
Abstract
Background
Mineral precipitates (scaling) from deep saline thermal waters often constitute a major problem during geothermal energy production. The occurrence of scale-fragments accumulating and clogging pipes, filters, and heat exchangers is of particular concern regarding an efficient energy extraction.
Methods
Carbonate scale-fragments from different sections of two geothermal power plants were collected and studied in a high-resolution scaling forensic approach comprising of microstructural characterization, elemental mapping, and stable carbon and oxygen isotope transects. The solid-phase analyses were evaluated in the context of natural environmental and technical (man-made) production conditions.
Results and discussion
Our results indicate an interaction of metal sulfide mineral layers mainly from H2S corrosion of the steel pipes and CaCO3 nucleation and crystal growth. A conceptual model of scale-fragment development addresses the relevance of two key interfaces: 1) the corrosion layer between the steel substrate and calcite scale and 2) the scale surface versus thermal fluid flow. The corrosion products constitute an attractive crystallization substrate of brittle and mechanically weak consistency. A rough carbonate scale surface tends to induce (micro) turbulences and increased flow resistance (frictional forces). These factors promote partial exfoliation, scale-fragment mobilization, and rapid clogging. This investigation highlights the potential of detailed petrographic and geochemical analyses of mineral precipitates for evaluating favorable versus unfavorable processes in geotechnical environmental settings.
Similar content being viewed by others


Background
Geothermal power gains in importance for heat and electricity production owing to its base-load capabilities compared to other renewable energy sources and promising technological developments (DiPippo 2015; Finster et al. 2015). Many geothermal installations (e.g., hydrothermal doublet systems), however, face major operational challenges, such as unwanted scaling (mineral precipitation) and corrosion of components accompanying the exploitation of typically high salinity, temperature, and gas content thermal waters from deep wells (Mundhenk et al. 2013; Klapper et al. 2016). The extent and site-specific characteristics of scaling and corrosion problems depend on geogenic as well as man-made environmental conditions, i.e., hydrogeochemistry and geology, water reservoir depth (temperature/pressure), flow rate, operating pressure, geometries, and composition of materials of the geothermal facilities.
Variable environmental (production) conditions and disturbed natural hydrogeochemical equilibria (temperature/pressure changes, CO2/H2O outgassing) can significantly promote scaling in production wells, downhole pumps, transport pipes, valves, and heat exchangers leading to inner diameter reduction, disturbed flow regimes, and clogging counteracting efficient energy extraction (Zarrouk et al. 2014). Similarly, mineral precipitation and mobilization (e.g., fine particles) can significantly reduce the reinjection efficiency of cooled thermal fluids depending on the respective physicochemical injection conditions, aquifer lithology, and hydraulic parameters (Rivera Diaz et al. 2016). Calcium carbonate (CaCO3) precipitation is widespread in low- to moderate enthalpy geothermal facilities tapping deep limestone/dolomite aquifers (e.g., Central European sedimentary basins) entailing elevated dissolved carbonate and carbon dioxide concentrations (Alt-Epping et al. 2013; Stober 2014). Silica/silicates, in contrast, dominate scale formation in high-enthalpy geothermal regions (e.g., Iceland, New Zealand; Gunnarsson and Arnórsson 2005; Zarrouk et al. 2014). Sulfates, sulfides, (hydro)oxides, or even chlorides can also form scale deposits (Corsi 1986; Regenspurg et al. 2015). Next to problematic mineral precipitation, corrosion of varying chemistry and degree can significantly harm carbon- and low-alloy steel components as well as concrete casings of geothermal installations (Mundhenk et al. 2013; Nogara and Zarrouk 2014). These material deterioration processes mostly depend on the prevailing electrochemical (redox) conditions and critical solution/gas constituents of the thermal fluid. Reducing conditions in aquifers and geothermal wells typically result in H2S-based corrosion and formation of various metal sulfides, while (intermittent) oxidizing conditions support the occurrence of iron-(oxy)hydroxides (Valdez et al. 2009; Bai et al. 2014). Steel corrosion and scaling processes may further be affected (e.g., catalyzed) by diverse microbial communities, i.e., playing an active (e.g., changing hydrochemistry) or passive (e.g., being a substrate) role for corrosion/scale mineral crystallization (Lanneluc et al. 2015; Würdemann et al. 2016).
The occurrence of scale-fragments (flakes) of variable dimensions constitutes a particular development of scaling in geothermal installations (Wolfgramm et al. 2011; Bauer et al. 2014). Typically thin (mm-range) scale deposits form in wells and transport pipes and are later mobilized (flaked off) in the course of thermal water flow. The growth and subsequent mobilization of such scale-fragments can cause specific and often serious problems, i.e., material damages and a rapid deterioration in the efficiency of hydrogeothermal heat extraction. Over short time intervals (e.g., weeks to few months in this study), critical accumulation, cementation, and clogging of filters and heat exchangers can occur. This leads to significantly reduced fluid flows and energy transfers. As a consequence, necessary maintenance intervals are shortened and impaired technical components (e.g., plate or tube bundle heat exchangers) have to be replaced unscheduled. In worst cases, the commercial viability of the geothermal infrastructure becomes a subject of debate.
In this study, we have conducted a scaling forensic approach. The scale-fragments themselves constitute a valuable chemical archive, i.e., their variable chemical composition and particular fabric (crystal growth) capture production-specific fluid–solid interaction. Applying high-resolution analytical techniques, the natural and man-made environmental conditions promoting unwanted carbonate scaling, corrosion, and fragmentation are evaluated comparable to scientific approaches in paleoenvironmental (paleoclimate) studies based on mineral precipitates (e.g., Boch et al. 2011; Dietzel et al. 2015). An increased process understanding of the particular carbonate scale growth dynamics and scale-fragment formation is targeted, i.e., elucidating the favorable versus unfavorable environmental and technical conditions that support scale-fragment evolution.
