
Abstract
Interferon-α2b (IFN-α2b) is a highly active cytokine that belongs to the interferon-α (IFN-α) family. IFN-α2b has beneficial antiviral, antitumour, antiparasitic and immunomodulatory activities. Direct and indirect antiproliferative effects of IFN-α2b have been found to occur via multiple pathways, mainly the JAK-STAT pathway, in certain cancers. This article reviews mechanistic studies and clinical trials on IFN-α2b. Potential regulators of the function of IFN-α2b were also reviewed, which could be utilized to relieve the poor response to IFN-α2b. IFN-α2b can function not only by enhancing the systematic immune response but also by directly killing tumour cells. Different parts of JAK-STAT pathway activated by IFN-α2b, such as interferon alpha and beta receptors (IFNARs), Janus kinases (JAKs) and IFN‐stimulated gene factor 3 (ISGF3), might serve as potential target for enhancing the pharmacological action of IFN-α2b. Despite some issues that remain to be solved, based on current evidence, IFN-α2b can inhibit disease progression and improve the survival of patients with certain types of malignant tumours. More efforts should be made to address potential adverse effects and complications.
Similar content being viewed by others

Background
Malignant tumour is a major public health problem worldwide and the second leading cause of death in the United States. Despite decreased total morbidity and mortality in recent years, the treatment of malignanttumours still faces great challenges, partly due to the high heterogeneity. Tumours of various tissue origins differ in degree of malignancy, and even neoplasms originating from the same tissue show different chemosensitivity. Additionally, due to the lack of specific early clinical symptoms, cancer patients have to face delayed diagnosis and treatment, resulting in an inevitable decline in the clinical diagnosis rate and cure rate [1,2,3]. Surgery is an option for cancer patients diagnosed at an early stage. Patients with advanced carcinoma and certain types of neoplastic lesions, e.g., neoplastic haematologic disorders and malignant bone tumours, have chemotherapy and radiotherapy as options. The increase in the clinical application of neoadjuvant chemotherapy, targeted therapy and immunotherapy in recent years has provided more options for patients and clinicians and revealed new directions for cancer research and drug development [4, 5].
Interferon-α2b (IFN-α2b) is a highly active cytokine protein that belongs to the interferon-α (IFN-α) family. It is an important form of IFN-α for clinical chemotherapy and immunotherapy (the other form is IFN-α2a). As a pleiotropic cytokine, IFN-α2b has beneficial antiviral, antitumour, antiparasitic and immunomodulatory activities [6, 7]. The Food and Drug Administration (FDA) has approved IFN-α2b as a treatment for hairy cell leukaemia, renal cell cancer and melanoma. A large number of clinical studies on IFN-α2b as therapy for leukaemia, melanoma, renal cell carcinoma and other diseases have been carried out. The combined use of IFN-α2b and surgical treatment, targeted therapy and traditional chemotherapy obviously improves the tumour cell killing effect and compensate for the defects of these treatments alone [8,9,10,11,12].
In this review, we focus on the antitumour effects of IFN-α2b, especially its direct and indirect effect on tumour cells. The pharmacological action of IFN-α2b, related pathway signallings and potential regulators are discussed. Furthermore, we review the findings of clinical studies of the application of IFN-α2b, especially effective compatibilities. Finally, we describe the current challenges of IFN-α2b therapy, including its administration route, side effects, related preparations and markers to provide recommendations for future clinical research.
Overview of IFN-α family proteins
In 1957, Isaacs, A. and Lindenmann, J. first discovered the interferon protein. This protein was named “interferon” because it interferes with the replication of the viral genome [13]. Interferons are divided into three groups, type I (IFN-I), type II (IFN-II) and type III (IFN-III), according to differences in molecular structure and antigenicity. IFN-I family includes IFN-β, IFN-ε, IFN-κ, IFN-ω and at least twenty-four subtypes of IFN-α. IFN-δ derived from pigs and IFN-τ derived from sheep are also listed in the IFN-I family. IFN-I plays an important biological role by binding to the common receptors, interferon alpha and beta receptors (IFNARs) [14]. IFN-α is encoded by fourteen different genes, which could explain the abundance of IFN-α subtypes. The molecular weight of each subtype varies from 16 to 26 kDa, and these subtypes have approximately 70% homologous amino acid sequences (Fig. 1A) [15,16,17,18]. Currently, two subtypes of IFN-α2 synthesized by DNA recombination technology, recombinant IFN-α2a and IFN-α2b, are widely used in clinical practice, and they were first tested in phase III clinical studies of certain tumours. Compared with wild-type IFN-α2, the N-terminal of both IFN-α2a and IFN-α2b is deleted. The difference between recombinant IFN-α2a and IFN-α2b is that the 23rd amino acid of IFN-α2a is lysine, while this amino acid is arginine in the IFN-α2b structure (Fig. 1B) [19, 20].

The result of protein sequence alignment. A The differences in protein sequences among IFN-α subtypes. B The differences in protein sequences among wild-type IFN-α2 and two variants (IFN-α2a and IFN-α2b). The sequences were downloaded from the Protein database (https://www.ncbi.nlm.nih.gov/protein/). Sequence alignment was performed by CLUSTALW and ESPript 3.0. Red box, conservative sequence. Yellow box, high similarity. White box, low similarity
Antitumour mechanism of IFN-α2b and relevant potential regulatory factors
Signalling pathways induced by IFN-α2b: the JAK-STAT pathway
The molecular structure of IFN-α2b was shown in Fig. 2. In vivo and in vitro, IFN-α2b can activate the JAK-STAT pathway (also known as IFN-I pathway) [21]. As mentioned above, IFN-α2b can bind to IFNARs. IFNARs are heterodimeric proteins that are ubiquitously expressed and mainly distribute in the cell membrane, and the two domains are encoded by two disparate genes, IFNAR1 and IFNAR2. Once suitable ligands, including IFN-I family members, bind to IFNARs with high affinity, dimerization of IFNAR1 and IFNAR2 is induced, and downstream kinases (also known as Janus kinases, JAKs) are activated, leading to phosphorylation of the substrate, signal transducers and activators of transcription (STATs). Currently, four JAKs (JAK1–3 and tyrosine kinase 2, TYK2) and seven STATs (STAT1–4, 5A, 5B, and 6) have been identified [22]. In the activated IFN-I signalling pathway, IFNAR1 binds to TYK2 and IFNAR2 binds to JAK1 in response to conformational changes of IFNARs (Fig. 3). As a result, JAK1 and TYK2 are activated, and STAT1 and STAT2 are phosphorylated [23, 24]. Then, phosphorylated STAT1 and STAT2 (p-STAT1 and p-STAT2) bind to interferon regulatory factor 9 (IRF9) and form a transcriptional complex named IFN‐stimulated gene factor 3 (ISGF3), which functions by being recruited to conserved genomic regions called the IFN-stimulated response elements (ISREs) and regulates the transcription of downstream IFN‐stimulated genes (ISGs) (Fig. 3) [25,26,27]. Upregulation of ISGs is one of the signs of IFN-I signalling pathway activation. In the context of certain tumours, autocrine and paracrine activity of IFN-α can amplify interferon signalling [28]. However, once the pathway is activated, some negative regulators also come into play to maintain homeostasis and normal cell activities. For example, suppressor of cytokine signalling 1 (SOCS1) can decrease the phosphorylation level of JAK1 and STAT1. Ubiquitin specific peptidase 18 (USP18) interferes with IFN-I signalling by inducing degradation of ISG15 and disrupting IFNAR2-JAK1 binding [29, 30].

The presentation of the 2D and 3D structures of IFN-α2b. The structures were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/)

The presentation of the pathways activated by IFN-α2b. The regulatory mechanisms targeting different parts of the pathways are shown in rounded rectangles
Other pathways
Previous studies on the antitumour mechanisms of IFN-α2b have not always focused on the traditional JAK-STAT pathway. For example, crosstalk between IFN-α2b and the Wnt/β-catenin signalling pathway has been found. Ceballos et al. pointed out that IFN-α2b represses this pathway by downregulating β-catenin and the FZD7 protein and blocking the interaction between β-catenin and TCF/LEF factors such as TCF4. As a result, IFN-α2b impedes the proliferation of hepatocellular carcinoma (HCC) cells and induces apoptosis [31]. Additionally, Parody et al. identified a new indirect molecular mechanism. In a rat preneoplastic liver model, IFN-α2b induced the disinhibition of FoxO3a activity. FoxO3a competed for nuclear β-catenin with TCF/LEF and inhibited the Wnt/β-catenin signalling pathway [32].
In addition, despite the limited amount of correlational research, IFN-α2b has been found to activate several pathways, such as the Beclin1 pathway and Notch pathway, involved in multiple intracellular events, such as growth arrest and autophagy, and these pathways are summarized in Table 1. However, it remains to be seen whether and how these pathways interact with the JAK-STAT pathway.
The direct and indirect effects of IFN-α and the function of ISGs in treating malignancy
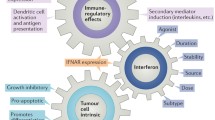
Upon IFN-α2b treatment, different biological effects can be induced depending on the type of targeted cells, and these effects can be direct or indirect. First, IFN-α2b can directly act on malignant cells and induce cell cycle arrest, apoptosis and angiogenesis inhibition, having a strong impact on tumour initiation and progression. Second, IFN-α2b can stimulate the indirect effects including immunomodulatory effects on the tumour microenvironment, including enhanced proliferation, maturation and antigen presentation of immune cells such as dendritic cells (DCs), macrophages (Mφ) and natural killer (NK) cells, which strengthens the innate and adaptive immunity to causative agents and malignancy [43, 44]. Third, direct and indirect effects can lead to the lysis of malignant cells and the release of exposed tumor antigen and further strength the antigen presentation and indirect effects. Fourth, more IFN-α can be secreted by these immune cells activated by indirect effects to strengthen the direct effects. As a result, both effects can exhibit synergy against cancerous lesions under ideal conditions.
For virus-infected cells, IFN-α2b treatment leads to a drastic increase in the expression of ISGs and initiates a signalling cascade. ISGs include hundreds of genes that have similar functions in protecting the host from virus entry, replication and spread, including the myxovirus resistance protein family (MXs) and 2'-5'-oligoadenylate synthetase family (OASs) [45]. The molecular mechanisms of antiviral activity include the following: 1. inhibiting the transport of viruses between intracellular and extracellular environments; 2. inhibiting the transcription and translation of the viral genome; and 3. inhibiting viral DNA replication. Interestingly, some ISGs seem to have tumour suppressor properties (reviewed in Table 2), which is in line with the anticancer effect of IFN-α2b. For example, interferon-induced protein with tetratricopeptide repeats 2 (IFIT2) can inhibit the invasion and metastasis of gallbladder carcinoma. MX1 protein can induce autophagy of prostate cancer cells [46, 47]. Nonetheless, the effect of some ISGs on tumours is still controversial. Apolipoprotein B mRNA editing enzyme catalytic subunit 3 (APOBEC3) was previously regarded as an oncogene due to its function in leading to somatic mutation in cervical cancer, but Green et al. demonstrated that some subtypes can increase the fragility of the leukaemia cell genome and increase the sensitivity to apoptosis induced by inhibitors [48, 49]. Eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) plays a dual role in activating the NF-κB signalling pathway and phosphorylating eukaryotic initiation factor 2α (eIF2α). Thus, its exact function is still unknown [50, 51]. Given this, it could be inferred that the anti-proliferation role of IFN-α2b is the result of overall effect of complex interactions between certain ISGs in the context of different tumours, called ISG portrait, so the carcinogenic function of single ISGs might not conflict with the conclusion. Even so, the relationship between IFN treatment and ISG function is worth investigating, and an understanding of this relationship will provide useful information for clinical treatment of malignancy.
Potential regulatory factors of IFN-α2b pharmacological action
In this section, we discuss the potential regulatory factors of the IFN-α2b-related pathway and the possibility of improving the local environment for proper activation of the IFN-I pathway. The regulatory mechanism is shown in Fig. 3.
IFNAR
The IFNAR family is composed of two distinct forms, IFNAR1 and IFNAR2, which include two different chains of the receptor for IFN-I and mediate receptor-ligand interactions and transduction of extracellular signals (e.g., IFNA2 and IFNW1) (Fig. 4). Similar to other receptor proteins, IFNARs have extracellular domains (ECDs) and intracellular domains (ICDs). The ECD of IFNAR1 comprises a tandem array of four fibronectin III (FNIII) subdomains, while the ECD of IFNAR2 is characterized by two FNIII-like domains. The IFNAR ICD participates in the interaction between TYK2 and IFNAR1 and between JAK1 and IFNAR2 [65].

The presentation of the proteins that interacting with different parts of the IFNAR complex, JAKs and ISGF3. The result is retrieved in STRING database (https://cn.string-db.org/) and visualized by Cytoscape. All the proteins are classified by different signalling pathways (black dotted box)
In contrast to IFNAR2, IFNAR1 has been a larger focus of mechanistic studies of IFN-I signalling. IFNAR1 is crucial for maintaining an active immune reaction, monitoring abnormal proliferation and maintaining hypersensitivity to IFN-I. Once mutation occurs or deficiency develops, the chances of promoting tumour progression increase. Castiello et al. demonstrated that knockout of IFNAR1 (Ifnar1−/−) was associated with earlier onset and marked vascularization in murine breast cancer [66]. The immune-privileged tumour microenvironment induced by IFNAR1 inactivation can be partly explained by interference of immune cell homeostasis. Deletion of IFNAR1 significantly reduces DC-dependent T-cell infiltration into the liver and lung and splenomegaly without altering the frequency of effector/memory-like CD4+T cells [67]. Zanker et al. pointed out that the degranulation of NK cells and lysis of breast cancer cells were impaired upon IFNAR1 knockout [68]. Therefore, IFNAR1 could serve as an important mediator of the direct and indirect effect of IFN-I therapy, which has been validated in the context of myeloid-derived suppressor cells and melanoma cells [69, 70].
