
Abstract
Background
The identification of banana cultivars genome is needed to provide a valid identity from the accession of bananas which are used as basic data in the management of in situ and ex situ banana conservation as well as for further breeding of banana cultivars.
Results
The size of PCR-amplified matK ranged from 844 to 860 bp and showed a high variability. The haplotype diversity was 0.9048 with nine haplotypes. Haplotype distribution map revealed the lineage patterns of banana cultivars from Java. Reconstruction of genetic relationships using the maximum likelihood, maximum parsimony and Bayesian inference algorithms produces tree topologies and classifications that are grouped according to their genomic groups, into three main clades, i.e., AA/AAA, AAB and ABB. Based on the previously derived age constraints and fossil data, we estimate (Musaceae) that genetic divergence times of all samples occurred during the Eocene (95% HPD: 51.9 Mya), Musa acuminata group (AA, AAA, AAB) with Musa balbisiana group (BB and ABB) occurred during the Oligocene (95% HPD: 26 Mya), and the separation on each banana cultivars occurred during the Middle Miocene to Pliocene (95% HPD: 16.5–2.5 Mya).
Conclusions
From this study, we conclude that all studied cultivars are closely related according to its genomic groups with high variation. Genetic variation among those cultivars creates nine haplotypes. The development of variety which leads to the formation of different banana cultivars had suggested to be occurred long ago along with human migration and domestication.
Similar content being viewed by others


Background
Banana cultivars (Musa spp., Family Musaceae) are horticultural commodities which are the fourth most important fruit crop in developing countries (Perrier et al. 2011; Kharadi et al. 2014). Banana cultivars are thought to have originated in Indo-Malesia especially Southeast Asia, which then spread to all tropical and subtropical countries (De Langhe et al. 2009; Häkkinen 2013; Sulistiyaningsih et al. 2014). Therefore, Southeast Asia was considered a central origin of banana (Valmayor et al. 2000; Perrier et al. 2011) since banana cultivars are widely distributed in Southeast Asia, including Indonesia (Ministry of Agriculture 2016). In Indonesia, more than 200 cultivars are well recognized and widely cultivated throughout main islands including Sulawesi, Sumatra, Java and Madura (BPS-Statistic Indonesia 2019). Some regions which contributed to banana production in Indonesia include East Java (29.08%), West Java (16.76%) and Lampung (16.61%) (BPS-Statistic Indonesia 2019).
Two types of banana have been characterized by Linneaus in 1953 and 1959, namely plantain (Musa paradisiaca Linn.) and dessert banana (Musa sapientum Linn.) (Valmayor et al. 2000). However, the application of both taxonomical names in Southeast Asia as the center origin of banana generated confusion (Valmayor et al. 2000; Singh et al. 2001). Another problem confronting taxonomists and horticulturists in Southeast Asia is the presence of many banana cultivar names and synonymies cultivars in different languages, dialects and regions (Valmayor et al. 2000; Wahyudi et al. 2020). Therefore, new classification scheme proposed by Simmonds and Shepherd (1955) that approved by a consensus in 1999 may be a breakthrough of this problem (Valmayor et al. 2000). The new taxonomical scheme of bananas or called three-tier system consists of the species name, followed by letter combinations indicating the ploidy and genome groups contributed by their ancestral and followed by local cultivars name (Simmonds and Shepherd 1955; Valmayor et al. 2000).
The three-tier system was adopted from the fact that banana cultivars in Southeast Asia were originated from hybridization between the two ancestors Musa acuminata Colla (contributors of A genome, x = 11) and Musa balbisiana Colla (contributors of B genome, x = 11) (Singh et al. 2001; De Langhe et al. 2009; Li et al. 2010, 2013; Häkkinen 2013). Hybridization followed by chromosome restitution has arisen of banana diversity with various ploidy level and genomic combinations such as AAA (Ambon, Berlin, Mas), AAB (Pisang Raja) and ABB (kapok, Ebung) (Sumardi and Wulandari 2010; Hapsari et al. 2015). The determination of the genomic groups of banana cultivars was assessed based on morphological characters (Hapsari et al. 2015). However, morphological approaches are sometimes inaccurate because it was very subjective; the process is time-consuming and can be influenced by environmental factors (Probojati et al. 2019).
The latest approach to identify genome composition and grouping of banana cultivars is by the molecular marker. The molecular approach based on the sequence of DNA is proven to have a higher level of accuracy and efficiency compared to morphological techniques and PCR-based analysis such as PCR–RFLP, random amplified polymorphism DNA (RAPD), amplified fragment length polymorphism (AFLP) and inter simple sequence repeat (ISSR) or microsatellite (Poerba and Ahmad 2010; de Jesus et al. 2013; Lamare and Rao 2015; Sundari et al. 2017; Poerba et al. 2019; Probojati et al. 2019; Wahyudi et al. 2020). Although the PCR-based technique can classify banana cultivars genome, most cultivars' maternal and parental donors are still uncertain (Li et al. 2013). Therefore, identification of banana cultivar genome based on DNA sequence from chloroplast genome such as rbcL, intron trnK, trnl-F and matK (maturase K) (Liu et al. 2010; Bieniek et al. 2015; Wahyudi et al. 2013; Janssens et al. 2016; Nikmah et al. 2016; Udensi et al. 2017) may cope the problem. Chloroplast DNA is structurally stable, non-recombinant, and it is inherited from the maternal (Costion et al. 2011; Yuan et al. 2015; Shekhar et al. 2019).
matK gene is widely used for identification because of its effectiveness, slow mutation rate in plants and a more specific accuracy level than other genes. In addition, the consortium barcode of life (CBOL) has also recommended matK gene as a marker for general plant identification (CBOL Plant Working Group et al. 2009). matK gene was even succeeded to reconstruct phylogenetic trees and estimated genetic divergence time (Liu et al. 2009; Udensi et al. 2017). To date, no report has been found about the genetic relationship and the estimated time of genetic divergence of bananas cultivars in Java based on the matK gene sequence.