Sites, samples, and methods
Mineral precipitates (scales) were collected from different sections of two geothermal power plants nearly identical in construction (henceforward referred to as KIR and DUR), suffering from major problems with scale-fragment formation, i.e., the mobilization and accumulation of scale material formed inside pipes and subsequently clogging the outer faces of heat exchangers. Located in southern Germany, the two hydrogeothermal doublet systems with Organic Rankine Cycles produce district heat and electric power (7 MWel max. each; ~31 GWh/a; Weber et al. 2016) from deep thermal water production and reinjection wells (KIR: GT1 4214 m, GT2 4452 m final measured depth; DUR: GT1 4393 m, GT2a 4530 m) tapping the regional Malm (Upper Jurassic) limestone/dolomite aquifer overlain by Cretaceous marls and sandstones followed by Paleogene to Late Miocene successions of marls, sands, and clays (Mayrhofer et al. 2014; Dussel et al. 2016; Goldbrunner and Vasvari 2016). Top of Malm at the location KIR is reached at 3447 m (GT1) and 3711 m (GT2) and at 3715 and 3641 m for GT1 and GT2a at DUR, respectively. The corresponding open hole sections range from 650 to 930 m. The thermal fluid temperatures reach 134 and 136 °C at the wellheads and maximum discharges are 135 and 115 l/s for KIR and DUR, respectively. Scale-fragment samples were recovered from dismounted well steel pipes of different depths (increasing sample numbers, e.g., 1–81) and from the outer (water inlet) sides of (tube bundle) heat exchangers in the plants (samples “WT”) during a major maintenance procedure of both facilities in September 2014 (Fig. 1). Individual up to 10-cm-long and typically a few millimeters thick fragments (flakes) were sampled from the interior surfaces of the conventional (low-alloy) steel pipes (Fig. 1b, d) afore being mounted vertically in different borehole depths. Accumulations of cemented scale-fragments were also sampled as impressive blocks of dm-size heavily blocking the outer heat exchanger surfaces (Fig. 1c, e). Scale-fragments recurrently assembling such massive blocks over time (typically within weeks to months), however, were deposited and mobilized within a relatively short spatial distance, i.e., they originate from a thermal water transport section after some upstream filter equipment of the uppermost production well water flow. After impacting and accumulation on the outer heat exchanger surface, the scale-fragments were successively cemented and compacted by ongoing mineral precipitation from the streaming thermal water.

Scale-fragments deposited in thermal water transport pipes of different depths as well as successively accumulated on heat exchangers of two geothermal power plants. a Steel pipes dismounted from a deep geothermal borehole during a major maintenance procedure in front of the geothermal facility. b Carbonate scale-fragments flaked off from the interior of a steel pipe. c Massive block of cemented carbonate scale-fragments clogging an outer heat exchanger surface within weeks to few months. d Rough growth surface of dark crystalline Mg-calcite scale-fragment precipitated inside a pipe (sample KIR33 collected from a deeper production well section). e View on agglomerated and cemented scale-fragments of variable sizes recovered from a heavily impaired heat exchanger in the power plant
Selected and representative scale-fragment samples from both study sites were investigated in detail via state-of-the-art mineralogical and geochemical methods. Solid-phase characterization included powder X-ray diffraction (XRD) using a PANalytical X’Pert PRO diffractometer equipped with a Co-Kα X-ray radiation source (40 mA, 40 kV) and an X’Celerator detector and applying measurement parameters 4°–85° 2θ range, ~0.01° step propagation (2θ), and 40 s/step counting time. Typical sample aliquots amounted to 0.5 g of powder pestled for bulk (carbonate) scale analyses and ca. 0.1 g for identifying the insoluble residuum after dissolving the (carbonate) scale matrix (ca. 10 g sample) in diluted nitric acid (12 vol.%, for 24 h). Elemental compositions of the scale-fragments were determined with a Philips PW 2404 X-ray fluorescence spectrometer (WDX-XRF) analyzing ca. 0.8 g sample aliquot in fused Li2B4O7 glass pellets for the major and minor elements (analytical error 0.5–5 wt.%) and ca. 12 g sample aliquot in pressed powder pellets for the trace elemental compositions (analytical error <10 wt.%). Loss-on-ignition (L.O.I.) was determined at 1050 °C. Chemical compositions were further analyzed by wet chemistry techniques. Parts of the scale-fragments were powdered and digested with HNO3 (supra-pure). Measurements of the elements were performed by optical emission spectrometry (ICP-OES, Perkin Elmer Optima 7300DV) and inductively coupled plasma mass spectrometry (ICP-MS, Perkin Elmer Sciex ELAN 6100). Total organic carbon (TOC) was measured with a Carlo-Erba EA11008 Elemental Analyzer. Microscopic (petrographic) characterization of scale samples was conducted based on polished, non-covered thin sections for transmitted and reflected light microscopy (Leica DMLP) connected to an Olympus DP26 camera. Scanning electron microscopy (SEM) was performed on carbon-coated samples using a Zeiss DSM 982 Gemini SEM operated at 2 kV.
Selected scale-fragments were further investigated by high spatial (temporal) resolution analytical techniques involving elemental mapping using the wavelength-dispersive analytical mode (WDX) and back-scattered electron imaging (BSE) using a JEOL JXA-8200 electron microprobe (EPMA). Semi-quantitative elemental distribution images of Ca, Mg, Fe, S, and Si were recorded with an accelerating voltage of 15 kV, a beam current of 30 nA, and ~1 µm electron beam resolution. 1000 × 800 point analyses, a step size of 0.44 µm, and a counting time of 15 ms/step yielded elemental distribution maps of 440 × 350 µm. Scale-fragments were also embedded in epoxy resin, cut, and polished to obtain a few millimeters thick cross-sections. Tiny sample amounts (0.25–0.60 mg) were extracted from two selected and embedded scale-fragments at 100 µm spatial resolution (KIR33: 39 samples; DUR34: 10 samples) using an ESI New Wave Research computer-controlled micromill and continuous sampling transects (Spötl and Mattey 2006; Boch et al. 2011). Additional samples from other fragments were recovered using a dentist drill. The stable carbon and oxygen isotopic compositions of the micromilled and drilled carbonate powders were subsequently measured by continuous-flow isotope ratio mass spectrometry (CF-IR-MS), i.e., a Thermo Fisher Scientific GasBench II connected to a Finnigan Deltaplus XP mass spectrometer, following the procedure reported in Dietzel et al. (2015). Measured isotope ratios are quoted relative to the VPDB standard and typical precision (2σ) is ±0.08 and ±0.1‰ for δ13C and δ18O values, respectively. In order to discuss fluid (thermal water/gases) versus solid (scale/corrosion) interaction, hydrochemical data from available reports of the two geothermal facilities were used. Mineral saturation states (SI values defined as ion activity product versus solubility product on a logarithmic scale, i.e., SI = log [IAP/Ksp], SI = 0 means saturation) and aqueous solution CO2 partial pressures were calculated based on these data applying the PHREEQC computer code for hydrogeochemical modeling (version 3.2.0, phreeqc.dat database, Parkhurst and Appelo 2013).