The regulatory mechanism related to IFNAR1 is shown in Fig. 3. One of the most studied molecular mechanisms is IFNAR1 protein degradation. The IFNAR1 level is strictly controlled by IFNAR1 ubiquitination and degradation facilitated by the SCF-β-TrCP E3 ligase complex, which can bind to phosphorylated sites [71]. PKR-like ER kinase (PERK) regulates the phosphorylation priming process, and subsequently, casein kinase 1 α (CK1α) induces amplification, leading to the recruitment of the E3 ligase complex [72]. Another mechanism is mediated by matrix metalloproteinase-2 (MMP2) in the context of melanoma. MMP2 is secreted from melanoma cells and then cleaves IFNAR1, leading to inactivation of DCs, induction of T helper 2 (Th2) cell polarization and low levels of STAT1 phosphorylation [73]. Both mechanisms could serve as alternative targets for malignancies. Additionally, IFNAR1 can also be regulated at the posttranscriptional level. miR-93-5p targets IFNAR1 and promotes gastric cancer metastasis. miR-208b and miR-499a-5p produced in response to Hepatitis C virus (HCV) infection destabilize IFNAR1 mRNA directly and antagonize IFN-I signalling [74, 75]. In summary, stabilization of IFNAR1 mRNA and blockade of abnormal protein metabolism might be viable strategies to combat the effects of interferon resistance.
JAK1 and TYK2
JAK1 and TYK2 belong to the JAK kinase family, a subgroup of the nonreceptor protein tyrosine kinases that possess similar molecular structures. The N-terminal moiety of JAK kinases includes a FERM (band 4.1, ezrin, radixin, moesin) domain and an Src-homology 2 domain (SH2), both of which are structurally important for binding to IFNAR. The C-terminal moiety of JAK constitutes the catalytic kinase domain, whose active conformation can be maintained by phosphorylation of certain tyrosine residues and blocked by an adjacent pseudokinase domain [76, 77]. The catalytic kinase domain exhibits a high degree of homology among the four members. Despite differences in amino acid sequence and molecular structure, JAK3 has 84% sequence homology with JAK1, 87% sequence homology with JAK2 and 80% sequence homology with TYK2 [78].
JAK1 and TYK2 play critical roles in catalysing the phosphorylation process and inducing the active form of STATs to provoke direct and indirect effects. Additionally. JAK1 could bind to IFNLR1, mediating the crosstalk with IFN-III signals (Fig. 4). These two kinases are involved in different pathways, not only the IFN-I pathway but also the JAK1-STAT3 pathway, a harmful carcinogenic signalling pathway in most cases, conflicting with the antitumour effects of JAK1-STAT1 signalling (Fig. 4) [79, 80]. JAKs are necessary for maintaining the numbers and maturation of NK cells, keeping the normal function of Mφ and overcoming immunotherapy resistance, indicating that the role of JAK1-STAT3 pathway in the parenchyma is worth investigating, as the proper immune response is suppressed in cancerous lesions [81,82,83]. Consistent with this view, JAK1 was found to be critical for inducing resistance to NK cells in melanoma and recurrence of HCC [84, 85]. Indeed, overexpression of both members has been observed in multiple cancer types, along with excessive activation of JAK1-STAT3 signalling. These phenomena may be partly explained by gain-of-function mutations associated with overexpression or hyperactivation. To address this problem, a variety of JAK inhibitors have been developed to block STAT3 signalling by disturbing the phosphorylation-associated activation loop and impairing the enzymatic activity of JAKs. JAK inhibitor (e.g., ruxolitinib and AZD1480) treatment significantly reduced the level of phosphorylated STAT3 and inhibited the proliferation of K-RAS-driven lung adenocarcinoma, small cell lung cancer and triple-negative breast cancer [86,87,88]. Cirsiliol binds with TYK2 and then suppresses the kinase activity, leading to growth arrest of oesophageal squamous cell carcinoma cells [89]. More exogenic factors are reviewed in Table 3. In the context of hypoxia, JAK1/STAT3 is activated, but transcriptional and translational downregulation of the participants in the IFN-I pathway is also observed. Although targeting JAKs has proven to be a promising strategy, this treatment can interfere with the proper activation of the IFN-I pathway because of the pivotal role of JAKs in the process [43, 90, 91]. The relationship between the two pathways remains to be validated.
In addition to inhibitors that are externally administered, JAKs can also be regulated by intracellular proteins in two main ways: 1. modifying the expression of JAKs; and 2. decreasing JAK phosphorylation levels (Fig. 3). First, the protein level of JAKs can be modulated by the degradation pathway. Cytokine-inducible SH2-containing protein (CIS) was found to interact with JAK1, induce proteasomal degradation in NK cells and then mediate resistance to melanoma and prostate and breast cancer metastasis [98]. Quick et al. demonstrated that JAK1 was a substrate for USP6, which leads to deubiquitination of JAK1 and amplification of STAT3 signal [92]. Second, phosphorylation of JAKs can be altered. For example, cytotoxic T lymphocyte-associated antigen 4 (CTLA4) can activate TYK2 by subsequent phosphorylation, resulting in the promotion of lymphoma cell proliferation [105]. Hypoxia inducible factor 1α (HIF-1α) has a similar cellular function to increase the phosphorylation level of JAK1 and promotes the initiation, progression, and recurrence of gliomas [90]. Third, similar to IFNAR1, noncoding RNA (ncRNA) can alter the stability of JAK mRNAs, for example, miR-3677-5p in liver cells, circRNA-9119 in HCC cells and miR-93 in breast stem cells [122, 123]. Similar intracellular factors are reviewed in Table 3, and despite the absence of sufficient evidence, these factors could have the potential to overcome resistance to interferon-based drugs, e.g., BRCA1 (BRCA1 DNA repair associated), a famous target for cancer therapy, significantly upregulates JAK1 expression and interacts with STAT1 dimers, which govern the IFN-dependent anti-proliferative response of breast cancer [96].
One of the reasons for the functional diversity of JAKs might be the nonspecific binding of JAKs and the abundance of ligands. As shown in Fig. 4, JAK1 can interact with multiple receptor proteins, such as CSF2RB, the receptor for IL-3 and IL-5; IL2RB, the receptor for IL-2; IL6ST, the receptor for IL-6 and IL10RA, the receptor for IL-10, indicating that competitive binding of JAKs might give rise to low efficacy of IFN-α2b. After all, most of these ligands have been reported to be principal culprits for abnormal JAK-STAT3 signalling and factors contributing to tumourigenesis. One of the most studied cytokine proteins is leukaemia inhibitory factor (LIF). LIF significantly upregulates the phosphorylation of JAK1 and sustains JAK1/STAT3 signalling by binding to the receptor LIFR, strongly preventing the differentiation of HCC cells and promoting the metastasis and an immunosuppressive phenotype of prostate cancer [124,125,126]. Targeting such cytokines or cytokine-associated signals might be an effective strategy to address the binding of JAKs (Fig. 3). For example, arsenic trioxide treatment reduces the expression of LIF in HCC [125]. Albrengues et al. found that LIF-induced constitutive phosphorylation of JAK1 was correlated with DNA methylation, which is a well-known, effective target for treating malignancy [124]. Previously, IL-6, IL-11, IL-15 IL-20, and IL-22 were reported to have similar effects on JAK1. IL-6 also contributes to phosphorylation of TYK2 [127,128,129,130,131,132]. However, in prostate cancer, IL-6 enhances the antiproliferative effect of IFN-α [133]. The exact relationship between interferons and these cytokines depends on the cellular context and needs to be further studied.
In conclusion, potential solutions for overcoming poor response to IFN-α2b include the following: 1. moderate regulation of JAK activity and drug compatibility; 2. targeting of intracellular JAK regulatory factors; and 3. the JAK by targeting harmful ligands and reversing the direction of signalling.
ISGF3
ISGF3 is composed of p-STAT1, p-STAT2 and IRF9. STAT1 and STAT2 belong to the STAT family which encodes seven transcription factor proteins characterized by structurally and functionally conserved regions, including the N-terminal domain (NTD), coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LD), SH2, tyrosine-phosphorylation (PY) site, and transcriptional activation domain (TAD) [134]. Among these domains, the SH2 domain is susceptible to many STAT inhibitors and mediates homodimerization and heterodimerization (e.g., the STAT1-STAT2 dimer in the activated IFN-I pathway and STAT1 homodimer in the activated IFN-II pathway) along with the NTD. The transcriptional activity of STATs can be regulated by serine phosphorylation of the TAD and recruitment of additional transcriptional activators [135]. CCD mediates the nuclear localization process by selectively binding to the IRF-association domain (IAD) of IRF9 [136].
As essential messengers and mediators of the IFN-I-associated direct and indirect effects, STAT1 and STAT2 can not only provoke STAT-mediated apoptosis, but also participate in enhancing the immune response by upregulating the amount of cytotoxic T lymphocytes (CTLs) and NK cells, both of which are involved in inflammation as well as oncogenesis [136]. STAT1 can act as a negative regulator of T-cell exhaustion and triggers immune responses in head and neck squamous cell carcinoma [137].
ISGF3 can also be regulated by intracellular molecules via an altered phosphorylation level, directly regulating the expression of key components in the complex. The serine threonine-specific protein kinase C-θ (PKC-θ) can increase STAT1 phosphorylation in NK cells in the context of lymphoma [138]. Adenosine deaminase acting on RNA (ADAR1) was reported to block the transport of ISGF3 into the nucleus by upregulating miRNA-302a and thereby targeting IRF9 and STAT1 in gastric cancer [139]. As mentioned above, BRCA1 can also increase the expression of STAT1 and STAT2, providing the evidence of the crosstalk between BRCA1 and IFN-I signalling [140]. Retinoic acid-inducible gene I (RIG-I), an important participant in innate immunity, contributes to IFN-I production and phosphorylation of STAT1 [141]. More factors are reviewed in Table 4. Interestingly, Lu et al. found that in diffuse large B-cell lymphoma, STAT3 could inhibit the expression of STAT1 and STAT2, and Li et al. demonstrated that RIG-I could also be repressed by STAT3 in melanoma.(141, 152) Both studies provide evidence for the antagonism between JAK-STAT3 and the IFN-I pathway, which could be a theoretical basis for the combination of STAT3 inhibitors and IFN-α2b.
The function of ISGF3 depends on proper corepressors and coactivators in transcription initiation complexes (TICs). As shown in Fig. 4, protein kinase C-δ (PKC-δ, encoded by PRKCD) influences the phosphorylation level of STAT1 and controls the activation of ISGF3, similar to PKC-θ [138, 158]. Yang et al. found that IFIT3, a known ISG, can bind to STAT1 and STAT2 and enhance the formation of STAT1-STAT2 heterodimers during IFN-α treatment in HCC, indicating the feedback loop between IFN-I signalling pathway and ISGs [149]. Other important factors include IRF1 and IFNGR1, indispensable participants in the IFN-II pathway; EGFR, the receptor of epidermal growth factor (EGF); PR, the receptor of progesterone and PIAS1, a specific inhibitor of p-STAT1 [146, 159,160,161]. It is worth noting that some histone acetyltransferases, such as EP300 and CREBBP, are also involved (Fig. 4), indicating the correlation between ISGF3 and epigenetic modification mediated by the chromatin remodelling complex. Indeed, Testoni et al. pointed out that upon IFN-α treatment in HCC, ISGF3 was recruited to the promoter of DNp73, an antiapoptotic protein and p53 repressor, along with histone deacetylases (HDACs) and EZH2, a component of polycomb repressive complex 2 (PRC2) [162]. HDACs can be further regulated by S-nitrosylation, as reported in melanoma [147]. In summary, in addition to differences in functional status and intracellular factors, individual differences in patients, which might be evaluated based on the level of endocrine factors, should also be considered. Additionally, targeting epigenetic modifications with inhibitors of downstream regulatory enzymes as an adjunct strategy to IFN-α2b treatment might be an optional worth studying (Fig. 3).
Clinical application of IFN-α2b in treating malignancy
Owing to its broad-spectrum antiviral roles, IFN-α2b has become an essential part of standard treatments for virus infection. IFN-α2b has been proven to be efficacious in relieving West Nile virus, influenza A virus (IAV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection [163,164,165,166]. Moreover, application of IFN-α2b in patients with some virus-related malignancies, such as HIV (human immunodeficiency virus)-related Kaposi sarcoma and HCV-related HCC, was shown to be successful [167, 168]. Progress in treating malignancies not related to virus infection has also been made.
Melanoma
The majority of clinical trials involving the application of IFN-α2b as adjuvant treatment for malignancy focus on melanoma. The regimens have included high-dose IFN-α2b (HDI) and low-dose IFN-α2b (LDI). LDI had limited effects in prolonging survival time and lowering potential toxicity. No obvious synergistic therapeutic effect was seen when LDI was combined with other medicines, e.g., bevacizumab [169, 170]. In contrast, the HDI regimen pioneered by Kirkwood et al. was proven to improve the prognosis of patients with high-risk melanoma or ulcerated melanoma [171]. The most utilized protocol is a combination of initial induction therapy (20 MU/m2 i.v. daily for 5 days each week for 4 weeks) and maintenance therapy (10 MU/m2 s.c. three times a week for 48 weeks) [172]. The adjuvant HDI regimen proved effective in prolonging the relapse-free survival (RFS) and overall survival (OS) of melanoma patients in some previous clinical trials, e.g., the E1684 trial in 1996 (RFS: 1 vs. 1.7 years, OS: 2.8 vs. 3.8 years, continuous disease-free rate: 26% vs. 37%) [171]. When the criterion of recruiting melanoma patients was updated in the E1690 trial in 2000 (e.g., patients with T4 primary tumours were included), significant improvement in RFS was still observed, but a difference in OS was not obvious, partly because of the better clinical support and OS improvement of the control group [173]. A meta-analysis combined four randomized clinical trials with large-scale cohorts and found that patients with resected high-risk melanoma could derive RFS and melanoma-specific survival (MSS) benefits from adjuvant HDI [174]. Although the value of HDI has been shown, the results might depend on stage and/or treatment type. IFN-α2b failed to strengthen the immune responses of patients with stage IV melanoma based on the results of enzyme-linked immunospot analysis [175].
