, Salvador-Montoya CA, Del-Bem LE, Badotti F, Azevedo VAC, Brenig B, Aguiar E, Drechsler-Santos ER, Fonseca PLC, Goes-Neto A (2021) Comparative mitogenomics of agaricomycetes: diversity, abundance, impact and coding potential of putative open-reading frames. Mitochondrion 58:1–13
Archibald JM (2015) Endosymbiosis and eukaryotic cell evolution. Curr Biol 25(19):R911-921
Barros L, Venturini BA, Baptista P, Estevinho LM, Ferreira IC (2008) Chemical composition and biological properties of Portuguese wild mushrooms: a comprehensive study. J Agric Food Chem 56(10):3856–3862
Barton N, Jones JS (1983) Evolutionary biology. Mitochondrial DNA: new clues about evolution. Nature 306(5941):317–318
Bar-Yaacov D, Blumberg A, Mishmar D (2012) Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim Biophys Acta 1819(9–10):1107–1111
Basse CW (2010) Mitochondrial inheritance in fungi. Curr Opin Microbiol 13(6):712–719
Belfort M, Lambowitz AM (2019) Group II Intron RNPs and reverse transcriptases: from retroelements to research tools. Cold Spring Harb Perspect Biol 11(4):a32375
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27(2):573–580
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, Sayers EW (2018) GenBank. Nucleic Acids Res 46(D1):D41–D47
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69(2):313–319
Bhanja SK, Rout D, Patra P, Nandan CK, Behera B, Maiti TK, Islam SS (2013) Structural studies of an immunoenhancing glucan of an ectomycorrhizal fungus Ramaria botrytis. Carbohydr Res 374:59–66
Bleasby AJ, Wootton JC (1990) Construction of validated, non-redundant composite protein sequence databases. Protein Eng 3(3):153–159
Boussau B, Brown JM, Fujita MK (2011) Nonadaptive evolution of mitochondrial genome size. Evolution 65(9):2706–2711
Burki F (2016) Mitochondrial evolution: going, going. Gone Curr Biol 26(10):R410-412
Cai N, Gomez-Duran A, Yonova-Doing E, Kundu K, Burgess AI, Golder ZJ, Calabrese C, Bonder MJ, Camacho M, Lawson RA, Li L, Williams-Gray CH, Group I-PS, Di Angelantonio E, Roberts DJ, Watkins NA, Ouwehand WH, Butterworth AS, Stewart ID, Pietzner M, Wareham NJ, Langenberg C, Danesh J, Walter K, Rothwell PM, Howson JMM, Stegle O, Chinnery PF, Soranzo N (2021) Mitochondrial DNA variants modulate N-formylmethionine, proteostasis and risk of late-onset human diseases. Nat Med 27(9):1564–1575
Caspermeyer J (2016) MEGA evolutionary software re-engineered to handle today’s big data demands. Mol Biol Evol 33(7):1888
Centko RM, Ramon-Garcia S, Taylor T, Patrick BO, Thompson CJ, Miao VP, Andersen RJ (2012) Ramariolides A-D, antimycobacterial butenolides isolated from the mushroom Ramaria cystidiophora. J Nat Prod 75(12):2178–2182
Chen C, Khaleel SS, Huang H, Wu CH (2014) Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med 9:8
Chen Y, Ye W, Zhang Y, Xu Y (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res 43(16):7762–7768
Chen C, Wang J, Li Q, Fu R, Jin X, Huang W, Lu D (2020a) Mitogenomes of two phallus mushroom species reveal gene rearrangement, intron dynamics, and basidiomycete phylogeny. Front Microbiol 11:573064
Chen K, Lu P, Beeraka NM, Sukocheva OA, Madhunapantula SV, Liu J, Sinelnikov MY, Nikolenko VN, Bulygin KV, Mikhaleva LM, Reshetov IV, Gu Y, Zhang J, Cao Y, Somasundaram SG, Kirkland CE, Fan R, Aliev G (2020b) Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin Cancer Biol 16:196
Chen C, Li Q, Fu R, Wang J, Deng G, Chen X, Lu D (2021) Comparative mitochondrial genome analysis reveals intron dynamics and gene rearrangements in two Trametes species. Sci Rep 11(1):2569