Results and discussion
Scale chemistry and fabric
Several scale-fragments of 2–10 cm size and 1–4 mm thickness were analyzed for their mineralogical and chemical composition. Our results showed that all of these samples deposited either on the outer side (thermal water influx and obstacle for scale-fragments) of the heat exchangers (e.g., KIR-WT, DUR-WT), at the top (e.g., KIR-GOK), or base (DUR1, KIR1) of the uppermost steel pipe in the borehole and thus close to the wellhead, or several hundred meters (<1000 m) deep in the borehole (KIR56, DUR81) are almost exclusively composed of Mg-calcite (~90 to 99 wt.%), i.e., crystalline CaCO3 solid solutions with 5–8 mol-% MgCO3 (Fig. 2). All of the scale-fragments analyzed revealed a very similar mineralogical and chemical composition. Acid digestion of the prominently dark-colored carbonate scales resulted in a black residuum (insoluble portion) composed of variable contents of metal sulfides and silica (Fig. 3). The sulfides are dominated by FeS (e.g., pyrrhotite, typically <2 wt.% in the scale), CuFeS2 (chalcopyrite, <1 wt. %), ZnS (sphalerite, <0.5 wt.%), and NiS (millerite, <0.1 wt.%; Fig. 3; Table 1, Additional file 1: Tables S1, S2). The SiO2 component (typically <1 wt%) represents quartz (cf. XRD peak in Fig. 2) and also adsorbed amorphous silica. In addition, elevated TOC concentrations of the scale material (up to ~4 wt.%) were measured. The observation of magnetic particles as part of the dark residuum supports significant contributions of pyrrhotite (Fe1−xS). Minor contents of Fe3O4 (magnetite; Fig. 3) in one sample might result from partial later stage oxidation of iron sulfide. The prominent occurrence of various metal sulfides reflects reducing redox conditions of the scale precipitating thermal waters. The dark color of the otherwise whitish carbonate scales likely originates from the organic content as well as typically dark-colored and finely disseminated metal sulfide particles. The latter is somewhat analogous to hydrothermal “black smoker” deposits of the deep ocean floors (Baker et al. 2016).

Mineralogical composition of KIR and DUR scale-fragment samples based on powder X-ray diffraction (XRD) spectra. The samples were recovered from heat exchangers (KIR-WT, DUR-WT), close to the wellhead (KIR-GOK, DUR1), or from pipes installed several hundred meters deep in the borehole (KIR56, DUR81). All samples measured are almost exclusively composed of Mg-calcite. Zincite (ZnO) was used as an internal standard. In the left XRD pattern, the determining crystal planes (Miller Indices) of Mg-calcite are indicated

Mineralogical composition of typically dark-colored KIR and DUR scale-fragment samples collected from the heat exchangers (WT) after acid digestion of the carbonate matrix based on powder XRD spectra. The black residua are mainly composed of variable metal sulfide and silica contents of variable crystallinity, i.e., FeS (pyrrhotite), CuFeS2 (chalcopyrite), ZnS (sphalerite), NiS (millerite), and a SiO2 component (quartz or adsorbed silica). Minor contents of Fe3O4 (magnetite) might result from partial later stage oxidation of iron sulfide
Detailed optical characterization of the scale material provided further insights into petrographic and genetic aspects. Macroscopically, the carbonate scales expose well-developed crystal faces on their growth surfaces supporting some prominent light reflection (brilliance) in spite of their dark coloring (Fig. 1d). The sparitic fabric entails an extra rough scale surface shaped by numerous individual crystal tips. Microscopic techniques (Fig. 4; Additional file 1: Figure S1) revealed a compact growth fabric with little intra- and intercrystalline porosity of these CaCO3 precipitates. The crystal shapes display idiomorphic (euhedral), rhombohedral calcite crystals frequently terminating as distinct crystal tips at the actual growth surface, i.e., the interface toward the thermal fluid (Fig. 4a, b, e). Abundant nucleation represented by numerous small crystallites gradually develops toward relatively large (mm-size), columnar Mg-calcite crystals showing rather smooth crystal (grain) boundaries (Fig. 4c, d, f). Such competitive crystal growth after numerous initial crystallites results in the dense interrelation of the scale assembling crystals (Frisia et al. 2000). Importantly, these petrographic and growth characteristics support a brittle consistency of the calcite scale material. Visible light and electron microscopy show the occurrence of various metal sulfides (variable shapes and colors) arranged (i) in distinct layers of alternating thickness at the interface between calcite scale and the underlying (steel) substrate (Fig. 4a, c) and (ii) as recurrent, typically much thinner sulfide layers within the compact carbonate scales (Fig. 4f; Additional file 1: Figure S1C). The omnipresent metal sulfide layers at the scale base most likely are of an in-situ chemical origin, i.e., from an early mineral-forming reaction of the corrosive thermal fluid at the exposed fresh steel surface. This is supported by some overall growth orientation of the sulfide minerals. In contrast, the thin layers within the scales possess a more particular (individual grains) character and also strongly variable thicknesses (Fig. 4b; Additional file 1: Figure S1C). This suggests a detrital origin for this type of sulfide layers, i.e., an event-like accumulation after transport in suspension. An important feature consists in the pronounced nucleation of CaCO3 on the basal sulfide layers (Fig. 4c, d) as well as re-nucleation on later stage (detrital) layers (Fig. 4b).