To reduce toxicity and improve tolerance, many attempts have been made to design new regimens in combination with IFN-α2b as adjuvant chemotherapy for patients with unresectable melanoma. Unfortunately, the result has not been satisfactory. Among the recently used monoclonal antibody preparations, ecromeximab and ipilimumab proved to be safe in combination with the HDI regimen, whereas no significant improvement in OS was observed [176,177,178]. The addition of anticancer cytokine preparations, e.g., IL-2, granulocyte-monocyte colony stimulating factor (GM-CSF) and endostatin, failed to induce the expected amplification of the immune response and increased the incidence of adverse reactions in metastatic melanoma patients [174, 179, 180]. Some putative therapeutic regimens composed of traditional chemotherapeutic drugs were found ineffective in clinical trials, e.g., the IFN-α2b + dacarbazine + cisplatin + fotemustine regimen and the IFN-α2b + dacarbazine + cisplatin + carmustine + tamoxifen regimen [181, 182]. Encouragingly, temozolomide, an alkylating agent, was shown to be beneficial for controlling multiple metastases in combination with IFN-α2b. This regimen was demonstrated to be relatively simple, safe and effective and even allows outpatient or home application [183, 184]. Even so, it could be speculated that IFN-α2b is unsuitable for the treatment of metastatic melanoma, partly due to severely impaired immune responses and blocked indirect effects.
Haematological tumours
The application of IFN-α2b in leukaemia mainly focuses on the treatment of chronic myelogenous leukaemia (CML). In addition to direct antitumour mechanisms, IFN-α2b was shown to eliminate CML cells by activating circulating PR1-specific CTLs, a subtype of T cells involved in cytogenetic remission and usually deleted in untreated CML [185]. IFN-α2b plus a low-dose cytarabine regimen was used to control chronic-phase myeloproliferation and delay progression to the accelerated phase. After application of the combined regimen, according to Lindauer et al., the survival rate at 3 years was 77%. Maloisel et al. reported a 3-year survival rate of 79% [186, 187]. However, at present, due to its toxicity and unclear application utility, the IFN-α2b regimen is considered a second-line chemotherapy strategy or alternative choice for maintenance therapy. Clinical trials with larger cohorts are needed. Even so, the combination of IFN-α2b with first-line targeted drugs, e.g., dasatinib and imatinib, was demonstrated to induce increased cytogenetic response rates and manageable toxicity [188, 189].
Defective immune surveillance as a result of Epstein–Barr virus (EBV) infection is an important inducer of proliferative disorders and even lymphoma, necessitating the application of IFN-α2b. IFN-α2b can be utilized for low-grade lymphomatoid granulomatosis (LYG), a rare nonmalignant lymphoproliferative disorder, and potentially prevent progression to overt lymphoma by augmenting the immune response to EBV. However, this regimen is not applicable for high-grade lesions that are characterized by insensitivity to immunopotentiators [190, 191]. For the treatment of follicular lymphoma, a kind of indolent B-cell lymphoma, the combination of IFN-α2b with traditional chemotherapeutic drugs was proven to induce a good response, such as the cyclophosphamide + vincristine + prednisone (CVP) + IFN-α2b regimen followed by IFN-α2b maintenance therapy [192, 193]. IFN-α2b therapeutic schedules can also be used for T-cell lymphomas. Geskin et al. argued that IFN-α2b can impair the abnormal immunosuppression mediated by T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) in Sézary syndrome patients, providing evidence for the medical compatibility of the above strategy [194]. For early-stage Sézary syndrome patients, the psoralen + ultraviolet A irradiation (PUVA) + IFN-α2b regimen is a common systemic combination therapy. The statistical results from three distinct centres indicated complete remission rates of 36%, 73% and 84% [195,196,197].
Digestive system tumours
Strong antiproliferation effects of IFN-α2b were observed in HCC cell lines and an HCC animal model, providing preclinical evidence for the next stage of clinical trials [198]. The combination of IFN-α2b and 5-fluorouracil (5-FU) was suitable for patients with advanced HCC [199]. For HCC patients who had already received radiofrequency ablation, long-term maintenance therapy with IFN-α2b was shown to remarkably inhibit recurrence and prolong survival time [200]. Notably, hepatitis viruses participate in not only the transformation of normal liver to cirrhotic liver, a precancerous lesion, but also further development into HCC. Even when HCC develops, patients can still benefit from antiviral treatment for the prevention of secondary systemic infection and hepatic insufficiency [201]. Hence, the application of IFN-α2b was extended to cases of chronic virus infection, in which it can function to suppress deterioration as a promising complementary therapy for antiviral treatment. The evidence is as follows. First, antiviral drugs, such as telbivudine and entecavir, were able to impair the proliferation of virus-infected HCC cells [202]. Second, the combination of IFN-α2b and antiviral drugs appeared to be more effective than monotherapies. Francesco et al. found that virus infection-related cirrhosis patients with a sustained response to IFN-α2b plus ribavirin tended to have a lower HCC incidence. This regimen overcame the disadvantage of IFN-α2b monotherapy, which failed to improve the clinical outcome, although a curative biochemical response was observed [167, 203, 204]. Third, for those with active hepatitis or cirrhosis, IFN-α2b might prevent the aggravation induced by other risk factors. Liu et al. found that IFN-α2b could significantly relieve ethanol-triggered HBV replication and liver damage [205].
Compared with HCC, gastrointestinal cancer has insufficient clinical data, partly due to poor response and unnecessary side effects. The IFN-α2b + 5-FU + leucovorin regimen showed limited clinical benefits and increased toxicity in patients with rectal carcinoma [206]. In gastric cancer, IFN-α2b administered in a complex regimen and via special delivery methods exerted therapeutic effects [207]. The role of IFN-α2b in the treatment of cholangiocarcinoma is controversial. Kasai et al. argued that the IFN-α2b plus 5-FU regimen was suitable for the treatment of advanced cholangiocarcinoma. Patt et al. added two agents, cisplatin and doxorubicin, to this regimen, yet the curative effects could not counteract the increase in toxicity. To date, a standard regimen containing IFN-α2b has not been determined [208, 209].
Other tumours
The application of IFN-α2b is relatively rare in other common cancers. IFN-α2b plus lanreotide was shown to be effective for relieving clinical symptoms and decreasing serum calcitonin levels in advanced medullary thyroid carcinoma patients [210]. The IFN-α2b + all-trans retinoic acid (ATRA) regimen, IFN-α2b + paclitaxel + concomitant radiation regimen and IFN-α2b + cisplatin + 5-FU + leucovorin + concomitant radiation regimen were proven to be effective in advanced renal cell carcinoma, advanced ovarian cancer and advanced nasopharyngeal cancer, respectively [211,212,213,214,215]. In some rare malignancies, such as cutaneous angiosarcoma and Langerhans cell sarcoma, the antitumour effect of IFN-α2b has been validated [216, 217].
Notably, instead of systematic therapy, topical delivery of IFN-α2b has been attempted in certain types of tumours whose primary sites are easy to access, especially ophthalmic neoplasms. Similar topical regimens, such as intravesical delivery of IFN-α2b plus Bacillus Calmete-Guerin (BCG), have been tried in patients with superficial bladder cancer, yet an actual curative effect is controversial [218]. Application of IFN-α2b in eye drops was shown to be successful in the treatment of ocular surface squamous neoplasia and conjunctival epithelial neoplasia [219,220,221,222]. For those with refractory epithelial neoplasia, addition of mitomycin C should be considered [221]. However, the decision to apply topical IFN-α2b should be made carefully after the neoplasm is diagnosed to be highly invasive. Although no significant differences in the recurrence rate between surgery and IFN-α2b were found by Nanji et al., the reduction of cancer cell burden induced by surgery was not achieved by IFN-α2b [222]. Selection of either treatment should be based on pathologic stage and grade.
The current challenges of IFN-α2b treatment
Many studies have shown the antineoplastic role of IFN-α2b, which has been approved by the FDA for the treatment of certain kinds of tumours. However, there are several factors that have prevented IFN-α2b from becoming an established therapy. First, due to tumour heterogeneity and differences in mutational burden, certain types of tumours have relatively poor responses to IFN-α2b treatment at the molecular mechanism level [223]. Second, the reports about alternative biomarkers for the inclusion of the patients suitable for IFN-α2b treatment and indicators for controllable therapeutic responses in long-term IFN-α2b therapy are greatly needed. Third, IFN-α commonly causes almost inevitable side effects in certain cases, probably because of the nonspecific stimulation and enhancement of inflammation pathways and off-target effects on non-neoplastic tissues. Fourth, various IFN-α2b formulations differ in bioavailability and tissue specificity.
Challenges in the poor response to IFN-α2b treatment caused by multiple factors
As mentioned above, the proper induction of IFN-I signalling pathway requires to satisfy many conditions, e.g., intact JAK-STAT pathway. However, in cancerous lesions, endogenous IFN-I signals are significantly severely suppressed, which could be partly explained by tumour heterogeneity and interactions among various cell subsets. Additionally, mutational burden arising from hypoxia stress and overgrowth can interrupt the IFN-I pathway, which should be taken seriously. JAK mutations, such as TYK2 mutations in malignant peripheral nerve sheath tumours (approximately 60%) and acute leukaemia (approximately 40%) and JAK1 mutations in acute leukaemia (27.3%) and certain subtypes of prostate cancer (68%) could inhibit the IFN-I signals and even reverse the biological effect [224,225,226,227]. Substantial dysfunction of ISGF3 is found as well. In addition to the downregulation of STATs caused by underlying molecular mechanisms, accumulated mutations (e.g., STAT1-Y701F and STAT2-Y690F) also play important roles in preventing ISGF3 formation and thereby blocking IFN signals, indicating that it is necessary to evaluate the functional status of the participants in the pathway before deciding on IFN-α2b treatment [228, 229].
Target therapies via virus vectors, called oncolytic virus therapies, were recently developed and hopeful for overcoming the resistance to IFN-I. Oncolytic virus (OV) refers to these modified virus vectors that can specifically infect cancer cells [230]. The replication of OV is more effective in tumour than normal tissues partly due to unique characteristics of malignant cells, such as abnormal signal activation (e.g., RAS/RAF1/MEK/ERK signalling pathway), overactive biosynthesis function and instable genome for the convenience of viral genetic recombination [231]. The antitumour mechanism of OV was summarized as follows: First, the replication and accumulation of OV can provoke the lysis of malignant cells. Second, the residual lysis and OV itself could stimulate the innate and adaptive immunity by cytokine secretion and antigen presentation. Third, the coding genes carried by genetically engineered OV vectors could facilitate the antitumour effects [232].
The relation between IFN-I and OV remains controversial. As OV derives from wild-type virus with pathogenicity and immunogenicity, the activation of IFN-I signals upon the systematic or intratumoural application of OV (e.g., induction of certain ISG portraits) is regarded as antiviral and protective responses, which limits the normal antitumour oncolytic process. On the contrary, the impact of OV could be maximized in those lesions with minimal T cell infiltrates, absence of IFN-I signatures, and the presence of immunosuppressive cells [233]. The percentage of infected benign prostate cells is lower than metastatic cancer cells under the same multiplicity of infection (MOI) indicating the amplification of OV genomes significantly slows down because of intact IFN-I signalling. In contrast, glioma tumour cells and melanoma cells, characterized by the restricted intrinsic IFN-I responses, show better sensibility to OV and worse cell viability [234,235,236]. Additionally, OV contributes to the specific pattern of immune cell infiltration (e.g., CD8+ T cells and CD4+T cells) especially in IFNAR-knockout tumours [237]. When IFN-I signal was blocked by JAK inhibitors, the progeny yield of OV significantly increased, indicating enhanced oncolysis process [238, 239]. Nevertheless, on the other hand, moderate activation of IFN-I response needs to be maintained endogenously or exogenously for the following reasons. First, OV and induced IFN-I responses have common antitumour mechanism, e.g., increasing the phosphorylation level IRF3 for sustained secretion of IFN-I [240]. Second, appropriate levels of IFN-I can sustain IFN-I-dependent stimulation to NK cells, necessitating the insertion of IFN-I or tumour antigen encoding genes into the structure of OV. Notably, excessively high dose of IFN-I may decrease the NK cells in peripheral circulation [230, 241, 242]. Third, in some cases, the cytotoxicity of OV seems to be independent of antitumour IFN-I signals. For example, in the context of pancreatic cancer, OV infection is not influenced by ISG upregulation and the activation of the stimulator of interferon genes (STING). In glioblastoma cells, knockdown of certain ISGs can even inhibit the proper function of OV [242,243,244]. Recently, the strategy that inhibits the IFN-I signalling during the initial phase of OV infection by small molecule chemical (BLT-1) was proved to be effective to induce efficient virus replication and strong immune response, which balanced both sides of IFN-I functions [245].
Challenges and advances in the inclusion of patients and indication of therapeutic effects
Long-term IFN-α2b therapy is often chosen to induce clinical remissions and prevent future recurrence. However, as mentioned above, it is far from a reasonable choice to apply IFN-α2b to those with exhausted immune systems and poor general conditions (e.g., elevation of aminotransferase, renal hypofunction and simultaneous peripheral blood cell reduction). The appropriate application parameters and indications are worth investigating, though these are common problems of all the toxic chemicals for chemotherapy. Except for traditional laboratory diagnosis, diagnostic biomarkers are one of many possible solutions for confirming the necessity and feasibility of IFN-α2b therapy in different patients. Tarhini et al. pointed out that IFN-α2b should be given to patients with a higher mortality risk to avoid unnecessary toxicity and cost. This risk could be evaluated by a prognostic model based on tumour necrosis factor alpha receptor II (TNF-RII), transforming growth factor alpha (TGF-α), tissue inhibitor of metalloproteinases 1 (TIMP-1) and C-reactive protein (CRP) levels [246].