This present study aims to analyze the genetic diversity, relationship and divergence time among local banana cultivars from Java Island based on maturase K (matK) gene sequence. The results of this study are expected to be useful not only ini agriculture but also in conservation and banana breeding activities. Furthermore, the genetic divergences time provides information about the relation to its distribution range and geological history of local banana cultivars in Java Island.
Methods
Plant materials
A total of 14 local banana cultivars scattered from Java Island were collected from banana germplasm garden of Yogyakarta, Indonesia (Table 1). Banana cultivars were originated from areas covering ten districts of Java Island (Fig. 1) and represented four genomic groups, i.e., AA, AAA, AAB and ABB (Table 1). Further, young leaves of banana cultivars were collected and dried with silica gel before be analyzed. In addition, two species from GenBank NCBI were also used as outgroup, i.e., Ensete glaucum and Musella lasiocarpa.

Map of banana cultivars locality from Jawa Island. 1. Yogyakarta, 2. Banyuwangi, 3. Pasuruan, 4. Purworejo, 5. DKI Jakarta, 6. Bantul, 7. Karanganyar, 8. Malang, 9. Ponorogo, 10. Banyumas
Molecular analysis
Whole genomic DNA isolation was carried out using Promega Wizard® Genomic DNA Purification Kit, following its manufacturer’s instructions. The total genomic DNA was confirmed both quantitatively and qualitatively. Quantitative examination of total DNA concentration was done using AE-Nano 200 Nucleic Acid Analyze version 2.0. The qualitative examination was carried out using electrophoresis separation on 1% agarose gels stained with 2 μg/ml ethidium bromide (Etbr) for 30 min at a voltage of 80 V and then photographed on GelDOC UV—Transilluminator (BioRAD). The estimated length of total genomic DNA was measured using a 1-Kb DNA ladder marker (Thermo Scientific, California, USA). Amplification of matK gene was accomplished using primer pairs of matK-1RKIM-f 5′-ACCCAGTCCATCTGGAAATCTTGGTTC-3′ and matK-3FKIM-r 5′-CGTACAGTACTTTTGTGTTTACGAG-3′ designed by K. J. Kim from School of Life Sciences and Biotechnology, Korea University, Seoul, Korea (Kuzmina et al. 2012). PCR reactions were conducted in 40 μl volume consisted of 20 μl of 2 × TopTaq Master Mix Kit (Qiagen), 1 μl of 5 pmol each of forward and reverse primers, 4 μl coral load, 8 μl RNA-se Free Water and 6 μl DNA template (100–150 ng/uL). PCR cycle consists of DNA pre-denaturation at 94 °C for 4 min, followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 48 °C for 40 s and extension at 72ºC for 60 s. Final extension carried out at 72 ºC for 10 min. PCR products were purified and sequenced at 1st BASE Laboratories Sdn Bhd, Malaysia.
Data analysis
Genetic diversity and haplotype analysis
The matK gene DNA sequence data were evaluated using ABI sequences Scanner v.10. Then, sequence alignment process was analyzed using Clustal W menu with MEGA X software (Kumar et al. 2018). The aligned sequences were further rechecked with the trimmed using Gblocks 0.91b (Castresana 2000) based on sequence alignment of the taxa Outgroup and default parameters of the program. Genetic diversity and haplotype diversity were analyzed with DnaSP version 5.10.01. Reconstruction of the haplotype distribution map was generated with Haplotype Network 4.6.1.2 (Paradis 2018).
Phylogenetic analysis
Phylogenetic analyses were reconstructed using maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI) approaches. The maximum parsimony (MP) and maximum likelihood (ML) analyses were implemented in PAUP*v4.0b1 software (Swofford 2002), using trees bisection-reconnection (TBR) branch swapping on 1000 bootstrap replicates. The evolutionary models for the ML and BI analyses were estimated using jModeltest 2.0 (Darriba et al. 2012), and a GTR (General Time Reversible) + G (Gamma) + I (Invariant Sites) substitution model was used. The Bayesian inference analysis was performed using MrBayes 3.0b4 software (Huelsenbeck and Ronquist 2001). Parameters for the best model were then applied in an MCMC (Monte Carlo Markov Chain), were run for 10,000,000 generations, and every 1000 generations were sampled. All sample points that occur red before stationarity of negative log likelihood (–lnL) scores were discarded as burn-in period and were retained in the 50% (burn-in = 500) majority rule consensus tree. Furthermore, each output file produced was visualized through FigTree v1.4.3 (Drummond and Rambaut 2007). A phylogeny tree has a support node, nodes with bootstrap value ≥ 70 for MLBS and MPBS, and ≥ 95 for BPP value (Huelsenbeck and Hilis 1993).