Cheng J, Luo Q, Ren Y, Luo Z, Liao W, Wang X, Li Q (2021) Panorama of intron dynamics and gene rearrangements in the phylum Basidiomycota as revealed by the complete mitochondrial genome of Turbinellus floccosus. Appl Microbiol Biotechnol 105(5):2017–2032
Coordinators NR (2017) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36:13–21
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45(4):e18
Dong M, Hou Y, Ding X (2020) Structure identification, antitumor activity and mechanisms of a novel polysaccharide from Ramaria flaccida (Fr.) Quel. Oncol Lett 20(3):2169–2182
Du F, Yu Q, Yan SS (2022) Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer's disease. Hum Mol Genet
Ferandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, Barroso G (2010) The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS ONE 5(11):e14048
Fonseca PLC, De-Paula RB, Araujo DS, Tome LMR, Mendes-Pereira T, Rodrigues WFC, Del-Bem LE, Aguiar E, Goes-Neto A (2021) Global characterization of fungal mitogenomes: new insights on genomic diversity and dynamism of coding genes and accessory elements. Front Microbiol 12:787283
Fu J, Zhang LL, Li W, Zhang Y, Zhang Y, Liu F, Zou L (2022) Application of metabolomics for revealing the interventional effects of functional foods on metabolic diseases. Food Chem 367:130697
Garcia-Souto D, Bruzos AL, Diaz S, Rocha S, Pequeno-Valtierra A, Roman-Lewis CF, Alonso J, Rodriguez R, Costas D, Rodriguez-Castro J, Villanueva A, Silva L, Valencia JM, Annona G, Tarallo A, Ricardo F, Bratos Cetinic A, Posada D, Pasantes JJ, Tubio JM (2022) Mitochondrial genome sequencing of marine leukaemias reveals cancer contagion between clam species in the Seas of Southern Europe. Elife 11:e66946
Giachini AJ, Hosaka K, Nouhra E, Spatafora J, Trappe JM (2010) Phylogenetic relationships of the Gomphales based on nuc-25S-rDNA, mit-12S-rDNA, and mit-atp6-DNA combined sequences. Fungal Biol 114(2–3):224–234
Gray MW, Burger G, Lang BF (2001) The origin and early evolution of mitochondria. Genome Biol 2(6):1018
Guan JY, Zhang ZY, Cao YR, Xu XD, Storey KB, Yu DN, Zhang JY (2021) The complete mitochondrial genome of Choroterpes (Euthralus) yixingensis (Ephemeroptera: Leptophlebiidae) and its mitochondrial protein-coding gene expression under imidacloprid stress. Gene 800:145833
Hayashi JI, Hashizume O, Ishikawa K, Shimizu A (2016) Mutations in mitochondrial DNA regulate mitochondrial diseases and metastasis but do not regulate aging. Curr Opin Genet Dev 38:63–67
Himmelstrand K, Olson A, Brandstrom Durling M, Karlsson M, Stenlid J (2014) Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of Agaricomycetes. Curr Genet 60(4):303–313
Huang L, Liu XL, Cao CS, Ying Q (2009) Outbreak of fatal mushroom poisoning with Amanita franchetii and Ramaria rufescens. BMJ Case Rep 2009:bcr0620080327
Huang W, Feng H, Tu W, Xiong C, Jin X, Li P, Wang X, Li Q (2021) Comparative mitogenomic analysis reveals dynamics of intron within and between tricholoma species and phylogeny of basidiomycota. Front Genet 12:534871
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20(4):1160–1166
Khaund P, Joshi SR (2014) DNA barcoding of wild edible mushrooms consumed by the ethnic tribes of India. Gene 550(1):123–130
Lambowitz AM, Zimmerly S (2004) Mobile group II introns. Annu Rev Genet 38:1–35
Lambowitz AM, Zimmerly S (2011) Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol 3(8):a003616