During the IFN-α2b treatment, indicators of curative effects can be helpful for later decisions. IFN-α2b can induce a state of autoimmunity that is beneficial to the prognosis, which can be detected by the measurement of autoantibodies [247]. Some components of the IFN-I pathway, such as p-STAT1 and ISGs, can also indicate the pharmacological response to IFN-α2b [21]. In a joint laboratory and clinical trial (E1690), modulation of intercellular adhesion molecule 1 (ICAM1) was observed by flow cytometry upon IFN-α2b treatment [248]. Additionally, two studies from distinct centres focused on the role of cytokines, which are relatively easy to detect and could be potential markers for monitoring disease progression during IFN-α2b therapy. Yurkovetsky et al. found that upon IFN-α2b treatment, the levels of proinflammatory cytokines, including serum tumour necrosis factor-α (TNF-α) and various ILs, were upregulated, while the levels of angiogenic factors, including vascular endothelial growth factor (VEGF) and EGF, were downregulated [249]. Hofmann et al. found that in stage II and III melanoma patients, a low level of TNF-α indicated a poor response and unfavourable RFS, while a high level of serum TNF-α correlated with toxicity [250]. Despite all the progress noted above, most conclusions have come from observational studies. The precise role of these markers in clinical practice remains unclear and needs to be validated by higher-level evidence.
Challenges in the adverse effects and complications triggered by IFN-α2b
One of the important factors limiting the large-scale application of IFN-α2b in treating malignancy is the prevailing adverse effects, although they are not life-threatening in most cases. In a retrospective study of melanoma, 71% of the patients were found to experience at least one kind of adverse effect, but no drug-related death was observed [251]. Another study on Sézary syndrome reported a similar percentage of patients who experienced a toxic response (60%) [196]. The common adverse effects in studies using IFN-α2b monotherapy have been reviewed. The most frequent side effects were fever and fatigue. In addition, adverse effects of the digestive system of different grades were also common, such as anorexia, diarrhoea, vomiting and hepatic dysfunction. Furthermore, haematologic toxicity could also be observed, e.g., neutropenia, thrombocytopenia and anaemia. Neurologic complications, e.g., anxiety, depression and cognitive disturbance, and metabolic complications (e.g., hyperglycaemia and hyperlipemia), were relatively unusual. The most common adverse effect was influenza-like symptoms, featuring a series of symptoms including fever, fatigue, rigors, arthralgia, myalgia, headache and sweating, indicating that this complication might be associated with nonspecific activation of the inflammatory response. Although most adverse effects were found to be moderate, they were undesirable and painful for many patients who received IFN-α2b, leading to a relatively high rate of treatment discontinuation, especially for those with poor overall health caused by primary disease. Most adverse effects could be relieved by dose reduction and even drug withdrawal, which was accompanied by a dose-dependent delay in a strong therapeutic response. In addition, poor compatibility often induced unnecessary complications, and the overall survival was not improved, which are challenges for the application of IFN-α2b in the treatment of multiple tumours [169]. Even so, most side effects can be relieved by symptomatic treatments, e.g., the application of non-steroid anti-inflammatory drugs (NSAIDs) and colony-stimulating factor preparations could relieve fever and leukocytopenia, respectively. Serious adverse effects, such as hypohepatia, proteinuria and myelodysplasia and neuropsychiatric symptoms, can be solved by medication discontinuation and convalescence in most cases.
Challenges and advances in the delivery and preparation of IFN-α2b
In clinical use, IFN-α2b is usually administered subcutaneously to exert antitumour effects [206, 211, 252]. Some studies have reported that intravenous administration is beneficial for promoting lymphatic absorption and targeting the lesion site [253]. For the treatment of ophthalmic tumours, subconjunctival injection and eye drops can be considered as well [254]. Additionally, IFN-α2b in combination with a melanoma vaccine can enhance the antigen presentation response to kill tumour cells in the treatment of melanoma. New vaccine preparations, such as HeberFERON, are available but are still controversial [174, 255,256,257]. In addition, the overexpression of IFN-α2b induced by adeno-associated virus vectors (AAV) can inhibit the proliferation of prostate cancer cells, which has been validated in animal studies [258,259,260,261].
Natural IFN-α2b has antiviral, anticancer, antiparasitic and immunoregulatory pharmacological effects. Nonetheless, it has disadvantages as well, such as poor stability and potential immunogenicity, which could lead to mostly uncontrollable biological degradation. For patients with cancer, this degradation necessitates an excessive dose to achieve ideal therapeutic effects, which increases the incidence of complications and limits wide usage [262, 263]. To address this problem, pharmaceutical advances to add poly (ethylene glycol) (PEG) modifications to natural IFN-α2b molecules, named PEG-IFN-α2b, have been made. This preparation maintains the advanced structures of IFN-α2b, inhibits drug metabolism, and decreases filtration by the kidney, resulting in a prolonged plasma half-life and a lower frequency of administration (once per week) [264, 265]. Although the modification decreases the biological activity of IFN-α2b and its binding ability with interferon receptors, there is no significant difference in antiviral and antitumour effects in vitro [266]. In recent years, more IFN-α2b preparations have been developed, such as IFN-α2b with polysarcosine modifications and cyclic chimeric IFN-α2b. However, the clinical efficacy has not been supported by sufficient evidence and needs to be tested in more clinical studies. Moreover, excessive modification of IFN-α2b might result in heterogeneous mixture of isomers and decrease efficiency, indicating that the balance should be achieved by more evidence of pharmacokinetics and clinical trials [267, 268].
Conclusions and future perspectives
IFN-α2b clearly plays a role in antitumour therapy, not only by enhancing the systematic immune response but also by directly killing tumour cells, just as a role serving as an archer as well as an arrow. Based on the current evidence, IFN-α2b inhibits disease progression in and improves the survival of patients with certain types of malignant tumours, such as melanoma, leukaemia, lymphoma and HCC. In addition, because compared with other antitumour agents, IFN-α2b preparations can be relatively easily and inexpensively delivered, IFN-α2b might be a convenient solution for outpatient medication and therapy in areas with lower economic status and without access to newly developed drugs.
Despite the advantages mentioned above, after reviewing all the literature, we found that most major breakthroughs were made before 2010. In addition, the extension of IFN-α2b treatment to other tumours has been unsatisfactory and beset with challenges. One of the critical reasons is the adverse effects and complications that, as mentioned above, lead to interruption of therapy plans and increase potential biases of clinical trials, such as reporting bias, diagnostic suspicion bias and observer bias. Therefore, addressing the drug resistance and nonspecific inflammatory responses caused by IFN-α2b should be a focus of future studies. According to the majority of available evidence, the following aspects may lead to solutions. First, there should be reasonable strategies for administering further IFN-α2b treatment in patients. The general conditions should be evaluated by strict physical examination and laboratory inspection. Gene detection can be conducted before IFN-α2b treatment to recognize potential alterations of the important participants in the JAK/STAT pathway. Second, evidence-based therapies combining pharmacotherapies with IFN-α2b should be developed, as some of the clinical trials seemed to have few or weak evidences in phase I clinical study. These combinations should be based on the animal studies and mechanism researches associated with IFN-α2b or members of related pathways, e.g., the JAK/STAT pathway, rather than simply on the clinical experience or historical documents. Third, a timely plan for dealing with potential adverse effects and complications of IFN-α2b should be developed, which deserve serious consideration by researchers and operators. Fourth, development of new preparations of IFN-α2b should be encouraged. In particular, agents with higher tissue targeting specificity and higher potency should be developed for advanced-phase clinical trials.
Availability of data and materials
Not applicable.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- AAV:
-
Adeno-associated virus vectors
- ADAR1:
-
Adenosine deaminase acting on RNA
- APOBEC3:
-
Apolipoprotein B mRNA editing enzyme catalytic subunit 3
- ATRA:
-
All-trans retinoic acid
- BCG:
-
Bacillus Calmete-Guerin
- BRCA1:
-
BRCA1 DNA repair associated
- CCD:
-
Coiled-coil domain
- CIS:
-
Cytokine-inducible SH2-containing protein
- CK1α:
-
Casein kinase 1 α
- CML:
-
Chronic myelogenous leukaemia
- CRP:
-
C-reactive protein
- CTL:
-
Cytotoxic T lymphocyte
- CTLA4:
-
Cytotoxic T lymphocyte-associated antigen 4
- CVP:
-
Cyclophosphamide + vincristine + prednisone regimen
- DBD:
-
DNA-binding domain
- DC:
-
Dendritic cell
- EB:
-
Epstein–Barr virus
- ECD:
-
Extracellular domain
- EGF:
-
Epidermal growth factor
- EIF2AK2:
-
Eukaryotic translation initiation factor 2 alpha kinase 2
- eIF2α:
-
Eukaryotic initiation factor 2α
- FDA:
-
The Food and Drug Administration
- FNIII:
-
Fibronectin III subdomain
- GM-CSF:
-
Granulocyte-monocyte colony stimulating factor
- HCC:
-
Hepatocellular carcinoma
- HCV:
-
Hepatitis C virus
- HDAC:
-
Histone deacetylase
- HDI:
-
High-dose IFN-α2b
- HIF-1α:
-
Hypoxia-induced hypoxia inducible factor 1α
- HIV:
-
Human immunodeficiency virus
- IAD:
-
IRF-association domain
- IAV:
-
Influenza A virus
- ICAM1:
-
Intercellular adhesion molecule 1
- ICD:
-
Intracellular domain
- IFIT2:
-
Interferon-induced protein with tetratricopeptide repeats 2
- IFNAR:
-
Interferon alpha and beta receptor
- IFN-α2b:
-
Interferon-α2b
- IFN-I, II, III:
-
Type I, II, III interferon
- IL:
-
Interleukin
- IRF:
-
Interferon regulatory factor
- ISG:
-
Interferon‐stimulated gene
- ISGF3:
-
Interferon‐stimulated gene factor 3
- ISRE:
-
Interferon-stimulated response element
- JAK:
-
Janus kinase
- LD:
-
Linker domain
- LDI:
-
Low-dose IFN-α2b
- LIF:
-
Leukaemia inhibitory factor
- LYG:
-
Lymphomatoid granulomatosis
- MDSC:
-
Myeloid-derived suppressor cell
- MMP2:
-
Matrix metalloproteinase-2
- MSS:
-
Melanoma-specific survival
- MX:
-
Myxovirus resistance protein family
- Mφ:
-
Macrophage
- ncRNA:
-
Noncoding RNA
- NSAID:
-
Non-steroid anti-inflammatory drug
- NK:
-
Natural killer cell
- NTD:
-
N-terminal domain
- OAS:
-
2'-5'-Oligoadenylate synthetase family
- OS:
-
Overall survival
- OV:
-
Oncolytic virus
- PEG:
-
Poly (ethylene glycol)
- PERK:
-
PKR-like ER kinase
- PKC:
-
Protein kinase C
- PRC2:
-
Polycomb repressive complex 2
- PUVA:
-
Psoralen + ultraviolet A irradiation regimen
- PY:
-
Tyrosine-phosphorylation site
- RFS:
-
Relapse-free survival
- RIG-I:
-
Retinoic acid-inducible gene I
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SH2:
-
Src-homology 2 domain
- SOCS1:
-
Suppressor of cytokine signalling 1
- STAT:
-
Signal transducers and activators of transcription
- STING:
-
The stimulator of interferon genes
- TAD:
-
Transcriptional activation domain
- TGF-α:
-
Transforming growth factor alpha
- Th2:
-
T helper 2 cell
- TIC:
-
Transcription initiation complex
- TIMP-1:
-
Tissue inhibitor of metalloproteinases 1
- TNF-RII:
-
Tumour necrosis factor alpha receptor II
- TNF-α:
-
Tumour necrosis factor-α
- Treg:
-
T regulatory cell
- TYK2:
-
Tyrosine kinase 2
- USP18:
-
Ubiquitin specific peptidase 18
- VEGF:
-
Vascular endothelial growth factor
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
Kuroki L, Guntupalli SR. Treatment of epithelial ovarian cancer. BMJ (Clinical research ed). 2020;371:m3773.
Joshi SS, Badgwell BD. Current treatment and recent progress in gastric cancer. CA Cancer J Clin. 2021;71(3):264–79.
Byrd DR, Brierley JD, Baker TP, Sullivan DC, Gress DM. Current and future cancer staging after neoadjuvant treatment for solid tumors. CA Cancer J Clin. 2021;71(2):140–8.
Hoelzer D. Chemotherapy-free treatment - a new era in acute lymphoblastic leukemia? N Engl J Med. 2020;383(17):1673–4.
Agarwala SS. An update on pegylated IFN-α2b for the adjuvant treatment of melanoma. Expert Rev Anticancer Ther. 2012;12(11):1449–59.
Sebina I, Haque A. Effects of type I interferons in malaria. Immunology. 2018;155(2):176–85.
Pfeffer LM, Dinarello CA, Herberman RB, Williams BR, Borden EC, Bordens R, et al. Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Can Res. 1998;58(12):2489–99.
Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. 2010;15(1):104–11.
Wada-Ohno M, Ito T, Furue M. Adjuvant Therapy for Melanoma. Curr Treat Options Oncol. 2019;20(8):63.
Pinilla-Ibarz J, Bello C. Modern approaches to treating chronic myelogenous leukemia. Curr Oncol Rep. 2008;10(5):365–71.
Hurley KE, Chapman PB. Helping melanoma patients decide whether to choose adjuvant high-dose interferon-alpha2b. Oncologist. 2005;10(9):739–42.
Isaacs A, Lindenmann J. Virus interference. I. the interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258–67.
Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15(6):407–22.
David M. Transcription factors in interferon signaling. Pharmacol Ther. 1995;65(2):149–61.
Li L, Sherry B. IFN-alpha expression and antiviral effects are subtype and cell type specific in the cardiac response to viral infection. Virology. 2010;396(1):59–68.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(Web Server issue):W320-4.
Dias PVS, Arthuso FS, Oliveira JE, Suzuki MF, Sousa JM, Ribela M, et al. Determination of recombinant Interferon-α2 in E. coli periplasmic extracts by reversed-phase high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1072:193–8.
