
Abstract
Mulberry bacterial wilt is a devastating disease that is difficult to control and causes serious economic losses to the sericulture industry. This disease is mostly caused by a diverse group of pathogenic and opportunistic bacteria including, Ralstonia pseudosolanacearum, Pantoea ananatis, Enterobacter cloacae complex (ECC), Klebsiella pneumoniae species complex (KpSC), and K. oxytoca complex (KoC). Due to the lack of a rapid and reliable test to simultaneously detect these complex pathogens of mulberry wilt, we developed a multiplex PCR (mPCR) assay to detect five virulence-related genes carried by the pathogenic bacteria of mulberry bacterial wilt disease. The primers were designed for the virulence-related genes: pleD (GGDF structural domain-containing protein), yjfP (esterase), pelY (peripheral pectate lyase), ampD (N-acetyl-anhydromuranmyl-L-alanine amidase), and ripW (type III effector). Overall, the developed mPCR assay showed highly specific, sensitive and reproducible detection of target pathogens. Briefly, the results showed that the mPCR was highly specific in individual reactions, and the lowest detection concentration of the five pathogenic bacteria was 1.87 × 103 CFU/mL (DNA = 2.45 pg/μL). From 46 natural mulberry wilt samples, the mPCR detection rates of P. ananatis, ECC, KpSC, KoC and R. pseudosolanacearum were 8.69, 91.3, 34.7, 23.9 and 65.21%, respectively. The traditional culture media isolation methods showed comparable results. The pathogenicity test of 84 suspected pathogenic bacteria revealed that the morbidity (average morbidity level) caused by the pathogenic bacteria detected by mPCR was ≥ 65.5%, while the morbidity of the undetected pathogenic bacteria was ≤ 35.5%. Based on these results, we believe that the mPCR developed in the present study will be useful in rapid, reproducible, and sensitive detection of the pathogenic bacteria causing mulberry bacterial wilt including, R. pseudosolanacearum, P. ananatis, ECC, KpSC, and KoC.
Graphical abstract

Similar content being viewed by others

Introduction
Mulberry is a tree or shrub of a significant economic value and is mainly distributed in Eurasia, Africa, and the Americas [83]. Mulberry can be used as raw material in food and beverage [53], medical [51], and animal husbandry [1] industries. Mulberry is now seen as a unique plant with a positive impact on environmental sustainability and ecological restoration [14, 66]. Several devastating diseases have been reported to affect the healthy growth of mulberry. From these, mulberry bacterial wilt, a devastating bacterial quarantine disease, seriously affects mulberry production both in terms of yield and quality [32]. Historically, the occurrence of mulberry bacterial wilt was first reported in 1969 in Shunde city of Guangdong province of China, and then it spread to most of the mulberry planting areas in the region [54]. At present, mulberry bacterial wilt is still endemic in the central sericulture-producing regions of Guangdong and Guangxi provinces and other places in China [15].
The casual pathogens of mulberry bacterial wilt have a complex diversity [81]. The pathogens such as Pantoea ananatis, Enterobacter cloacae complex (ECC), Klebsiella pneumoniae species complex (KpSC), K. oxytoca complex (KoC) and Ralstonia pseudosolanacearum have been reported to cause mulberry wilt disease, (see Additional file 1: Figure S1) [81]. Initially, R. solanacearum complex (RSSC) was identified as a pathogen causing mulberry bacterial wilt [40], however, it was later re-classified as R. pseudosolanacearum. Previously, ECC polyphyletic group was also identified to cause mulberry wilt [72, 88]. Similarly, several strains of mulberry wilt pathogens including E. cloacae (ECC), E. asburiae (ECC), K. pneumoniae (KpSC), K. variicola (KpSC), and K. oxytoca (KoC) were identified in Guangdong and Guangxi provinces of China [10]. Recently, we have identified P. ananatis strain LCFJ-001 [79], E. roggenkampii strain KQ-01 (ECC) [87], and K. michiganensis strain AKKL-001 (KoC) [49] as causal pathogens of mulberry bacterial wilt. Interestingly, these bacteria have been reported to cause bacterial wilt in other crop species as well [24, 45, 61].
The host range of R. pseudosolanacearum is broad, including mulberry. This pathogen infects the roots or stems, eventually causing the host to wither and die [67]. ECC is a commonly reported plant pathogen belonging to the genus Enterobacter [61]. It has been reported to cause wilt and bulb rot of onions [84], ginger rot [86], rot of garlic bulbs [41], necrosis of pepper seedlings [21], bacterial wilt of rice [77], and yellowing and decay of papaya internals [33]. KpSC and KoC also cause bacterial diseases in other crops. For instance, K. variicola (KpSC) can cause rhizome rot of banana [45], sorghum leaf spot [52], plantain soft rot [20], papaya soft rot disease [25], and carrot bacterial soft rot [6]. K. pneumonia (KpSC) can cause onion bulb disease [44] and bacterial top rot in maize [30]. K. oxytoca (KoC) causes wetwood disease of Morus trees [4]. P. ananatis is an important plant pathogen of the genus Pantoea [8]. It was first identified as a pathogen of pineapple fruit rot in Philippines in 1928 [56]. It was found to cause bacterial brown rot of maize [56], center and bulb rot of onions [2], wilt and collapse of eucalyptus [12], bacterial leaf streaking of sweet corn [85], and bacterial diseases of rice [78]. Evidence from these reports, therefore, shows that P. ananatis, ECC, KpSC, KoC and RSSC can cause bacterial diseases in a wide range of plant hosts.
To prevent the spread of mulberry bacterial wilt, there is a need of a sensitive molecular assay to rapidly and simultaneously detect different wilt pathogens. This is particularly relevant due to inherent limitation of the traditional culture methods which are labor-intense and time-consuming. Currently, the methods of molecular detection of mulberry wilt pathogens in practice include PCR [71], RT‒PCR [47], LAMP [27], and IMSA-LAMP [80]. However, there is no practical rapid test for the detection of compound infections of bacterial wilt pathogens of mulberry. Given that the multiplex PCR (mPCR) technique involves the addition of two or more primer pairs to the same reaction system, therefore, it enables rapid and simultaneous detection of multiple pathogens, with significant reduction in detection time and cost of material used [7, 31].
Based on our previous results [81], the highly pathogenic flora include: R. pseudosolanacearum, E. cloacae complex (ECC,E. asburia, E. roggenkampi, and E. cloacae), K. pneumoniae species complex (KpSC; K. quasipneumoniae, K. pneumoniae, and K. variicola), K. oxytoca complex (KoC; K. michiganensis, and K. oxytoca) and P. ananatis [81]. Therefore, in keeping with the foregoing evidence and to achieve the targeted detection of pathogenic bacteria, in this study we included important virulence-related genes (Hydrolase, effectors, drug resistance and diguanylate cyclase genes, etc.) of the pathogenic bacteria of plants, and established the mPCR assay to detect these bacteria. We believe that this mPCR assay will provide a reliable technical means for rapid diagnosis, prevention and control of mulberry bacterial wilt.
Results
Verification of 10 strains of mulberry bacterial wilt pathogens based on the Koch's postulates
The verification test results are shown in Fig. 1. The virulence level of each strain was observed as follows: XCqk = KQ-01 > YDKL > XCYG > AKKL > MBWS14. (14) > LCKL = MBWS6. (10) > MBWS31. (7) > LCFJ > CK (Fig. 1). The morbidity levels of KQ-01, XCqk, YDKL and XCYG were significantly higher (P < 0.05) than those of AKKL, MBWS14. (14), LCKL, MBWS6. (10) and MBWS31. (7). The morbidity level of LCFJ was significantly lower (P < 0.05) than that of the other strains. Meanwhile, all 10 strains of pathogenic bacteria were isolated from the diseased mulberry stems. While the mulberry seedlings in the control group showed no symptoms, and no bacteria were isolated from the mulberry stems. This finding indicated that these 10 strains of pathogenic bacteria (R. pseudosolanacearum XCqk, E. asburiae XCYG, E. roggenkampii KQ-01, E. cloacae MBWS6. (10), K. pneumoniae MBWS31. (7), K. variicola MBWS14. (14), K. quasipneumoniae YDKL, K. michiganensis AKKL, K. oxytoca LCKL and P. ananatis LCFJ) were all pathogenic bacteria of mulberry bacterial wilt.

Verification of 10 strains of mulberry bacterial wilt pathogens based on the Koch’s postulates. a The morbidity level of strains of mulberry bacterial wilt pathogens (KQ-01, XCqk, YDKL, XCYG, MBWS6. (10), MBWS14. (14), AKKL, LCKL, MBWS31. (7) and LCFJ) infecting mulberry seedlings at different points of time. b The state of mulberry seedlings infected with mulberry bacterial wilt pathogens (KQ-01, XCqk, YDKL, XCYG, MBWS6. (10), MBWS14. (14), AKKL, LCKL, MBWS31, (7) and LCFJ) after 12 days of inoculation. “d”: day. Bars with different letters indicate a significant difference between means by one-way analysis of variance (ANOVA) and least significant difference (LSD) tests (P < 0.05). Error bars indicate ± standard deviation. Morbidity level: 0: no symptoms; 1: one to two leaves yellow or withered; 2: 3–4 leaves yellow or withered; 3: all leaves yellow or withered; 4: whole plant died. In addition, the morbidity level 0 was represented as I; the morbidity level 0 ~ 1 was represented as HR; the morbidity level 1 ~ 2 was represented as MR; the morbidity level 2 ~ 3 was represented as MS; and the morbidity level 3 ~ 4 was represented as HS. XCqk: R. pseudosolanacearum XCqk; XCYG: E. asburiae XCYG; KQ-01: E. roggenkampii KQ-01; MBWS6. (10): E. cloacae MBWS6. (10); MBWS14. (14): K. variicola MBWS14. (14); MBWS31. (7): K. pneumoniae MBWS31. (7); YDKL: K. quasipneumoniae YDKL; AKKL: K. michiganensis AKKL; LCKL: K. oxytoca LCKL; LCFJ: P. ananatis LCFJ; CK: CK: control group (sterile MS medium)
Analysis of conservation of target genes based on NCBI datasets
After downloading the genome sequences of selected pathogenic bacteria, we compared them between the strains with the whole genome in the NCBI, and counted the distribution of different target genes in the strains. The results are shown in Table 1. The gene yjfP (esterase YjfP) was contained in 95.65% of E. asburiae, 100% of E. roggenkampii, 15.82% of E. cloacae, 11.06% of E. kobei and 0.08% of E. hormaechei. The pelY (periplasmic pectate lyase) gene was present in 99.31% of K. michiganensis and 87.37% of K. oxytoca. ampD (N-acetyl-dehydromuramoyl-L-alanine amidase) gene was present in 99.73% of K. quasipneumoniae, 24.77% of K. variicola, 0.71% of K. aerogenes and 0.10% of K. pneumoniae. 99.62% of P. ananatis contained pleD (diguanylate cyclase with sensor domain). ripW (type III effector RipW) gene was present in 85.93% of R. pseudosolanacearum, and 49.83% of R. solanacearum.