Microscopic characterization of the carbonate scale-fragment fabric. a Reflected light (RL) cross-section showing a basal sulfide corrosion layer (red arrows), columnar Mg-calcite crystals, and rhombohedral crystal tips forming a rough scale growth surface (blue arrows). b RL image showing a layer of detrital particles (variable thickness, yellow arrows) on the former calcite growth surface (blue arrow) and subsequent re-nucleation of numerous calcite crystallites (white arrows). c Typical metal sulfide layer of variable thickness at the scale-fragment base (red arrow). d Transmitted light (TL) image of basal corrosion layer being an attractive substrate for abundant calcite nucleation and ongoing competitive crystal growth. e Scanning electron microscope (SEM) image of rough scale growth surface showing a dense fabric of rhombohedral calcite crystals. f Back-scattered electron (BSE) image illustrating typical metal sulfide layers at the scale-fragment base (partially fractured, red arrow) and as thin layers within the compact carbonate scales (yellow arrows)
Carbonate scaling versus corrosion
Our mineralogical and petrographic results suggest an intimate connection at the basal sulfide layer and calcite scale interface. High spatial resolution electron microprobe elemental maps measured in this study (Fig. 5) support the results from bulk scale chemical analysis, i.e., the layers at the scale base toward the underlying steel pipe substrate primarily consist of Fe-sulfide minerals and some associated SiO2. Variable iron and sulfur concentrations in these basal layers represent a gradual development from more or less pure Fe (steel) toward metal sulfides of varying stoichiometric composition. Minor later stage alteration of the sulfides (e.g., oxidation toward goethite, magnetite) might also have occurred according to the microprobe (Fig. 5) and XRD (Fig. 3) results. Regarding the basal layer chemistry and texture, the analyses suggest an origin primarily from H2S steel corrosion (Choi et al. 2011; Bai et al. 2014), e.g., represented by the overall chemical reactions:

High spatial resolution electron microprobe back-scattered electron images and elemental maps (Ca, Mg, Fe, S, Si) from the basal sections of two different scale-fragment samples (a, b). The precipitated scale material consists of homogeneous Mg-calcite indicated by the measured Ca and Mg concentrations. The basal corrosion layer primarily consists of iron sulfide minerals (Fe and S concentrations) resulting from gradual alteration of the conventional steel (pink color indicating high elemental concentration on relative color range). Elevated silica contents (Si) are typically associated with the Fe-sulfide layers. Note the frequent occurrence of cracks displacing sulfide corrosion products and related scale carbonate (red arrow in BSE image)
Natural (hydrochemistry) as well as technical (conventional low-alloy steel pipes) environmental conditions support a high corrosion potential at both geothermal facilities. Hydrochemical data from the local limestone/dolomite aquifer and production wells of the geothermal installations (Eichinger 2010; Seibt 2010; Wolfgramm et al. 2011; Mayrhofer et al. 2014; Stober 2014; Goldbrunner and Vasvari 2016) reveal strongly reducing redox conditions (up to −360 mV) at pH ~6, increased dissolved (ΣS ~ HS− + S2− ≤35 mg/l) and gaseous (H2S up to 8.7 vol.% in gas phase and odor of thermal water) sulfide concentrations, as well as relatively high chloride (up to 200 mg/l) and gaseous CO2 (up to 90 vol.% of gas phase) contents. Next to these corrosive constituents, the high thermal fluid temperatures (reaching 141 °C in the aquifer) and high fluid discharges (up to 135 l/s) promote efficient and rapid steel corrosion. In the carbonate scales, we find no distinct evidence (e.g., dendritic filaments) of microbially influenced Fe corrosion and/or CaCO3 precipitation (Fan et al. 2013; Lanneluc et al. 2015) likely precluded by the hostile high fluid temperatures (Takai et al. 2008; Lerm et al. 2013). An origin of the sulfide layers from steel corrosion is further supported by the relatively low dissolved metal concentrations of the respective thermal waters (e.g., Fe ≤380 µg/l, Cu ≤2, Zn ≤17, Ni ≤26 µg/l; Seibt 2010). In contrast, aqueous silicic acid concentrations are relatively high (H4SiO4 ~80 mg/l) supporting adsorption and co-precipitation of silica with the basal corrosion products (Figs. 2, 5) further promoted by cooling during thermal water ascent (decreasing solubility of silica). The hydrochemical data consequently suggest a principal origin from steel corrosion for the typically thin but variable metal sulfide layers within the carbonate scales (Fig. 4f; Additional file 1: Figure S1C). They could be the manifestation of flushing events in the course of restarts after major production cessations favoring sustained corrosion and detrital particle transport in suspension. Possibly, the particulate sulfide minerals are mobilized from deeper corroded pipe sections (below downhole pump) and/or from corroded steel surfaces of upper borehole sections accessible after some scale-fragment mobilization. However, some proportion of the metal sulfides detected could also originate from the anoxic aquifer, i.e., heavy metals mobilized from deep sediments as, e.g., Cl− or HS− aquo-complexes (Barnes 2015) and subsequently precipitated with the carbonate scale.
Calcite (CaCO3) scale precipitation from the deep thermal waters and at the sulfide corrosion layers is represented by the overall reactions:
The chemical equilibria and finally CaCO3 mineralization in the aqueous carbonate system are mainly controlled by pressure-, temperature-, and pH (H+ activity)-dependent dissolution and degassing of CO2 in the thermal waters, as well as calcium (and magnesium) and carbonate mobilization from the deep limestone/dolomite aquifer. Our results, however, also suggest a critical role of the underlying, corroded steel substrate. Abundant and competitive CaCO3 crystallites at the scale versus corrosion mineral (metal sulfide) interface characterize an attractive substrate for initial nucleation and ongoing crystal growth (Figs. 4, 5). Such an enhancing effect is supported by the overall lower crystallinity, high specific surface areas, and common defect sites of typical corrosion mineral layers (Zimer et al. 2013; Bai et al. 2014). In this context, electrostatic forces could also play a role (Kashkovskiy et al. 2012; Fan et al. 2013). Regarding the overall progress of steel corrosion, the precipitation of carbonate and corrosion products might reduce the determining electrochemical reactions, i.e., reduce ongoing corrosion at places of significant scale deposition (cf. Nogara and Zarrouk 2014). Such an effect of scaling versus corrosion is also likely in the case presented here. Steel corrosion favoring initial calcite nucleation and thereby influencing further crystal growth, however, is in line with the observed extensive scaling progress in spite of relatively low total dissolved solid contents measured in the thermal waters (≤700 mg/l, Seibt 2010; Mayrhofer et al. 2014; Dussel et al. 2016). In addition, hydrochemical calculations (using PHREEQC) reveal that calcite saturation states of thermal fluids sampled near the wellhead are close to equilibrium (only slightly supersaturated or even undersaturated). The calculated CO2 partial pressures are prominently high (log pCO2 = −0.9 to −0.1 atm). It is important to note that the production wells of both geothermal facilities operate at a relatively high working pressure (~18 bar) in order to avoid outgassing. Several factors therefore counteract an increased calcite precipitation and support the potential role of some surficial trigger inducing the initiation of scale nucleation. Regarding the recurrent metal sulfide layers within the carbonate scales, such effects on crystallization may strongly depend on the local thickness of the attractive substrate (Fig. 4b; Additional file 1: Figure S1C), i.e., both continued (unperturbed) calcite growth or re-nucleation might occur.