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34(3):772–773
Li H (2017) Extraction, purification, characterization and antioxidant activities of polysaccharides from Ramaria botrytis (Pers.) Ricken. Chem Cent J 11:24
Li H, Wu S, Ma X, Chen W, Zhang J, Duan S, Gao Y, Kui L, Huang W, Wu P, Shi R, Li Y, Wang Y, Li J, Guo X, Luo X, Li Q, Xiong C, Liu H, Gui M, Sheng J, Dong Y (2018a) The genome sequences of 90 mushrooms. Sci Rep 8(1):9982
Li Q, Chen C, Xiong C, Jin X, Chen Z, Huang W (2018b) Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl Microbiol Biotechnol 102(14):6143–6153
Li Q, Ren Y, Shi X, Peng L, Zhao J, Song Y, Zhao G (2019a) Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum agaricomycotina. Int J Mol Sci 20(20):5167
Li Q, Xiang D, Wan Y, Wu Q, Wu X, Ma C, Song Y, Zhao G, Huang W (2019b) The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int J Biol Macromol 139:397–408
Li Q, He X, Ren Y, Xiong C, Jin X, Peng L, Huang W (2020a) Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and Asymbiotic amanita species. Front Microbiol 11:1382
Li Q, Ren Y, Xiang D, Shi X, Zhao J, Peng L, Zhao G (2020b) Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11:12
Li Q, Yang L, Xiang D, Wan Y, Wu Q, Huang W, Zhao G (2020c) The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int J Biol Macromol 145:974–984
Li X, Li L, Bao Z, Tu W, He X, Zhang B, Ye L, Wang X, Li Q (2020d) The 287,403 bp mitochondrial genome of ectomycorrhizal fungus tuber calosporum reveals intron expansion, tRNA loss, and gene rearrangement. Front Microbiol 11:591453
Li Q, Li L, Feng H, Tu W, Bao Z, Xiong C, Wang X, Qing Y, Huang W (2021a) Characterization of the complete mitochondrial genome of basidiomycete yeast Hannaella oryzae: intron evolution, gene rearrangement, and its phylogeny. Front Microbiol 12:646567
Li Q, Wu P, Li L, Feng H, Tu W, Bao Z, Xiong C, Gui M, Huang W (2021b) The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int J Biol Macromol 172:560–572
Li Q, Bao Z, Tang K, Feng H, Tu W, Li L, Han Y, Cao M, Zhao C (2022) First two mitochondrial genomes for the order Filobasidiales reveal novel gene rearrangements and intron dynamics of Tremellomycetes. IMA Fungus 13(1):7
Lin Y, Xu X, Wang W, Liu F, Zhao D, Li D, Ji K, Li W, Zhao Y, Yan C (2021) A mitochondrial myopathy-associated tRNA(Ser(UCN)) 7453G>A mutation alters tRNA metabolism and mitochondrial function. Mitochondrion 57:1–8
Liu K, Wang J, Zhao L, Wang Q (2013) Anticancer, antioxidant and antibiotic activities of mushroom Ramaria flava. Food Chem Toxicol 58:375–380
Liu DZ, Li JG, Zhang MW, Liu G (2014) Two new alkaloids from the edible macrofungus Ramaria madagascariensis. J Basic Microbiol 54(Suppl 1):S70-73
Liu DZ, Li JG, Zhang MW, Liu G (2015) New bicyclic hemiacetals from the edible mushroom Ramaria madagascariensis. J Antibiot (tokyo) 68(2):137–138
Lohse M, Drechsel O, Kahlau S, Bock R (2013) Organellar genome DRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res 41:575–581
Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44(W1):W54-57
Luchetti A, Plazzi F (2021) Molecular phylogenetics and mitochondrial evolution. Life (basel) 12(1):4
Martin WF, Garg S, Zimorski V (2015) Endosymbiotic theories for eukaryote origin. Philos Trans R Soc Lond B Biol Sci 370(1678):20140330
Megarioti AH, Kouvelis VN (2020) The coevolution of fungal mitochondrial introns and their homing endonucleases (GIY-YIG and LAGLIDADG). Genome Biol Evol 12(8):1337–1354