Using yjfP, pelY, ampD, pleD and ripW as target genes, this design was able to detect more than 85% of E. asburiae, E. roggenkampii, K. michiganensis, K. oxytoca, K. quasipneumoniae, P. ananatis, and R. pseudosolanacearum. This design had the ability to partly detect E. cloacae, E. kobei, K. variicola, and R. solanacearum, and could also detect a small number of E. hormaechei, K. aerogenes, and K. pneumoniae. However, it could not detect E. mori in the samples.
Specificity of mPCR primers
The pleD gene was used to detect P. ananatis; yjfP was used to detect ECC; pelY was used to detect K. michiganensis and K. oxytoca; ampD was used to detect K. pneumoniae, K. quasipneumoniae, and K. variicol; ripW was used to detect R. pseudosolanacearum. Primer-Blast was used to design the corresponding primers, and the primer specificity was evaluated (Table 2).
The primers were analyzed in Primer-Blast and the results are shown in Additional file 1: Figure S2. Briefly, EyjfPF/EyjfPR detected E. cloacae and E. roggenkampii in ECC (Additional file 1: Figure S2a). KpelYF/KpelYR detected K. michiganensis, K. oxytoca, and a small number of K. pasturii (Additional file 1: Figure S2b). KampDF/KampDR detected K. pneumoniae, and K. quasipneumoniae (Additional file 1: Figure S2c). RripwF/RripwR detected R. solanacearum (Additional file 1: Figure S2d). PapleDF/PapleDR detected P. ananatis (Additional file 1: Figure S2e).
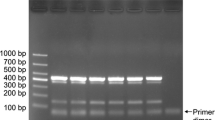
Next, the specificity of five pairs of primers was verified and all 10 target strains were detected effectively (see Fig. 2). In addition, no bands for other non-target bacteria were observed (Fig. 2: lanes 11–34). There was no amplification in the negative control group containing healthy mulberry DNA. These results show that the primers EyjfPF/EyjfPR, KampDF/KampDR, KpelYF/KpelYR, RripwF/RripwR and PpleDF/PpleDR designed in this study had reasonable specificity.

Gels representing result of analysis of mPCR primer specificity. M: 1000 marker; 1: R. pseudosolanacearum XCqk DNA; 2: E. asburiae XCYG DNA; 3: E. cloacae MBWS6. (10) DNA; 4: E. roggenkampii KQ-01 DNA; 5: K. quasipneumoniae YDKL DNA; 6: K. pneumoniae MBWS31. (7) DNA; 7: K. variicola MBWS14. (14) DNA; 8: K. michiganensis AKKL DNA; 9: K. oxytoca LCKL DNA; 10: P. ananatis LCFJ DNA; 11: P. samygadali pv. mori ZJDX-003 DNA; 12: Curtobacterium sp. YDDX-001 DNA; 13: B. subtilis YD-003 DNA; 14: P. aeruqinasa YD-001 DNA; 15: P. polymyxa YD-002 DNA; 16: P. mori L40-14 DNA; 17: Acinetobacter sp. KG-2 DNA; 18: Bacillus sp. ME-003 DNA; 19: Enterobacter sp. LJ-5 DNA; 20: Enterobacter sp. LJ-12 DNA; 21: Enterobacter sp. L40H-77 DNA; 22: Enterobacter sp. S2H-107 DNA; 23: Enterobacter sp. T10H-85 DNA; 24: Escherichia sp. L-G-93 DNA; 25: Glutamicibacter sp. KJ-15 DNA; 26: Mesorhizobium sp. KG-15 DNA; 27: Pantoea sp. L-G-95 DNA; 28: Pantoea sp. L40H-91 DNA; 29: Pantoea sp. S2H-99 DNA; 30: Pantoea sp. T10H-16 DNA; 31: Pantoea sp. L40H-43 DNA; 32: Pseudomonas sp. ME-001 DNA; 33: Staphylococcus sp. KG-23 DNA; 34: Stenotrophomonas sp. KG-24 DNA; NC: Healthy Mulberry samples DNA. “Pa”: P. ananatis; “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex
In summary, the primers designed in this study were effective in detecting the targeted pathogens of mulberry bacterial wilt including, R. pseudosolanacearum, E. cloacae complex (ECC; E. asburiae, E. roggenkampi, and E. cloacae), K. pneumoniae species complex (KpSC; K. quasipneumoniae, K. pneumoniae, and K. variicola), K. oxytoca complex (KoC; K. michiganensis, and K. oxytoca) and P. ananatis. Table 2 presents the information of the specific genes and primers used for PCR identification of ECC, KoC, KpSC, R. pseudosolanacearum and P. ananatis.
Establishment of optimal mPCR amplification system
Screening of optimal amount of primer
Five pairs of primers (RripwF/RripwR, EyjfPF/EyjfPR, KpelYF/KpelYR, KampDF/KampDR, and PpleDF/PpleDR) were subjected to an orthogonal experiment with five factors and four levels. The optimal addition amount of 16 combinations of primers was determined by scoring (Additional file 2: Table S1). The results (Fig. 3 and Additional file 1: Figure S3) revealed that the five pairs of primers had P values lesser than 0.05, indicating that this orthogonal design was reliable. The best combination (Compute group) of the five pairs of primers was as follows: RripwF/RripwR: 0.25 μM, EyjfPF/EyjfPR: 0.25 μM, KpelYF/KpelYR: 1 μM, KampDF/KampDR: 0.5 μM, PpleDF/PpleDR: 0.25 μM.

Screening of the best primers for mPCR assay. M: 1000 marker. 1–16: The results after mixing 5 pairs of primers (RripwF/RripwR, EyjfPF/EyjfPR, KpelYF/KpelYR, KampDF/KampDR, and PpleDF/PpleDR) according to primer group 1 to 16 and performing PCR amplification (see Additional file 2: Table S1); 17: Primer group 1; 18: Primer group 2; 19: Primer group 9; 20: Primer group 20; 21: Compute group. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex
To further validate the results, we compared the Compute group with the primer groups that scored higher than 90 in the orthogonal experiment. These included: primer group 1 (Fig. 3: lane 17), primer group 2 (Fig. 3: lane 18), primer group 9 (Fig. 3: lane 19), primer group 15 (Fig. 3: lane 20). The results are shown in Fig. 3 and Additional file 1: Figure S3: Lanes 17–21. The Compute group (Fig. 3: Lane 21) obtained the target bands at 929 bp, 624 bp, 476 bp, 357 bp and 258 bp, respectively. Except for K. quasipneumoniae YDKL, the amplified bands of R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. michiganensis AKKL, and P. ananatis LCFJ were clear and bright. Meanwhile, no amplification band was observed in the negative control (Additional file 1: Figure S3: lane 21). The comparison between these primer groups showed that the Compute group had the highest score of 95 points. These results further validated the optimal amount of primers used in this study.
Screening of optimal amount of 10 mM dNTP
Next, we evaluated the optimal amount of 10 mM dNTP in the mPCR reaction system (see Fig. 4a and Additional file 1: Figure S4a). The results showed that when the amount of 10 mM dNTP added was 0.4 μL (Fig. 4a: lane 3), five clear target bands appeared, and no false positives were seen (Additional file 1: Figure S4a: lane 3). When the amount of 10 mM dNTP was kept to 0 μL (Fig. 4a: Lane 1) and 0.2 μL (Fig. 4a: Lane 2), the five bright bands were still observed, but false positives were also seen (Additional file 1: Figure S4a: Lane 1 and 2). When the addition amount of 10 mM dNTP was kept to 0.6 μL, the amplification efficiency decreased significantly (Fig. 4a: lane 4). Likewise, when the added amount of 10 mM dNTP was increased to 0.8 μL and above, no amplification band was produced (Fig. 4a and Additional file 1: Figure S4a: lanes 5 ~ 11). Therefore, we selected 0.4 μL as an optimal addition amount of 10 mM dNTP.

Effect of different 10 mM dNTP (a), and 25 mM MgCl2 dNTP (b) addition amounts, amplification cycle numbers (c), annealing temperatures (d) on mPCR efficiency. Note: M: 1000 marker. a1-a11: 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 μL, 1.4, 1.6, and 2.0 μL of 10 mM dNTPs, respectively. b1-b11: Add 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 μL, 1.4, 1.6, and 2.0 μL of 25 mM MgCl2, respectively. c1-c6: represent cycle numbers: 24, 26, 28, 30, 32 and 34, respectively. d1 ~ d11: respectively, represent the annealing temperature: 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63 °C. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex
Screening of optimal amount of 25 mM MgCl2 dNTP
The results of screening of optimal amount of 25 mM MgCl2 in the mPCR reaction system are shown in Fig. 4b and Additional file 1: figure S4b. Briefly, it was observed that when the amount of 25 mM MgCl2 was kept to 0.4 μL (Fig. 4b: Lane 3), five clear target bands appeared, and the false positives were not obvious (Additional file 1: Figure S4b: Lane 3). However, when 25 mM MgCl2 was added at 0 μL (Fig. 4b: lane 1) and 0.2 μL (Fig. 4b: lane 2), the bands did appear but they were not bright. When the amount of 25 mM MgCl2 was increased to 0.6 μL and above, the false positive bands were obvious (Additional file 1: Figure S4b: lanes 4 ~ 11). Therefore, 0.4 μL was selected as an optimal addition amount of 25 mM MgCl2.