Vecchiarello N, Timmick SM, Goodwine C, Crowell LE, Love KR, Love JC, et al. A combined screening and in silico strategy for the rapid design of integrated downstream processes for process and product-related impurity removal. Biotechnol Bioeng. 2019;116(9):2178–90.
Suarez-Kelly LP, Levine KM, Olencki TE, Del Campo SEM, Streacker EA, Brooks TR, et al. A pilot study of interferon-alpha-2b dose reduction in the adjuvant therapy of high-risk melanoma. Cancer Immunol Immunother. 2019;68(4):619–29.
Clere-Jehl R, Mariotte A, Meziani F, Bahram S, Georgel P, Helms J. JAK-STAT targeting offers novel therapeutic opportunities in sepsis. Trends Mol Med. 2020;26(11):987–1002.
Carson WE. Interferon-alpha-induced activation of signal transducer and activator of transcription proteins in malignant melanoma. Clin Cancer Res. 1998;4(9):2219–28.
Dedoni S, Olianas MC, Onali P. Interferon-β induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J Neurochem. 2010;115(6):1421–33.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49.
Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of Type I and Type III interferons. Immunity. 2019;50(4):907–23.
Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev. 2013;255(1):25–39.
Zhang L, Jiang X, Pfau D, Ling Y, Nathan CF. Type I interferon signaling mediates mycobacterium tuberculosis-induced macrophage death. J Exp Med. 2021;218(2):e20200887.
Luan J, Fu J, Wang D, Jiao C, Cui X, Chen C, et al. miR-150-based RNA interference attenuates tubulointerstitial fibrosis through the SOCS1/JAK/STAT pathway In Vivo and In Vitro. Mol Ther Nucleic Acids. 2020;22:871–84.
Osei Kuffour E, König R, Häussinger D, Schulz WA, Münk C. ISG15 deficiency enhances HIV-1 infection by accumulating misfolded p53. mBio. 2019;10(4):e01342-19.
Ceballos MP, Parody JP, Alvarez Mde L, Ingaramo PI, Carnovale CE, Carrillo MC. Interferon-α2b and transforming growth factor-β1 treatments on HCC cell lines: Are Wnt/β-catenin pathway and Smads signaling connected in hepatocellular carcinoma? Biochem Pharmacol. 2011;82(11):1682–91.
Parody JP, Ceballos MP, Quiroga AD, Frances DE, Carnovale CE, Pisani GB, et al. FoxO3a modulation and promotion of apoptosis by interferon-α2b in rat preneoplastic liver. Liver Int. 2014;34(10):1566–77.
Romerio F, Riva A, Zella D. Interferon-alpha2b reduces phosphorylation and activity of MEK and ERK through a Ras/Raf-independent mechanism. Br J Cancer. 2000;83(4):532–8.
Zhao J, Wang ML, Li Z, Gao DM, Cai Y, Chang J, et al. Interferon-alpha-2b induces autophagy in hepatocellular carcinoma cells through Beclin1 pathway. Cancer Biol Med. 2014;11(1):64–8.
de Luján AM, Ronco MT, Ochoa JE, Monti JA, Carnovale CE, Pisani GB, et al. Interferon alpha-induced apoptosis on rat preneoplastic liver is mediated by hepatocytic transforming growth factor beta(1). Hepatology (Baltimore, MD). 2004;40(2):394–402.
Quiroga AD, Alvarez Mde L, Parody JP, Ronco MT, Francés DE, Pisani GB, et al. Involvement of reactive oxygen species on the apoptotic mechanism induced by IFN-alpha2b in rat preneoplastic liver. Biochem Pharmacol. 2007;73(11):1776–85.
de Luján AM, Cerliani JP, Monti J, Carnovale C, Ronco MT, Pisani G, et al. The in vivo apoptotic effect of interferon alfa-2b on rat preneoplastic liver involves Bax protein. Hepatology (Baltimore, MD). 2002;35(4):824–33.
Su Y, Cheng R, Zhang J, Qian J, Diao C, Ran J, et al. Interferon-α2b gene-modified human bone marrow mesenchymal stem cells inhibit hepatocellular carcinoma by reducing the Notch1 levels. Life Sci. 2015;143:18–26.
van Koetsveld PM, Vitale G, de Herder WW, Feelders RA, van der Wansem K, Waaijers M, et al. Potent inhibitory effects of type I interferons on human adrenocortical carcinoma cell growth. J Clin Endocrinol Metab. 2006;91(11):4537–43.
Cheriyath V, Glaser KB, Waring JF, Baz R, Hussein MA, Borden EC. G1P3, an IFN-induced survival factor, antagonizes TRAIL-induced apoptosis in human myeloma cells. J Clin Investig. 2007;117(10):3107–17.
Quesada P, Malanga M, Di Meglio S, De Lorenzo S, Fabbrocini A, Garbi C, et al. Recombinant IFN-alpha2b treatment activates poly (ADPR) polymerase-1 (PARP-1) in KB cancer cells. Eur J Cancer. 2003;39(14):2103–9.
Bae SI, Cheriyath V, Jacobs BS, Reu FJ, Borden EC. Reversal of methylation silencing of Apo2L/TRAIL receptor 1 (DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28 melanoma cells to interferons (IFNs) or Apo2L/TRAIL. Oncogene. 2008;27(4):490–8.
Lukhele S, Boukhaled GM, Brooks DG. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin Immunol. 2019;43: 101277.
Blaauboer A, Sideras K, van Eijck CHJ, Hofland LJ. Type I interferons in pancreatic cancer and development of new therapeutic approaches. Crit Rev Oncol Hematol. 2021;159:103204.
Tan JMJ, Garner ME, Regeimbal JM, Greene CJ, Márquez JDR, Ammendolia DA, et al. Listeria exploits IFITM3 to suppress antibacterial activity in phagocytes. Nat Commun. 2021;12(1):4999.
Shen H, Zhan M, Zhang Y, Huang S, Xu S, Huang X, et al. PLZF inhibits proliferation and metastasis of gallbladder cancer by regulating IFIT2. Cell Death Dis. 2018;9(2):71.
Ortiz E, Sanchis P, Bizzotto J, Lage-Vickers S, Labanca E, Navone N, et al. Myxovirus resistance protein 1 (MX1), a Novel HO-1 interactor, tilts the balance of endoplasmic reticulum stress towards pro-death events in prostate cancer. Biomolecules. 2020;10(7):1005.
Revathidevi S, Murugan AK, Nakaoka H, Inoue I, Munirajan AK. APOBEC: a molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021;496:104–16.
Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, et al. Cytosine deaminase APOBEC3A sensitizes leukemia cells to inhibition of the DNA replication checkpoint. Can Res. 2017;77(17):4579–88.
Li X, Wu Z, An X, Mei Q, Bai M, Hanski L, et al. Blockade of the LRP16-PKR-NF-κB signaling axis sensitizes colorectal carcinoma cells to DNA-damaging cytotoxic therapy. eLife. 2017;6:e27301.
Mounir Z, Krishnamoorthy JL, Robertson GP, Scheuner D, Kaufman RJ, Georgescu MM, et al. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal. 2009;2(102):ra85.
Chen L, Zhai W, Zheng X, Xie Q, Zhou Q, Tao M, et al. Decreased IFIT2 expression promotes gastric cancer progression and predicts poor prognosis of the patients. Cell Physiol Biochem. 2018;45(1):15–25.
Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, et al. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012;8(5):e1002712.
Jia H, Song L, Cong Q, Wang J, Xu H, Chu Y, et al. The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene. 2017;36(19):2655–66.
Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science (New York, NY). 2016;352(6284):463–6.
Rentoft M, Lindell K, Tran P, Chabes AL, Buckland RJ, Watt DL, et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc Natl Acad Sci USA. 2016;113(17):4723–8.
Kodigepalli KM, Li M, Liu SL, Wu L. Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell cycle (Georgetown, Tex). 2017;16(2):179–88.
Rossi D. SAMHD1: a new gene for CLL. Blood. 2014;123(7):951–2.
Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, et al. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci USA. 2013;110(35):14272–7.
Leonard B, Starrett GJ, Maurer MJ, Oberg AL, Van Bockstal M, Van Dorpe J, et al. APOBEC3G expression correlates with T-Cell infiltration and improved clinical outcomes in high-grade serous ovarian carcinoma. Clin Cancer Res. 2016;22(18):4746–55.
Losada A, Muñoz-Alonso MJ, Martínez-Díez M, Gago F, Domínguez JM, Martínez-Leal JF, et al. Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumour cell survival. Br J Cancer. 2018;119(11):1410–20.
Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, et al. An integrated stress response via PKR suppresses HER2+ cancers and improves trastuzumab therapy. Nat Commun. 2019;10(1):2139.
Lu D, Di S, Zhuo S, Zhou L, Bai R, Ma T, et al. The long noncoding RNA TINCR promotes breast cancer cell proliferation and migration by regulating OAS1. Cell death discovery. 2021;7(1):41.
Schwartz SL, Park EN, Vachon VK, Danzy S, Lowen AC, Conn GL. Human OAS1 activation is highly dependent on both RNA sequence and context of activating RNA motifs. Nucleic Acids Res. 2020;48(13):7520–31.
Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell. 2011;146(4):621–32.
Castiello L, Sestili P, Schiavoni G, Dattilo R, Monque DM, Ciaffoni F, et al. Disruption of IFN-I signaling promotes HER2/Neu tumor progression and breast cancer stem cells. Cancer Immunol Res. 2018;6(6):658–70.
Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med. 2017;214(5):1493–507.
Zanker DJ, Owen KL, Baschuk N, Spurling AJ, Parker BS. Loss of type I IFN responsiveness impairs natural killer cell antitumor activity in breast cancer. Cancer Immunol Immunother. 2021;70(8):2125–38.
Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12(1):1717.
Gatti G, Nuñez NG, Nocera DA, Dejager L, Libert C, Giraudo C, et al. Direct effect of dsRNA mimetics on cancer cells induces endogenous IFN-β production capable of improving dendritic cell function. Eur J Immunol. 2013;43(7):1849–61.
Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, et al. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell. 2017;31(2):194–207.
Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, et al. Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene. 2013;32(36):4214–21.
Godefroy E, Bhardwaj N. Dysregulation of anti-tumor immunity by the matrix metalloproteinase-2. Oncoimmunology. 2012;1(1):109–11.
Ma DH, Li BS, Liu JJ, Xiao YF, Yong X, Wang SM, et al. miR-93-5p/IFNAR1 axis promotes gastric cancer metastasis through activating the STAT3 signaling pathway. Cancer Lett. 2017;408:23–32.
Jarret A, McFarland AP, Horner SM, Kell A, Schwerk J, Hong M, et al. Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat Med. 2016;22(12):1475–81.
Springuel L, Renauld JC, Knoops L. JAK kinase targeting in hematologic malignancies: a sinuous pathway from identification of genetic alterations towards clinical indications. Haematologica. 2015;100(10):1240–53.
Lupardus PJ, Ultsch M, Wallweber H, Bir Kohli P, Johnson AR, Eigenbrot C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc Natl Acad Sci USA. 2014;111(22):8025–30.
Danese S, Argollo M, Le Berre C, Peyrin-Biroulet L. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut. 2019;68(10):1893–9.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809.
Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Can Res. 2005;65(8):3447–53.
Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci Transl Med. 2020;12(565):eabb0152.
Shen L, Kang L, Wang D, Xun J, Chen C, Du L, et al. Legumain-deficient macrophages promote senescence of tumor cells by sustaining JAK1/STAT1 activation. Cancer Lett. 2020;472:40–9.
Witalisz-Siepracka A, Klein K, Prinz D, Leidenfrost N, Schabbauer G, Dohnal A, et al. Loss of JAK1 Drives Innate Immune Deficiency. Front Immunol. 2018;9:3108.
Ding X, He M, Chan AWH, Song QX, Sze SC, Chen H, et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology. 2020;157(6):1630-1645.e6.
Freeman AJ, Vervoort SJ, Ramsbottom KM, Kelly MJ, Michie J, Pijpers L, et al. natural killer cells suppress t cell-associated tumor immune evasion. Cell Rep. 2019;28(11):2784-94.e5.
Mohrherr J, Haber M, Breitenecker K, Aigner P, Moritsch S, Voronin V, et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int J Cancer. 2019;145(12):3376–88.
Lee JH, Park KS, Alberobello AT, Kallakury B, Weng MT, Wang Y, et al. The Janus kinases inhibitor AZD1480 attenuates growth of small cell lung cancers in vitro and in vivo. Clin Cancer Res. 2013;19(24):6777–86.
Stover DG, Gil Del Alcazar CR, Brock J, Guo H, Overmoyer B, Balko J, et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ breast cancer. 2018;4:10.
Jia X, Huang C, Hu Y, Wu Q, Liu F, Nie W, et al. Cirsiliol targets tyrosine kinase 2 to inhibit esophageal squamous cell carcinoma growth in vitro and in vivo. J Exp Clin Cancer Res. 2021;40(1):105.
Almiron Bonnin DA, Havrda MC, Lee MC, Liu H, Zhang Z, Nguyen LN, et al. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene. 2018;37(8):1107–18.
Miar A, Arnaiz E, Bridges E, Beedie S, Cribbs AP, Downes DJ, et al. Hypoxia induces transcriptional and translational downregulation of the Type I IFN pathway in multiple cancer cell types. Can Res. 2020;80(23):5245–56.
Quick L, Young R, Henrich IC, Wang X, Asmann YW, Oliveira AM, et al. Jak1-STAT3 signals are essential effectors of the USP6/TRE17 oncogene in tumorigenesis. Can Res. 2016;76(18):5337–47.
Zhang HX, Xu ZS, Lin H, Li M, Xia T, Cui K, et al. TRIM27 mediates STAT3 activation at retromer-positive structures to promote colitis and colitis-associated carcinogenesis. Nat Commun. 2018;9(1):3441.
Zhu Z, Yang Q, Zhang B, Wu W, Yuan F, Zhu Z. miR-106b promotes metastasis of early gastric cancer by targeting ALEX1 in Vitro and in Vivo. Cell Physiol Biochem. 2019;52(3):606–16.
Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–42.
Welcsh PL, Lee MK, Gonzalez-Hernandez RM, Black DJ, Mahadevappa M, Swisher EM, et al. BRCA1 transcriptionally regulates genes involved in breast tumorigenesis. Proc Natl Acad Sci USA. 2002;99(11):7560–5.
Kuang Y, Guo W, Ling J, Xu D, Liao Y, Zhao H, et al. Iron-dependent CDK1 activity promotes lung carcinogenesis via activation of the GP130/STAT3 signaling pathway. Cell Death Dis. 2019;10(4):297.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17(7):816–24.
Rojas A, Zhang P, Wang Y, Foo WC, Muñoz NM, Xiao L, et al. A Positive TGF-β/c-KIT Feedback Loop Drives Tumor Progression in Advanced Primary Liver Cancer. Neoplasia (New York, NY). 2016;18(6):371–86.
Zhang XD, Baladandayuthapani V, Lin H, Mulligan G, Li B, Esseltine DW, et al. Tight junction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma via EGFR/JAK1/STAT3 signaling. Cancer Cell. 2016;29(5):639–52.
Tactacan CM, Phua YW, Liu L, Zhang L, Humphrey ES, Cowley M, et al. The pseudokinase SgK223 promotes invasion of pancreatic ductal epithelial cells through JAK1/Stat3 signaling. Mol Cancer. 2015;14:139.
Jung YY, Ko JH, Um JY, Chinnathambi A, Alharbi SA, Sethi G, et al. LDL cholesterol promotes the proliferation of prostate and pancreatic cancer cells by activating the STAT3 pathway. J Cell Physiol. 2021;236(7):5253–64.
Ahmad R, Rajabi H, Kosugi M, Joshi MD, Alam M, Vasir B, et al. MUC1-C oncoprotein promotes STAT3 activation in an autoinductive regulatory loop. Sci Signal. 2011;4(160):ra9.
Maschler S, Gebeshuber CA, Wiedemann EM, Alacakaptan M, Schreiber M, Custic I, et al. Annexin A1 attenuates EMT and metastatic potential in breast cancer. EMBO Mol Med. 2010;2(10):401–14.
Herrmann A, Lahtz C, Nagao T, Song JY, Chan WC, Lee H, et al. CTLA4 Promotes Tyk2-STAT3-dependent B-cell oncogenicity. Can Res. 2017;77(18):5118–28.
Gasparyan M, Lo MC, Jiang H, Lin CC, Sun D. Combined p53- and PTEN-deficiency activates expression of mesenchyme homeobox 1 (MEOX1) required for growth of triple-negative breast cancer. J Biol Chem. 2020;295(34):12188–202.
She S, Zhao Y, Kang B, Chen C, Chen X, Zhang X, et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020;11(9):724.
Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. 2010;9(4):953–62.
Zi J, Yuan S, Qiao J, Zhao K, Xu L, Qi K, et al. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am J Cancer Res. 2017;7(4):869–80.
Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)–>signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Can Res. 2008;68(8):2920–6.
Kumamoto T, Fujii M, Hou DX. Myricetin directly targets JAK1 to inhibit cell transformation. Cancer Lett. 2009;275(1):17–26.
Tsai CF, Chen JH, Wu CT, Chang PC, Wang SL, Yeh WL. Induction of osteoclast-like cell formation by leptin-induced soluble intercellular adhesion molecule secreted from cancer cells. Ther Adv Med Oncol. 2019;11:1758835919846806.
Wang Z, Jin H, Xu R, Mei Q, Fan D. Triptolide downregulates Rac1 and the JAK/STAT3 pathway and inhibits colitis-related colon cancer progression. Exp Mol Med. 2009;41(10):717–27.
Wu RY, Kong PF, Xia LP, Huang Y, Li ZL, Tang YY, et al. Regorafenib promotes antitumor immunity via inhibiting PD-L1 and IDO1 expression in melanoma. Clin Cancer Res. 2019;25(14):4530–41.
Jung YY, Shanmugam MK, Narula AS, Kim C, Lee JH, Namjoshi OA, et al. Oxymatrine attenuates tumor growth and deactivates STAT5 signaling in a lung cancer xenograft model. Cancers. 2019;11(1):49.
Kim HJ, Lotan R. Identification of retinoid-modulated proteins in squamous carcinoma cells using high-throughput immunoblotting. Can Res. 2004;64(7):2439–48.
Selvendiran K, Tong L, Bratasz A, Kuppusamy ML, Ahmed S, Ravi Y, et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol Cancer Ther. 2010;9(5):1169–79.
Xing Y, Mi C, Wang Z, Zhang ZH, Li MY, Zuo HX, et al. Fraxinellone has anticancer activity in vivo by inhibiting programmed cell death-ligand 1 expression by reducing hypoxia-inducible factor-1α and STAT3. Pharmacol Res. 2018;135:166–80.
Kim C, Lee SG, Yang WM, Arfuso F, Um JY, Kumar AP, et al. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018;431:123–41.
Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009;106(20):8368–73.
Wang Y, Ma X, Yan S, Shen S, Zhu H, Gu Y, et al. 17-hydroxy-jolkinolide B inhibits signal transducers and activators of transcription 3 signaling by covalently cross-linking Janus kinases and induces apoptosis of human cancer cells. Can Res. 2009;69(18):7302–10.
Servais FA, Kirchmeyer M, Hamdorf M, Minoungou NWE, Rose-John S, Kreis S, et al. Modulation of the IL-6-signaling pathway in liver cells by miRNAs targeting gp130, JAK1, and/or STAT3. Mol Ther Nucleic Acids. 2019;16:419–33.
Yang L, Xue H, Sun Y, Zhang L, Xue F, Ge R. CircularRNA-9119 protects hepatocellular carcinoma cells from apoptosis by intercepting miR-26a/JAK1/STAT3 signaling. Cell Death Dis. 2020;11(7):605.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204.
Zhang X, Hu B, Sun YF, Huang XW, Cheng JW, Huang A, et al. Arsenic trioxide induces differentiation of cancer stem cells in hepatocellular carcinoma through inhibition of LIF/JAK1/STAT3 and NF-kB signaling pathways synergistically. Clin Transl Med. 2021;11(2):e335.
Zhang W, Shi X, Chen R, Zhu Y, Peng S, Chang Y, et al. Novel long non-coding RNA lncAMPC promotes metastasis and immunosuppression in prostate cancer by stimulating LIF/LIFR expression. Mol Ther. 2020;28(11):2473–87.
Yang H, Geng YH, Wang P, Yang H, Zhou YT, Zhang HQ, et al. Extracellular ATP promotes breast cancer invasion and chemoresistance via SOX9 signaling. Oncogene. 2020;39(35):5795–810.
Jiang M, Chen J, Zhang W, Zhang R, Ye Y, Liu P, et al. Interleukin-6 trans-signaling pathway promotes immunosuppressive myeloid-derived suppressor cells via suppression of suppressor of cytokine signaling 3 in breast cancer. Front Immunol. 2017;8:1840.
Liang M, Ma Q, Ding N, Luo F, Bai Y, Kang F, et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019;10(5):353.
Marra P, Mathew S, Grigoriadis A, Wu Y, Kyle-Cezar F, Watkins J, et al. IL15RA drives antagonistic mechanisms of cancer development and immune control in lymphocyte-enriched triple-negative breast cancers. Can Res. 2014;74(17):4908–21.
Gao W, Wen H, Liang L, Dong X, Du R, Zhou W, et al. IL20RA signaling enhances stemness and promotes the formation of an immunosuppressive microenvironment in breast cancer. Theranostics. 2021;11(6):2564–80.
Abikhair Burgo M, Roudiani N, Chen J, Santana AL, Doudican N, Proby C, et al. Ruxolitinib inhibits cyclosporine-induced proliferation of cutaneous squamous cell carcinoma. JCI insight. 2018;3(17):e120750.
Erb HH, Langlechner RV, Moser PL, Handle F, Casneuf T, Verstraeten K, et al. IL6 sensitizes prostate cancer to the antiproliferative effect of IFNα2 through IRF9. Endocr Relat Cancer. 2013;20(5):677–89.
Lee CJ, An HJ, Cho ES, Kang HC, Lee JY, Lee HS, et al. Stat2 stability regulation: an intersection between immunity and carcinogenesis. Exp Mol Med. 2020;52(9):1526–36.
Verhoeven Y, Tilborghs S, Jacobs J, De Waele J, Quatannens D, Deben C, et al. The potential and controversy of targeting STAT family members in cancer. Semin Cancer Biol. 2020;60:41–56.
Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux N, Panne D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc Natl Acad Sci USA. 2018;115(4):E601–9.
Ryan N, Anderson K, Volpedo G, Hamza O, Varikuti S, Satoskar AR, et al. STAT1 inhibits T-cell exhaustion and myeloid derived suppressor cell accumulation to promote antitumor immune responses in head and neck squamous cell carcinoma. Int J Cancer. 2020;146(6):1717–29.
Comet NR, Aguiló JI, Rathoré MG, Catalán E, Garaude J, Uzé G, et al. IFNα signaling through PKC-θ is essential for antitumor NK cell function. Oncoimmunology. 2014;3(8):e948705.
Jiang L, Park MJ, Cho CJ, Lee K, Jung MK, Pack CG, et al. ADAR1 suppresses interferon signaling in gastric cancer cells by MicroRNA-302a-mediated IRF9/STAT1 regulation. Int J Mol Sci. 2020;21(17):6195.
Buckley NE, Hosey AM, Gorski JJ, Purcell JW, Mulligan JM, Harkin DP, et al. BRCA1 regulates IFN-gamma signaling through a mechanism involving the type I IFNs. Mol Cancer Res. 2007;5(3):261–70.
Li Y, Song Y, Li P, Li M, Wang H, Xu T, et al. Downregulation of RIG-I mediated by ITGB3/c-SRC/STAT3 signaling confers resistance to interferon-α-induced apoptosis in tumor-repopulating cells of melanoma. J Immunother Cancer. 2020;8(1):e000111.
Tisserand J, Khetchoumian K, Thibault C, Dembélé D, Chambon P, Losson R. Tripartite motif 24 (Trim24/Tif1α) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor α (Rarα) inhibition. J Biol Chem. 2011;286(38):33369–79.
Shang Y, Baumrucker CR, Green MH. The induction and activation of STAT1 by all-trans-retinoic acid are mediated by RAR beta signaling pathways in breast cancer cells. Oncogene. 1999;18(48):6725–32.
Yan M, Sun L, Li J, Yu H, Lin H, Yu T, et al. RNA-binding protein KHSRP promotes tumor growth and metastasis in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38(1):478.
Yu N, Xue M, Wang W, Xia D, Li Y, Zhou X, et al. RNF168 facilitates proliferation and invasion of esophageal carcinoma, possibly via stabilizing STAT1. J Cell Mol Med. 2019;23(2):1553–61.
Goodman ML, Trinca GM, Walter KR, Papachristou EK, D’Santos CS, Li T, et al. Progesterone RECEPTOR Attenuates STAT1-mediated IFN signaling in breast cancer. J Immunol. 2019;202(10):3076–86.
Xu P, Ye S, Li K, Huang M, Wang Q, Zeng S, et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J Exp Clin Cancer Res. 2019;38(1):483.
Amalraj J, Cutler SJ, Ghazawi I, Boyle GM, Ralph SJ. REST negatively and ISGF3 positively regulate the human STAT1 gene in melanoma. Mol Cancer Ther. 2013;12(7):1288–98.
Yang Y, Zhou Y, Hou J, Bai C, Li Z, Fan J, et al. Hepatic IFIT3 predicts interferon-α therapeutic response in patients of hepatocellular carcinoma. Hepatology (Baltimore, MD). 2017;66(1):152–66.
Lee CJ, An HJ, Kim SM, Yoo SM, Park J, Lee GE, et al. FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation. Proc Natl Acad Sci USA. 2020;117(1):584–94.
Feng HH, Zhu ZX, Cao WJ, Yang F, Zhang XL, Du XL, et al. Foot-and-mouth disease virus induces lysosomal degradation of NME1 to impair p53-regulated interferon-inducible antiviral genes expression. Cell Death Dis. 2018;9(9):885.
Lu L, Zhu F, Zhang M, Li Y, Drennan AC, Kimpara S, et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2018;115(3):E498-e505.
Peng D, Chen L, Sun Y, Sun L, Yin Q, Deng S, et al. Melanoma suppression by quercein is correlated with RIG-I and type I interferon signaling. Biomed Pharmacother. 2020;125:109984.
Bing Y, Zhu S, Yu G, Li T, Liu W, Li C, et al. Glucocorticoid-induced S-adenosylmethionine enhances the interferon signaling pathway by restoring STAT1 protein methylation in hepatitis B virus-infected cells. J Biol Chem. 2014;289(47):32639–55.
Thomas M, Finnegan CE, Rogers KM, Purcell JW, Trimble A, Johnston PG, et al. STAT1: a modulator of chemotherapy-induced apoptosis. Can Res. 2004;64(22):8357–64.
Hung WC, Chuang LY. Sodium butyrate enhances STAT 1 expression in PLC/PRF/5 hepatoma cells and augments their responsiveness to interferon-alpha. Br J Cancer. 1999;80(5–6):705–10.
Jiang GM, Wang HS, Du J, Ma WF, Wang H, Qiu Y, et al. Bortezomib relieves immune tolerance in nasopharyngeal carcinoma via STAT1 suppression and indoleamine 2,3-dioxygenase downregulation. Cancer Immunol Res. 2017;5(1):42–51.
Fielhaber JA, Han YS, Tan J, Xing S, Biggs CM, Joung KB, et al. Inactivation of mammalian target of rapamycin increases STAT1 nuclear content and transcriptional activity in alpha4- and protein phosphatase 2A-dependent fashion. J Biol Chem. 2009;284(36):24341–53.
Tong J, Taylor P, Moran MF. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteomics. 2014;13(7):1644–58.