Scale-fragment development
Discussing scale formation and fragmentation, two interfaces are considered to be of crucial importance: (i) the interface between calcite scale and the sulfide corrosion products and (ii) the interface between thermal fluid and calcite scale. The metal sulfide corrosion products frequently evolving from the corrosion of carbon- and low-alloy steels interacting with H2S-rich thermal solutions possess distinct chemical and mechanical characteristics (Kashkovskiy et al. 2012; Zheng et al. 2015). Different sulfide corrosion mechanisms are distinguished, e.g., sulfide stress cracking (SSC; Bai et al. 2014; Liu et al. 2014) or pitting corrosion (Zimer et al. 2013; Nogara and Zarrouk 2014). In essence, these corrosion mechanisms result in localized but ongoing steel deterioration allowing corrosive constituents of the thermal fluid to further penetrate into the unaltered steel surface. This is also the case for hydrogen atoms and molecules emerging from sulfide corrosion [see chemical reactions (1)] and leading to widespread hydrogen-induced cracking (HIC) and hydrogen embrittlement favored at high temperatures (Liu et al. 2014; Bai et al. 2014). Increased H2 gas concentrations also measured in the fluid flow of the geothermal facilities investigated in this study (Seibt 2010) are recognized as an indicator of significant corrosion (Smith et al. 2011; Alt-Epping et al. 2013). As a major consequence, steel surfaces become increasingly brittle and show a weakened mechanical resistance against (shearing) stress and strain, e.g., from the streaming thermal fluid. Macroscopic, microscopic, and high spatial resolution electron microprobe imaging support the common occurrence of small cracks and related flakes being closely associated with the corrosion product layers, i.e., (hairline) cracks are a common visible feature affecting the coherence of the sulfide corrosion layer and often also the adjacent calcite scale base (Figs. 4, 5).
Another prominent feature of the scale-fragments studied is their typically rough growth surface, i.e., the interface between thermal fluid flow and calcite scale. This was directly related to calcite nucleation, competitive and euhedral (rhombohedral) crystal growth determining the actual scale fabric. Importantly, a rough scale surface topography results in an increased flow resistance and higher frictional forces at the interface, probably causing the occurrence of (micro)turbulences. The latter would entail increased CO2 and H2O outgassing from aqueous solution and such degassing would strongly enhance local supersaturation and further CaCO3 precipitation considering the respective chemical equilibria reported in the previous Eq. (2). High carbonate scale deposition rates and the resulting saturation states close to calcite equilibrium observed support this mechanism. Moreover, the occurrence of (micro)turbulences at the interface can explain rapid calcite precipitation in spite of the typically high production well operating pressures (~18 bar) applied in order to counteract outgassing. In this context, variable thermal water production rates will also affect the processes at the interface. The prominently high solution CO2 partial pressures calculated further support a potential susceptibility with regard to a trigger of CO2 degassing. Increased but variable degrees of CO2 outgassing and steam (bubble) formation during thermal fluid flow are also strongly suggested by the analyzed C and O isotopic distribution and evolution in the carbonate scale-fragments (Fig. 6; Additional file 1: Tables S3, S4). The stable isotope measurements of the calcite precipitates show overall high δ13C values ranging from −1.5 to −0.5‰ (VPDB) primarily reflecting dissolution of the Malm aquifer limestone/dolomite and overall low δ18O values of −28.3 to −26.6‰ (VPDB) primarily reflecting the meteoric origin and high temperatures of the thermal waters. A comparison of the corresponding carbonate δ13C versus δ18O values does not show a significant correlation. The natural isotopic variability of deep thermal waters and consequently of carbonate formed over short time intervals (e.g., scale formation in this study) is typically considered to be very low. Micromilled (continuous) C and O isotope profiles show δ13C amplitudes of ~0.7‰ and δ18O amplitudes of ~1.5 and 1.3‰ for samples from KIR and DUR, respectively (Fig. 6). Importantly, these variations occur within short spatial (1-4 mm) and temporal (scaling rate >1 mm/month) intervals. Oxygen isotope fractionation between water and calcite is known to strongly depend on the prevailing temperature (Epstein et al. 1953; O’Neil et al. 1969). Based on published O isotope thermometers, it is possible to translate the measured calcite δ18O values (−28.1 to −26.6‰ VPDB for KIR33, Fig. 6, this study) and thermal water δ18O values (−11.7‰ VSMOW for KIR; Seibt 2010) into absolute temperature changes. Applying well-established fractionation factors, the lowest carbonate δ18O values (−28.1‰) result in calcite formation temperatures of (i) 136 °C (Friedman and O’Neil 1977) or (ii) 138 °C (Coplen 2007), i.e., reflecting the actual KIR thermal water temperatures in the production well between Malm aquifer (141 °C max.) and wellhead (134 °C typically) very well. In contrast, the higher carbonate δ18O values (−26.6‰) result in temperatures of (i) 118 °C (Friedman and O’Neil 1977) or (ii) 124 °C (Coplen 2007), i.e., much lower temperatures. Thus, relative water-temperature variations of (i) 18 °C or (ii) 14 °C should occur based on these calculations. This is not supported by typical ranges of temperature variations in these deep and high-discharge geothermal wells (few °C at wellhead). Consequently, temperature changes alone cannot explain the observed oxygen isotope fractionation. Rather, processes such as rapid CO2 degassing and H2O vapor formation from solution are well known to kinetically enrich the isotopically light carbon dioxide (12C16O2) and water (1H 162 O) molecules in the separated gas phase resulting in higher but variable δ13C versus δ18O values of the precipitating calcite (Mickler et al. 2006; Dreybrodt and Romanov 2016). These processes of kinetic isotope fractionation vary temporally and spatially depending on the amount and proportion of partial gas separation of CO2 and H2O. Altogether, we interpret the distinct isotope variations analyzed in the scales to primarily reflect physicochemical processes such as variable CO2 and H2O (steam) outgassing, a variable degree of (micro)turbulences in the pipes, some contribution from temperature changes and minor natural isotopic variability. These combined effects are further closely linked to production rate and thus operational conditions, which can be monitored temporally and spatially by the above stable isotope signatures of the calcite considering a small impact by natural isotope variations. Importantly, the amount of carbonate scale deposition (scaling progress) is intimately connected with the variable release of CO2 and H2O from the streaming thermal water [see chemical equilibria reported in Eq. (2)].

