
Abstract
Background
Despite modern antiretroviral therapy (ART), people living with HIV (PLWH) have increased relative risk of inflammatory-driven comorbidities, including cardiovascular disease (CVD). The gut microbiome could be one of several driving factors, along with traditional risk factors and HIV-related risk factors such as coinfections, ART toxicity, and past immunodeficiency.
Results
PLWH have an altered gut microbiome, even after adjustment for known confounding factors including sexual preference. The HIV-related microbiome has been associated with cardiometabolic comorbidities, and shares features with CVD-related microbiota profiles, in particular reduced capacity for short-chain fatty acid (SCFA) generation. Substantial inter-individual variation has so far been an obstacle for applying microbiota profiles for risk stratification. This review covers updated knowledge and recent advances in our understanding of the gut microbiome and comorbidities in PLWH, with specific focus on cardiometabolic comorbidities and inflammation. It covers a comprehensive overview of HIV-related and comorbidity-related dysbiosis, microbial translocation, and microbiota-derived metabolites. It also contains recent data from studies in PLWH on circulating metabolites related to comorbidities and underlying gut microbiota alterations, including circulating levels of the SCFA propionate, the histidine-analogue imidazole propionate, and the protective metabolite indole-3-propionic acid.
Conclusions
Despite recent advances, the gut microbiome and related metabolites are not yet established as biomarkers or therapeutic targets. The review gives directions for future research needed to advance the field into clinical practice, including promises and pitfalls for precision medicine.
Video Abstract
Similar content being viewed by others

Introduction
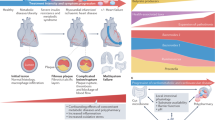
Although modern antiretroviral therapy (ART) reduces detectable HIV virus levels to a minimum in people living with HIV (PLWH), a higher morbidity and a shorter life expectancy remain [1, 2]. In particular, PLWH have increased relative risk of inflammatory-driven comorbidities including cardiovascular disease, cancer, kidney, liver, bone, and neurocognitive disease [3].
From 2015, WHO has recommended treatment of all PLWH. Recent data from the Antiretroviral Therapy Cohort Collaboration (ATCC) showed improvements in life expectancy for PLWH that started ART from 2015 and onwards compared to those PLWH that started ART 1999–2014. However, despite these improvements, some groups of PLWH, most notably women living with HIV and PLWH with lower CD4 T cell counts, still have not achieved life expectancy comparable to that in the background population, despite suppressed viral load and no prior AIDS [4]. It is well established that PLWH have a higher burden of comorbidity, and comorbidities occur at a younger age in PLWH. [5] This was elegantly shown in the Dutch AGEhIV study where PLWH were compared to population controls matched on lifestyle including sexual behavior [6]. Importantly, this cohort was followed prospectively for 5.9 years, and number of comorbidities at baseline was associated with an increased risk of death (hazard ratio 3:33 per additional comorbidity) [7] indicating that comorbidities are likely to contribute to excess mortality in PLWH. Likewise, in a study from the Danish HIV Cohort, the probability of survival was dramatically reduced in PLWH with comorbidites [8]. Importantly, despite universal rollout of ART from 2015, PLWH still have fewer years without comorbidity than controls from the general population [9]. Worldwide populations of PLWH are aging, and a recent study from the United States estimated that 23% of ART users will be aged ≥ 65 years in 2030 [10]. Since the incidence of comorbidities increases with increasing age, the absolute burden of comorbidities in PLWH is likely to increase.
Cardiovascular disease (CVD) and diabetes both rank among the top 10 causes of disability-adjusted life years (DALYs), while HIV ranks 11 [11]. Hence, any adverse interaction between HIV and these diseases is likely to have a major impact on health in PLWH, and PLWH do seem to be disproportionately affected by comorbidities. CVD is probably the most well-studied comorbidity in PLWH, and in a recent systematic review across 80 studies that included nearly 800,000 PLWH and a total follow-up of 3.5 million person-years, the crude rate of CVD was 61.8 per 10,000 person-years. Importantly, in comparison with persons without HIV, the risk ratio for cardiovascular disease was just above two. Given the increased risk of ischemic CVD [12], it is not surprising that PLWH also have increased risk of heart failure [13] (HF) with the highest risk among PLWH with lower CD4 T-cell counts or ongoing viral replication [14, 15]. PLWH also have high prevalence of electrocardiographic alterations and seem to be at higher risk of sudden cardiac death [16,17,18,19]. Other manifestations of CVD that may be more prevalent among PLWH include aortic aneurysms and peripheral artery diseases [12, 20,21,22,23,24], although these findings are not entirely consistent. Across several studies and different manifestations of CVD, lower CD4 T-cell counts and/or ongoing viral replication is associated with higher risk.
The main risk factor for CVD in both the general population and in PLWH is smoking. Unfortunately, PWLH are more likely to smoke than persons without HIV [25], and smoking is associated with higher risk of myocardial infarction in PLWH than in the general population [26]. Another important risk factor for CVD in PLWH is inflammation, including elevated levels of interleukin (IL)-1 and IL-6 [27, 28]. The Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) found that anti-inflammatory therapy with canakinumab, a monoclonal antibody blocking IL-1β, led to a lower rate of CVD than placebo [29], providing evidence to the role of inflammation in the pathogenesis leading to CVD. Chronic inflammation and immune activation are hallmarks of HIV infection, and even well-treated PLWH have higher levels of inflammation and immune activation [30], which is associated with higher CVD risk [31, 32].