Optimization of mPCR amplification cycle numbers
The results of optimization of number of amplification cycles in the mPCR reaction system are shown in Fig. 4c and Additional file 1: figure S4c. The optimal results [5 clear and obvious bands (Fig. 4c: Lane 3) and no amplification band in the blank group (Additional file 1: Figure S4c: Lane 3)] were obtained when the number of cycles was kept to 28. The amplification bands were not bright enough, when the cycle number was kept to 24 (Fig. 4c: lane 1) and 26 (Fig. 4c: lane 2). When the number of cycles was kept to 30 and above, the false positives were more obvious (Additional file 1: Figure S4c: lanes 4–6). Therefore, 28 cycles were regarded as an optimal number for the mPCR assay.
Screening of optimal annealing temperature for mPCR reaction
The screening results of annealing temperature for the mPCR reaction are shown in Fig. 4. The optimal results were obtained when annealing temperature was set at 61 °C (Fig. 4d: Lane 9): 5 clear and most obvious bands appeared, and no amplification band was recorded in the blank group (Additional file 1: Figure S4d: Lane 9). However, when the annealing temperature was set to the range of 53–60 °C (Fig. 4d: lanes 1–8), the bands were not clear and bright enough. When the annealing temperature was raised to 62 °C (Fig. 4d: Lane 10) and 63 °C (Fig. 4d: Lane 11), the amplification efficiency started to decrease and the amplified bands began to darken. Therefore, 61 °C was selected as an optimal annealing temperature.
Practical/reproducibility testing of mPCR assay
Validation of optimal reaction system and amplification conditions for mPCR assay
To further validate the efficiency of the developed system, 10 ng/μL genomic DNA of R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCF was randomly combined in different proportions as templates. These templates were then detected by mPCR (Fig. 5a). The amplification results were consistent with the theoretical values, indicating that the developed mPCR assay can effectively detect different combinations of the target bacteria.

Practical and Reproducibility testing of mPCR assay. a Efficiency of different combinations of genomic DNA of five pathogenic bacteria used in mPCR; b mPCR detection limit of pathogenic bacteria in pure culture; c Anti-interference tests of mPCR detection of pathogenic bacteria with endophytic bacteria at different mixing proportion. M: 1000 Maker. a:1—a:18: mPCR detection results at different mixing proportion of DNA of R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCFJ. b1: 2.46 ng/1.87 × 106 CFU/mL; b2: 1.87 × 105 CFU/mL; b3: 1.87 × 104 CFU/mL; b4: 1.87 × 103 CFU/mL; b5; 1.87 × 102 CFU/mL;b 6: 1.87 × 101 CFU/mL; b7: 1.87 CFU/m; c1- c5: Enterobacter sp. LJ-12,; c6- c10: Pseudomonas sp. ME-001; c11- c15: Staphylococcus sp. KG-23; c16- c20: Bacillus sp. ME-003; c21- c25: Stenotrophomonas sp. KG-24; c26- c30: Acinetobacter sp. KG-2; the ratio of target bacteria to interfering bacteria at 1:102, 1:10, 1:1, 10:1, and 102:1. PC: template mixed in equal proportions (1:1:1:1:1) with E. asburiae XCYG, K. michiganensis AKKL, K. quasipneumoniae YDKL, R. pseudosolanacearum XCqk, and P. ananatis LCFJ genomes at 10 ng/μL as positive control. NC: the healthy mulberry genome as a negative control. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex
Detection limit of mPCR
The results of sensitivity of mPCR assay are shown in Fig. 5. Briefly, when the bacterial concentration was kept to 1.87 × 103 -1.87 × 107 CFU/mL (Fig. 5b: Lane 1–4), the bands of 929 bp (P. ananatis), 624 bp (K. michiganensis), 476 bp (ECC), 357 bp (R. pseudosolanacearum) and 258 bp (K. quasipneumoniae) were obtained. Therefore, it was noted that the lowest detection limit of mPCR for P. ananatis, KpSC, ECC, R. pseudosolanacearum and KoC was 1.87 × 103 CFU/mL (DNA = 2.45 pg/μL).
Interference testing of mPCR assay
The sensitivity of mPCR assay to non-target DNA interference was determined by mixing target pathogens (E. asburiae XCYG, K. michiganensis AKKL, K. quasipneumoniae YDKL, R. pseudosolanacearum XCqk, and P. ananatis) and non-target bacteria in different ratios (Fig. 5c). The mixture of all strains tested produced five clear bands whose brightness/intensity was comparable to those obtained by analyzing the pure cultures of the target bacteria. This result indicated that the presence of non-target endophytes Enterobacter sp. LJ-12, Pseudomonas sp. ME-001, Staphylococcus sp. KG-23, Bacillus sp. ME-003, Stenotrophomonas sp. KG-24, and Acinetobacter sp. KG-2 had no impact on detection of the target pathogenic bacteria.
Application of mPCR assay for analysis of natural mulberry wilt disease samples
Next, we compared the efficiency of the developed mPCR with the traditional triphenyl tetrazolium chloride (TTC) culture medium detection method. The results are shown in Fig. 6 and Additional file 2: Table S2. From 46 samples tested for the suspected mulberry bacterial wilt disease, the mPCR detection rates of Pantoea sp. (P. ananatis), Enterobacter sp. (E. cloacae complex, ECC), Ralstonia sp. (R. pseudosolanacearum), and Klebsiella spp. (KpSC, and KoC) were 8.69, 91.3, 65.21, and 41.3%, respectively (Additional file 2: Table S2 and Fig. 6). As for the Klebsiella spp., the detection rates of KpSC and KoC were 34.7 and 23.9%, respectively (Fig. 6). Whereas the detection rates of the pathogenic bacteria Pantoea sp., Enterobacter sp., Ralstonia sp., and Klebsiella spp. isolated from the traditional TTC culture media were 26.08, 86.95, 36.95, and 32.60%, respectively (Additional file 2: Table S2). These results showed a difference between the traditional TTC medium isolation and the mPCR assay.

mPCR-based detection of the suspected mulberry bacterial wilt in the field. M: 1000 Maker; 1 ~ 46: suspected mulberry bacterial wilt disease-like DNA in the field. PC: the template mixed in equal proportions (1:1:1:1:1) with E. asburiae XCYG, K. michiganensis AKKL, K. quasipneumoniae YDKL, R. pseudosolanacearum XCqk, and P. ananatis LCFJ genomes at 10 ng/μL as positive control; NC: the healthy mulberry genome as a negative control. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex
Application of mPCR assay for isolation of pathogenic bacteria
Using the mPCR assay, we further tested Ralstonia sp., Enterobacter sp., Klebsiella spp., and Pantoea sp. isolated from field samples of mulberry bacterial wilt. The pathogenicity test was also performed concurrently. The results are shown in Additional file 1: figure S5 and Additional file 2: Table S3. Briefly, a total of 12 strains of Ralstonia sp. were detected in this study. From these, 10 strains showed a 357 bp band by mPCR (Additional file 1: figure S5: Lane 1–12). The concurrent pathogenicity tests showed that the detected Ralstonia sp. caused morbidity of ≥ 70.5% to mulberry branches, while the undetected Ralstonia sp. caused 0% morbidity to mulberry branches. From 32 Enterobacter strains, 10 strains showed a 476 bp band when subjected to mPCR assay (Additional file 1: Figure S5: Lane 13–44). The morbidity rate caused by the detected Enterobacter sp. was > 65.5%, and the morbidity rate caused by the undetected Enterobacter sp. was ≤ 35.5%. From 13 Klebsiella strains, 8 strains showed 258 bp bands by mPCR, and 2 strains obtained 624 bp bands (Additional file 1: Figure S5: Lane 45–57). The morbidity caused by the detected Klebsiella spp. was ≥ 65.5%, while that of the undetected Klebsiella spp. was 0%. From 12 strains of Pantoea sp., one strain showed 929 bp band by mPCR (Additional file 1: Figure S5: Lane 58–69). The morbidity caused by the detected Pantoea sp. was ≥ 65.5%, while the morbidity of undetected ones was ≤ 16.5%. These results show that the mPCR assay developed in the present study can effectively detect the pathogenic bacteria causing mulberry wilt disease.
In summary, we designed an mPCR detection method based on important virulence-related genes of plant pathogenic bacteria: Hydrolase (Esterase yjfP and periplasmic pectate lyase pelY), effector (Type III effector ripW), drug resistance (N-acetyl-anhydromuranmyl-L-alanine amidase ampD) and virulence regulatory gene (GGDEF domain-containing protein pleD). The mPCR was performed in a 25 µL volume. The reaction mixture contained 12.5 µL 2 × Taq PCR Master Mix (Sangon Biotechnology (Shanghai) Co., Ltd.), primers (0.25 µM RripwF, 0.25 µM RripwR, 0.25 µM EyjfPF, 0.25 µM EyjfPR, 1 µM KpelYF, 1 µM KpelYR, 0.5 µM KampDF, 0.5 µM KampDR, 0.25 µM PpleDF, and 0.25 µM PpleDR), 1 µL of the above genomic DNA as template, and ddH2O was added to 25 µL. mPCR thermal cycle: 94 °C for 5 min, followed by 28 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, extension step at 72 °C for 10 min. The mPCR products were analyzed by electrophoresis on a 1.2% agarose gel at 110 V for 30 min under UV light irradiation. It can simultaneously detect ECC (476 bp), KoC (624 bp), KpSC (258 bp), R. pseudosolanacearum (357 bp), and P. ananatis (929 bp). The developed mPCR assay is summarized in graphical abstract.
Discussion
As a devastating disease, bacterial wilt is classified as an acute quarantine disease [16], which is difficult to control due to its complex etiology. Traditionally used isolation methods are tedious, time consuming and prone to human bias in the results [73]. At present, the molecular detection methods for mulberry bacterial wilt include PCR [71], RT‒PCR [47], LAMP [27] and IMSA-LAMP [80]. However, all of these methods can only detect one pathogen at a time. In keeping with this operational limitation, there is a dire need to develop a rapid and effective method to simultaneously detect the complex pathogens of mulberry bacterial wilt. The mPCR molecular assays using more than one pair of primers are becoming increasingly popular, as they can detect multiple targets simultaneously in a single PCR [17]. The mPCR-based species identification methods have been developed for many organisms, including nematodes [29], fungi [64], bacteria [37], and viruses [28]. The development of mPCR in the present study provides rapid and cost-effective means for simultaneous detection of the five mulberry bacterial wilt pathogens including P. ananatis, ECC, KpSC, KoC and R. pseudosolanacearum.
