
Abstract
Background
The resistome describes the array of antibiotic resistant genes (ARGs) present within a microbial community. Recent research has documented the resistome in the rumen of ruminants and revealed that the type and abundance of ARGs could be affected by diet and/or antibiotic treatment. However, most of these studies only assessed ARGs using metagenomics, and expression of the resistome and its biological function within the microbiome remains largely unexplored.
Results
We characterized the pools of ARGs (resistome) and their activities in the rumen of 48 beef cattle belonging to three breeds (Angus, Charolais, Kinsella composite hybrid), using shotgun metagenomics and metatranscriptomics. Sixty (including 20 plasmid-associated) ARGs were expressed which accounted for about 30% of the total number of ARGs (187) identified in metagenomic datasets, with tetW and mefA exhibiting the highest level of expression. In addition, the bacterial hosts of 17 expressed ARGs were identified. The active resistome was less diverse in Kinsella composite hybrid than Angus, however, expression of ARGs did not differ among breeds. Although not associated with feed efficiency, the total abundance of expressed ARGs was positively correlated with metabolic pathways and ‘attenuation values’ (a measurement of stability) of the active rumen microbiome, suggesting that ARGs expression influences the stability and functionality of the rumen microbiome. Moreover, Ruminococcus spp., Prevotella ruminicola, Muribaculaceae spp. and Collinsella aerofaciens were all identified as hosts of expressed ARGs, possibly promoting the dominance of these carbohydrate degraders within the rumen microbiome.
Conclusions
Findings from this study provide new insight into the active rumen resistome in vivo, which may inform strategies to limit the spread of ubiquitously found ARGs from the rumen to the broader environment without negatively impacting the key functional outcomes of the rumen microbiome.
Similar content being viewed by others

Background
Antimicrobials have been widely used in food producing animals since the 1950s to enhance feed efficiency, accelerate growth, and control infectious diseases [1]. It is estimated that antimicrobials used to prevent/treat disease and/or promote growth in chickens, pigs, and cattle will increase from 63,151 tonnes in 2010 to 105,596 tonnes in 2030 [2]. Antimicrobial consumption or administration in livestock has been proposed to select for antimicrobial resistant bacteria within the digestive tract of livestock and in aquatic/soil environments [3, 4]. The development of antimicrobial resistant bacteria in food-producing animals not only reduces the therapeutic efficacy of antimicrobials against infectious disease, but also selects for reservoirs of antimicrobial resistant genes (ARGs) that could be transferred to bacteria and infect humans via the food production chain or through the environment [5,6,7]. Therefore, reducing antimicrobial resistance and preventing antimicrobial residues from entering the food production chain is a priority for the livestock sectors to address food security, food safety, and public health concerns [8].
In fact, antimicrobial resistance (AMR) in bacteria is an ancient phenomenon and was present long before the widespread clinical and agricultural use of antimicrobials [9]. Numerous ARGs encode for resistance to an array of antimicrobials that microbes produce to compete and survive within complex ecological systems [10]. Recent research has documented the resistome in the rumen of ruminants including beef cattle [11,12,13], dairy cattle [13, 14], and sheep [15], and revealed that the profiles and abundance of ARGs within the resistome can be affected by diet [12] and/or antibiotic treatment [11]. However, most of these studies only assessed these profiles at a genomic level using metagenomics and rumen samples collected from a limited number of animals. Compared with the mere presence of a gene, its expression is a better proxy for gauging functional activity within biological ecosystems [16]. A recent study assessed the resistome within wastewater at both metagenomic and metatranscripomic levels and revealed that the abundance of ARGs and their transcripts were not highly correlated [17]. However, the extent to which ARGs are expressed in the gut of mammals such as rumen of cattle is largely unknown, especially when it is not under the selective pressure of antimicrobials.
In this study we assessed the presence (metagenomic profiling) and expression (metatranscriptomic profiling) of ARGs in the rumen of 48 beef steers raised without antimicrobials used in human medicine. To date, read-based and assembly-based approaches are two main methods for profiling the resistome, with no consensus on a single approach, as both have trade-offs [18]. Therefore, we applied both approaches in this study with the aim to comprehensively describe the ARGs profiles within resistomes as well as their expression in the rumen. We hypothesized that (1) rumen resistome may be affected by host factors such as breed; (2) not all ARGs are expressed, and those expressed may play a role in the function or stability of the rumen microbiome; (3) expression of ARGs could be associated with feed efficiency in beef cattle, as we previously reported that active rumen microbiome was linked to feed efficiency in beef cattle [19].
Results
Analysis of the rumen metagenomic datasets
The average number of metagenomic reads was higher in KC (59,835,594) than CH (49,746,190) (P = 0.009, d = 0.40; Additional file 1: Table S1). The proportion of metagenomic reads aligned to bovine genome was 0.13% ± 0.004% (average ± standard deviation), which did not differ among breeds (P = 0.134, d = 0.52). The average number of ARG-like reads (KC: 54,385; AN: 47,319; CH: 49,880) and their proportion of total metagenomic reads (KC: 0.090; AN: 0.088; CH: 0.098) did not differ among breeds (P = 0.407, d = 0.15 and P = 0.749, d = 0.40; Additional file 1: Table S1). After metagenomic assembly, a total of 2,776,208 contigs with an average length of 3515 bp (max 446,148 bp) and a N50 length of 6065 bp were generated. An average of 70.4%, 78.4%, and 75.2% of reads were mapped to their assemblies for KC, AN, and CH, respectively. The proportion of mapped metagenomic reads was higher in AN than KC (P = 0.002, d = 0.26). The average number of metagenomic contigs (P < 0.001; d = 0.06) and ARG-like contigs was higher (P < 0.001; d = 0.26), while the proportion of ARG-like contigs was lower (P < 0.001; d = 0.20) in KC compared with the other two breeds (Additional file 1: Table S1).
Profiles of ARGs in the rumen microbiome and plasmids
In total, we identified 183 individual ARGs belonging to 18 classes using a read-based approach, and 104 individual ARGs belonging to 16 classes using assembly-based methods (Table 1). The abundances (calculated as reads/contigs per million of total reads/contigs) of individual ARGs identified using the two approaches are reported in Additional file 1: Table S2 and S3. Eighty-six ARGs were identified using both approaches (Additional file 2: Fig. S1a), and most were more often identified using read- vs assembly-based approach (Additional file 2: Fig. S1b). Of the 17 unique ARGs identified using an assembly-based approach, most were only identified in less than 5 samples except for vanU (Additional file 2: Fig. S1c). Based on these findings, we concluded that results from a read-based approach were more comprehensive and as a result this approach was used for downstream analysis.
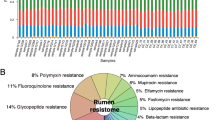
Tetracycline, macrolide-lincosamide-streptogramin (MLS), and aminoglycoside classes accounted for the majority of ARGs within all samples (Fig. 1a). Eight ARGs belonging to tetracycline, MLS, and aminoglycoside classes were identified in all samples, and represented 90% of the abundance of ARGs (Fig. 1b). The PCR analysis confirmed the prevalence of tetQ, tetW, and mefA in extracted DNA (Additional file 2: Fig. S2). We further found that 90 ARGs, representing 15 classes were plasmid-associated (Additional file 1: Table S4), which accounted for 6.25% ± 0.30% of total ARGs (Fig. 1c). Similarly, tetracycline, aminoglycoside, and MLS were the most abundant classes (Additional file 2: Fig. S3a), and 7 ARGs (tetW, aadA, tetO, tet44, tetM, vatB, and tet32) accounted for 85% of the total plasmid-associated ARGs (Additional file 2: Fig. S3b).