Divergence time estimation
Divergence time estimation analyses were conducted if the tree topology was considered credible in explaining the separation between taxa. Based on the matK genes, the divergence times of banana cultivars were estimated using the Monte Carlo Markov Chain (MCMC) method, and each analysis was run for 10 million generations with parameters sampled every 1,000 generations, which was implemented in BEAST (Bayesian Evolutionary Analysis Sampling Trees) v2.4.1 package software (Drummond and Rambaut 2007), under an uncorrelated lognormal-relaxed clock model of rate variation among lineages. External calibration was obtained from Janssens et al. (2016), i.e., the time of genetic divergence between Ensete glaucum and Musella lasiocarpa was occured at 53 Mya (CI 46.1– 61.4), while genetic divergence time among Genus Ensete, Musella and Musa was occured at 53 Mya (CI 46.1–61.4). The convergence of the stationary distribution of prior and posterior was checked through the likelihood and parameter sample plots using Tracer software ver1.4 (Drummond and Rambaut 2007). The output results in the form of log and tree files were combined using Logcombiner v2.4.1 software (Drummond and Rambaut 2007). Tree results of 100,001 were ignored until there is only one tree (burnin = 1,000,000) and trees were summarized with TreeAnnotator v2.4.1 software (Drummond and Rambaut 2007). The final reconstruction chronogram and divergence times were visualized using Figtree v.1.4.3 software (Drummond and Rambaut 2007) and were compared to supporting literature.
Results
Amplification and DNA sequences of matK genes
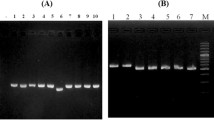
Amplification using primers of matK-1RKIM-f and matK-3FKIM-r was successfully carried out on the 14 banana cultivars examined, resulted in 800–900 bp DNA fragments (Fig. 2). Sequencing of this amplicons produced 844–860 bp length fragments (Table 2). Based on the BLAST analysis, all of those fragments were homologues to the Musa acuminata (MF68180.4), Musa balbisiana (KC904686.1) and matK sequences (similarity 73–100%).

Electropherogram amplicon of matK gene of 14 banana cultivars. M = marker 1 kb. Lane 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, are Rejang, Mas, Berlin, Kojo Santen, Ambon Hong, Morosebo, Raja Seribu, Triolin, Brentel Warangan, Saba Awu, Ebung, Raja Bandung, Kluthuk Ijo, Kluthuk Wulung
Nucleotides composition of matK gene in 14 banana cultivars examined was dominated by T(U) and A bases content than C and G (Table 2). The average composition value of each base was 36.5% T (U) bases, 29.3% in A bases, 15.8% in G bases and 18.3% in C (Table 2). The A/T bases content value of 14 banana cultivars was 58.4–67.1%, while the value of GC bases content was 32.6–41.6%. These matK sequences produced 962 characters of nucleotide sites. As much as 360 (37.42%) out of 962 characters were identified as conserved region sites (invariable or monomorphic), 468 (48.65%) were variable sites (polymorphic), and 134 (13.93%) were alignment gaps or missing data. Around 210 (21.82%) was singleton variable sites, and 258 (26.81%) has the potential to parsimony informative sites.
Haplotype diversity
Analysis of haplotype diversity using Ensete genus as out-group resulted in 9 haplotypes with high haplotype diversity values (Hd = 1000 < 0.9048 < 0.5000) (Fig. 3). This study revealed that the studied cultivars lineaged from several regions in Java Island, which were interconnected and classified according to their genomics into 3 groups.

Haplotype network of 14 banana cultivars from Java based on the matK gene
The haplotype network was formed by outgroup (H9), namely Ensete glaucum, then flows into the haplotype 8 network (H_8) that consists of 4 cultivars, which was genome ABB and BBw. Then, it flows into the haplotype networks H_3, H_6 and H_7, which all consist of bananas AAB-type. Furthermore, H_1, H_2, H_4 and H_5, where all haplotypes consist of diploid AA banana and AAA triploid.
Banana cultivars of AA and AAA genomes were grouped into one haplotype which previously thought to be result hybrid between the gene flow from haplotype 1 (H_1) network and haplotype 2 (H_2), so it becomes haplotype 4 and haplotype 5 which were AAA genomes. This result could support the finding in the previous study by Simmonds and Shepherd (1955) that triploids originated from pollination of haploids from diploid female cells. For example, triploid genome AAA was originally from the combination of diploid AA female cells with haploid A male cells, so that it will produce AAA triploid (M. acuminata).
Unlike the haplotype 3 (H_3) which consists of Kojo Santen (AAA), Brentel Warangan (AAB) and Saba Awu (ABB), these three banana cultivars have a genomes and origin different. The AAB and ABB genomes are possible from the results of hybridization between M. acuminata and M. balbisiana. Almost all triploid bananas origin from one or two species of M. acuminata (A) and M. balbisiana (B) which will be classified into AA, AAA, AAB and ABB according to the relative participation of each genome in the genotype (De Langhe et al. 2009).