From these five pathogens of mulberry bacterial wilt, in addition to R. pseudosolanacearum, P. ananatis [11], ECC [62], KpSC [22, 57, 65], and KoC [22, 57, 65] have all been reported to cause human diseases. They are human opportunistic pathogens known for their high drug resistance and play complex roles in ecology. P. ananatis, ECC, KpSC, and KoC are emerging pathogens that, unlike most plant pathogenic microorganisms, can also infect humans [8, 19]. The pathogens such as P. ananatis, ECC, KpSC, and KoC can not only infect humans and plants as opportunistic pathogens, but also act as beneficial bacteria that promote plant growth and disease resistance [35, 36, 48, 50, 63].
The phenomenon by which these microorganisms can change their lifestyles from pathogen to endophytes and vice versa are much complex than they appear to be. Comparative genomic studies have revealed that the colonization mechanisms used by endophytes are also the same as used by plant pathogens [46, 58]. Interestingly, besides their beneficial roles in plant health, endophytes can increase the severity of disease in plants, raising new questions regarding plant disease ecology and management [5]. Recently, the endophytic Pantoea agglomerans (DAPP-PG 734) was reported to indirectly enhance the virulence of P. savastanoi pv. savastanoi (DAPP-PG 722) in olive knots, potentially through its active hypersensitive response and pathogenicity (Hrp) type III secretion system (T3SS) [59]. This evidence raises new questions for researchers to understand the role of endophytes in plant–pathogen interactions. It has been said that harmless bacteria acquire virulence traits through horizontal gene transfer (discussed in [46]). It is also believed that the disease process can also be triggered by the interaction between microbial communities [68]. A decade ago, an emerging idea of “pathobiome” was proposed, pointing to the paradigm shift from “pathogens” to “pathobiome [70]. It was argued that the scenario “a microbe + virulence factors = disease” is probably far from reality. This was largely based on the argument that “actual pathogens have extremely broad biological diversity and that their pathogenicity is an outcome of strong and highly specific interactions with their microbial environment and hosts” [70]. In support of this, recently it was shown that the endophytic bacteria including several strains of Enterobacter and Pantoea genera share a wide number of virulence-related genes within their genomes and that the distribution of gene patterns were comparable to that of pathogenic strains [46]. It was also proposed that the endophytic and pathogenic lifestyles are composed of a base core virulence genome that might be used and expressed differentially. Unfortunately, so far our understanding of these virulence-related complex interactions is still poor, and many outstanding questions need to be resolved to draw a definitive line between pathogens and endophytes [46].
As discussed above, mulberry bacterial wilt pathogens P. ananatis, ECC, KpSC, and KoC also play complex roles in plant ecology. In the present study, we therefore selected several virulence-related genes related to these bacteria as targets to develop a rapid, efficient, and cost-effective molecular detection (mPCR) technique. Currently, the targets of nucleic acid detection methods for mulberry bacterial wilt are LpxC [71], ropB [47], and MG67 [27] genes. In the present study, we selected esterase YjfP synthesis yjfP (CP058637.1) gene to detect ECC (E. asburiae, E. roggenkampi, and E. cloacae), periplasmic pectate lyase synthesis pelY (CP060111.1) gene for KoC (K. michiganensis and K. oxytoca), N-acetyl-anhydromuranmyl-L-alanine amidase ampD (CP068237.1) gene for KpSC (K. quasipneumoniae, K. pneumoniae and K. variicola), Type III effector RipW synthetic ripW (WP_197360060.1) gene for R. pseudosolanacearum and GGDEF domain-containing protein pleD (CP066803.1) gene for P. ananatis.
In this case, given the complexity of the casual pathogens of mulberry bacterial wilt disease, we conducted several tests on collected samples to ensure that the detected bacteria are pathogenic. Meanwhile, to verify the feasibility of the selected target genes, we conducted mPCR assay using the target pathogenic strains and some endophytic strains. The results showed that healthy plant-derived Enterobacter sp. (LJ-5, LJ-12, L40H-77, S2H-107, T10H-85), and Pantoea sp. (L-G-95, L40H-91, S2H-99, T10H- 16, L40H-43) and other non-target strains could not be detected (see Fig. 2). In addition, we also performed mPCR assay on isolated Ralstonia sp., Enterobacter sp., Klebsiella spp., and Pantoea sp. We found that the morbidity caused by the pathogenic bacteria detected by mPCR was ≥ 65.5%, while the morbidity of the undetected pathogenic bacteria was ≤ 35.5%. In addition, during pathogenicity test, Ralstonia was found to be the most pathogenic, followed by Enterobacter, Klebsiella, while Pantoea was the least pathogenic. This finding is in conformity with our recent study [81]. This shows that the target genes selected in this study and the primers designed can specifically detect the pathogenic bacteria of mulberry wilt including, R. pseudosolanacearum, P. ananatis, ECC, KpSC, and KoC.
In the present study, P. ananatis, ECC, KpSC, KoC and R. pseudosolanacearum were detected at a lowest concentration of 1.87 × 103 CFU/mL (DNA = 2.45 pg/μL) (Fig. 5). Previously, bacterial pathogens P. etewartii and Rathayibacter tritici were detected in grains using PCR with a minimum detection limit of 8.8 × 103 CFU/mL and DNA = 2 pg/μL [3]. Our result showed that the minimum detection limit of mPCR assay developed in the present study was closer to that of the previously reported PCR detection methods [3]. In addition, Enterobacter sp. LJ-12, Pseudomonas sp. ME-001, Staphylococcus sp. KG-23, Bacillus sp. ME-003, Stenotrophomonas sp. KG-24, and Acinetobacter sp. KG-2, endophytic strains of healthy mulberry, did not affect the detection of target strains, indicating that the mPCR assay developed in the present study was reliable. This result is supported by a recent study [41] in which it was reported that the non-target Listeria strains (S. enteritidis CMCC 50335, S. aureus ATCC 25923, and E. coli ATCC 25922) did not interfere with the detection of target pathogenic Listeria spp. while using the mPCR assay.
In the present study, subtle differences were observed between the results of the traditional TTC culture medium isolation and mPCR detection of pathogens, when 46 suspected mulberry wilt samples collected in the field were tested. This result perhaps points to the underlying shortcoming of the traditional TTC culture isolation method, which has significant limitations [9]. For example, R. pseudosolanacearum can only be isolated from fresh samples [69]. Both methods showed that ECC and R. pseudosolanacearum were the main pathogenic groups of bacterial wilt of mulberry, followed by Klebsiella (KpSC and KoC), and P. ananatis [74, 75]. This shows that the mPCR molecular detection method is reliable, has short operation time and is highly efficient. Meanwhile, the detection rates of R. pseudosolanacearum, ECC, KpSC, KoC, and P. ananatis were found to be 83.33, 31.23, 61.53, 15.38, and 8.3%, respectively. This finding indicates that R. pseudosolanacearum is an important pathogen causing bacterial wilt of mulberry, while ECC, KpSC, KoC, and P. ananatis might be opportunistic pathogens. This further augments our recently published study [81], in which we have reported a similar finding.
At present, mulberry is also used as food for humans. Its leaves and fruits are used as food, and its roots and bark are used as medicine [13, 26, 55]. While P. ananatis, ECC, KpSC, and KoC are considered as opportunistic pathogens of mulberry, these pathogenic bacteria are also opportunistic pathogens of humans, and have antimicrobial resistance (AMR) [18]. It remains to be explored whether consumption of mulberry products, particularly raw products, contaminated with such antibiotic-resistant bacteria (ARB) can lead to the direct acquisition of ARB and the spread of resistant bacteria in the food chain [18]. But, evidence shows that the plant endophytic bacteria, as opportunistic pathogens, can as well be pathogenic to mammalian hosts. For instance, Pseudomonas aeruginosa BP35, an endophyte of black pepper, exhibited resistance to antibiotics and was pathogenic in a murine model, with a similar level of virulence to its clinical strain [38]. This finding raised a significant concern related to human food safety, particularly related to use of endophytic bacteria in sustainable plant protection strategies [38]. Given that mulberry products and byproducts are extensively consumed by humans, the mPCR assay developed in the present study could be useful in rapid detection of these complex pathogenic bacteria, which might as well have a potential threat to food safety in cultural settings where mulberry is consumed by humans.
Conclusion
In this study, using five virulence-related genes of bacteria as target, we developed an mPCR assay that showed high specificity, reproducibility, and sensitivity during testing. The assay results showed a comparable trend as those of the traditional methods of pathogen isolation. The mPCR assay developed in the present study can be used for rapid, sensitive, and simultaneous molecular detection of five virulence-related genes carried by different pathogens of mulberry wilt disease including P. ananatis, ECC, KpSC, KoC and R. pseudosolanacearum.
Material and methods
Sterile plant material and growing conditions
In this study, aseptic seedlings were obtained by cultivating Guisang 12 (M. atropurpurea) seeds, which were then used in subsequent experiments. The cultivation process for sterile mulberry seedlings is depicted in Additional file 1: figure S6. Briefly, Guisang 12 seeds were initially treated with 75% alcohol for 1 min, rinsed six times with sterile water, treated with 0.05% mercuric chloride for 10 min, and finally rinsed six times with sterile water. Seeds were planted on sterile MS sugar agar medium [60]. The seedlings were then grown for 60 days in an artificial climate of 12 h/d light and 25 °C. The sterile mulberry seedlings lacked bacterial and fungal growth in the culture bottles [79, 81, 82].