Profiles of resistome in the rumen of 48 beef cattle. a The rumen resistome is predominant by ARGs belonging to tetracycline, MLS, and aminoglycoside classes, and ‘others’ include other 15 classes. b Proportion of 8 predominant ARGs and ‘others’ include other 179 individual ARGs. c Proportion of plasmid-associated ARGs in the total ARGs. ARG, antimicrobial resistant gene; MLS, macrolide-lincosamide-streptogramin
Analysis of the rumen metatranscriptomic datasets
The average number of metatranscriptomic reads (KC: 6,277,349; AN: 5,649,606; CH: 5,464,699), ARG-like transcripts (KC: 308; AN: 324; CH: 259) and their proportion of total metagenomic reads (KC: 0.005; AN: 0.006; CH: 0.005) was not different among breeds (P = 0.161, d = 0.55; P = 0.442, d = 0.10; P = 0.668, d = 0.44, respectively; Additional file 1: Table S5). The proportion of metatranscriptomic reads aligned to bovine genome was 0.05% ± 0.0008%, which did not differ among breeds (P = 0.937, d = 0.18). After the assembly of the metatranscriptome data, a total of 28,005 contigs with an average length of 1611 bp (max 9788 bp) and a N50 length of 1520 bp were generated. Mapped reads did not differ (P = 0.401, d = 0.01) among breeds, with an average of 82.6%, 84.0%, and 84.0% of reads for KC, AN, and CH, respectively. The average number of metatranscriptomic contigs (P = 0.065, d = 0.44) and ARG-like contigs (P = 0.062, d = 0.45) tended to be higher in AN than CH (Additional file 1: Table S5). The proportion of ARG-like metatranscriptomic contigs of total metatranscriptomic contigs did not differ (P = 0.591, d = 0.05) among breeds and was 0.145% ± 0.046% (SD), 0.159% ± 0.038%, and 0.155% ± 0.035% for KC, AN, and CH respectively (Additional file 1: Table S5).
Profiles of expressed ARGs in the active rumen microbiome and plasmids
Sixty and 37 expressed ARGs belonging to 10 classes were identified using read- and assembly-based approaches, respectively (Table 1). Notably, the same number (60) of expressed ARGs were identified using either the SARG database or the ARG-like sequences identified in metagenomic datasets as the reference database. Abundances of expressed ARGs identified using these two approaches are shown in Additional file 1: Table S6 and S7. Twenty-three expressed ARGs were identified using both approaches (Additional file 2: Fig. S4a), with more being identified in samples using read- than assembly-based approaches (Additional file 2: Fig. S4b). Of the 14 unique expressed ARGs identified using the assembly-based method, only tetP were expressed in more than half of the samples (Additional file 2: Fig. S4c). Consequently, we used the read-based approach in subsequent downstream analysis of expressed ARGs.
Tetracycline (tetW, tet40, tetQ, tetM) and MLS (mefA) classes made up the majority of expressed ARGs from all samples (Fig. 2a and b). Indeed, only 0.61% ± 0.03% (calculated as the number of ARG-like reads of a sample identified in metatranscriptomic data/number of ARG-like reads of the same sample identified in metagenomic data) of ARGs were expressed, with multidrug (MDR, n = 28), MLS (n = 48), tetracycline (n = 48), and aminoglycoside (n = 37) classes being most prevalent (Fig. 2c). Of the expressed ARGs, tet40, mefA, tetM, tetW, and tetQ were the most prevalent in all samples (Fig. 2d). The PCR analysis confirmed the expression of tetQ, tetW, and mefA in the cDNA of the majority of samples (Additional file 2: Fig. S2). Twenty of the expressed ARGs were plasmids-associated and represented 7 different classes (Additional file 1: Table S8). Transcripts associated with tetracycline ARGs were the most prevalent (Additional file 2: Fig. S5a), with plasmid-associated tetW being identified in all samples (Additional file 2: Fig. S5b).

Profiles of active resistome in the rumen of 48 beef cattle. a The active rumen resistome is predominant by ARGs belonging to tetracycline and MLS classes, and ‘others’ include other 8 classes. b Proportion of 5 predominant expressed ARGs and ‘others’ include other 55 individual expressed ARGs. c Proportion of major expressed ARG classes. d Proportion of major expressed ARGs. ARG, antimicrobial resistant gene; MDR, multidrug; MLS, macrolide-lincosamide-streptogramin
Effect of breed on diversity and abundances of ARGs and expressed ARGs in the rumen microbiome
The ARGs belonging to tetracycline and MLS classes were predominant in the rumen, regardless of breeds (Fig. 3a). The Chao1 index of ARGs was lower in KC than AN and CH (P = 0.007 and 0.002, d = 0.24), while the Simpson’s index of ARGs did not differ among breeds (P = 0.512, d = 0.29; Fig. 4a). Principle Coordinate Analysis (PCoA) showed that the profiles of ARGs also did not separate by breed (PERMANOVA P = 0.241; Fig. 4b). The KC (125) harbored fewer subtypes of ARGs compared with AN (153) or CH (136) (Additional file 1: Table S9). The abundance of total ARGs in KC was also lower than that in the other two breeds (Fig. 4c). Specifically, the abundance of several ARGs belonging to aminoglycoside (aadA, aadE, ant(9)-I), bacitracin (bacA), MLS (ermA, ermB, ermG, lsa, macB), tetracycline (tet40), and vancomycin (vanG, vanS) (Fig. 4d) classes in KC were lower than in AN or CH.

Abundance (normalized into reads per million of reads) of classes of ARGs and ARG transcripts in the rumen of KC, CH, and AN beef steers. a Abundance of 13 classes of ARGs. b Abundance of 8 classes of expressed ARGs. ARG, antimicrobial resistant gene; KC, Kinsella composite hybrid; AN, Angus; CH, Charolais; MLS, macrolide-lincosamide-streptogramin; MDR, multidrug

Difference in diversity annd abundance (normalized into reads per million of reads) of ARGs in the rumen of KC (in purple), CH (in green), and AN (in yellow) beef steers. a Difference in Chao1 index and Simpson’s index among breeds. b PCoA based on Bray–Curtis distance. c Difference in the abundance of total ARGs among breeds. d Difference in the abundance of 12 ARGs belonging to aminoglycoside, bacitracin, MLS, tetracycline, and vancomycin classes among breeds. ARG, antimicrobial resistant gene; KC, Kinsella composite hybrid; AN, Angus; CH, Charolais; MLS, macrolide-lincosamide-streptogramin; MDR, multidrug. PCoA, principal coordinate analysis. **P < 0.05 by Kruskal–Wallis test followed by multiple pairwise comparisons using Dunn’s test with Benjamini–Hochberg method for false discovery rate control
The KC (64) harbored fewer subtypes of plasmid-associated ARGs compared with AN (74) or CH (67) (Additional file 1: Table S10). No difference was observed in the abundance of total plasmid-associated ARGs among breeds. The KC had a higher abundance of aadE as well as vatB than AN, and ant(9)-I, aph(3″)-I, as well as tet44 than CH (Additional file 2: Fig. S6). In contrast, the abundance of ermB, ermG, and lnuA was lower in KC than the other two breeds (Additional file 2: Fig. S6).