Phylogeny of banana cultivars from Java Island
In order to analyze the relationships among 14 banana cultivars in this study, phylogenetic analyses were performed by the 3 methods, which has a different bootstrap support value of each branch point/node. Nodes were considered well supported if value maximum likelihood bootstrap support (MLBS) ≥ 70%, maximum parsimony bootstrap support (MPBS) ≥ 70% and Bayesian inference posterior probabilities ≥ 95% (Huelsenbeck and Ronquist 2001). Although some nodes of 3 methods phylogenetic topology exhibited low bootstrap support, the phylogenetic topology this study consisted of three major clades, according to the genome which well supported (MLBS = 100%, MPBS = 100%, BPP = 100%) (Fig. 4).


Phylogenetic trees of 14 banana cultivars infered from matK gene using different algorithms. (a) Maximum likelihood (ML). (b) Maximum parsimony (MP). (c) Bayesian inference (BI). Numbers associated with branches are bootstrap percentages of ML and MP higher than 50% and Bayesian posterior probabilities greater than 0.90, respectively. Each cultivar name was followed genome composition of the cultivar which the previously recognized. Ensete glaucum used as outgroup
Clade I consists of AA genome groups (Rejang, Mas, Berlin) and AAA genome groups (Morosebo, Ambon Hong). However, AAB groups (Raja Seribu) were nested in the clade 1 together with AA and AAA groups, but the clades itself had weak categorized bootstrap support (MLBS = 61%, BPP = 55%) and moderate categorized for (MPBS = 74%) (Fig. 4). Although this result of Raja Seribu (AAB) is nested, the AA/AAA group's relationship with the AAB group had tended to separate into two groups based on the haplotype distribution map (Fig. 3). Clade II mainly consists of AAB groups (Triolin, Brentel Warangan, Saba Awu) with polytomy topology and weak support (MLBS = 66%, BPP = 93%) (Fig. 4a, c), and moderate support for MPBS = 77% (Fig. 4b). However, there are also Kojo Santen (AAA group) and Saba Awu (ABB group) nested in this clade. Their relationship had tended into low bootstrap support in its topology separation. In clade II, the triploid genome group (AAA, AAB, ABB) is interconnected and becomes a clade within it. This result was supported by previous studies which likely that the triploid genome has the same paternal and maternal origins. Clade III consists of ABB and BBw group with polytomy topology and strong supported bootstrap for (MLBS = 90%, MPBS = 94%) (Fig. 4a, b) and relatively weak support for BPP = 93% (Fig. 4c). These results suggest that ABB groups (Ebung, Raja Bandung) and BBw (Kluthuk Ijo, Kluthuk Wulung) are closely related, so it is thought to be sister species group (100% identical). ABB and BBw genome groups show the same relationship patterns and belonging to one group with genetic distance value of 0,000. These findings suggest multiple inter-specific hybridization origins for the B genome in different cultivars of domesticated bananas. Based on this finding, we suggest that M. balbisiana might donate B genome.
Divergence time estimations
The divergence time estimation of 14 studied cultivars based on matK using BEAST maximum clade credibility tree is depicted in Fig. 5. The mean ages and 95% HPD are shown in Table 3. The divergence time for Musaceae (including all samples of this study) was estimated at 51.9 Mya (CI 60.5–44.8 Mya 95% highest posterior density (HPD)) (node 1), indicating that the origin was started in the early Eocene. Likewise, the separation between genera Ensete and Musella is also formed in the early Eocene at 45 Mya (48–42.5 Mya 95% HPD; calibration point used was 43 Mya) (node 2). The diversification of genus Musa containing species of M. acuminata and M. balbisiana groups started during the Oligocene estimated average age 26 Mya (95% HPD: 37–16 Mya; node 3). However, the initial separation of each cultivars containing Ambon Hong, Raja Seribu, Morosebo, Mas and Rejang occurred in the middle of Miocene estimated average age 16.5 Mya (95% HPD: 26–7.5 Mya; node 4). Furthermore, cultivars Raja Seribu and Mas, Rejang, Morosebo diversified in the late Miocene with an estimated divergence time of 13 Mya (95% HPD: 22–5.9 Mya; node 5). Likewise, the split between cultivars Mas and Rejang at 7 Mya (95% HPD: 11–3 Mya; node 6), and separation between Berlin cultivar and Triolin, Kojo Santen, Brentel Warangan and others occurred at 12.5 Mya (95% HPD: 29.8–5 Mya; node 7) also situated in the late Miocene. Estimated time of divergence of cultivars Triolin, Kojo Santen, Brentel Warangan, Saba Awu and Kluthuk IJo, Ebung, Kluthuk Wulung, Raja Bandung at 4 Mya (95% HPD: 6–2.1 Mya; node 8) in the Pliocene and cultivar Kojo Santen, Brentel Warangan, Saba Awu/Triolin is also situated in the Pliocene at 2.5 Mya (95% HPD: 2.1–1 Mya; node 9).