Cultivation of mulberry branches
Healthy 15-year-old M. atropurpurea cultivar Lun40 (susceptible to bacterial wilt) obtained from the South China Agricultural University mulberry field (Guangzhou, Guangdong, China; 113.35º, 23.17º) was used as plant material in this study. The samples were collected in May 2023. The old branches of Lun40 with a diameter of 0.5–0.75 cm were selected and cut into stem segments of 10–12 cm in length and containing three lateral shoots. The stems were washed with soapy water to remove surface dust and soaked in 0.5% sodium hypochlorite solution for five hours. The stems were then put into the sterile MS liquid medium and placed in an artificial climate incubator at 25 °C, 12 h/d light, and 85% humidity. The culture was incubated for 25 days until the lateral shoots sprouted and exhibited 2–3 leaves. During this period, the sterile MS liquid medium was changed every day [81, 82].
Verification by Koch’s postulates
Ten (10) pathogenic bacteria were identified in our laboratory: R. pseudosolanacearum XCqk, E. asburiae XCYG, E. roggenkampii KQ-01, E. cloacae MBWS6. (10), K. pneumoniae MBWS31. (7), K. variicola MBWS14. (14), K. quasipneumoniae YDKL, K. michiganensis AKKL, K. oxytoca LCKL and P. ananatis LCFJ (Additional file 2: Table S4). Ten strains of pathogenic bacteria were placed in casamino acid, peptone, and glucose (CPG) broth [34], and 12 h of shaking at 28 °C and 140 rpm yielded a bacterial suspension. The bacterial suspension was diluted to OD600 nm value of 0.1 (1 × 108 CFU/mL) with MS inorganic salt liquid medium. Five mL of the diluted bacterial suspension was added to sterile mulberry seedlings. They were then placed in an artificial climate incubator at 28 °C with 12 h of light every day and grown for 12 days. In addition, a control group containing only MS inorganic salt sterile liquid medium was established. Twenty sterile mulberry seedlings were used in each group (Additional file 1: figure S6) [81, 82].
The level of disease of mulberry seedlings in each group was recorded at 0 and 30 min, and days 1, 3, 6, 9, 12. On day 12, the stems of mulberry seedlings in each group were removed for aseptic grinding, and then separated according to the dilution coating plate method. The mulberry seedlings cultured by the sterile MS were used as the control group. If bacteria were isolated again from the diseased mulberry seedlings, it proved that those bacteria were pathogens of mulberry bacterial wilt. While if they did not produce the disease symptoms in mulberry seedlings and neither they were isolated from mulberry stems, those bacteria were not regarded to be pathogens of mulberry bacterial wilt (Additional file 1: Figure S6) [81, 82].
Morbidity level was scored as follows: 0: no symptoms; 1: one to two leaves faded or withered; 2: three to four leaves became yellow or withered; 3: all leaves withered or turned yellow; 4: the entire plant died. In addition, the morbidity level 0 was represented as I, 0–1 was represented as HR, 1–2 was represented as MR, 2–3 was represented as MS, and 3–4 was represented as HS [39, 81, 82].
Average morbidity level = (∑A)/20; A: the morbidity level value of mulberry seedlings [39, 81, 82].
Extraction of bacterial and plant genomic DNA
Bacterial strains used in the present study were obtained from the Guangdong Provincial Key Lab of Agro-Animal Genomics and Molecular Breeding, College of Animal Science, South China Agricultural University (Additional file 2: Table S4). For bacterial genomic DNA extraction, all strains were inoculated in nutrient broth (NB) medium (Guangdong Huankai Co., Ltd., Guangzhou, China) and incubated for 12–24 h at 28 °C. Bacterial genomic DNA was extracted using the Ezup Column Bacteria Genomic DNA Purification Kit (Sangon Biotech (Shanghai) Co., Ltd. China). Plant genomic DNA was extracted using Dzup (Plant) genomic DNA isolation reagent (Sangon Biotech (Shanghai) Co., Ltd. China) according to the manufacturer’s instructions. Extracted DNA was stored at − 20 °C for PCR analysis [81, 82].
Screening of specific target genes and primers design
A total of 22,913 genome sequences of pathogenic bacteria were downloaded from the National Library of Medicine (NCBI, https://www.ncbi.nlm.nih.gov/; accessed on January 20, 2023) and analyzed. These included ECC (276 E. asburiae, 335 E. cloacae, 2267 E. hormaechei, 244 E. kobei, 30 E. mori, 317 E. roggenkampii), KoC (442 K. michiganensis, and 293 K. oxytoca), KpSC (16833 K. pneumoniae, 377 K. quasipneumoniae, and 440 K. variicola), 418 K. aerogenes, RSSC (64 R. pseudosolanacearum, and 309 R. solanacearum), and 269 P. ananatis. The specific information for the sequences is provided in Additional file 2: Table S5. We then selected specific plant-associated pathogenic genes that can identify R. pseudosolanacearum XCqk, E. asburiae XCYG, E. roggenkampii KQ-01, E. cloacae MBWS6. (10), K. pneumoniae MBWS31. (7), K. variicola MBWS14. (14), K. quasipneumoniae YDKL, K. michiganensis AKKL, K. oxytoca LCKL and P. ananatis LCFJ, including: pleD (GGDF structural domain-containing protein), yjfP (esterase), pelY (peripheral pectate lyase), ampD (N-acetyl-anhydromuranmyl-L-alanine amidase) and ripW (type III effector). The conservation of a gene in the target strain was judged according to the following criteria: the target gene nucleic acid sequence identifies > 85% and the coverage > 90% of the target strain [41].
Then, the target gene sequences were obtained using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) to design species-specific primers for target genes, and Oligo 7.0 software was used for primer evaluation (Table 1). For species-specific targets, the candidate primers were evaluated using the Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) database [41]. The candidate primer sets were synthesized by Sangon Biotechnology (Shanghai, China) Co., Ltd. Primer specificity was tested by performing PCR analysis on strains maintained in our laboratory (Additional file 1: Table S4: 1–34), and healthy mulberry genomic DNA was used as a negative control.
The mPCR was performed in a volume of 25 µL. The reaction mixture contained 12.5 µL 2 × Taq PCR Master Mix (Sangon Biotech (Shanghai Co., Ltd. China), 0.5 µM each of the primers (RripwF, RripwR, EyjfPF, EyjfPR, KpelYF, KpelYR, KampDF, KampDR, PpleDF, and PpleDR), 1 μL of the above genomic DNA as a template, and ddH2O added to 25 μL. mPCR thermal cycling involved an initial denaturation step at 94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, and a final extension step at 72 °C for 10 min. The mPCR products were analyzed after electrophoresis on a 1.2% agarose gel under ultraviolet light illumination. The obvious band at the target proved that the bacteria were detected specifically, while the absence of bands proved that the bacteria were not detected [81, 82].
Evaluation of optimal reaction system for mPCR
Five primer pairs were amplified with the highest efficiency in the same amplification system. 10 ng/μL DNA of R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCFJ in equal proportions (1:1:1:1:1) were used as template mix to explore the optimal reaction conditions for mPCR. In the above PCR amplification system, the amount of primers, the amount of Mg2+ added, the amount of dNTP added, the reaction annealing temperature and the amplification cycles were optimized [41].
To explore the optimal addition amount of five pairs of primers, we used an orthogonal experiment design consisting of five factors and four levels (Table 3). Based on this design, a total of 16 sets of primers were prepared and added to the reaction system and 1 μL template mix was taken for mPCR reaction. For other reaction systems and amplification conditions, see sub-heading “Screening of specific target genes and primer design”.
After electrophoresis, the clarity of each band in gel imaging was used as the evaluation standard. Evaluation criteria were as follows: each pair of primers scored 5 points for each band, 5 points each for the correct size of the band fragments, 5 points each for clear and bright bands, 5 points each if no false positives were recorded. This meant that for each pair of primers we had 20 points and that the five pairs of primers had a total score of 100 points. The added amount of primers in each group was scored based on the total score of bands amplified by the corresponding primers. Each set of experiments was independently repeated three times, and then the total score of each set of primer additions was analyzed using Statistical Package for the Social Sciences (SPSS) software version 25.0 (IBM Corp., Armonk, NY). The optimal addition amount of each pair of primers was calculated and then the verification experiment was conducted again. Finally, the primer addition amount with the highest score was used as the optimal addition amount [42].
After determining the optimal primer volume, we tested different 10 mM dNTP addition volumes (0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, and 2.0 μL), different amounts of 25 mM Mg2+ added (0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, and 2.0 μL), different annealing temperatures (50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60 and 61 °C), and different amplification cycle numbers (24, 26, 28, 30, 32, and 34 cycles) to get their optimal values. The PCR products were detected by 1.2% agarose gel electrophoresis, and the optimal parameters were selected in a fully automatic digital gel imaging system according to the brightness, clarity, and presence or absence of false positives of the bands [41].
After determining the optimal mPCR reaction system and conditions, we further verified them for accuracy. Firstly, 10 ng/μL of 10 ng/μL DNA of R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCFJ was mixed in different combinations and ratios as a template, then mPCR was used for detection of these templates to verify the accuracy and practicability of the assay [41].
mPCR practical testing
To validate the accuracy and scope for interference in the mPCR assay, Enterobacter sp. LJ-12, Pseudomonas sp. ME-001, Staphylococcus sp. KG-23, Bacillus sp. ME-003, Stenotrophomonas sp. KG-24, and Acinetobacter sp. KG-2, the endophytes of mulberry, were used as interference strains. Meanwhile, R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCFJ mixed in equal proportions (1:1:1:1:1) were used as pathogen mix cultures. The strains were cultured in TTC broth overnight and serially diluted (tenfold) with normal saline. The density of bacterial strains was adjusted to 106 CFU/mL. The pathogen mix cultures were individually mixed with the interference testing strain in ratios of 1:102, 1:10, 1:1, 10:1, and 102:1. The genomic DNA was extracted from the mixtures. Meanwhile, the genomic DNA from pathogen mix cultures without interference strains was used as a template for the positive control. The scope of mPCR assay to overcome interference was evaluated by 1.2% agarose gel electrophoresis [41].