The Chao1 index of expressed ARGs was lower in KC than AN (P = 0.024, d = 0.55), with no difference in the Simpson’s diversity among breeds (P = 0.541, d = 0.24; Additional file 2: Fig. S7a). The PCoA based on Bray–Curtis distance showed no clustering of expressed ARGs within the resistome among breeds (PERMANOVA P = 0.423; Additional file 2: Fig. S7b).
Metatranscriptomic analysis also showed that ARGs belonging to tetracycline and MLS classes were predominant in the expressed rumen resistome of all three breeds (Fig. 3b). A similar number of subtypes of expressed ARGs were identified in KC (44, including 26 plasmid-associated), AN (42, including 31 plasmid-associated), and CH (42, including 26 plasmid-associated), among which 30 (including 18 plasmid-associated) were identified in all breeds (Additional file 1: Table S11 and S12; Additional file 2: Fig. S7a and S7b). Breed did not impact the abundance of expressed ARGs (including plasmid-associated).
Investigation of the bacterial hosts of expressed ARGs
The ARG-containing contigs generated by metatranscriptomic assembly were used to predict the bacterial origin of expressed ARGs. A total of 420 ARG-containing contigs were assigned to a customized reference database (see Methods) and the bacterial hosts of 342 contigs were identified as belonging to Firmicutes (32.4%), Bacteroidetes (22.1%), Actinobacteria (16.4%), and Proteobacteria (8.1%) (Fig. 5a). For the bacterial hosts identified in Actinobacteria, the genus Collinsella (52.2%) accounted for more than half of the abundance, followed by Bifidobacterium (13.0%) (Fig. 5a). Muribaculaceae bacterium DSM 108,610 (14.0%) was the most abundant genus in the Bacteroidetes, followed by Bacteroides (10.7%) and Prevotella (9.7%) (Fig. 5a). In the Firmicutes, Bacillus, Ruminococcus, and Turicibacter were the most abundant genera (Fig. 5a). The 342 assembled-genomes were found to contain 17 expressed ARGs, among which mefA was the most abundant, followed by tetW and tetQ (Additional file 1: Table S13). Notably, we identified 8 bacterial species/strains that could potentially express bacA, bcrA, macB, mefA, tet40, and tetW (Fig. 5b). For example, Muribaculaceae bacterium DSM 108,610, Bacteroides fragilis, Bacillus cereus, and Turicibacter sp. H121 potentially expressed mefA, while Collinsella aerofaciens and Prevotella ruminicola 23 potentially expressed tetW.

The bacterial origin of active resistome. a Proportion of predicted bacterial hosts of expressed ARGs at phylum level. b Predicted bacterial hosts of 6 expressed ARGs including tetW, tet40, mefA, macB, bcrA, and bacA and their relative abundance. ARG, antimicrobial resistant gene
Using only the flanking regions of ARGs from the ARG-containing metatranscriptomic contigs, a total of 97 contigs were assigned to a customized reference database and the bacterial hosts were identified as belonging to Firmicutes (37.1%), Actinobacteria (25.8%), Bacteroidetes (19.6%), and Proteobacteria (4.1%) (Additional file 2: Fig. S8). For the bacterial hosts identified in Actinobacteria, the genus Collinsella (84.0%) accounted for more than half of the abundance, followed by Bifidobacterium (8.0%) (Additional file 2: Fig. S8). Muribaculaceae bacterium (26.3%) was the most abundant genus in the Bacteroidetes, followed by Muribaculum (21.0%) and Phocaeicola (15.8%) (Additional file 2: Fig. S8). In the Firmicutes, Bacillus, Ruminococcus, and Turicibacter were the most abundant genera (Additional file 2: Fig. S8). The 97 metatranscriptomic-assembled genomes were found to contain 13 expressed ARGs, among which mefA was the most abundant, followed by tetW (Additional file 1: Table S14). Notably, we identified 6 bacterial genera/species (Muribaculaceae, Bacteroides fragilis, Ruminococcus spp, Bacillus cereus, and Turicibacter spp., and Collinsella aerofaciens) that could potentially express mefA or tetW (Additional file 1: Table S14). In summary, bacterial species that were identified using ARG flanks were all included in the list of species identified using full-length contigs.
Functional potentials of expressed ARGs in the rumen microbiome
We first investigated if expressed ARGs contribute to the annotated function of the active rumen microbiome. Based on Spearman correlation analysis, we found significant correlations between the abundance of 19 KEGG pathways mainly related to ‘Metabolism’, ‘Cellular Processes’ as well as ‘Genetic Information Processing’ and the abundance of 8 subtypes of expressed ARGs, including mefA, tetW, tet40, tetO, tetM, tetracycline resistance protein (trp), tetO, and vatB (Fig. 6). Notably, positive correlations were observed between the abundance of most subtypes of expressed ARGs and ‘Metabolism’ pathways including ‘Two component system’, ‘Oxidative phosphorylation’, and ‘Purine metabolism’, while negative correlations were identified between those and ‘Salmonella infection’, ‘Ribosome’, as well as the ‘HIF-1 signaling pathway’ (Fig. 6). We then analyzed if the expression of the resistome was associated with feed efficiency in beef cattle. However, no significant correlation was observed between the abundance of total expressed ARGs or any individual ARGs and residual feed intake (RFI) or feed conversion ratio.

Heatmap showing the Spearman correlations between the abundance (normalized into reads per million) of KEGG pathways and the abundance of 8 subtypes of expressed ARGs. ARG, antimicrobial resistant gene; trp, tetracycline resistance protein
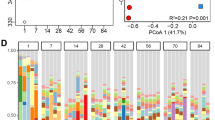
To test our hypothesis that an active resistome may play a role in conferring stability to the rumen microbiome, we calculated the attenuation value, a measurement of microbial stability. The Chao1 index of the active resistome was positively correlated with the attenuation value (rho = 0.46, P < 0.05; Fig. 7a). In addition, we also found that the abundance of expressed tetracycline class (rho = 0.47, P < 0.05; Fig. 7b) and tetW (rho = 0.47, P < 0.05; Fig. 7c) was positively correlated with the attenuation values of the active rumen microbiome, indicating that it may also contribute to functional stability.