Liao J, Fu Y, Shuai K. Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of activated signal transducer and activator of transcription (STAT) 1 (PIAS1) in cytokine-induced PIAS1-Stat1 interaction. Proc Natl Acad Sci USA. 2000;97(10):5267–72.
Ouaked N, Mantel PY, Bassin C, Burgler S, Siegmund K, Akdis CA, et al. Regulation of the foxp3 gene by the Th1 cytokines: the role of IL-27-induced STAT1. J Immunol. 2009;182(2):1041–9.
Testoni B, Schinzari V, Guerrieri F, Gerbal-Chaloin S, Blandino G, Levrero M. p53-paralog DNp73 oncogene is repressed by IFNα/STAT2 through the recruitment of the Ezh2 polycomb group transcriptional repressor. Oncogene. 2011;30(23):2670–8.
Gea-Banacloche J, Johnson RT, Bagic A, Butman JA, Murray PR, Agrawal AG. West Nile virus: pathogenesis and therapeutic options. Ann Intern Med. 2004;140(7):545–53.
Du Y, Tu L, Zhu P, Mu M, Wang R, Yang P, et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan. a retrospective observational study. Am J Respir Crit Care Med. 2020;201(11):1372–9.
Lim HK, Huang SXL, Chen J, Kerner G, Gilliaux O, Bastard P, et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J Exp Med. 2019;216(9):2038–56.
Pandit A, Bhalani N, Bhushan BLS, Koradia P, Gargiya S, Bhomia V, et al. Efficacy and safety of pegylated interferon alfa-2b in moderate COVID-19: a phase II, randomized, controlled, open-label study. Int J Infect Dis. 2021;105:516–21.
Imai Y, Kawata S, Tamura S, Yabuuchi I, Noda S, Inada M, et al. Relation of interferon therapy and hepatocellular carcinoma in patients with chronic hepatitis C. Osaka Hepatocellular Carcinoma Prevention Study Group. Ann Intern Med. 1998;129(2):94–9.
Krown SE, Lee JY, Lin L, Fischl MA, Ambinder R, Von Roenn JH. Interferon-alpha2b with protease inhibitor-based antiretroviral therapy in patients with AIDS-associated Kaposi sarcoma: an AIDS malignancy consortium phase I trial. J Acquir Immune Defic Syndr. 2006;41(2):149–53.
Kleeberg UR, Suciu S, Bröcker EB, Ruiter DJ, Chartier C, Liénard D, et al. Final results of the EORTC 18871/DKG 80–1 randomised phase III trial. rIFN-alpha2b versus rIFN-gamma versus ISCADOR M versus observation after surgery in melanoma patients with either high-risk primary (thickness >3 mm) or regional lymph node metastasis. Eur J Cancer. 2004;40(3):390–402.
Varker KA, Biber JE, Kefauver C, Jensen R, Lehman A, Young D, et al. A randomized phase 2 trial of bevacizumab with or without daily low-dose interferon alfa-2b in metastatic malignant melanoma. Ann Surg Oncol. 2007;14(8):2367–76.
Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7–17.
Kefford RF. Adjuvant therapy of cutaneous melanoma: the interferon debate. Ann Oncol. 2003;14(3):358–65.
Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE, Ernstoff MS, et al. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000;18(12):2444–58.
Najjar YG, Puligandla M, Lee SJ, Kirkwood JM. An updated analysis of 4 randomized ECOG trials of high-dose interferon in the adjuvant treatment of melanoma. Cancer. 2019;125(17):3013–24.
Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, et al. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/-granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin Cancer Res. 2009;15(4):1443–51.
Tarhini AA, Lee SJ, Li X, Rao UNM, Nagarajan A, Albertini MR, et al. E3611-A randomized phase II study of Ipilimumab at 3 or 10 mg/kg alone or in combination with high-dose Interferon-α2b in advanced melanoma. Clin Cancer Res. 2019;25(2):524–32.
Tarhini AA, Moschos SJ, Lin Y, Lin HM, Sander C, Yin Y, et al. Safety and efficacy of the antiganglioside GD3 antibody ecromeximab (KW2871) combined with high-dose interferon-α2b in patients with metastatic melanoma. Melanoma Res. 2017;27(4):342–50.
Tarhini A, Lin Y, Lin H, Rahman Z, Vallabhaneni P, Mendiratta P, et al. Neoadjuvant ipilimumab (3 mg/kg or 10 mg/kg) and high dose IFN-α2b in locally/regionally advanced melanoma: safety, efficacy and impact on T-cell repertoire. J Immunother Cancer. 2018;6(1):112.
Middleton M, Hauschild A, Thomson D, Anderson R, Burdette-Radoux S, Gehlsen K, et al. Results of a multicenter randomized study to evaluate the safety and efficacy of combined immunotherapy with interleukin-2, interferon-{alpha}2b and histamine dihydrochloride versus dacarbazine in patients with stage IV melanoma. Ann Oncol. 2007;18(10):1691–7.
Moschos SJ, Odoux C, Land SR, Agarwala S, Friedland D, Volker KM, et al. Endostatin plus interferon-alpha2b therapy for metastatic melanoma: a novel combination of antiangiogenic and immunomodulatory agents. Melanoma Res. 2007;17(3):193–200.
Daponte A, Ascierto PA, Gravina A, Melucci MT, Palmieri G, Comella P, et al. Cisplatin, dacarbazine, and fotemustine plus interferon alpha in patients with advanced malignant melanoma. a multicenter phase II study of the Italian Cooperative Oncology Group. Cancer. 2000;89(12):2630–6.
Stark JJ, Dillman RO, Schulof R, Wiemann MC, Barth NM, Honeycutt PJ, et al. Interferon-alpha and chemohormonal therapy for patients with advanced melanoma: final results of a phase I-II study of the Cancer Biotherapy Research Group and the Mid-Atlantic Oncology Program. Cancer. 1998;82(9):1677–81.
Agarwala SS, Kirkwood JM. Temozolomide in combination with interferon alpha-2b in patients with metastatic melanoma: a phase I dose-escalation study. Cancer. 2003;97(1):121–7.
Weber RW, O’Day S, Rose M, Deck R, Ames P, Good J, et al. Low-dose outpatient chemobiotherapy with temozolomide, granulocyte-macrophage colony stimulating factor, interferon-alpha2b, and recombinant interleukin-2 for the treatment of metastatic melanoma. J Clin Oncol. 2005;23(35):8992–9000.
Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6(9):1018–23.
Lindauer M, Domkin D, Döhner H, Kolb HJ, Neubauer A, Huhn D, et al. Efficacy and toxicity of IFN-alpha2b combined with cytarabine in chronic myelogenous leukaemia. Br J Haematol. 1999;106(4):1013–9.
Maloisel F, Guerci A, Guyotat D, Ifrah N, Michallet M, Reiffers J, et al. Results of a phase II trial of a combination of oral cytarabine ocfosfate (YNK01) and interferon alpha-2b for the treatment of chronic myelogenous leukemia patients in chronic phase. Leukemia. 2002;16(4):573–80.
Baccarani M, Martinelli G, Rosti G, Trabacchi E, Testoni N, Bassi S, et al. Imatinib and pegylated human recombinant interferon-alpha2b in early chronic-phase chronic myeloid leukemia. Blood. 2004;104(13):4245–51.
Hjorth-Hansen H, Stentoft J, Richter J, Koskenvesa P, Höglund M, Dreimane A, et al. Safety and efficacy of the combination of pegylated interferon-α2b and dasatinib in newly diagnosed chronic-phase chronic myeloid leukemia patients. Leukemia. 2016;30(9):1853–60.
Wilson WH, Kingma DW, Raffeld M, Wittes RE, Jaffe ES. Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b. Blood. 1996;87(11):4531–7.
Melani C, Jaffe ES, Wilson WH. Pathobiology and treatment of lymphomatoid granulomatosis, a rare EBV-driven disorder. Blood. 2020;135(16):1344–52.
Koster A, van Krieken JH, Mackenzie MA, Schraders M, Borm GF, van der Laak JA, et al. Increased vascularization predicts favorable outcome in follicular lymphoma. Clin Cancer Res. 2005;11(1):154–61.
Koster A, Tromp HA, Raemaekers JM, Borm GF, Hebeda K, Mackenzie MA, et al. The prognostic significance of the intra-follicular tumor cell proliferative rate in follicular lymphoma. Haematologica. 2007;92(2):184–90.
Geskin LJ, Akilov OE, Kwon S, Schowalter M, Watkins S, Whiteside TL, et al. Therapeutic reduction of cell-mediated immunosuppression in mycosis fungoides and Sézary syndrome. Cancer Immunol Immunother. 2018;67(3):423–34.
Olisova OY, Megna M, Grekova EV, Zaslavsky DV, Gorenkova LG, Sidikov AA, et al. PUVA and interferon α2b combined therapy for patients with mycosis fungoides at different stages of the disease: a seven-year retrospective study in Russia. J Eur Acad Dermatol Venereol. 2019;33(2):e72–4.
Rupoli S, Goteri G, Pulini S, Filosa A, Tassetti A, Offidani M, et al. Long-term experience with low-dose interferon-alpha and PUVA in the management of early mycosis fungoides. Eur J Haematol. 2005;75(2):136–45.
Rupoli S, Barulli S, Guiducci B, Offidani M, Mozzicafreddo G, Simonacci M, et al. Low dose interferon-alpha2b combined with PUVA is an effective treatment of early stage mycosis fungoides: results of a multicenter study. Cutaneous-T Cell Lymphoma Multicenter Study Group. Haematologica. 1999;84(9):809–13.
Yano H, Ogasawara S, Momosaki S, Akiba J, Kojiro S, Fukahori S, et al. Growth inhibitory effects of pegylated IFN alpha-2b on human liver cancer cells in vitro and in vivo. Liver Int. 2006;26(8):964–75.
Patt YZ, Hassan MM, Lozano RD, Brown TD, Vauthey JN, Curley SA, et al. Phase II trial of systemic continuous fluorouracil and subcutaneous recombinant interferon Alfa-2b for treatment of hepatocellular carcinoma. J Clin Oncol. 2003;21(3):421–7.
Kudo M, Sakaguchi Y, Chung H, Hatanaka K, Hagiwara S, Ishikawa E, et al. Long-term interferon maintenance therapy improves survival in patients with HCV-related hepatocellular carcinoma after curative radiofrequency ablation. a matched case-control study. Oncology. 2007;72(Suppl 1):132–8.
Mazzaferro V, Brunetto MR, Pasquali M, Regalia E, Pulvirenti A, Baratti D, et al. Preoperative serum levels of wild-type and hepatitis B e antigen-negative hepatitis B virus (HBV) and graft infection after liver transplantation for HBV-related hepatocellular carcinoma. J Viral Hepatitis. 1997;4(4):235–42.
Zhang S, Gao S, Zhao M, Liu Y, Bu Y, Jiang Q, et al. Anti-HBV drugs suppress the growth of HBV-related hepatoma cells via down-regulation of hepatitis B virus X protein. Cancer Lett. 2017;392:94–104.
Azzaroli F, Accogli E, Nigro G, Trere D, Giovanelli S, Miracolo A, et al. Interferon plus ribavirin and interferon alone in preventing hepatocellular carcinoma: a prospective study on patients with HCV related cirrhosis. World J Gastroenterol. 2004;10(21):3099–102.
Valla DC, Chevallier M, Marcellin P, Payen JL, Trepo C, Fonck M, et al. Treatment of hepatitis C virus-related cirrhosis: a randomized, controlled trial of interferon alfa-2b versus no treatment. Hepatology (Baltimore, MD). 1999;29(6):1870–5.
Liu Z, Wang J, Yuan H, Liu L, Bu Y, Zhao M, et al. IFN-α2b inhibits the ethanol enriched-HBV cccDNA through blocking a positive feedback loop of HBx/MSL2/cccDNA/HBV/HBx in liver. Biochem Biophys Res Commun. 2020;527(1):76–82.
Gennatas C, Dardoufas C, Mouratidou D, Tsavaris N, Pouli A, Androulakis G, et al. Surgical adjuvant therapy of rectal carcinoma: a controlled evaluation of leucovorin, 5-fluorouracil and radiation therapy with or without interferon-alpha2b. Ann Oncol. 2003;14(3):378–82.
Liu Q, Zhang D, Qian H, Chu Y, Yang Y, Shao J, et al. Superior antitumor efficacy of IFN-α2b-incorporated photo-cross-linked hydrogels combined with T cell transfer and low-dose irradiation against gastric cancer. Int J Nanomed. 2020;15:3669–80.
Kasai K, Kooka Y, Suzuki Y, Suzuki A, Oikawa T, Ushio A, et al. Efficacy of hepatic arterial infusion chemotherapy using 5-fluorouracil and systemic pegylated interferon α-2b for advanced intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2014;21(11):3638–45.
Patt YZ, Hassan MM, Lozano RD, Waugh KA, Hoque AM, Frome AI, et al. Phase II trial of cisplatin, interferon alpha-2b, doxorubicin, and 5-fluorouracil for biliary tract cancer. Clin Cancer Res. 2001;7(11):3375–80.
Vitale G, Tagliaferri P, Caraglia M, Rampone E, Ciccarelli A, Bianco AR, et al. Slow release lanreotide in combination with interferon-alpha2b in the treatment of symptomatic advanced medullary thyroid carcinoma. J Clin Endocrinol Metab. 2000;85(3):983–8.
Alvarez RD, Huh WK, Khazaeli MB, Meredith RF, Partridge EE, Kilgore LC, et al. A phase I study of combined modality (90)Yttrium-CC49 intraperitoneal radioimmunotherapy for ovarian cancer. Clin Cancer Res. 2002;8(9):2806–11.
Goldberg JS, Vargas M, Rosmarin AS, Milowsky MI, Papanicoloau N, Gudas LJ, et al. Phase I trial of interferon alpha2b and liposome-encapsulated all-trans retinoic acid in the treatment of patients with advanced renal cell carcinoma. Cancer. 2002;95(6):1220–7.