Evaluation of detection limit of mPCR assay
R. pseudosolanacearum XCqk, E. roggenkampii KQ-01, K. quasipneumoniae YDKL, K. michiganensis AKKL, and P. ananatis LCFJ grown overnight in TTC medium were diluted to OD600 nm = 0.1 with sterile water. After mixing the diluted bacterial suspension in equal proportions (1:1:1:1:1) as the target bacterial mixture, sterile water was used to dilute the mixed bacterial suspension 8 times according to the tenfold dilution method. Then, 1 mL of mixed bacterial suspension of each gradient was taken as a detection object to test the detection limit of mPCR. The total DNA of mixed bacterial suspension was extracted according to the strain genomic DNA extraction method, and finally the concentration of the extracted total DNA was detected using an ultramicron spectrophotometer. Two microliters of each were added to the optimal reaction system, and the reaction was carried out under the optimal amplification conditions. The sensitivity of mPCR was determined by the presence or absence of the target band using the automated digital gel imaging system [41].
Detection of suspected mulberry bacterial wilt disease samples by mPCR
A total of 46 samples of the suspected mulberry bacterial wilt disease were collected from Guangdong and Guangxi provinces of China. Bacterial strain and plant genomic DNA extraction kits were used to extract the genomic DNA of the sample, and 1 μL of genomic DNA liquid from the extracted sample for mPCR detection. Template mix was used as a positive control and the genomic DNA of healthy mulberry was used as a negative control. After gel electrophoresis, the position of the amplified band was evaluated. At the same time, 1 g of severely diseased parts of the roots was taken from each diseased sample, and the bacteria were isolated from the tissue by traditional methods for simultaneous detection. The 16S rDNA of the isolated strain was amplified with 27F/1492R, the PCR products were sent to Sangon Biotech, Shanghai, China for sequencing. The NCBI-BLAST was used to analyze the sequencing results [76]. All bacterial 16S rDNA sequences generated in this study have been submitted to the NCBI. The accession numbers OP989957-OP990607 are bacterial 16S rDNA sequences derived from the diseased samples.
mPCR detection and pathogenicity test of pathogenic bacteria isolated from mulberry bacterial wilt disease samples
To further verify the correlation between bacteria detected by the mPCR and their pathogenicity, 12 strains of Ralstonia, 32 strains of Enterobacter, 13 strains of Klebsiella and 12 strains of Pantoea isolated from 46 samples were extracted for mPCR detection.
A total of 69 bacterial strains were simultaneously tested for pathogenicity (Additional file 2: Table S4: 1–10 and 35–93). To investigate the pathogenicity of Ralstonia, Enterobacter, Klebsiella, and Pantoea, the following procedures were adopted: (1) the pure cultures of all the bacteria (Additional file 2: Table S4: 1–10 and 35–93) were placed in nutrient broth medium overnight. The overnight cultured bacterial solution was adjusted to OD600 nm = 0.1 with sterile MS liquid medium. (2) After cultivation, the Lun40 mulberry branch with 2 ~ 3 leaves was placed into the diluted bacterial solution. The sterile MS liquid medium was set as a healthy group. (3) The inoculated Lun40 mulberry branches were cultured for 12 days in an artificial climate incubator at 28 °C, 12 h/d light, and 85% humidity, and the disease incidence in plants was observed. (4) Morbidity rate = (A−B)/C × 100%. A: the total number of diseased mulberry branches in the experimental group; B: the total number of diseased mulberry branches in the control group; C: the total number of mulberry branches [81, 82].
Analysis of variance (ANOVA) was performed using Excel software. Each set of experiments for each pathogen species was tested using six healthy mulberry branches. With the sterile MS liquid medium as a control, each group had three replicates [81, 82].
Data analysis
The Statistical Package for the Social Sciences (SPSS) software version 25.0 (IBM Corp., Armonk, NY) was used for analyses. Data analysis was performed using a one-way analysis of variance (Levene’s test was used to evaluate the equality of variance before analysis), and the least significant difference test was used to determine the significant difference between the means as a post hoc analysis. P < 0.05 was considered as significant difference. Data were analyzed using Excel 2016 software (Microsoft, Redmond, WA, USA) and Origin 2019b 64 bit.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional information files.
References
Ali S, Saha S, Kaviraj A. Fermented mulberry leaf meal as fishmeal replacer in the formulation of feed for carp Labeo rohita and catfish Heteropneustes fossilis-optimization by mathematical programming. Trop Anim Health Prod. 2020;52:839–49.
Asselin JAE, Bonasera JM, Helmann TC, Beer SV, Stodghill PV. Complete genome sequence resources for the onion pathogen, Pantoea ananatis OC5a. Phytopathology. 2021;111:1885–8.
Baek KY, Lee HH, Son GJ, Lee PA, Roy N, Seo YS, Lee SW. Specific and sensitive primers developed by comparative genomics to detect bacterial pathogens in grains. Plant Pathol J. 2018;34:104–12.
Basavand E, Khodaygan P, Rahimian H, et al. Identification and characterization of Klebsiella oxytoca strains associated with wetwood disease of Morus trees. Ind Phytopathol. 2021;74:1123–7.
Busby PE, Peay KG, Newcombe G. Common foliar fungi of Populus trichocarpa modify Melampsora rust disease severity. New Phytol. 2016;209(4):1681–92.
Chandrashekar BS, Prasannakumar MK, Puneeth ME, Teli K, Priyanka K, Mahesh HB, Desai RU. First report of bacterial soft rot of carrot caused by Klebsiella variicola in India. New Dis Rep. 2018;37:21.
Claustres M, Kjellberg P, Desgeorges M, Bellet H, Sarda P, Bonnet H, Boileau C. Détéction des délétions par amplification d’exons (PCR multiplex) dans la myopathie de Duchenne [Detection of deletions by the amplification of exons (multiplex PCR) in Duchenne muscular dystrophy]. J Genet Hum. 1989;37:251–7.
Coutinho TA, Venter SN. Pantoea ananatis: an unconventional plant pathogen. Mol Plant Pathol. 2009;10:325–35.
Crisafi F, Denaro R, Genovese M, Cappello S, Mancuso M, Genovese L. Comparison of 16SrDNA and toxR genes as targets for detection of Vibrio anguillarum in Dicentrarchus labrax kidney and liver. Res Microbiol. 2010;162:223–30.
Dai F, Luo G, Wang Z, Li L, Tang C, Kuang Z, Zhao X, Ye M, Xiao J. Isolation and molecular identification of the pathogen causing mulberry wilt disease in Southern sericultural areas of China. Science of Sericulture. 2012;2011(38):981–7.
De Baere T, Verhelst R, Labit C, Verschraegen G, Wauters G, Claeys G, Vaneechoutte M. Bacteremic infection with Pantoea ananatis. J Clin Microbiol. 2004;42(9):4393–5.
De Maayer P, Chan WY, Venter SN, Toth IK, Birch PRJ, Joubert F, Coutinho TA. Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J Bacteriol. 2010;192:2936–7.
de Pádua LK, Rabelo ACS, Araújo CM, Brandão GC, de Souza GHB, da Silva RG, de Souza DMS, Talvani A, Bezerra FS, Cruz Calsavara AJ, Costa DC. Anti-inflammatory and antioxidant properties of black mulberry (Morus nigra L.) in a model of LPS-induced sepsis. Oxid Med Cell Longev. 2018;2018:5048031.
Dhanyalakshmi KH, Nataraja KN. Mulberry (Morus spp.) has the features to treat as a potential perennial model system. Plant Signal Behav. 2018;13:e1491267.
Dong Z, Guo Y, Yu C, Zhixian Z, Rongli M, Deng W, Li Y, Hu X. The dynamics in rhizosphere microbial communities under bacterial wilt resistance by mulberry genotypes. Arch Microbiol. 2021;203(3):1107–21.
EFSA Panel on Plant Health (EFSA PLH Panel); Bragard C, Dehnen-Schmutz K, Di Serio F, Gonthier P, Jaques Miret JA, Justesen AF, MacLeod A, Magnusson CS, Milonas P, Navas-Cortes JA, Parnell S, Potting R, Reignault PL, Thulke HH, Van der Werf W, Vicent Civera A, Yuen J, Zappalà L, Van der Wolf J, Kaluski T, Pautasso M, Jacques MA. Pest categorisation of the Ralstonia solanacearum species complex. EFSA J. 2019;17(2):e05618.
Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE. Multiplex PCR: optimization and application in diagnostic virology. Clin Microbiol Rev. 2000;13(4):559–70.
Esteban-Cuesta I, Dorn-In S, Drees N, Hölzel C, Gottschalk C, Gareis M, Schwaiger K. Antimicrobial resistance of E. cloacae complex isolates from the surface of muskmelons. Int J Food Microbiol. 2019;301:19–26.
Feng S, Guo Z, Mai Y, Mo Q, Liang J, Qi X, Yue S, Wang G. First Report of Enterobacter roggenkampii and E. cloacae complex sp. Caused tomato pith necrosis in Guangdong province, China. Plant Dis. 2023;107:2212.
Fulton J, Bec S, Fayette J, Ploetz R, Harmon C. First report of plantain soft rot caused by Klebsiella variicola in Haiti. Plant Dis. 2020;104(6):1851.
García-González T, Sáenz-Hidalgo HK, Silva-Rojas HV, Morales-Nieto C, Vancheva T, Koebnik R, Ávila-Quezada GD. Enterobacter cloacae, an Emerging Plant-Pathogenic Bacterium Affecting Chili Pepper Seedlings. Plant Pathol J. 2018;34(1):1–10.
Ghasemian A, Mohabati Mobarez A, Najar Peerayeh S, Talebi Bezmin Abadi A, Khodaparast S, Mahmood SS. Expression of adhesin genes and biofilm formation among Klebsiella oxytoca clinical isolates from patients with antibiotic-associated haemorrhagic colitis. J Med Microbiol. 2019;68(7):978–85.
Guérin F, Lallement C, Goudergues B, Isnard C, Sanguinetti M, Cacaci M, Torelli R, Cattoir V, Giard JC. Landscape of in vivo fitness-associated genes of E. cloacae complex. Front Microbiol. 2020;11:1609.
Hofmeister DL, Thoden JB, Holden HM. Investigation of a sugar N-formyltransferase from the plant pathogen Pantoea ananatis. Protein Sci. 2019;28(4):707–16.
Hossain S, Khan AA, Rahman MM, Iiyama K, Furuya N. First Report of Soft Rot Disease of Papaya Caused by Klebsiella variicola in Bangladesh. J Fac Agric Kyushu Univ. 2018;63(2):201–5.