Functions of active resistome. a Spearman correlation between the Chao1 index of expressed ARGs and attenuation values of active rumen microbiota. b Spearman correlation between the abundance of expressed ARGs in tetracycline class and attenuation values of active rumen microbiota. c Spearman correlation between the abundance of expressed tetW and attenuation values of active rumen microbiota. ARG, antimicrobial resistant gene
Discussion
Metagenomic analysis revealed that the abundance of ARGs in the rumen of beef steers that did not receive any antimicrobials other than monensin, was predominated by tetracycline, MLS, and aminoglycosides. We detected broader ARG profiles using a read-based approach (187 ARGs belonging to 18 classes) in the rumen, than that observed in a previous study where ARGs in beef cattle receiving monensin and tylosin were estimate using an assembly-based approach [11]. These researchers failed to detect aminoglycoside or β-lactam ARGs in any of the 5 ruminal samples examined. While Auffret et al. [12] detected macrolide, chloramphenicol, β-lactam, and aminoglycoside ARGs in the rumen of antimicrobial-free beef cattle under similar feeding condition to our study, they did not detect genes conferring resistance to vancomycin. More recently, Xue et al. [14] described a resistome containing 26 classes of ARGs using rumen digesta samples collected from 49 dairy cows that did not receive antimicrobials, with tetracycline being the most predominant, followed by glycopeptide and fluoroquinolone ARGs. The variation of ARGs profiles detected among studies could be due to difference in animal, environment, diet, antimicrobial use (AMU) as well as metagenome data nature and bioinformatic resources/tools. In addition, the detected ARGs are similar to those reported in fecal samples of beef cattle administered in feed antimicrobials (ionophores, chlortetracycline, or tylosin) [20] or not [21]. This suggests that the profiles of ARGs in the rumen are consistent with those found in feces. It should be noted that tetracycline and macrolide are the most frequently used antimicrobials in beef industry in Canada [22]. Such a practice could have resulted in these ARGs being the key components of the core resistome in the rumen, even in the absence of direct selective pressure as result of AMU.
Considering that the presence of genes does not necessarily directly correlate with their expression in biological systems, direct measurement of transcripts based on metatranscriptomics is an important complementary approach to describing the functionality of microbiomes. In our study, the abundance of expressed ARGs was about 30% of the whole resistome, suggesting that most ARGs were not transcribed and functional in the rumen of these cattle at the time of sampling. In fact, the expression of ARGs is not directly linked to the presence of ARGs as previously shown in wastewater microbiomes [17]. The expression of tet40, mefA, tetM, tetW, and tetQ was observed in all samples, suggesting that these ARGs may constitute the ‘core’ active resistome in the rumen of the steers investigated in our study. However, it is not clear if and how this expression pattern may change if antimicrobials are administered. To our knowledge, few studies have examined the presence and expression of ARGs simultaneously in microbiomes within food-producing animals. Although Sabino et al. [23] analyzed the expression of rumen ARGs, only 15 samples were used (5 dairy cattle, 5 beef cattle, and 5 sheep) and their aim was to confirm the expression of ARGs found in 435 ruminal bacteria and archaea reference genomes in silico. These researchers did not link the expression of ARGs back to the presence of those ARGs in the same rumen microbome using metagenomic data. More recently, the resistome in the chicken and pig gut were analyzed using both metagenomic and metatranscriptomic data, but only 6 fecal samples were used to define the gut resistome of each species [24]. In this regard, more effort is needed to detect and validate our findings based on both metagenomic and metatranscriptomic analysis. In addition, it should be noted that the average insert size varied between the DNA and cDNA libraries due to the fact that with an average gene length of ~ 1 kb in prokaryotes, and with selective expression, the overall transcriptome is more fragmented with essentially smaller average fragment size as compared to genomic DNA fragments that are generated due to DNA shearing. This difference may potentially lead to smaller average insert size of the cDNA-based library compared to a metagenomics DNA-based library. Smaller insert sizes may also impact the quality of reads and the downstream analysis of functional profiles (such as resistome) of Illumina sequencing output [25, 26]. Therefore, comparing communities across multiple experiments may require additional covariates (e.g., library preparation technique) in statistical design to account for these known differences and avoid potential biases. It should also be noted that there is still a lack of direct comparison of metagenomic and metatranscriptomic datasets from the same sample, and our results advanced the knowledge on what are key factors (e.g., sequencing depth, N50 length, library size) in affecting the data analysis outcomes as a whole.
Plasmids are mobile genetic elements that are abundant in the bacterial populations of bovine rumen [27], and play a major role in the spread of AMR through horizontal gene transfer [28]. Plasmid-mediated transfer of ARGs is the most common route for their acquisition by bacteria [29]. Metagenomic approaches have been used to characterize plasmid encoded ARGs in several non-biological habitats such as activated sludge [30, 31] as well as in the human gut [32]. However, the profiles and expressions of plasmid-associated ARGs have not been examined in food-producing animals. The identification of aadA and tetW being the most abundant plasmid-associated ARGs and the expression of tetW highest among all ruminal plasmid-associated ARG transcript, suggests that plasmid associated ARGs should be a focus of investigations on the impact of AMU in livestock on the resistome. The aadA gene has been detected on IncA/C2 plasmids in both Escherichia coli and Salmonella enteric [33]. More specifically, aadA14 was first identified as plasmid borne in a bovine respiratory disease-associated pathogen, Pasteurella multocida [34]. It has been reported that many of the tetracycline resistant genes are associated with mobile plasmids [35], among which tetW has been proven to be transferable among the ruminal bacteria, i.e., Butyrivibrio fibrosolvens, Selemonas ruminitanium, and Mitsuokella multiacidus [36]. It has been proven that rumen plasmidome, which represents all detectable plasmids in rumen, encodes a wide array of genes with roles in carbohydrate, protein and amino acids metabolism [27]. Therefore, we speculate that the plasmids that carry expressed ARGs likely also carry genes that play a role in fermentation. Recently, linking a wide range of bacterial plasmids to the microbiome in wastewater samples has been undertaken using the Hi-C method [37], which can link plasmids back to their bacterial host [38]. These approaches could also be applied to link plasmid-associated ARG to their rumen bacteria, which may help elucidate the contribution of plasmids to the transmission of ARGs within the rumen.
It has been reported that the prevalence and abundance of ARGs in the gut of cattle is affected by diet and host. For example, dietary transition from milk replacer to a starter diet led to alternation in the fecal resistome of dairy calves [7]. In addition, the diversity and abundance of total ARGs were higher in the rumen of beef cattle fed high concentrate than those fed high forage diet, with chloramphenicol and aminoglycoside resistance genes being predominant in forage- and concentrate-fed cattle, respectively [12]. On the other hand, several studies have shown that the gut microbiome is heavily influenced by the host animal [39,40,41], suggesting that host may also play a key role in shaping the rumen resistome. In this study, all beef steers were raised under the same dietary and environmental conditions, suggesting that the prevalence and expression of ARGs could have been influenced by host genetics. Contrary to the findings of Auffret et al. [11], who did not observe a breed effect on the abundance of microbiota or ARGs in the rumen of beef cattle, we observed a reduced prevalence of ARGs in the rumen of crossbred (KC) compared to purebred (AN or CH) cattle. Cohen’s d is a common way to measure the size of an effect, which is classified as small (d = 0.2), medium (d = 0.5), and large (d ≥ 0.8) [42]. Although there is significant difference in the average number of metagenomic reads and Chao1 index of ARG, only small to medium effect size was identified, suggesting that sequencing depth may not be a potential confounding factor in the comparison of the breed effect on diversity and abundance of ARGs. To our knowledge, there is no study reporting how host genetic factors may impact the gut resistome in mammalian species. In beef cattle, understanding the ‘mammalian host-resistome’ association may be a prerequisite to select breeds with high feed efficiency and a lower risk of transmission ARG into the environment.