Chronogram of the divergence time estimation of 14 banana cultivars based on matK gene using BEAST. Median ages of nodes are shown, with horizontal bars indicating the 95% highest posterior density (HPD) intervals
Discussion
Genetic diversity of banana cultivars
To evaluate the germplasm of domesticated bananas, several studies have investigated the genetic diversity of cultivated and wild Musa accessions using various molecular marker, i.e., AFLP, RAPD, PCR–RFLP, ISSR and microsatellite (Babu et al. 2018; Das et al. 2018; Poerba et al. 2019; Probojati et al. 2019; Wahyudi et al. 2020). Currently, molecular systematics studies in plants based on matK gene have been widely conducted as a potential candidate to determine genetic diversity and perform species identification or plant DNA barcoding (Costion et al. 2011; Hilu et al. 2014; Yuan et al. 2015; Shekhar et al. 2019). This study also unveiled that banana cultivars matK sequences were longer than of Poaceae (Das et al. 2013) and Dipterocarpaceae families matK (Harnelly et al. 2018) which in turn have shown that banana cultivars harbor high levels of genetic diversity.
Our present study has evaluated the nucleotide diversity of 14 banana cultivars cultivated in various regions of Java Island, Indonesia, by assessing the variable sites (polymorphic) of matK gene. In this study, we have successfully obtained the value of characters nucleotide sites which suggest that the variable sites (polymorphic) are exist. High variations among matK gene sequences in all of the bananas we studied show its high genetic diversity. This high nucleotide diversity in banana cultivars implies that it may have had a historically large population size and did not undergo any severe genetic bottleneck during the domestication process (Li et al. 2013). matK gene in studied banana also showed high parsimony informative sites, where there were changes in at least two types of nucleotides, and two of them appeared with a minimum frequency of two (Hapsari et al 2018). This gene also demonstrated a high average of GC bases content in our banana samples. It is consistent with what had been reported in plant studies including in Musaceae (Christelová et al. 2011) and Poaceae (Bieniek et al. 2015).
The relationship among banana cultivars in Java is evaluated from the haplotype distribution and phylogenetic
Haplotype network construction analysis was used to visualize the genealogical relationships among DNA sequences within a population or at the intraspecific level or to make inference about biogeography and history of populations. Haplotype network considered more informative than conventional phylogenetic trees to display intraspecific DNA sequence variation (Paradis 2018; Hapsari et al. 2020).
Our present study has successfully obtained the haplotype distribution map which showed that the banana cultivars AA-type nested with the AAA genome group. Furthermore, the banana cultivars of AA and AAA groups are directly connected to the banana cultivars AAB group, and AAB group is directly connected to the banana cultivars ABB group. Therefore, it is possible for AAB group bananas to function as intermediate bananas connecting the two genomes B and genome A. These finding further confirmed a previous study by De Langhe et al. (2009) that hybridization between cultiwild populations, accompanying human migration and still partly fertile clones from different origins led to the generation of more sterile diploids AA genome and the more vigorous and nearly sterile triploids (AAA first, then AAB and later on ABB).
In this study, to analyze the phylogenetics relationship we used 3 models, i.e., ML, MP and Bayesian inference, which are largely congruent in the formed tree topologies. The slight difference in the 3 methods is mainly in support at the species level (Fig. 4). This phylogenetic relationship analysis of banana cultivars differentiated each cultivar into 3 clades according to the genome characters. These findings suggest that matK gene sequences can be used as a recommendation for banana cultivars grouping based on its genomes.
There are 2 clades that form a polytomy topology as of also found in the population of Musa troglodytarum based on rbcL (Hiariej et al. 2015). Polytomy topology is the separation of branches that cannot distinguish or separate one species from another. It is probably caused by evolution that occurs simultaneously, thus causing uncertainty in phylogenetic topology (Kuhn et al. 2011). That is supported by previous genetic evolution studies in Poales (Hochbach et al. 2018), which revealed that the matK gene has an evolutionary pattern and tempo that distinguishes it from other genes yet less accurate to distinguish species at low taxa level. Another reason is that it is possible to require specific primers due to the location of matK in the trnK intron and its proximity to psbA which may provide a nearly universal primer for amplification. An effective sorting strategy is needed for matK in the Musaceae group. It is required to conduct further evaluation and analysis for this study.
There is the importance of genetic relationship analysis for genetic conservation of banana cultivars. Protection of banana genetic variation is needed to be done for both in situ and ex situ conservation. If conservation resources are limited, each cultivar which has a similarity and close relationship must be chosen, one of them as a representative. In our study, based on matK, we need to preserve both Saba Awu Banana (ABB) and Kaja Santen (AAA) since these two cultivars possess different types of genomes yet genetically are closely related (Fig. 4).
Divergence times and biogeographical events of banana cultivars in Java Island
The divergence time in this study is very helpful to interpret the temporal evolution of banana cultivars. Our present study provides divergence time estimation for banana cultivars which originated from Java Island. The estimated ages among Musa, Ensete and Musella formed in the early Eocene (Fig. 5); it is resembled to which reported in the previous study (Janssens et al. 2016). Based on the unearthed fossils, the earliest diversification of Musa occurred in northern Indo-Burma during the Late Eocene. It then spread from the north of Indo-Burma toward all area of Southeast Asia (including Malesia) followed by local diversification (Janssens et al. 2016). Based on these reports and the diverse genus distribution, it seems to be reasonable to assume that the genus Musa evolved and diversified in tropical Asia, especially in Southeast Asia region.