Hu QR, Lai PW, Chen F, Yu YF, Zhang B, Li H, Liu R, Fan Y, Deng ZY. Whole mulberry leaves as a promising functional food: From the alteration of phenolic compounds during spray drying and in vitro digestion. J Food Sci. 2022;87(3):1230–43.
Huang W, Zhang H, Xu J, Wang S, Kong X, Ding W, Xu J, Feng J. Loop-Mediated Isothermal Amplification Method for the Rapid Detection of Ralstonia solanacearum Phylotype I Mulberry Strains in China. Front Plant Sci. 2017;8:76. Erratum in: Front Plant Sci. 2017;8:325.
Huang HS, Tsai CL, Chang J, Hsu TC, Lin S, Lee CC. Multiplex PCR system for the rapid diagnosis of respiratory virus infection: systematic review and meta-analysis. Clin Microbiol Infect. 2018;24(10):1055–63.
Huang D, Yan G, Gudmestad N, Ye W, Whitworth J, Frost K, Crow W, Hajihassani A. Developing a one-step multiplex PCR assay for rapid detection of four stubby-root nematode species, Paratrichodorus allius, P. minor, P. porosus, and Trichodorus obtusus. Plant Dis. 2019;103(3):404–10.
Huang M, He P, Munir S, Wu Y, Li X, He P, He Y. Ecology and etiology of bacterial top rot in maize caused by Klebsiella pneumoniae KpC4. Microb Pathog. 2020;139:103906. https://doi.org/10.1016/j.micpath.2019.103906.
Imbert S, Portejoie L, Pfister E, Tauzin B, Revers M, Uthurriague J, Hernandez-Grande M, Lafon ME, Jubert C, Issa N, Dumas PY, Delhaes L. A Multiplex PCR and DNA-sequencing workflow on serum for the diagnosis and species identification for invasive Aspergillosis and Mucormycosis. J Clin Microbiol. 2023;61(1): e0140922.
Jiang G, Wei Z, Xu J, Chen H, Zhang Y, She X, Macho AP, Ding W, Liao B. Bacterial wilt in China, history, current status, and future perspectives. Front Plant Sci. 2017;8:1549.
Keith RC, Nishijima KA, Keith LM, Fitch MM, Nishijima WT, Wall MM. Atypical internal yellowing of papaya fruit in Hawaii caused by E. sakazakii. Plant Dis. 2008;92:487.
Kelman A. The relationship of pathogenicity in Pseudomonas solanacearum to colony appearance on a tetrazolium medium. Phytopathology. 1954;44(12):693–5.
Khalifa AY, Alsyeeh AM, Almalki MA, Saleh FA. Characterization of the plant growth promoting bacterium, Enterobacter cloacae MSR1, isolated from roots of non-nodulating Medicago sativa. Saudi J Biol Sci. 2016;23(1):79–86.
Kim HJ, Lee JH, Kang BR, Rong X, McSpadden Gardener BB, Ji HJ, Park CS, Kim YC. Draft genome sequence of Pantoea ananatis B1–9, a nonpathogenic plant growth-promoting bacterium. J Bacteriol. 2012;194(3):729.
Kini K, Agnimonhan R, Dossa R, Silué D, Koebnik R. Genomics-Informed multiplex PCR scheme for rapid identification of rice-associated bacteria of the genus Pantoea. Plant Dis. 2021;105:2389–94.
Kumar A, Munder A, Aravind R, et al. Friend or foe: genetic and functional characterization of plant endophytic Pseudomonas aeruginosa. Environ Microbiol. 2013;15(3):764–79.
Lai R, Ikram M, Li R, Xia Y, Yuan Q, Zhao W, Zhang Z, Siddique KHM, Guo P. Identification of novel quantitative trait nucleotides and candidate genes for bacterial wilt resistance in tobacco (Nicotiana tabacum L.) using genotyping-by-sequencing and multi-locus genome-wide association studies. Front Plant Sci. 2021;12: 744175.
Lai W, Zeng X, Tan B, Wu G, Chen J, Guan W, Fan H. Preliminary Identification of Pathogenic Bacteria of Mulberry Bacterial Wilt. Guangdong Silk Newsletter. 1979;21–24.
Li F, Ye Q, Chen M, Zhang J, Xue L, Wang J, Wu S, Zeng H, Gu Q, Zhang Y, Wei X, Ding Y, Wu Q. Multiplex PCR for the identification of pathogenic listeria in flammulina velutipes plant based on novel specific targets revealed by pan-genome analysis. Front Microbiol. 2021;11: 634255.
Li X, Wang X, Chen Y, Yang X, Cui Z. Optimization of combined phytoremediation for heavy metal contaminated mine tailings by a field-scale orthogonal experiment. Ecotoxicol Environ Saf. 2019;168:1–8.
Li E, Tian X, Zhao R, Wang Y, Wang G. First report of E. cloacae causing bulb decay on garlic in China. Plant Dis. 2021. https://doi.org/10.1094/PDIS-05-21-0972-PDN.
Liu S, Lv M, Gu Y, Zhou J. First report of bulb disease of onion caused by Klebsiella pneumonia in China. Plant Dis. 2015;99:5–15.
Loganathan M, Thangavelu RPP, Kalimuthu M, Ramesh R, Selvarajan R, Uma S. First report of rhizome rot of banana caused by Klebsiella variicola in India. Plant Dis. 2021. https://doi.org/10.1094/PDIS-10-20-2316-PDN.
Lòpez-Fernàndez S, Sonego P, Moretto M, Pancher M, Engelen K, Pertot I, Campisano A. Whole-genome comparative analysis of virulence genes unveils similarities and differences between endophytes and other symbiotic bacteria. Front Microbiol. 2015;6:419.
Lou MM, Jin GL, Tian WX, Zhang GQ, Fan XY, Wang F, Zhu B, Xie GL. Specific and sensitive detection of Enterobacter mori using reliable RT-PCR. Plant Dis. 2011;95(9):1070–4.
Lu L, Chang M, Han X, Wang Q, Wang J, Yang H, Guan Q, Dai S. Beneficial effects of endophytic Pantoea ananatis with ability to promote rice growth under saline stress. J Appl Microbiol. 2021;131(4):1919–31.
Luo L, Huang Y, Liu J. Genome sequence resources of Klebsiella michiganensis AKKL-001, which causes bacterial blight of mulberry. Mol Plant Microbe Interact. 2022;35(4):349–51.
Macedo-Raygoza GM, Valdez-Salas B, Prado FM, Prieto KR, Yamaguchi LF, Kato MJ, Canto-Canché BB, Carrillo-Beltrán M, Di Mascio P, White JF, Beltrán-García MJ. Enterobacter cloacae, an Endophyte That Establishes a Nutrient-Transfer Symbiosis With Banana Plants and Protects Against the Black Sigatoka Pathogen. Front Microbiol. 2019;10:804.
Mahboubi M. Morus alba (mulberry), a natural potent compound in management of obesity. Pharmacol Res. 2019;146: 104341.
Malik VK, Singh M, Sangwan P, Kumari P, Sharma BL, Kumari P, Verma P, Yadav P, Sheoran N, Singh A, Singh DP, Arya SK, Ahalawat N, Malik J. First report of Klebsiella leaf streak on Sorghum caused by Klebsiella variicola in Haryana, India. Plant Dis. 2023. https://doi.org/10.1094/PDIS-09-22-2200-PDN.
Manzoor MF, Hussain A, Tazeddinova D, Abylgazinova A, Xu B. Assessing the Nutritional-Value-Based Therapeutic Potentials and Non-Destructive Approaches for Mulberry Fruit Assessment: An Overview. Comput Intell Neurosci. 2022;2022:6531483. Retraction in: Comput Intell Neurosci. 2023; 2023: 9860875.
Mao K. Occurrence and control of mulberry bacterial wilt in Guangdong. Guangdong Canye. 1996;04:19–20.
Maqsood M, Anam Saeed R, Sahar A, Khan MI. Mulberry plant as a source of functional food with therapeutic and nutritional applications: a review. J Food Biochem. 2022;46(11):e14263
Miller AM, Figueiredo JE, Chaves CL, Ruas EA, Balbi-Peña MI, Colauto NB, Paccola-Meirelles LD. Genomic variability of Pantoea ananatis in maize white spot lesions assessed by AFLP markers. Genet Mol Res. 2016. https://doi.org/10.4238/gmr15049452.
Mineau S, Kozak R, Kissoon M, Paterson A, Oppedisano A, Douri F, Gogan K, Willey BM, Mceer A, Poutanen SM. Emerging antimicrobial resistance among Escherichia coli strains in bloodstream infections in Toronto, 2006–2016: a retrospective cohort study. Can Med Assoc Open Access J. 2018;6:580–6.
Mitter B, Petric A, Shin MW, Chain PS, Hauberg-Lotte L, Reinhold-Hurek B, Nowak J, Sessitsch A. Comparative genome analysis of Burkholderia phytofirmans PsJN reveals a wide spectrum of endophytic lifestyles based on interaction strategies with host plants. Front Plant Sci. 2013;4:120. https://doi.org/10.3389/fpls.2013.00120.
Moretti C, Rezzonico F, Orfei B, Cortese C, Moreno-Pérez A, van den Burg HA, Onofri A, Firrao G, Ramos C, Smits THM, Buonaurio R. Synergistic interaction between the type III secretion system of the endophytic bacterium Pantoea agglomerans DAPP-PG 734 and the virulence of the causal agent of olive knot Pseudomonas savastanoi pv. savastanoi DAPP-PG 722. Mol Plant Pathol. 2021;22(10):1209–25.
Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15(3):473–97.
Mustafa A, Ibrahim M, Rasheed MA, Kanwal S, Hussain A, Sami A, Ahmed R, Bo Z. Genome-wide analysis of four enterobacter cloacae complex type strains: insights into virulence and niche adaptation. Sci Rep. 2020;10(1):8150.
Pallitto CR, Feuerstein JL, Baer AZ, Bishara JD, Bhavsar SM. Enterobacter cloacae complex meningitis in a full-term neonate. Clin Pediatr. 2023;62(10):1150–4.