The positive correlation between the abundance of multiple subtypes of expressed ARGs and ‘Metabolism’ pathways suggest that ARGs may play a role in regulating bacterial metabolism. Indeed, ARGs, such as those encoding for efflux pumps may have roles in regulating microbial physiology and metabolism of amino acids, fatty acids or nucleotides, in addition to their intrinsic role in antimicrobial resistance [43,44,45]. Bacteria belonging to Muribaculaceae spp. encode enzymes that degrade plant hemicellulose and pectin [46]. Collinsella aerofaciens has been reported to be abundant in the rumen of low-methane yield sheep [47], which could ferment a range of different carbohydrates such as starch [48]. Prevotella ruminicola [49] and Ruminococcus spp. [50, 51] are also prevalent fiber degraders within the rumen. Our findings raise the possibility that expression of ARGs could impact the relative contribution of these species to ruminal fiber and starch digestion. Knowledge of the bacterial hosts of expressed ARGs could play an essential role in designing strategies to limit the spread of ARGs via manipulation of the rumen microbiome. Despite these findings, it should be noted that a large proportion of hosts of expressed ARGs could not be taxonomically classified or could only be identified at the phylum/family level. Considering the fact that when expressed, the transcript of a gene does not contain extensive flanking regions of a gene coding sequence (cds) except for the 5′ untranslated region (5′UTR) until the transcription start site upstream of an individual gene or the first cds in an operon where the member genes of the operon are transcribed together, and a small 3′ termination (rho-dependent or independent) signal downstream of an individual gene cds or the last cds of an operon, it is challenging to predict bacterial origin from metatranscriptomic assemblies. However, for certain genomic loci in prokaryotes where gene/operon transcripts overlap and if those loci happen to carry an ARG, the resulting assembled long ARG-carrying contigs may lead to a better prediction of bacterial species. This could be the case of the 97 ARG-containing metatranscriptomic contigs identified as active bacterial hosts using only the flanking regions that were not mobile genetic elements or partial ARGs. Conversely, although metagenomic assembled ARG-carrying contigs potentially harboring contiguous sequences of the host bacteria could provide better prediction of bacterial species, ARGs present in those contigs may or may not be expressed making the functionality of ARGs within the rumen microbiome questionable. In this regard, more effort is needed to develop parallel metagenomics and metatranscriptomic approaches to identify functional ARGs and their host genomic context.
We did predict that the expression of tetracycline class, especially tetW, was associated with the stability of active rumen microbiota. Stability refers to the ability of a microbial community to return to its original state after facing perturbations [52]. Higher stability (higher attenuation value in our study) indicates that a microbial community is more resilient to external perturbations and its functional profile is therefore less likely to change. Our findings suggest that a rumen microbial community with higher diversity or expression of tetW may be more resistant to external perturbations such as use of antimicrobials or changes in diet compositions. However, as the correlation analysis is not able to imply causation, the exact role that expressed ARGs play in regulating these functions requires further investigation.
Conclusions
We characterized ARGs profiles including the ‘active’ resistome within the rumen of beef steers that were not administered antimicrobials other than monensine. Our findings demonstrated that the presence and expression of ARGs in the rumen are not necessarily associated with AMU. Although a diversity of ARGs were found in the rumen, about 30% (60/183) were expressed, with multiple tetracycline ARG subtypes and mefA constituting the ‘core’ of the active rumen resistome. Individual PCR analysis further validated the prevalence and expression of the core ARGs identified using metagenomic and metatranscriptomic sequencing. We also revealed that Ruminococcus spp., Prevotella ruminicola, Muribaculaceae spp. and Collinsella aerofaciens were the primary hosts of expressed ARGs. In addition, both breed and feed efficiency exhibited a weak effect on the abundance of expressed ARGs, suggesting that in the absence of antimicrobial use, the rumen resistome is unlikely to be altered by breeding programs that select cattle based on improved feed efficiency. We also showed that the active resistome in the rumen may play a role in bacterial metabolism as well as maintaining the stability of active rumen microbiome. One potential limitation of the current study is that rumen resistome originated from cattle that only received monensin. It is unclear whether the overall rumen resistome or its expression would have been if antimicrobials such as tetracyclines or macrolides had been administered. In addition, variations in sequencing depth and library construction technique between metagenomic and metatranscritpomic datasets in our study may also impact the profiles of expressed resistome. Comparative analysis of transcriptional profiles of rumen samples originating from larger herds of beef or dairy cattle at different growth stages, raised with and without antimicrobials could provide further insight on the presence and expression of ARGs within the rumen microbiome. Considering that the N50 length for metatranscriptomic-assembled contigs were relatively short, appropriate method to identify active bacterial host of expressed ARGs based on metatranscriptomic sequencing needs further investigation. This study provides new insight into the active rumen resistome in the absence of antimicrobial selective pressure, generating information that could be used to develop strategies to limit the spread of ubiquitously found antimicrobial resistance from the rumen to the broader environment.
Methods
Animal experiment and sample collection
The datasets used in the current study originated from the study of Li et al. [53]. Briefly, ruminal digesta samples were collected from 48 beef steers at slaughter (range from 429 to 554 days of age, on average 502 ± 33 days of age) and snap-frozen in liquid nitrogen. The beef steers included three breeds: Kinsella composite hybrid (KC, n = 16), Angus (AN, n = 16), and Charolais (CH, n = 16). All steers were raised in the same feedlot conditions and fed the same diet, consisting of 80% barley grain, 15% barley silage, and 5% supplement. Cattle did not receive any antimicrobials other than monensin at 33 ppm in the diet, an ionophore that is not used in human medicine.
Metagenome and metatranscriptome sequencing
Total DNA and RNA were isolated from rumen digesta of 48 beef steers using the methods of Yu and Morrison [54] and Li et al. [55], respectively. After quality and quantity checks, metagenomic libraries of the DNA were constructed using TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, USA) and subjected to Illumina (HiSeq 2000) sequencing. After measurement of RNA yield and quality, samples with RNA integrity number (RIN) > 7.0 were used to construct metatranscriptome libraries using the TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA). All metagenomic and metatranscriptomic libraries (n = 16 for each breed) were sequenced (1 μg of input DNA or cDNA library) at the McGill University and Génome Québec Innovation Centre (Montréal, QC, Canada) using an Illumina HiSeq 2000 platform (100 bp paired-end sequencing of ~ 350 bp inserts for metagenome, and of ~ 140 bp cDNA fragments for metatranscriptome). Quality control (QC) of each dataset was performed as described by Li et al. [53]. Briefly, Trimmomatic (version 0.35) [56] was used to trim artificial sequences (adapters), cut low quality bases (quality scores < 20), and remove short reads (< 50 bp). In addition, Tophat2 (version 2.0.9) [57] was used to remove potential host DNA and RNA contamination by mapping trimmed reads to the bovine genome (UMD 3.1).