Mendoza H, Perlin MH, Schirawski J (2020) Mitochondrial Inheritance in phytopathogenic fungi-everything is known, or is it? Int J Mol Sci 21(11):3883
Michel F, Ferat JL (1995) Structure and activities of group II introns. Annu Rev Biochem 64:435–461
Michel F, Umesono K, Ozeki H (1989) Comparative and functional anatomy of group II catalytic introns—a review. Gene 82(1):5–30
Miyauchi S, Kiss E, Kuo A, Drula E, Kohler A, Sanchez-Garcia M, Morin E, Andreopoulos B, Barry KW, Bonito G, Buee M, Carver A, Chen C, Cichocki N, Clum A, Culley D, Crous PW, Fauchery L, Girlanda M, Hayes RD, Keri Z, LaButti K, Lipzen A, Lombard V, Magnuson J, Maillard F, Murat C, Nolan M, Ohm RA, Pangilinan J, Pereira MF, Perotto S, Peter M, Pfister S, Riley R, Sitrit Y, Stielow JB, Szollosi G, Zifcakova L, Stursova M, Spatafora JW, Tedersoo L, Vaario LM, Yamada A, Yan M, Wang P, Xu J, Bruns T, Baldrian P, Vilgalys R, Dunand C, Henrissat B, Grigoriev IV, Hibbett D, Nagy LG, Martin FM (2020) Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat Commun 11(1):5125
Mukhopadhyay J, Hausner G (2021) Organellar introns in fungi, algae, and plants. Cells 10(8):2001
Mullineux ST, Costa M, Bassi GS, Michel F, Hausner G (2010) A group II intron encodes a functional LAGLIDADG homing endonuclease and self-splices under moderate temperature and ionic conditions. RNA 16(9):1818–1831
Nie Y, Wang L, Cai Y, Tao W, Zhang YJ, Huang B (2019) Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl Microbiol Biotechnol 103(3):1379–1391
Nouhra ER, Horton TR, Cazares E, Castellano M (2005) Morphological and molecular characterization of selected Ramaria mycorrhizae. Mycorrhiza 15(1):55–59
Perez-Moreno J, Ferrera-Cerrato R (1995) A review of mushroom poisoning in Mexico. Food Addit Contam 12(3):355–360
Pickett SJ, Deen D, Pyle A, Santibanez-Koref M, Hudson G (2022) Interactions between nuclear and mitochondrial SNPs and Parkinson’s disease risk. Mitochondrion 63:85–88
Povea-Cabello S, Villanueva-Paz M, Suarez-Rivero JM, Alvarez-Cordoba M, Villalon-Garcia I, Talaveron-Rey M, Suarez-Carrillo A, Munuera-Cabeza M, Sanchez-Alcazar JA (2020) Advances in mt-tRNA mutation-caused mitochondrial disease modeling: patients’ brain in a dish. Front Genet 11:610764
Ren LY, Zhang S, Zhang YJ (2021) Comparative mitogenomics of fungal species in stachybotryaceae provides evolutionary insights into hypocreales. Int J Mol Sci 22(24):13341
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302
Scheid HV, Viegas Sallis ES, Riet-Correa F, Schild AL (2022) Ramaria flavo-brunnescens mushroom poisoning in South America: a comprehensive review. Toxicon 205:91–98
Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9:88
Slater GS, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinform 6:31
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28(6):1102–1104
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Vaidya G, Lohman DL, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27(2):171–180
Valach M, Burger G, Gray MW, Lang BF (2014) Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res 42(22):13764–13777
Wang X, Song A, Wang F, Chen M, Li X, Li Q, Liu N (2020a) The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Int J Biol Macromol 162:209–219
Wang X, Wang Y, Yao W, Shen J, Chen M, Gao M, Ren J, Li Q, Liu N (2020b) The 256 kb mitochondrial genome of Clavaria fumosa is the largest among phylum Basidiomycota and is rich in introns and intronic ORFs. IMA Fungus 11(1):26