Pavlova AS, Leontieva MR, Smirnova TA, Kolomeitseva GL, Netrusov AI, Tsavkelova EA. Colonization strategy of the endophytic plant growth-promoting strains of Pseudomonas fluorescens and Klebsiella oxytoca on the seeds, seedlings and roots of the epiphytic orchid, Dendrobium nobile Lindl. J Appl Microbiol. 2017;123(1):217–32.
Qian Y, Wei S, Zhang N, Yang Q, Chen W, Zhang X, Zhao H. Rapid detection of 5 fungal diseases in sunflower (Helianthus annuus) using dual priming oligonucleotide system-based multiplex PCR and capillary electrophoresis. SLAS Technol. 2022;27(4):253–60.
Ribeiro MG, de Morais ABC, Alves AC, Bolaños CAD, de Paula CL, Portilho FVR, de Nardi JG, Lara GHB, de Souza Araújo Martins L, Moraes LS, Risseti RM, Guerra ST, Bello TS, Siqueira AK, Bertolini AB, Rodrigues CA, Paschoal NR, de Almeida BO, Listoni FJP, Sánchez LFG, Paes AC. Klebsiella-induced infections in domestic species: a case-series study in 697 animals (1997–2019). Braz J Microbiol. 2022;53(1):455–64.
Rohela GK, Muttanna PS, Kumar R, Chowdhury SR. Mulberry (Morus spp.): an ideal plant for sustainable development. Trees Forests and People. 2020;2:100011.
Safni I, Cleenwerck I, De Vos P, Fegan M, Sly L, Kappler U. Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii subsp. syzygii subsp. nov., R. solanacearum phylotype IV strains as Ralstonia syzygii subsp. indonesiensis subsp. nov., banana blood disease bacterium strains as Ralstonia syzygii subsp. celebesensis subsp. nov. and R. solanacearum phylotype I and III strains as Ralstonia pseudosolanacearum sp. nov. Int J Syst Evol Microbiol. 2014;64(9):3087–103.
Shankar V, Hamilton MJ, Khoruts A, Kilburn A, Unno T, Paliy O, Sadowsky MJ. Species and genus level resolution analysis of gut microbiota in Clostridium difficile patients following fecal microbiota transplantation. Microbiome. 2014;2:13.
van Elsas JD, Kastelein P, de Vries PM, van Overbeek LS. Effects of ecological factors on the survival and physiology of Ralstonia solanacearum bv, 2 in irrigation water. Can J Microbiol. 2001;47(9):842–54.
Vayssier-Taussat M, Albina E, Citti C, Cosson JF, Jacques MA, Lebrun MH, Le Loir Y, Ogliastro M, Petit MA, Roumagnac P, Candresse T. Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta-omics. Front Cell Infect Microbiol. 2014;4:29.
Villa J, Tsuchiya K, Horita M, Natural M, Opina N, Hyakumachi M. DNA Analysis of Ralstonia solanacearum and related bacteria based on 282-bp PCR-amplified fragment. Plant Dis. 2003;87(11):1337–43.
Wang GF, Praphat K, Xie GL, Zhu B, Li B, Liu B, Zhou Q. Bacterial wilt of mulberry (Morus alba) caused by Enterobacter cloacae in China. Plant Dis. 2008;92(3):483.
Wang HY, Kang CZ, Wang S, Jiang DQ, Peng Z, Xu Y, Du YX, Zhang Y, Liu DH, Guo LP. Research strategies for endophytes in medicinal plants based on high-throughput sequencing and traditional culture and isolation methods. Zhongguo zhongyao zazhi. 2021;46:1910–9.
Wang J. Isolation, Identification and Control of Pantoea in Wilt of Mulberry. South China Agricultural University. 2019. In China
Wang J, Sun X, Luo L, Liu J. Isolation and identification of biocontrol bacteria against 4 soil-borne pathogens of mulberry. Sci Sericult. 2020;46:31–6.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173(2):697–703.
Xue Y, Hu M, Chen S, Hu A, Li S, Han H, Lu G, Zeng L, Zhou J. Enterobacter asburiae and Pantoea ananatis Causing Rice Bacterial Blight in China. Plant Dis. 2021;105(8):2078–88.
Yu L, Yang C, Ji Z, Zeng Y, Liang Y, Hou Y. Complete genomic data of Pantoea ananatis strain TZ39 associated with new bacterial blight of rice in China. Plant Dis. 2022;106(2):751–3.
Yuan T, Huang Y, Luo L, Wang J, Li J, Chen J, Qin Y, Liu J. Complete Genome sequence of Pantoea ananatis strain LCFJ-001 isolated from bacterial wilt mulberry. Plant Dis. 2023;107(8):2500–5.
Yuan T, Luo LH, Zhang XY, Liu JP. Establishment of IMSA-LAMP detection method for Ralstonia solanacearu. J South China Agric Univ. 2022;43(04):89–98.
Yuan T, Qazi IH, Li J, Yang P, Yang H, Zhang X, Liu W, Liu J. Analysis of changes in bacterial diversity in healthy and bacterial wilt mulberry samples using metagenomic sequencing and culture-dependent approaches. Front Plant Sci. 2023;14:1206691.
Yuan T, Qazi IH, Yang P, Zhang X, Li J, Liu J. Analysis of endophytic bacterial flora of mulberry cultivars susceptible and resistant to bacterial wilt using metagenomic sequencing and culture-dependent approach. World J Microbiol Biotechnol. 2023;39(6):163.
Yuan Q, Zhao L. The mulberry (Morus alba L.) fruit-A review of characteristic components and health benefits. J Agric Food Chem. 2017;65(48):10383–94.
Zaid AM, Bonasera JM, Beer SV. First report of enterobacter bulb decay of onions caused by Enterobacter cloacae in New York. Plant Dis. 2011;95(12):1581.
Zhang N, Li X, Li Y, Lu W, Xie L, Li G. First report of bacterial leaf streak in sweet corn caused by Pantoea ananatis in southeast China. Plant Dis. 2022. https://doi.org/10.1094/PDIS-06-22-1465-PDN.
Zhao N, Yang J, Liu H, Li L, Yan H, Liu D. Ginger rhizome rot caused by the Enterobacter cloacae in Tangshan. China Plant Dis. 2022;106:1293.
Zhou Y, Yang H, Liu J. Complete genome sequence of Enterobacter roggenkampii strain KQ-01, isolated from bacterial wilt-resistant mulberry cultivar YS283. Plant Dis. 2021;105(3):688–90.
Zhu B, Lou MM, Xie GL, Wang GF, Zhou Q, Wang F, Fang Y, Su T, Li B, Duan YP. Enterobacter mori sp. nov., associated with bacterial wilt on Morus alba L. Int J Syst Evol Microbiol. 2011;61(Pt 11):2769–74.
Acknowledgements
We thank all lab mates who supported during this project, but are not listed as co-authors.
Funding
The research was financially supported by the China Agriculture Research System (CARS-18-ZJ0304).
Author information
Authors and Affiliations
Contributions
T. Yuan wrote the initial draft of manuscript, conceived experiment design, performed experiments, data analysis and implementation; I.H. Qazi participated in data analysis, interpretation of the results and revised and edited the draft, and provided supervision; X.P. Huang collected materials, and assisted in the experiment and data analysis. J.P. Liu provided experimental platform and support, project supervision, and funding. All the authors read and agreed on submission of final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Schematic of the pathogenic flora of mulberry bacterial wilt disease [81]. Figure S2. Species detection range of five pairs of primers based on Primer-Blast analysis. (a): EyjfPF/EyjfPR; (b): KampDF/KampDR; (c): KpelYF/KpelYR; (d): RripwF/RripwR; (e): PpleDF/PpleDR. Figure S3. Screening of the best primers (no template added). M: 1000 marker. 1-16: The results after mixing 5 pairs of primers (RripwF/RripwR, EyjfPF/EyjfPR, KpelYF/KpelYR, KampDF/KampDR, and PpleDF/PpleDR) according to primer group 1 to 16 and performing PCR amplification (see Supplementary Table 1); 17: Primer group 1; 18: Primer group 2; 19: Primer group 9; 20: Primer group 20; 21: Compute group. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex. Figure S4. Effect of different 10mM dNTP (a), and 25mM MgCl2 dNTP (b) addition amounts, amplification cycle numbers (c), annealing temperatures (d) on mPCR efficiency (no template added). M: 1000 marker. a:1-a:11: 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 μL, 1.4, 1.6, and 2.0 μL of 10mM dNTPs, respectively. b:1-b:11: Add 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2 μL, 1.4, 1.6, and 2.0 μL of 25mM MgCl2, respectively. c:1-c:6: represent cycle numbers: 24, 26, 28, 30, 32 and 34, respectively. d:1~d:11: respectively represent the annealing temperature: 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63°C. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex. Figure S5. Detection of pathogenic bacteria isolated from field disease samples using the mPCR assay. M: 1000 Maker; 1-69: represents the detection of pathogenic bacteria of Ralstonia sp. (1-12), Enterobacter sp. (13-44), Klebsiella spp. (45-57), and Pantoea sp. (58-69). NC: the healthy mulberry genomic DNA as a negative control. “Pa”: P. ananatis “Rp”: R. pseudosolanacearum; “ECC”: E. cloacae complex; “KpSC”: K. pneumoniae species complex; “KoC”: K. oxytoca complex. Figure S6. Schematic of sterile mulberry seedling culture and verification of pathogenic bacteria by Koch’s postulates.
Additional file 2:
Table S1. Results of the primer orthogonal design. Table S2. Testing of natural mulberry wilt disease samples using mPCR and Traditional isolation methods. Table S3. mPCR detection and pathogenicity test of 69 bacterial strains isolated from mulberry bacterial wilt disease samples. Table S4. Bacterial strains used in this study. Table S5. Identification and coverage values of target genes in different strains based on BLAST (NCBI).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, T., Qazi, I.H., Huang, X. et al. Rapid detection of virulence-related genes by multiplex PCR in five pathogenic bacteria of mulberry bacterial wilt. Chem. Biol. Technol. Agric. 11, 72 (2024). https://doi.org/10.1186/s40538-024-00583-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-024-00583-z