Identification of ARGs using read- and assembly-based approaches
The prevalence and abundance of the rumen resistome of each metagenomic dataset was determined using read-based approach according to the instructions of ARG-OAP 2.0 pipeline [58]. Briefly, post-QC reads (paired-end) from each sample were blasted using BLASTX against the Structured ARG database (SARG) [59], comprised of the Antibiotic Resistance Genes Database (ARDB), the Comprehensive Antibiotic Resistance Database (CARD), and the National Center for Biotechnology Information Non-Redundant Protein Sequence Database (NCBI-NR), to extract ARG-like reads. Reads were subsequently annotated as ARG-like reads at the cut-off of E value of 10−10, sequence identity of 80% and alignment length more than > 25 amino acids (six-frame translation) using the default settings. By using this cut-off, the identification accuracy was estimated at 99.5% [60]. To evaluate the expression of the resistome, both SARG database [59] and extracted ARG-like sequences identified in metagenomic datasets were used as reference database to extract ARG-like transcripts from the metatranscriptome dataset using an ARG-OAP 2.0 pipeline [58]. Reads were annotated as ARG-like transcripts using the same cutoffs as described above.
In addition, the prevalence and abundance of the rumen resistome was also determined using assembly-based approach. Briefly, metagenomic and metatranscriptomic assemblies were generated with post-QC reads for each sample using MEGAHIT (version 1.2.9) [61] with default parameters. Quality of metagenomic and metatranscriptomic assemblies were evaluated using Quast (http://cab.cc.spbu.ru/quast/) and contigs with lengths less than 1000 bp in all of the assemblies were filtered out. The contigs were then aligned with SARG database using ‘diamond blastx’ [62] at the cut-off of E value of 10−10 and sequence identity of 80% for both metagenomic and metatranscriptomic sequencing.
PCR analysis of the prevalence and expression of tetQ, tetW, and mefA genes
All the DNA isolates (50 ng/μl) and cDNA were tested for the presence of antigen resistance genes by PCR. Individual PCRs were performed for tetW, tetQ and mefA. The primers and PCR conditions used were those described in Additional file 3: Table S15. The PCR assays were conducted in a 20-μl mixture containing 2 ul 10 × PCR buffer, 0.5 μl, 10 mM dNTP, 0.25 ul, 5 U/μl of Taq polymerase (Life Technologies, Foster City, CA), 1ul, 20 pmol/μl of each primer and nuclease-free water. Reactions were conducted using an Vetiri 96 well thermal cycler (Life Technologies, Foster City, CA). TetW, tetQ and mefA were amplified by subjecting DNA and cDNA template to the following conditions: an initial denaturation for 5 min at 94℃; then 30 cycles of 94℃ for 30 s, annealing for 30 s at different temperatures (Additional File 1: Table S13), and 72℃ for 30 s; and a final elongation for 7 min at 72℃. Finally, the PCR products were analyzed by gel electrophoresis using 1% (w/v) agarose in 1 × TBE buffer under 150 V. The agarose gel was steamed in Sybr safe and scanned under Azure c200 image system (Azure Biosystems Inc, Dublin, CA).
Identification of plasmid-associated ARGs
Plasmid-associated ARG were determined using a modified ARG-OAP 2.0 pipeline. Instead of SARG database, post-QC reads (paired-end) from each sample were blasted using BLASTX against the latest A CLAssification of Mobile genetic Elements (ACLAME) database, which contains information of mobile genetic elements including 457 bacteriophage genomes, 1109 plasmids and 760 prophages [63]. Plasmid associated reads and transcripts were then annotated at a cut-off E value of ≤ 10−7 criteria with amino acid identity ≥ 80% and coverage ≥ 70%.
Identification of bacterial origin of expressed ARGs
The bacterial origins of the active resistome were predicted by assigning taxonomy to metatranscriptomic-assembled contigs harboring ARGs using kraken2 [64]. First, the complete genomes for the bacterial domain in NCBI RefSeq (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/) [65] was downloaded with ‘kraken2-build –download-library bacteria –db RefSeq’ script. Then, the 4941 rumen-related metagenome-assembled genomes [66] were added to the RefSeq database with ‘kraken2-build –add-to-library 4941.fa –db RefSeq’ script. The metatranscriptomic-assembled contigs with ARGs were subject to the ‘kraken2 –db RefSeq –threads 32 –classified-out sample_classified –unclassified-out sample_unclassified -confidence 0.9 –fasta-input sample.contigs.fa –output sample.kraken’ script, where ‘RefSeq’ is the customized database, ‘0.9’ is the confidence score (between 0 and 1). To verify the bacterial host of an ARG, using a parallel approach, the full ARG-containing contigs, as well as their counterparts containing the left-over flanking regions following the SARG database BLAST-based removal of ARGs from each contig, were annotated using kraken2. The output files were further analyzed using Bracken [67] with ‘bracken -d RefSeq -i sample.kraken.txt -o sample.bracken.txt -l S’ script to calculate the relative abundance of bacteria at the species level. In addition, the flanking regions were further blasted against Gypsy Database [68] (https://gydb.org/index.php/Blast) to detect potential mobile genetic elements with E-value lower than 0.01.
Functional profiling of active ruminal microbiome
To analyze the functional profiles of the active ruminal microbiome, metatranscriptomic-assembled contigs were also annotated using Prodigal [69]. The output was then uploaded to KofamKOALA, which assigns K numbers by HMMER/HMMSEARCH against KOfam (a customized HMM database of KEGG Orthologs (KOs)) with an e-value lower than 0.01, to annotate the KEGG functional pathways [70]. The abundance of active KEGG functional pathways was normalized into counts per million.
Stability of active rumen microbiome
Attenuation value is a measurement of microbial stability, which evaluates the expected rate at which increases in the taxonomic perturbation magnitude are expected to increase functional profile shifts [71]. For example, our previous study showed that the attenuation values of fecal microbiota of neonatal calves increased over age, suggesting an increase in stability is a feature of gut microbiota in early life [72]. The attenuation value was calculated according to the method described by Eng and Borenstein [71]. As this method applies 16S rRNA dataset as input, we extracted 16S rRNA sequences from metatranscriptomic datasets using SILVA database of kraken2 [64]. Then the extracted 16S rRNA sequences were demultiplexed with “demux” plugin and subjected to quality control (QC) using “dada2” plugin [73] of QIIME2 (version 2020.2) [74] to generate a table containing the taxonomic composition of a sample. Finally, varying fold of perturbations of each community’s taxonomic composition was simulated and the average shift in the functional profile of a community as a power function of the taxonomic perturbation magnitude was established as:
where t indicates the magnitude of simulated taxonomic perturbation, f indicates the expected shift in functional capacity, a (attenuation) is defined as inversely proportional to the response curve slope, indicating the expected rate at which increases in the taxonomic perturbation magnitude are expected to increase functional profile shifts, b (buffering) indicates how large a perturbation must be before a functional profile shift becomes noticeable and approaches the expected shift magnitude defined by attenuation [71]. As the buffering values did not correlate with the diversity or abundance of expressed ARGs, they were not reported.