High-resolution micromilled stable carbon and oxygen isotope profiles analyzed across typical growth intervals (from base to growth surface, yellow rectangles) of carbonate scale-fragments deposited inside borehole pipes of the two geothermal facilities (KIR and DUR). The distinct spatiotemporal variations mostly reflect variable CO2 and H2O outgassing (affecting δ13C and δ18O) probably associated with variable (micro)turbulences at the rough calcite scale versus thermal fluid interface, as well as some contribution from temperature changes (mainly affecting δ18O) associated with the thermal water production conditions. See text for details
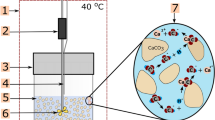
Regarding a conceptual understanding of scale-fragment development, the partial exfoliation within the vertical or horizontal transport pipes strongly relies on the preconditions at the two interfaces discussed (Fig. 7). Mobilization is favored by (i) the chemically and mechanically weak basal (sulfide) corrosion interface and (ii) increased flow resistance and associated (micro)turbulences at the rough calcite scale growth surface. In addition, flaking off might be promoted by some thermal contraction, e.g., from cooling of steel pipes during major production cessations. Also, additional mechanical stress on the rough scale surface could result from particle transport in suspension and abrasion. Significantly increased particle concentrations were observed during restart procedures after production cessations (Wolfgramm et al. 2011). After mobilization within thermal water flow (up to 135 l/s), the scale-fragments impact and eventually damage the relatively fragile outer surfaces of the pounded heat exchangers. Finally, severe clogging of heat exchangers or filters (obstacles) is accomplished by successive accumulation, agglomeration, and CaCO3 cementation during ongoing but deteriorating thermal fluid flow and heat/energy extraction (Fig. 7).

Schematic and stepwise (1–5) illustration of the principle mechanisms leading to scale-fragment development in pipes (vertical or horizontal) and finally resulting in heavy blockage of heat exchangers or other obstacles (e.g., filters). This conceptual model might also be applied in other cases of problematic scale-fragment evolution
Conclusions and environmental and technical considerations
Our scaling forensic approach highlights the critical interaction of natural and technical (operational) conditions with regard to an increased carbonate scaling and steel corrosion potential and associated problematic scale-fragment development. The exploitation of Upper Jurassic carbonate aquifer H2S-containing Na–Ca–HCO3–Cl thermal water (Mayrhofer et al. 2014; Stober 2014; Dussel et al. 2016; Goldbrunner and Vasvari 2016) entails a significant hydrogeochemical potential for both carbonate scaling and steel corrosion. This natural environmental potential consists in the initial versus disturbed dissolved carbonate equilibria leading to carbonate scaling and several corrosive constituents (H2S, CO2, Cl−, H2) in combination with high temperatures and high flow rates (component supply) effecting steel deterioration. Evaluating the source of H2S delivered with the regional thermal waters, thermochemical sulfate reduction (TSR) at sufficiently high temperatures (100–140 °C) and in association with various organic materials was discussed (Mayrhofer et al. 2014). These redox reactions would also contribute to the dissolved (inorganic) carbon budget, although the high aqueous solution (δ13CDIC = −0.2‰ VPDB; Seibt 2010) and high calcite scale (δ13C = −1.5 to −0.5‰ VPDB; this study) stable carbon isotopic compositions argue against a major organic contribution with regard to dissolved carbonate species and potential carbonate precipitation. Based on the available hydrochemical data and calculations performed, it further seems possible that the scaling problems encountered at the study sites are partially the result of physicochemical conditions associated with a deep fault zone. This is supported by prominently high aqueous solution pCO2 values, high water temperatures, and eventually other (e.g., sulfur isotopic) indicators. Fault zones are considered a primary target of productive geothermal wells in the region although being difficult to constrain spatially (Wolfgramm et al. 2009; Stober 2014; Dussel et al. 2016; Goldbrunner and Vasvari 2016, Przybycin et al. 2017). Deep faults delivering abundant CO2 might further be associated with hypogenic karstification, i.e., providing an efficient mechanism for aquifer host rock dissolution and potential secondary carbonate precipitation. In essence, fault zones could entail favorable (high discharge and high temperature) as well as unfavorable (chemical constituents such as CO2) production conditions. Considering the technical (operational) potential of the two studied geothermal doublets with regard to carbonate scaling and corrosion in the production wells, the selection of materials in contact with the thermal fluid is likely of major importance. The occurrence of metal sulfide corrosion layers favoring scale-fragment development and probably being a partial source of the dark scale coloring should diminish when more corrosion resistant (alloyed or coated surface) steels are used. A major effect of corrosion layers enhancing carbonate precipitation in contrast to plastic coatings reducing the overall scale thicknesses was supported by observations and simulations of a recent study (Wanner et al. 2017). We are aware, however, that the application of specific materials might not always be viable due to other circumstances (e.g., costs, abrasive effects, temperature resistance). In order to avoid accumulation and critical cementation of scale-fragments, these should be filtered/removed timely and near the heat exchanger surface or other obstacles during water flow. Thermal water production cessations and restarts most likely have a significant effect on scale-fragment mobilization, i.e., causing additional mechanical stress from pressure and temperature changes, increased (micro)turbulences, and probably elevated (abrasive) particle concentrations. To summarize, scaling forensics can help to better understand the site-specific favorable and unfavorable physicochemical processes determining scaling and/or corrosion of affected geothermal power plants.