Geographic distributions of genus Musa containing species of M. acuminata and M. balbisiana groups (Nodes 3, Fig. 5) started during in the Oligocene. Based on the study by Janssens et al. (2016), the diversification of Ingentimusa, Callimusa and Australimusa started to spread from the northern Indo-Burmese region during the Oligocene, whereas the first dispersal of each M. acuminata and M. balbisiana groups in Java Island (Nodes 4) is distributed from the West region now known as Jakarta (including Raja seribu), toward Yogyakarta, Central Java (including Ambon Hong, Morosebo, Mas and Rejang). It occurred in the middle Miocene to late Miocene. At this time, there is a diversification of Eumusa and Rhodochlamys, which was followed by speciation events (Janssens et al. 2016).
Further spreading into and diversification in East Java (including Berlin, Kojo Santen, Brentel Warangan, Saba Awu, Kluthuk Wulung, Ebung and Kluthuk Ijo) also occurred during the Late Miocene. It is different from that originated from Bantul, Central Java, which occurred in Pliocene (Nodes 8 and 9, Fig. 5). Based on this finding, the spreading of those banana cultivars is supposed to be collateral with human migration both in and out, and became isolated to the regions. Domestication due to human selection and migration is one aspect of evolution (Kantar et al. 2017), especially in banana cultivars. However, we realize that it is still challenging to determine the exact cause of the distribution of existing banana cultivars due to the complexity between phylogenetic patterns and geographical distribution.
Conclusion
Analysis of genetic relationships produced trees which grouped the studied cultivars according to their genomic groups with nine haplotypes. This will allow local banana genetic diversity data to be retained and also to provide intellectual property protection. The estimated divergence time of the formation of all samples (Musaceae) supposed to be occurred during the Eocene (95% HPD: 51.9 Mya), Musa acuminata group (AA, AAA, AAB) with Musa balbisiana group (BB and ABB) occurred during the Oligocene (95% HPD: 26 Mya), and the separation on each banana cultivars occurred during the Middle Miocene to Pliocene (95% HPD: 16.5–2.5 Mya). This study recommends that an analysis of estimated divergence times requires more calibration data by comparing several taxa to determine more specific divergence times. Furthermore, genetic conservation for all 14 studied cultivars from Java is strongly needed to preserve its genetic resources.
Availability of data and materials
All data on which abstracted of the study have been drawn are presented in the main manuscript.
Abbreviations
- A:
-
Adenine
- AFLP:
-
Amplified fragment length polymorphism
- BI:
-
Bayesian inference
- Bp:
-
Base pair
- BPP:
-
Bayesian inference bootstrap support
- C:
-
Cytosine
- CBOL:
-
Consortium barcode of life
- CI:
-
Confidence interval
- G:
-
Guanine
- GTR:
-
General time reversible
- Hd:
-
Haplotype diversity
- HPD:
-
Highest posterior density
- ISSR:
-
Inter simple sequence repeat
- matK:
-
Maturase K
- MCMC:
-
Monte Carlo Markov chain
- ML:
-
Maximum likelihood
- MLBS:
-
Maximum likelihood bootstrap support
- MP:
-
Maximum parsimony
- MPBS:
-
Maximum parsimony bootstrap support
- Mya:
-
Millions of years ago
- PCR:
-
Polymerase chain reaction
- RAPD:
-
Random amplified polymorphic DNA
- rbcL:
-
Ribulose bisphosphate carboxylase large subunit
- RFLP:
-
Restriction fragment length polymorphism
- T:
-
Thymine
- TBR:
-
Tress bisection reconnection
References
Babu AG, Prabhuling G, Karani RS, Satish D, Patil RK, Mulla SR, Raghavendra G, Jagadeesha RC (2018) Genetic diversity analysis among banana cultivars through ISSR markers. J Pharmacog Phytochem 7(6):1576–1580
BPS-Statistic Indonesia (2019) Horticultural statistic. BPS-Statistic Indonesia, Jakarta
Bieniek W, Mizianty M, Szklarczyk M (2015) Sequence variation at the three chloroplast loci (matK, rbcL, trnH-psbA) in the Triticeae tribe (Poaceae): comments on the relationships and utility in DNA barcoding of selected species. Plant Syst Evol 301:1275–1286
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
CBOL Plant Working Group, Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, et al (2009) A DNA barcode for land plants. PNAS. 106(31): 12794–7.