Wolfsberger WW, Ayala NM, Castro-Marquez SO, Irizarry-Negron VM, Potapchuk A, Shchubelka K, Potish L, Majeske AJ, Oliver LF, Lameiro AD, Martinez-Cruzado JC, Lindgren G, Oleksyk TK (2022) Genetic diversity and selection in Puerto Rican horses. Sci Rep 12(1):515
Wu P, Bao Z, Tu W, Li L, Xiong C, Jin X, Li P, Gui M, Huang W, Li Q (2021a) The mitogenomes of two saprophytic Boletales species (Coniophora) reveals intron dynamics and accumulation of plasmid-derived and non-conserved genes. Comput Struct Biotechnol J 19:401–414
Wu P, Yao T, Ren Y, Ye J, Qing Y, Li Q, Gui M (2021b) Evolutionary insights into two widespread ectomycorrhizal fungi (Pisolithus) from comparative analysis of mitochondrial genomes. Front Microbiol 12:583129
Xu JP, Wang PF (2015) Mitochondrial inheritance in basidiomycete fungi. Fungal Biol Rev 29(3–4):209–219
Ye J, Cheng J, Ren Y, Liao W, Li Q (2020) The first mitochondrial genome for geastrales (Sphaerobolus stellatus) reveals intron dynamics and large-scale gene rearrangements of basidiomycota. Front Microbiol 11:1970
Zhang S, Bai X, Ren LY, Sun HH, Tang HP, Vaario LM, Xu J, Zhang YJ (2021a) Dynamic evolution of eukaryotic mitochondrial and nuclear genomes: a case study in the gourmet pine mushroom Tricholoma matsutake. Environ Microbiol 23(11):7214–7230
Zhang X, Yang L, Hou L, Li H, Xiang H, Zhao X (2021b) Ancient mitogenomes reveal the domestication and distribution of cattle during the Longshan culture period in North China. Front Genet 12:759827
Zhong G, Madry H, Cucchiarini M (2022) Mitochondrial genome editing to treat human osteoarthritis-a narrative review. Int J Mol Sci 23(3):1467
Zimorski V, Ku C, Martin WF, Gould SB (2014) Endosymbiotic theory for organelle origins. Curr Opin Microbiol 22:38–48
Zou L, Wu D, Ren G, Hu Y, Peng L, Zhao J, Garcia-Perez P, Carpena M, Prieto MA, Cao H, Cheng KW, Wang M, Simal-Gandara J, John OD, Rengasamy KRR, Zhao G, Xiao J (2021) Bioactive compounds, health benefits, and industrial applications of Tartary buckwheat (Fagopyrum tataricum). Crit Rev Food Sci Nutr 8:1–17
Zubaer A, Wai A, Hausner G (2018) The mitochondrial genome of Endoconidiophora resinifera is intron rich. Sci Rep 8(1):17591
Acknowledgements
Not applicable.
Funding
This study was funded by the open project of Key Laboratory of Integrated Pest Management on Crops in Southwest, Ministry of Agriculture (2021).
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: QL, LZ, and CC. Analyzed the data: LL, QW, ZB, TZ, PX, and WT. Wrote and reviewed the paper: QL and CC. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Adherence to national and international regulations
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1. Comparison on mitogenomes among six Phallomycetidae species. Table S2. Characterization and annotation of the two Ramaria mitogenomes. Table S3. Start and stop codons analysis of 6 species from Phallomycetidae. Table S4. Local BLAST analysis of the two Ramaria mitogenomes against themselves. Table S5 Tandem repeats detected in the two Ramaria mitogenomes. Table S6. Aligned fragments between mitogenomes and nuclear genomes of the two Ramaria species. Table S7. Species information used for phylogenetic analysis in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Li, L., Zhang, T. et al. The first two mitochondrial genomes for the genus Ramaria reveal mitochondrial genome evolution of Ramaria and phylogeny of Basidiomycota. IMA Fungus 13, 16 (2022). https://doi.org/10.1186/s43008-022-00100-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43008-022-00100-7