References
Alt-Epping P, Waber HN, Diamond LW, Eichinger L. Reactive transport modeling of the geothermal system at Bad Blumau, Austria: implications of the combined extraction of heat and CO2. Geothermics. 2013;45:18–30.
Bai P, Zheng S, Zhao H, Ding Y, Wu J, Chen C. Investigations of the diverse corrosion products on steel in a hydrogen sulfide environment. Corros Sci. 2014;87:397–406.
Baker ET, Resing JA, Haymon RM, Tunnicliffe V, Lavelle JW, Martinez F, Ferrini V, Walker SL, Nakamura K. How many vent fields? New estimates of vent field populations on ocean ridges from precise mapping of hydrothermal discharge locations. Earth Planet Sc Lett. 2016;449:186–96.
Barnes HL. Hydrothermal processes. Geochem Perspect. 2015;4:1–93.
Bauer M, Freeden W, Jacobi H, Neu Th, editors. Handbuch tiefe geothermie—prospektion, exploration, realisierung, nutzung. Berlin & Heidelberg: Springer Spektrum; 2014.
Boch R, Spötl C, Frisia S. Origin and palaeoenvironmental significance of lamination in stalagmites from Katerloch Cave, Austria. Sedimentology. 2011;58:508–31.
Choi YS, Nesic S, Ling S. Effect of H2S on the CO2 corrosion of carbon steel in acidic solutions. Electrochim Acta. 2011;56:1752–60.
Coplen TB. Calibration of the calcite-water oxygen-isotope geothermometer at Devils Hole, Nevada, a natural laboratory. Geochim et Cosmochim Acta. 2007;71:3948–57.
Corsi R. Scaling and corrosion in geothermal equipment: problems and preventive measures. Geothermics. 1986;15:839–56.
Dietzel M, Schön F, Heinrichs J, Deditius AP, Leis A. Tracing formation and durability of calcite in a Punic-Roman cistern mortar (Pantelleria Island. Italy): Isot Environ Healt S; 2015. doi:10.1080/10256016.2015.1016430.
DiPippo R. Geothermal power plants: evolution and performance assessments. Geothermics. 2015;53:291–307.
Dreybrodt W, Romanov D. The evolution of 13C and 18O isotope composition of DIC in a calcite depositing film of water with isotope exchange between the DIC and a CO2 containing atmosphere, and simultaneous evaporation of the water. Implication to climate proxies from stalagmites: a theoretical model. Geochim et Cosmochim Acta. 2016;195:323–38.
Dussel M, Lüschen E, Thomas R, Agemar T, Fritzer T, Sieblitz S, Huber B, Birner J, Schulz R. Forecast for thermal water use from Upper Jurassic carbonates in the Munich region (South German Molasse Basin). Geothermics. 2016;60:13–30.
Eichinger L. Ergebnisse der hydrochemischen, isotopenhydrologischen und gasphysikalischen Untersuchungen während des Langzeitpumpversuchs an der geothermischen Förderbohrung in Kirchstockach. Hydroisotop Report; 2010. p 28.
Epstein S, Buchsbaum R, Lowenstam H, Urey HC. Revised carbonate-water isotopic temperature scale. Bull Geol Soc Am. 1953;64:1315–26.
Fan MM, Liu HF, Dong ZH. Microbiologically influenced corrosion of X60 carbon steel in CO2-saturated oilfield flooding water. Mater Corros. 2013;64:242–6.
Finster M, Clark C, Schroeder J, Martino L. Geothermal produced fluids: characteristics, treatment technologies, and management options. Renew Sust Energ Rev. 2015;50:952–66.
Friedman I, O’Neil JR. Compilation of stable isotope fractionation factors of geochemical interest. In: Fleischer M, editor. Data of geochemistry, U.S. Geological Survey Professional Paper 440-KK; 1977. pp 1–12.
Frisia S, Borsato A, Fairchild IJ, McDermott F. Calcite fabrics, growth mechanisms, and environments of formation in speleothems from the Italian alps and southwestern Ireland. J Sediment Res. 2000;70:1183–96.
Goldbrunner JE, Vasvari V. Hydrogeology and geothermic simulation of the geothermal doublet at Waldkraiburg (Bavaria). Austrian J Earth Sci. 2016;109:99–113.
Gunnarsson I, Arnórsson S. Impact of silica scaling on the efficiency of heat extraction from high-temperature geothermal fluids. Geothermics. 2005;34:320–9.
Kashkovskiy RV, Kuznetsov YI, Kazansky LP. Inhibition of hydrogen sulfide corrosion of steel in gas phase by tributylamine. Corros Sci. 2012;64:126–36.
Klapper HS, Baker MM, Baker MP. Scaling and corrosion behavior of metallic materials after long-term exposure to the geothermal fluid of the north German basin. NACE Int Corros Conf Series. 2016;5:3470–8.
Lanneluc I, Langumier M, Sabot R, Jeannin M, Refait P, Sablé S. On the bacterial communities associated with the corrosion product layer during the early stages of marine corrosion of carbon steel. Int Biodeter Biodegr. 2015;99:55–65.
Lerm S, Westphal A, Miethling-Graff R, Alawi M, Seibt A, Wolfgramm M, Würdemann H. Thermal effects on microbial composition and microbiologically induced corrosion and mineral precipitation affecting operation of a geothermal plant in a deep saline aquifer. Extremophiles. 2013;17:311–27.
Liu ZY, Wang XZ, Liu RK, Du CW, Li XG. Electrochemical and sulfide stress corrosion cracking behaviors of tubing steels in a H2S/CO2 annular environment. J Mater Eng Perform. 2014;23:1279–87.
Mayrhofer C, Niessner R, Baumann T. Hydrochemistry and hydrogen sulfide generating processes in the Malm aquifer, Bavarian Molasse Basin, Germany. Hydrogeol J. 2014;22:151–62.
Mickler PJ, Stern LA, Banner JL. Large kinetic isotope effects in modern speleothems. Geol Soc Am Bull. 2006;118:65–81.