Christelová P, Valárik M, Hřibová E, De Langhe E, Doleže J (2011) A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol Biol 11:103
Costion C, Ford A, Cross H, Crayn D, Harrington M, Lowe A (2011) Plant DNA barcodes can accurately estimate species richness in poorly known floras. PLoS ONE 6(11):e26841
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9(8):772
Das MM, Mahadani P, Singh R, Karmakar K, Ghosh SK (2013) MATK sequence based plant DNA barcoding failed to identify bambusa (family: Poaceae) species from Northeast India. J Environ Sociobiol 10(1):49–54
Das SC, Balamohan TN, Poornima K, Bergh IVD (2018) Evaluation of genetic diversity in some banana hybrids using ISSR markers. Intl J Curr Microbiol App Sci 7(1):146–157
de Jesus ON, Silva SO, Amorim EP, Ferreira CF, de Campos JMS, Silva GG, Figueira A (2013) Genetic diversity and population structure of Musa accessions in ex situ conservation. BMC Plant Biol 13:41
De Langhe E, Vrydaghs L, de Maret P, Perrier X, Denham T (2009) Why bananas matter: an introduction to the history of banana domestication. Ethnobot Res Appl 7:165
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Espino RRC, Jamaludin SH, Silayoi, Bechamas, Nasution RE (1992) Musa L. (edible cultivars). In: Verheis EWM, Coronel RE (Eds): Plant resources of South-East Asia No. 2 edible fruits and nuts. Prosea Indonesia, Bogor
Häkkinen M (2013) Reappraisal of sectional taxonomy in Musa (Musaceae). Taxon 62(4):809–813
Hapsari L, Wahyudi D, Azrianingsih R, Arumingtyas EL (2015) Genome identification of bananas (Musa L.) from East Java Indonesia assessed with PCR-RFLP of the internal transcribed spacers nuclear ribosomal DNA. Intl J Biosci 7(3):42–52
Hapsari L, Azrianingsih R, Arumingtyas EL (2018) Genetic variability and relationship of banana cultivars (Musa L.) from East Java, Indonesia based on the internal transcribed spacer region nrDNA sequences. J Trop Biol Con 15:101–120
Hapsari L, Lestari DA, Probojati RT (2020) Haplotype network analysis of wild banana relatives Ensete glaucum, and Musa balbisiana based on cpDNA rbcL sequences in ex-situ collection. Indian J. Genet 80(3):1–7
Harnelly E, Thomy Z, Fathiya N (2018) Phylogenetic analysis of Dipterocarpaceae in ketambe research station, gunung leuser national park (Sumatra, Indonesia) based on rbcL and matK genes. Biodiversitas 19(3):1074–1080
Hiariej A, Arumingtyas EL, Widoretno W, Azrianingsih, R (2015) Genetic kinship of tongkat langit banana (Musa troglodytarum L.) from moluccas based on rbcL gene sequence. Indian J Gen Plant Breed. 75(4): 526
Hilu KW, Black CM, Oza D (2014) Impact of gene molecular evolution on phylogenetic reconstruction: a case study in the Rosids (superorder Rosanae, Angiosperms). PLoS ONE 9(6):e99725
Hochbach A, Linder HP, Röser M (2018) Nuclear genes, matK and the phylogeny of the Poales. Taxon 67(3):521–536
Huelsenbeck JP, Hillis DM (1993) Success of phylogenetic methods in the four-taxon case. Syst Biol 42(3):247–264
Huelsenbeck JP, Ronquist FR (2001) MrBayes: bayesian inference of phylogenetic trees. Bioinfo 17(8):754–755
Janssens SB, Vandelook F, Langhe ED, Verstraete B, Smets E, Vandenhouwe I, Swennen R (2016) Evolutionary dynamics and biogeography of Musaceae reveal a correlation between the diversification of the banana family and the geological and climatic history of Southeast Asia. New Phyto 210(4):1453–1465
Kuhn TS, Mooers AØ, Thomas GH (2011) A simple polytomy resolver for dated phylogenies: resolving polytomies under a birth-death model. Met Ecol Evol 2(5):427–436
Kharadi CS, Pandey M, Chaudhary A, Sharma MC, Chikara SK (2014) Analysis of genetic diversity amongst banana cultivars prevalent in Gujarat region of India using ISSR markers. Indian J Biotech Pharm Res 2(2):14–20
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–2154
Kuzmina ML, Johnson KL, Barron HR, Herbert PDN (2012) Identification of vascular plants of Churchill, Manitoba, using a DNA barcode library. BMC Ecol 12:25
Lamare A, Rao SR (2015) Efficacy of RAPD, ISSR and DMD markers in assessment of genetic variability and population structure of wild Musa acuminata Colla. Physiol Mol Biol Plants 21(3):349–358
Li LF, Häkkinen M, Yuan YM, Hao G, Ge XJ (2010) Molecular phylogeny and systematics of the banana family (Musaceae) inferred from multiple nuclear and chloroplast DNA fragments, with a special reference to the genus Musa. Mol Phylo Evol 57(1):1–10
Li LF, Wang HY, Zhang C, Wang XF, Shi FX, Chen WN, Ge XJ (2013) Origins and domestication of cultivated banana inferred from chloroplast and nuclear genes. PLoS ONE 8(11):e80502
Liu N, Zhu Y, Wei Z, Chen J, Wang Q, Jian S, Zhou D, Shi J, Yang Y, Zhong Y (2009) Phylogenetic relationships and divergence times of the family Araucariaceae based on the DNA sequences of eight genes. Sci Bull 54(15):2648–2655
Liu AZ, Kress WJ, Li DZ (2010) Phylogenetic analyses of the banana family (Musaceae) based on nuclear ribosomal (ITS) and chloroplast (trnL-F) evidence. Taxon 59(1):20–28
Kantar MB, Nashoba AR, Anderson JE, Blackman BK, Rieseberg LH (2017) The genetics and genomics of plant domestication. BioSci 67(11):971–982
Ministry of Agriculture (2016) The outlook of agricultural commodities in the horticulture (banana) sub-sector. Agricultural Information System and Data Center of the Ministry of Agriculture, Jakarta
Nikmah IA, Azrianingsih R, Wahyudi D (2016) Genetic diversity of porang populations (Amorphophallus muelleri Blume) in Central Java and West Java based on LEAFY second intron marker. J Trop Life Sci 6(1):23–27
Paradis E (2018) Analysis of haplotype networks: the randomized minimum spanning tree method. Methods Ecol Evol 9:1308–1317
Perrier X, Langhe ED, Donohue M, Lentfer C, Vrydaghs L, Bakry F, Carreel F, Hippolyite I, Horry JP, Jenny C, lebot V, Risterucci AM, Tomekpe K, Doutrelepont H, Ball T, Manwaring J, Maret Pd, Denham T (2011) Multidisciplinary perspectives on banana (Musa Spp.) domestication. PNAS. 108(28): 11311–18
Poerba YS, Ahmad F (2010) Genetic variability among 18 cultivars of cooking bananas and plantains by RAPD and ISSR markers. Biodiversitas 11(3):118–123
Poerba YS, Martanti D, Ahmad F (2019) Genetic variation of wild Musa acuminata Colla from Indonesia. Biotropia 26(2):115–126
Probojati RT, Wahyudi D, Hapsari L (2019) Clustering analysis and genome inference of pisang raja local cultivars (Musa Spp.) from Java Island by random amplified polymorphic DNA (RAPD) marker. J Trop Biodiv Biotechnol. 4(2): 42–53.