Calculations and statistical analyses
The abundance of ARG classes, total ARGs, and individual ARG were calculated as ‘number of reads per million of post-QC metagenomic/metatranscriptomic reads’ or ‘number of assemblies per million of post-QC metagenomic/metatranscriptomic assemblies. Difference in the proportion of post-QC metagenomic/metatranscriptomic reads aligned to bovine genome, the abundance of ARGs and ARG transcripts among breeds were analyzed using the Kruskal–Wallis test in R (version 3.6.1). The P-value of multiple comparison of breed effect was adjusted into false discovery rate (FDR) using the Benjamini–Hochberg algorithm using ‘dunnTest’ function in R. The effect size of multiple comparisons was conducted using ‘Cohen’s d’ function in R. Circos plot analysis was performed in R using the RCircos package [75]. Alpha diversity indices, including Chao1 and Simpson’s index, was performed using ‘chao1’ and ‘diversity’ functions in R, respectively. Principle Coordinate Analysis (PCoA) based on Bray–Curtis distance was performed for the ARGs and ARG trasncripts using ‘betadisper’ and the results were visualized using ‘plot’ function in R. Spearman correlation analysis was performed for the abundance of expressed ARGs and RFI, functional profiles, and attenuationn value of active microbiome and the results were visulized using ‘ggscatter’ and ‘ggplot2’ functions in R Studio. The P-value of Spearman correlations were corrected for multiple inference using Holm's method using ‘rcorr.adjust’ function in R. Significant difference was declared at P ≤ 0.05 and tendencies at 0.05 < P ≤ 0.10. The Spearman’s correlation coefficient, known as rho (ρ), ranges from − 1.00 (a perfect negative correlation) to + 1.00 (a perfect positive correlation). A rho value higher than 0.40 or lower than − 0.40 as well as P value ≤ 0.05 is considered as significant correlation.
Availability of data and materials
Rumen metagenome and total-RNA-based metatranscriptome sequences were deposited into NCBI Sequence Read Archive (SRA) with accession number SRS4217479, and SRS4211817-SRS4211863 under bioproject number PRJNA448333.
References
Cogliani C, Goossens H, Greko C. Restricting antimicrobial use in food animals: lessons from Europe: banning nonessential antibiotic uses in food animals is intended to reduce pools of resistance genes. Microbe. 2011;6:274–9.
Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, et al. Global trends in antimicrobial use in food animals. Proc Natl Acad Sci. 2015;112:5649–54.
Campagnolo ER, Johnson KR, Karpati A, Rubin CS, Kolpin DW, Meyer MT, et al. Antimicrobial residues in animal waste and water resources proximal to large-scale swine and poultry feeding operations. Sci Total Environ. 2002;299:89–95.
Heuer H, Solehati Q, Zimmerling U, Kleineidam K, Schloter M, Müller T, et al. Accumulation of sulfonamide resistance genes in arable soils due to repeated application of manure containing sulfadiazine. Appl Environ Microbiol. 2011;77:2527–30.
Davies J, Davies D. Origins and Evolution of Antibiotic Resistance. Microbiol Mol Biol Rev. 2010;74:417–33.
Hao H, Cheng G, Iqbal Z, Ai X, Hussain HI, Huang L, et al. Benefits and risks of antimicrobial use in food-producing animals. Front Microbiol. 2014;5:288.
Liu J, Taft DH, Maldonado-Gomez MX, Johnson D, Treiber ML, Lemay DG, et al. The fecal resistome of dairy cattle is associated with diet during nursing. Nat Commun. 2019;10:4406.
Gao P, Ma C, Sun Z, Wang L, Huang S, Su X, et al. Feed-additive probiotics accelerate yet antibiotics delay intestinal microbiota maturation in broiler chicken. Microbiome. 2017;5:91.
Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–86.
D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–7.
Thomas M, Webb M, Ghimire S, Blair A, Olson K, Fenske GJ, et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Rep. 2017;7:12257.
Auffret MD, Dewhurst RJ, Duthie CA, Rooke JA, Wallace RJ, Freeman TC, et al. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome. 2017;5:159.
Bickhart DM, Watson M, Koren S, Panke-Buisse K, Cersosimo LM, Press MO, et al. Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation. Genome Biol. 2019;20:1–8.
Xue MY, Xie YY, Zhong YF, Liu JX, Guan LL, Sun HZ. Ruminal resistome of dairy cattle is individualized and the resistotypes are associated with milking traits. Anim Microbiome. 2021;3:18.
Hitch TCA, Thomas BJ, Friedersdorff JCA, Ougham H, Creevey CJ. Deep sequence analysis reveals the ovine rumen as a reservoir of antibiotic resistance genes. Environ Pollut. 2018;235:571–5.
Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet. 2010;11:9–16.
Ju F, Beck K, Yin X, Maccagnan A, McArdell CS, Singer HP, et al. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019;13:346–60.
Carr R, Borenstein E. Comparative analysis of functional metagenomic annotation and the mappability of short reads. PLoS ONE. 2014;9: e105776.
Li F, Guan LL. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl Environ Microbiol. 2017;83:e00061-e117.
Zaheer R, Lakin SM, Polo RO, Cook SR, Larney FJ, Morley PS, et al. Comparative diversity of microbiomes and resistomes in beef feedlots, downstream environments and urban sewage influent. BMC Microbiol. 2019;19:197.
Rovira P, McAllister T, Lakin SM, Cook SR, Doster E, Noyes NR, et al. Characterization of the microbial resistome in conventional and “raised without antibiotics” beef and dairy production systems. Front Microbiol. 2019;10:1980.
Canadian Integrated Program for Antimicrobial Resistance Surveillance. 2018. http://www.phac-aspc.gc.ca/cipars-picra/pubs-eng.php#ar.
Sabino YN, Santana MF, Oyama LB, Santos FG, Moreira AJ, Huws SA, et al. Characterization of antibiotic resistance genes in the species of the rumen microbiota. Nat Commun. 2019;10:5252.
Wang Y, Hu Y, Liu F, Cao J, Lv N, Zhu B, et al. Integrated metagenomic and metatranscriptomic profiling reveals differentially expressed resistomes in human, chicken, and pig gut microbiomes. Environ Int. 2020;138: 105649.
Kircher M, Heyn P, Kelso J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genomics. 2011;12:382.
Gaulke CA, Schmeltzer ER, Dasenko M, Tyler BM, Thurber RV, Sharpton TJ. Evaluation of the effects of library preparation procedure and sample characteristics on the accuracy of metagenomic profiles. mSystems. 2021;6:e00440-21.
Kav AB, Sasson G, Jami E, Doron-Faigenboim A, Benhar I, Mizrahi I. Insights into the bovine rumen plasmidome. Proc Natl Acad Sci. 2012;109:5452–7.
Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol. 2003;20:1598–602.
Munita JM, Arias CA. Mechanisms of antibiotic resistance Microbiol Spect. 2016;4:1–37.
Parsley LC, Consuegra EJ, Kakirde KS, Land AM, Harper WF, Liles MR. Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl Environ Microbiol. 2010;76:3753–7.
Zhang T, Zhang XX, Ye L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS ONE. 2011;6: e26041.
Lee K, Kim D-W, Lee D-H, Kim Y-S, Bu J-H, Cha J-H, et al. Mobile resistome of human gut and pathogen drives anthropogenic bloom of antibiotic resistance. Microbiome. 2020;8:2.