Mundhenk N, Huttenloch P, Sanjuan B, Kohl T, Steger H, Zorn R. Corrosion and scaling as interrelated phenomena in an operating geothermal power plant. Corros Sci. 2013;70:17–28.
Nogara J, Zarrouk SJ. Evaluation of corrosion resistant alloys as construction material for acidic geothermal wells. In: Proceedings 36th New Zealand Geothermal Workshop. Auckland, New Zealand; 2014. p 14.
O’Neil JR, Clayton RN, Mayeda TK. Oxygen isotope fractionation in divalent metal carbonates. J Chem Phys. 1969;51:5547–58.
Parkhurst DL, Appelo CAJ. Description of input and examples for PHREEQC version 3—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. US Geol Surv Tech Methods. 2013;6:497.
Przybycin AM, Scheck-Wenderoth M, Schneider M. The origin of deep geothermal anomalies in the German Molasse Basin: results from 3D numerical models of coupled fluid flow and heat transport. Geotherm Energy. 2017;5:1. doi:10.1186/s40517-016-0059-3.
Regenspurg S, Feldbusch E, Byrne J, Deon F, Driba DL, Henninges J, Kappler A, Naumann R, Reinsch T, Schubert C. Mineral precipitation during production of geothermal fluid from a Permian Rotliegend reservoir. Geothermics. 2015;54:122–35.
Rivera Diaz A, Kaya E, Zarrouk SJ. Reinjection in geothermal fields—a worldwide review update. Renew Sust Energ Rev. 2016;53:105–62.
Seibt A. Tiefbohrung Kirchstockach—Gt KST 1—Langzeitpumptest. BWG Report; 2010. p 22.
Smith P, Roy S, Swailes D, Maxwell S, Page D, Lawson J. A model for the corrosion of steel subjected to synthetic produced water containing sulfate, chloride and hydrogen sulfide. Chem Eng Sci. 2011;66:5775–90.
Spötl C, Mattey D. Stable isotope microsampling of speleothems for palaeoenvironmental studies: a comparison of microdrill, micromill and laser ablation techniques. Chem Geol. 2006;235:48–58.
Stober I. Hydrochemical properties of deep carbonate aquifers in the SW German Molasse basin. Geotherm Energy. 2014;2:13. doi:10.1186/s40517-014-0013-1.
Takai K, Nakamura K, Toki T, Tsunogai U, Miyazaki M, Miyazaki J, Hirayama H, Nakagawa S, Nunoura T, Horikoshi K. Cell proliferation at 122°C and isotopically heavy CH4 production by a hyperthermophilic methanogen under high-pressure cultivation. Proc Natl Acad Sci USA. 2008;105:10949–54.
Valdez B, Schorr M, Quintero M, Carrillo M, Zlatev R, Stoytcheva M, De Dios Ocampo J. Corrosion and scaling at Cerro Prieto geothermal field. Anti-Corros Method Mater. 2009;56:28–34.
Wanner C, Eichinger F, Jahrfeld T, Diamond LW. Unraveling the formation of large amounts of calcite scaling in geothermal wells in the Bavarian Molasse Basin: a reactive transport modeling approach. Procedia Earth Planet Sci. 2017;17:344–7.
Weber, J, Ganz, B, Sanner, B, Moeck, I. Geothermal energy use, country update for Germany. Paper presented at the European geothermal congress, Strasbourg, 19–24 September 2016; 2016.
Wolfgramm M, Obst K, Beichel K, Brandes J, Koch R, Rauppach K, Thorwart K. Produktivitätsprognosen geothermischer Aquifere in Deutschland. Der Geothermiekongress 2009, Bochum; 2009. p 13.
Wolfgramm M, Rauppach K, Thorwart K. New mineral formations and transport of particles in the thermal water loop of geothermal plants in Germany. Z Geol Wiss. 2011;39:213–39.
Würdemann H, Westphal A, Kleyböcker A, Miethling-Graff R, Teitz S, Kasina M, Seibt A, Wolfgramm M, Eichinger F, Lerm S. Microbial metabolic processes affect the operation of geothermal plants and the success of countermeasures. Grundwasser. 2016;21:93–106.
Zarrouk SJ, Woodhurst BC, Morris C. Silica scaling in geothermal heat exchangers and its impact on pressure drop and performance: Wairakei binary plant, New Zealand. Geothermics. 2014;51:445–59.
Zheng Y, Ning J, Brown B, Nesic S. Electrochemical model of mild steel corrosion in a mixed H2S/CO2 aqueous environment in the absence of protective corrosion product layers. Corros Sci Sect. 2015;71:316–25.
Zimer AM, De Carra MAS, Rios EC, Pereira EC, Mascaro LH. Initial stages of corrosion pits on AISI 1040 steel in sulfide solution analyzed by temporal series micrographs coupled with electrochemical techniques. Corros Sci. 2013;76:27–34.
Authors’ contributions
The manuscript was written through contributions of all authors. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Otmar Plank for his help with sampling of scale material at the geothermal power plants and Andrea Shirbaz for providing hydrochemical and operational data. M.Sc. student Sascha Speil is acknowledged for providing photographs of the investigated installations, as well as for his help during sampling and electron microscope analysis. Our laboratory technicians Maria Hierz, Andrea Wolf, Judith Jernej, and Sylvia Perchthold are acknowledged for their dedicated work comprising state-of-the-art (geo)chemical and mineralogical analytical techniques at NAWI Graz Central Lab for Water, Minerals and Rocks. The general as well as detailed suggestions of two anonymous reviewers helped to improve our manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional information. Figures of additional microscopic characterization of scale-fragment fabrics based on reflected and transmitted light and electron microscopy; Tables of minor and trace elemental compositions of the mineral precipitates based on XRF, ICP-OES, and ICP-MS analysis; and stable carbon and oxygen isotopic compositions of selected micromilled and drilled carbonate scales are provided.
Funding
This study was funded by the Austrian Research Promotion Agency (FFG) and the Climate & Energy Fund in the course of the e!Mission program, project “NoScale” (Nr. 843827).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Boch, R., Leis, A., Haslinger, E. et al. Scale-fragment formation impairing geothermal energy production: interacting H2S corrosion and CaCO3 crystal growth. Geotherm Energy 5, 4 (2017). https://doi.org/10.1186/s40517-017-0062-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40517-017-0062-3