Shekhar R, Bhavya G, Prakash HS, Geetha N (2019) DNA barcoding bsed authentication of Musa acuminata var. nanjangud rasabale. Jetir. 6(3): 5.
Sulistiyaningsih LD, Megia R, Widjaya EA (2014) Two new records of wild bananas (Musa balbisiana and Musa itinerans) from Sulawesi. Makara J Sci 18(1):1–6
Simmonds NW, Shepherd K (1955) The taxonomy and origins of the cultivated bananas. J Linn Soc London Bot 55(359):302–312
Singh, H, Uma, S, Sathiamoorthy, S (2001) A tentative key for identification and classification of indian bananas. National Research Centre for Banana (NRCB), Tiruchirapalli, India
Sumardi I, Wulandari M (2010) Anatomy and morphology character of five Indonesian banana cultivars (Musa spp.) of different ploidy level. Biodiversitas. 11(4): 167–75.
Sundari, Arumingtyas EL, Hakim L, Azrianingsih R, Wahyudi D (2017) Genetic variability of local durian (Durio zibethinus Murr.) in Ternate Island based on RAPD markers. Plant Cell Biotechnol Mol Biol 18:68–75
Swofford DL (2002) PAUP*.phylogenetic analysis using parsimony (*and other methods) version 4.Sinauer Associates, Sunderland, MA
Udensi OU, Ita EE, Ikpme EV, Ubi G, Emeagi LI (2017) Sequence analysis of maturase K (matk): a chloroplast-encoding gene in some selected pulses. Glob JPAS 23:213–230
Valmayor R, Jamaluddin SH, Silayoi B, Kusumo S, Danh LD, Pascua OC, Espino RRC (2000) Banana cultivar names and synonyms in southeast asia. In: banana cultivar names and synonyms in Southeast Asia. INIBAP, Los Banos, Laguna, Philippines
Wahyudi D, Azrianingsih R, Mastuti R (2013) Genetic variability of porang populations (Amorphophallus muelleri) in West Java and Central Java based on trnL intron sequences. J Biodiv Environ Sci 3(9):31–41
Wahyudi D, Rifliyah K, Uslan (2020) Genome evaluation of banana cultivars based on morphological character and inter-simple sequence repeat (issr) molecular marker. Biodiversitas 21(7):2982–2990
Yuan, Qing-Jun, Bin Z, Dan J, Wen-Jing Z, Tsai-Yun L, Nian-He W, Shu-Jiau C, Lu-Qi H (2015) Identification of species and Materia medica within Angelica L. (Umbelliferae) based on phylogeny inferred from DNA barcodes. Mol Ecol Res 15(2): 358–71.
Acknowledgements
The authors would like to thank the Banana Germplasm Garden of Yogyakarta, at Special Region of Yogyakarta for providing the sample of plant materials examined in this study and Genetics and Molecular Laboratory, Biology Department of UIN Maulana Malik Ibrahim Malang and Laboratory of Biotechnology, Mineral Central Laboratory and Advanced Materials, FMIPA, State University of Malang for molecular and genetic facilities.
Funding
The authors declare that this research was funded by DRPM DIKTI Hibah Tesis research grant of fiscal year 2020 (Contract No. 2020.24-LT-DRPM-01-2020) to D.L., R.T.P. and S.
Author information
Authors and Affiliations
Contributions
RTP and DW completed the sample collection in the field and laboratory experiment work, the data analysis and the writing of the draft of the manuscript. DL was designed the research work and involved in the result interpretations and reviewed the manuscript. S was involved in reviewing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Probojati, R.T., Listyorini, D., Sulisetijono, S. et al. Phylogeny and estimated genetic divergence times of banana cultivars (Musa spp.) from Java Island by maturase K (matK) genes. Bull Natl Res Cent 45, 33 (2021). https://doi.org/10.1186/s42269-021-00492-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-021-00492-3