Call DR, Singer RS, Meng D, Broschat SL, Orfe LH, Anderson JM, et al. blaCMY-2-positive IncA/C plasmids from Escherichia coli and Salmonella enterica are a distinct component of a larger lineage of plasmids. Antimicrob Agents Chemother. 2010;54:590–6.
Kehrenberg C, Catry B, Haesebrouck F, de Kruif A, Schwarz S. Novel spectinomycin/streptomycin resistance gene, aadA14, from Pasteurella multocida. Antimicrob Agents Chemother. 2005;49:3046–9.
Chopra I, Roberts M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. 2001;65:232–60.
Barbosa TM, Scott KP, Flint HJ. Evidence for recent intergenic transfer of a new tetracycline resistance gene, tet(W), isolated from Butyrivibrio fibrisolvens, and the occurrence of tet(O), in ruminal bacteria. Environ Microbiol. 1999;1:53–64.
Stalder T, Press MO, Sullivan S, Liachko I, Top EM. Linking the resistome and plasmidome to the microbiome. ISME J. 2019;13:2437–46.
Burton JN, Liachko I, Dunham MJ, Shendure J. Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3 (Bethesda). 2014;4:1339–46.
Henderson G, Cox F, Ganesh S, Jonker A, Young W, Global Rumen Census Collaborators, et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567.
Zhou M, Peng YJ, Chen Y, Klinger CM, Oba M, Liu JX, et al. Assessment of microbiome changes after rumen transfaunation: implications on improving feed efficiency in beef cattle. Microbiome. 2018;6:62.
Li F, Li C, Chen Y, Liu J, Zhang C, Irving B, et al. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome. 2019;7:92.
Cohen J. Statistical Power Analysis for the Behavioral Sciences. New York: Routledge Academic; 1988.
Martínez JL. Antibiotics and antibiotic resistance genes in natural environments. Science. 2008;321:365–6.
Martínez JL, Sánchez MB, Martínez-Solano L, Hernandez A, Garmendia L, Fajardo A, et al. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol Rev. 2009;33:430–49.
Aminov RI. The role of antibiotics and antibiotic resistance in nature. Environ Microbiol. 2009;11:2970–88.
Ormerod KL, Wood DL, Lachner N, Gellatly SL, Daly JN, Parsons JD, Dal’Molin CGO, Palfreyman RW, Nielsen LK, Cooper MA, Morrison M, Hansbro PM, Hugenholtz P. Genomic characterization of the uncultured Bacteroidales family S24–7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4:36.
Kamke J, Kittelmann S, Soni P, Li Y, Tavendale M, Ganesh S, Janssen PH, Shi W, Froula J, Rubin EM, Attwood GT. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterised by lactic acid formation and utilisation. Microbiome. 2016;4:56.
Kageyama A, Benno Y, Nakase T. Phylogenetic and phenotypic evidence for the transfer of Eubacterium aerofaciens to the genus Collinsella as Collinsella aerofaciens gen. nov., comb. nov. Int J Syst Bacteriol. 1999;49:557–65.
Purushe J, Fouts DE, Morrison M, White BA, Mackie RI, North American Consortium for Rumen Bacteria, et al. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol. 2010;60:721–9.
Suen G, Stevenson DM, Bruce DC, Chertkov O, Copeland A, Cheng J-F, et al. Complete genome of the cellulolytic ruminal bacterium Ruminococcus albus 7. J Bacteriol. 2011;193:5574–5.
McCoun M, Oyebade A, Estrada-Reyes ZM, Pech-Cervantes AA, Ogunade IM. Effects of multi-species direct-fed microbial products on ruminal metatranscriptome and carboxyl-metabolome of beef steers. Animals. 2021;11:72.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;13:220–30.
Li F, Hitch TC, Chen Y, Creevey CJ, Guan LL. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome. 2019;7:6.
Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–12.
Li F, Henderson G, Sun X, Cox F, Janssen PH, Guan LL. Taxonomic assessment of rumen microbiota using total RNA and targeted amplicon sequencing approaches. Front Microbiol. 2016;7:987.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Yin X, Jiang XT, Chai B, Li L, Yang Y, Cole JR, et al. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics. 2018;34:2263–70.
Yang Y, Jiang X, Chai B, Ma L, Li B, Zhang A, et al. ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics. 2016;32:2346–51.
Yang Y, Li B, Ju F, Zhang T. Exploring variation of antibiotic resistance genes in activated sludge over a four-year period through a metagenomic approach. Environ Sci Technol. 2013;47:10197–205.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010;38:D57-61.
Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken2. Genome Biol. 2019;20:257.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–45.
Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol. 2019;37:953–61.
Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. Peerj Comput Sci. 2017;3: e104.
Llorens C, Futami R, Covelli L, Dominguez-Escriba L, Viu JM, Tamarit D, et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011;39(suppl 1):D70–4.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2.
Eng A, Borenstein E. Taxa-function robustness in microbial communities. Microbiome. 2018;6:45.
Ma T, Villot C, Renaud D, Skidmore A, Chevaux E, Steele M, et al. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: prediction of diarrhea. ISME J. 2020;14:2223–35.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat Biotechnol. 2019;37:852–7.
Zhang H, Meltzer P, Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics. 2013;14:244.
Olfert ED, Cross BM, McWilliam AA. Guide to the Care and Use of Experimental Steers. Ottawa: Canadian Council on Animal Care; 1993.
Acknowledgements
The authors are sincerely grateful to Dr. C. Fitzsimmons, Mr. B. Irving and staff at Roy Berg Kinsella Ranch for animal trial and providing all phenotypic records of beef steers phenotypic data collection. We appreciate Y. Chen, X. Sun, and F. Cox for their assistance on the DNA/RNA isolation and sequencing library construction. We thank all members from Guan lab for their help on sampling. We also appreciate Mr. Yingnan Ju from Indiana University for his inputs in data processing. We thank all members from Guan lab for their help on sampling.
Funding
This work was financially supported by Ministry of Alberta Agriculture and Forestry (2015P008R and 2018F095R) as well as the NSERC Discovery Grant and the Genome Alberta “Omics” project held by LLG and TAM. In addition, TM, YT, and QD was supported by the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2017-FRI-04) and Central Public-interest Scientific Institution Basal Research Fund (Y2022QC10).
Author information
Authors and Affiliations
Contributions
TM and LLG designed this study. LLG provided funding. FL performed the DNA/RNA isolation, mRNA enrichment, and sequencing of library construction. TM, WG and FL conducted bioinformatics and statistical analyses. TM, RZ, TAM, YT, QD and LLG were responsible for the data interpretation and manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study received research ethics approval from the Livestock Care Committee of the University of Alberta (Protocol No.: AUP00000882), following the guideline of the Canadian Council on Animal Care [76].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1–S14.
Additional file 2.
Fig. S1–S8.
Additional file 3: Table S15.
The primers used for PCR analysis of tetW, tetQ, and mefA genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, T., Zaheer, R., McAllister, T.A. et al. Expressions of resistome is linked to the key functions and stability of active rumen microbiome. anim microbiome 4, 38 (2022). https://doi.org/10.1186/s42523-022-00189-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-022-00189-6