Oh JL, Vokes EE, Kies MS, Mittal BB, Witt ME, Weichselbaum RR, et al. Induction chemotherapy followed by concomitant chemoradiotherapy in the treatment of locoregionally advanced nasopharyngeal cancer. Ann Oncol. 2003;14(4):564–9.
Mantovani G, Maccio A, Massa E, Mulas C, Mudu MC, Massidda S, et al. Phase II study of induction chemotherapy followed by concomitant chemoradiotherapy in advanced head and neck cancer: clinical outcome, toxicity and organ/function preservation. Int J Oncol. 2000;16(6):1227–33.
Boorjian SA, Milowsky MI, Kaplan J, Albert M, Cobham MV, Coll DM, et al. Phase 1/2 clinical trial of interferon alpha2b and weekly liposome-encapsulated all-trans retinoic acid in patients with advanced renal cell carcinoma. J Immunother. 2007;30(6):655–62.
Schneider B, Fukunaga A, Murry D, Yoder C, Fife K, Foster A, et al. A phase I, pharmacokinetic and pharmacodynamic dose escalation trial of weekly paclitaxel with interferon-alpha2b in patients with solid tumors. Cancer Chemother Pharmacol. 2007;59(2):261–8.
Lalevee S, Ortonne N, Hotz C, Schlemmer F, Beldi-Ferchiou A, Delfau-Larue MH, et al. Febrile ulceronecrotic Mucha Habermann disease mimicking aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma: a diagnostic challenge. Eur J Dermatol. 2018;28(6):834–5.
Lam JS, Benson MC, O’Donnell MA, Sawczuk A, Gavazzi A, Wechsler MH, et al. Bacillus Calmete-Guérin plus interferon-alpha2B intravesical therapy maintains an extended treatment plan for superficial bladder cancer with minimal toxicity. Urol Oncol. 2003;21(5):354–60.
Galor A, Karp CL, Chhabra S, Barnes S, Alfonso EC. Topical interferon alpha 2b eye-drops for treatment of ocular surface squamous neoplasia: a dose comparison study. Br J Ophthalmol. 2010;94(5):551–4.
Herwig-Carl MC, Grossniklaus HE, Müller PL, Atzrodt L, Loeffler KU, Auw-Haedrich C. Pyogenic granuloma associated with conjunctival epithelial neoplasia: report of nine cases. Br J Ophthalmol. 2019;103(10):1469–74.
Holcombe DJ, Lee GA. Topical interferon alfa-2b for the treatment of recalcitrant ocular surface squamous neoplasia. Am J Ophthalmol. 2006;142(4):568–71.
Nanji AA, Moon CS, Galor A, Sein J, Oellers P, Karp CL. Surgical versus medical treatment of ocular surface squamous neoplasia: a comparison of recurrences and complications. Ophthalmology. 2014;121(5):994–1000.
Kataoka I, Shinagawa K, Shiro Y, Okamoto S, Watanabe R, Mori T, et al. Multiple sclerosis associated with interferon-alpha therapy for chronic myelogenous leukemia. Am J Hematol. 2002;70(2):149–53.
Hirbe AC, Kaushal M, Sharma MK, Dahiya S, Pekmezci M, Perry A, et al. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer. 2017;123(7):1194–201.
Dawson NA, Zibelman M, Lindsay T, Feldman RA, Saul M, Gatalica Z, et al. An emerging landscape for canonical and actionable molecular alterations in primary and metastatic prostate cancer. Mol Cancer Ther. 2020;19(6):1373–82.
Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14(12):3716–21.
Waanders E, Scheijen B, Jongmans MC, Venselaar H, van Reijmersdal SV, van Dijk AH, et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia. 2017;31(4):821–8.
Clifford JL, Yang X, Walch E, Wang M, Lippman SM. Dominant negative signal transducer and activator of transcription 2 (STAT2) protein: stable expression blocks interferon alpha action in skin squamous cell carcinoma cells. Mol Cancer Ther. 2003;2(5):453–9.
Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8.
LaRocca CJ, Salzwedel AO, Sato-Dahlman M, Romanenko MV, Andrade R, Davydova J, et al. Interferon alpha-expressing oncolytic adenovirus for treatment of esophageal adenocarcinoma. Ann Surg Oncol. 2021;28(13):8556–64.
Noser JA, Mael AA, Sakuma R, Ohmine S, Marcato P, Lee PW, et al. The RAS/Raf1/MEK/ERK signaling pathway facilitates VSV-mediated oncolysis: implication for the defective interferon response in cancer cells. Mol Ther. 2007;15(8):1531–6.
Sonzogni O, Zak DE, Sasso MS, Lear R, Muntzer A, Zonca M, et al. T-SIGn tumor reengineering therapy and CAR T cells synergize in combination therapy to clear human lung tumor xenografts and lung metastases in NSG mice. Oncoimmunology. 2022;11(1):2029070.
Annels NE, Simpson GR, Denyer M, Arif M, Coffey M, Melcher A, et al. Oncolytic reovirus-mediated recruitment of early innate immune responses reverses immunotherapy resistance in prostate tumors. Mol Ther Oncolytics. 2021;20:434–46.
García-Romero N, Palacín-Aliana I, Esteban-Rubio S, Madurga R, Rius-Rocabert S, Carrión-Navarro J, et al. Newcastle Disease Virus (NDV) oncolytic activity in human glioma tumors is dependent on CDKN2A-type I IFN gene cluster codeletion. Cells. 2020;9(6):1405.
Kedarinath K, Parks GD. Differential In Vitro growth and cell killing of cancer versus benign prostate cells by oncolytic parainfluenza virus. Pathogens. 2022;11(5):493.
Muster T, Rajtarova J, Sachet M, Unger H, Fleischhacker R, Romirer I, et al. Interferon resistance promotes oncolysis by influenza virus NS1-deletion mutants. Int J Cancer. 2004;110(1):15–21.
Fend L, Yamazaki T, Remy C, Fahrner C, Gantzer M, Nourtier V, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Can Res. 2017;77(15):4146–57.
Escobar-Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther. 2013;20(10):582–9.
Panagioti E, Kurokawa C, Viker K, Ammayappan A, Anderson SK, Sotiriou S, et al. Immunostimulatory bacterial antigen-armed oncolytic measles virotherapy significantly increases the potency of anti-PD1 checkpoint therapy. J Clin Invest. 2021;131(13):e141614.
Oosenbrug T, van den Wollenberg DJM, Duits EW, Hoeben RC, Ressing ME. Induction of robust type I interferon levels by oncolytic reovirus requires both viral replication and interferon-α/β receptor signaling. Hum Gene Ther. 2021;32(19–20):1171–85.
Parrish C, Scott GB, Migneco G, Scott KJ, Steele LP, Ilett E, et al. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia. 2015;29(9):1799–810.
Wantoch M, Wilson EB, Droop AP, Phillips SL, Coffey M, El-Sherbiny YM, et al. Oncolytic virus treatment differentially affects the CD56(dim) and CD56(bright) NK cell subsets in vivo and regulates a spectrum of human NK cell activity. Immunology. 2022;166(1):104–20.
Lipatova AV, Soboleva AV, Gorshkov VA, Bubis JA, Solovyeva EM, Krasnov GS, et al. Multi-omics analysis of glioblastoma cells’ sensitivity to oncolytic viruses. Cancers. 2021;13(21):5268.
Morimoto D, Matsumura S, Bustos-Villalobos I, Sibal PA, Ichinose T, Naoe Y, et al. C-REV retains high infectivity regardless of the expression levels of cGAS and STING in cultured pancreatic cancer cells. Cells. 2021;10(6):1502.
Vasquez M, Fioravanti J, Aranda F, Paredes V, Gomar C, Ardaiz N, et al. Interferon alpha bioactivity critically depends on Scavenger receptor class B type I function. Oncoimmunology. 2016;5(8):e1196309.
Tarhini AA, Lin Y, Yeku O, LaFramboise WA, Ashraf M, Sander C, et al. A four-marker signature of TNF-RII, TGF-α, TIMP-1 and CRP is prognostic of worse survival in high-risk surgically resected melanoma. J Transl Med. 2014;12:19.
Tarhini AA, Shin D, Lee SJ, Stuckert J, Sander CA, Kirkwood JM. Serologic evidence of autoimmunity in E2696 and E1694 patients with high-risk melanoma treated with adjuvant interferon alfa. Melanoma Res. 2014;24(2):150–7.
Kirkwood JM, Richards T, Zarour HM, Sosman J, Ernstoff M, Whiteside TL, et al. Immunomodulatory effects of high-dose and low-dose interferon alpha2b in patients with high-risk resected melanoma: the E2690 laboratory corollary of intergroup adjuvant trial E1690. Cancer. 2002;95(5):1101–12.
Yurkovetsky ZR, Kirkwood JM, Edington HD, Marrangoni AM, Velikokhatnaya L, Winans MT, et al. Multiplex analysis of serum cytokines in melanoma patients treated with interferon-alpha2b. Clin Cancer Res. 2007;13(8):2422–8.
Hofmann MA, Kiecker F, Küchler I, Kors C, Trefzer U. Serum TNF-α, B2M and sIL-2R levels are biological correlates of outcome in adjuvant IFN-α2b treatment of patients with melanoma. J Cancer Res Clin Oncol. 2011;137(3):455–62.
Espinosa E, Soriano V, Malvehy J, Berrocal A, Martínez de Prado P, Quindós M, et al. Treatment patterns of adjuvant interferon-α2b for high-risk melanoma: a retrospective study of the Grupo Español Multidisciplinar de Melanoma - Prima study. Melanoma Res. 2016;26(3):278–83.
Conill C, Jorcano S, Domingo-Domènech J, Marruecos J, Vilella R, Malvehy J, et al. Toxicity of combined treatment of adjuvant irradiation and interferon alpha2b in high-risk melanoma patients. Melanoma Res. 2007;17(5):304–9.
Yadav P, McLeod VM, Nowell CJ, Selby LI, Johnston APR, Kaminskas LM, et al. Distribution of therapeutic proteins into thoracic lymph after intravenous administration is protein size-dependent and primarily occurs within the liver and mesentery. J Control Release. 2018;272:17–28.
Giaconi JA, Karp CL. Current treatment options for conjunctival and corneal intraepithelial neoplasia. Ocul Surf. 2003;1(2):66–73.
Sheng L, Chen X, Wang Q, Lyu S, Li P. Interferon-α2b enhances survival and modulates transcriptional profiles and the immune response in melanoma patients treated with dendritic cell vaccines. Biomed Pharmacother. 2020;125:109966.
Spaner DE. Amplifying cancer vaccine responses by modifying pathogenic gene programs in tumor cells. J Leukoc Biol. 2004;76(2):338–51.
Bello-Rivero I, Garcia-Vega Y, Duncan-Roberts Y, Vazquez-Blomquistc D, Santana-Milian H, Besada-Perez V, et al. HeberFERON, a new formulation of IFNs with improved pharmacodynamics: Perspective for cancer treatment. Semin Oncol. 2018;45(1–2):27–33.
Ahmed CM, Sugarman BJ, Johnson DE, Bookstein RE, Saha DP, Nagabhushan TL, et al. In vivo tumor suppression by adenovirus-mediated interferon alpha2b gene delivery. Hum Gene Ther. 1999;10(1):77–84.
Ahmed CM, Wills KN, Sugarman BJ, Johnson DE, Ramachandra M, Nagabhushan TL, et al. Selective expression of nonsecreted interferon by an adenoviral vector confers antiproliferative and antiviral properties and causes reduction of tumor growth in nude mice. J Interferon Cytokine Res. 2001;21(6):399–408.
Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39(1):47–55.
Iqbal Ahmed CM, Johnson DE, Demers GW, Engler H, Howe JA, Wills KN, et al. Interferon alpha2b gene delivery using adenoviral vector causes inhibition of tumor growth in xenograft models from a variety of cancers. Cancer Gene Ther. 2001;8(10):788–95.
Aghemo A, Rumi MG, Colombo M. Pegylated interferons alpha2a and alpha2b in the treatment of chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2010;7(9):485–94.
García-Martínez E, Smith M, Buqué A, Aranda F, de la Peña FA, Ivars A, et al. Trial Watch: Immunostimulation with recombinant cytokines for cancer therapy. Oncoimmunology. 2018;7(6):e1433982.
Hodgson DJ, Aubin Y. Assessment of the structure of pegylated-recombinant protein therapeutics by the NMR fingerprint assay. J Pharm Biomed Anal. 2017;138:351–6.
Wang Y, Liu D, Crowell LE, Love KR, Wu SL, Hancock WS. The application of HPLC/MS analysis with a multi-enzyme digest strategy to characterize different interferon product variants produced from Pichia pastoris. Amino Acids. 2019;51(9):1353–63.
Vyas K, Brassard DL, DeLorenzo MM, Sun Y, Grace MJ, Borden EC, et al. Biologic activity of polyethylene glycol12000-interferon-alpha2b compared with interferon-alpha2b: gene modulatory and antigrowth effects in tumor cells. J Immunother. 2003;26(3):202–11.
Hu Y, Hou Y, Wang H, Lu H. Polysarcosine as an alternative to PEG for therapeutic protein conjugation. Bioconjug Chem. 2018;29(7):2232–8.
Blank VC, Peña C, Roquin LP. A cyclic chimeric interferon-alpha2b peptide induces apoptosis in tumor cells. Cancer Biol Ther. 2007;6(11):1787–93.
Acknowledgements
Not applicable.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (#81502108, #81974438 and #82173069). The authors declare that there was no financial relationship with the organizations that sponsored this study, and the funding body didn’t participate in study design, data collection, analysis and writing of this study.
Author information
Authors and Affiliations
Contributions
FX wrote the initial manuscript. QW, GHW, WZL, BW and YJC revised the manuscript. All of the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiong, F., Wang, Q., Wu, Gh. et al. Direct and indirect effects of IFN-α2b in malignancy treatment: not only an archer but also an arrow. Biomark Res 10, 69 (2022). https://doi.org/10.1186/s40364-022-00415-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00415-y