Immune activation and inflammation, in turn, may be driven by a number of factors including lifestyle as indicated by a study from the Comorbidity in Relation to AIDS (COBRA) cohort [33]. Furthermore, the prevalence of obesity is increasing in PLWH, and several inflammatory pathways are shared between obesity and treated HIV infection [34]. In the Copenhagen Comorbidity in HIV (COCOMO) study, it was found that abdominal obesity is more common in PLWH than in the general population [35], and abdominal obesity was closely linked to inflammation [36]. Unsurprisingly, the prevalence of metabolic syndrome and diabetes in PLWH is high [37, 38], and a recent meta-analysis found the pooled incidence rate of overt diabetes to be 13.7 per 1000 person-years of follow-up. At present, it is still debated if HIV is an independent risk factor for diabetes [38], but HIV and diabetes are both associated with increased inflammation as manifest by increased levels of proinflammatory markers and monocyte activity as well as an increased risk of CVD.
As such, cardiometabolic comorbidities are common in PLWH and associated with inflammation. Identifying modifiable risk factors is therefore of utmost biomedical importance. The microbiome could be one of several driving factors, along with viral replication, ART toxicity, lipodystrophy, traditional risk factors, coinfections, and past and present immunodeficiency [3]. This review will cover updated knowledge and recent advances in our understanding of the gut microbiome and comorbidities in PLWH, with specific focus on cardiometabolic comorbidities and inflammation. It also contains recent data on circulating metabolites related to comorbidities and underlying gut microbiota alterations.
The gut microbiota and disease-related dysbiosis
Gut microbiota alterations in PLWH: confounders and context
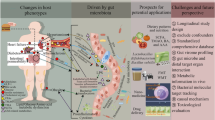
The advent of 16S rRNA-based microbiome characterization enabled comprehensive investigations of associations between gut microbiome composition and features of HIV infection. After several early studies investigating the microbiome in PWH had been completed, it was discovered that sexual behavior has a significant impact on gut microbiome composition including, most notably, an increased abundance of Prevotella in men who have sex with men (MSM) [39, 40]. As MSM comprise the predominant population of PLWH in many sites in Western Europe and the United States, and as the general population is predominantly non-MSM, comparison of random samplings of PLWH and random samplings of the general population is prone to be confounded by sexual behavior. Indeed, several early HIV microbiome studies were not intentionally matched for sexual behavior, and these studies uniquely reported an increased abundance of Prevotella in PLWH [41]. Studies in which PLWH and population controls were matched for sexual behavior have not consistently found enrichment of Prevotella in PLWH [42,43,44,45]. Within MSM, this taxon was enriched in those who took part in recent anal receptive intercourse as compared to those that did not [46], suggesting Prevotella is linked with sexual behavior. As sexual behavior has been shown to have a dominant impact on the microbiome that is greater than HIV serostatus itself [39, 46, 47], microbiome studies addressing hypotheses related to HIV face challenges in circumventing the role of sexual behavior in driving microbiome composition patterns.
Other known confounding variables that influence the microbiome include age, body mass index (BMI), sex, alcohol intake, and certain dietary intake patterns [48]. PLWH can have unique alcohol intake distributions [49], suggesting this variable ought to be captured in future studies, and that intake should be adjusted for or matched between comparison groups to mitigate confounding effects. Studies in which dietary intake was measured have not found significant differences between PLWH and people without HIV [50, 51], though further exploration is warranted. However, even when PLWH and people without HIV are matched for the aforementioned microbiota-confounding variables such as in the AGEhIV cohort study, significant differences in gut microbiome composition have been observed [46, 52]. Due to widely varying methods of microbiome analysis (including differences in quality filtering, read processing, beta-diversity assessment, and statistical analyses), it can be difficult to compare the magnitude of microbiome compositional differences between cases and unaffected controls across studies. Hence, putting such microbiome differences in context of what is observed for other human diseases is challenging. Using the American Gut Project [48, 53], which encompasses individuals that self-reported 19 different diseases as well as healthy control subjects, we applied an identical analytical pipeline as that employed in the AGEhIV cohort studies [46, 52]. We found that HIV-associated dysbiosis was among the strongest disease-associated dysbioses, second to inflammatory bowel disease and stronger than the remaining 18 diseases (Fig. 1).

Gut microbiota differences in cases versus unaffected controls across 20 common human diseases. A For diseases assessed using the American Gut Project, differences between cases and controls in distribution of confounding variables were assessed as previously described by comparing cases to randomly selected controls [48]. For PLWH, previously reported differences between PLWH and the HIV-uninfected population are represented for alcohol [54], diet [50, 51], sex, and anal receptive intercourse [55]. B Sequencing data were collated from the American Gut Project and prior analyses of the AGEhIV cohort and were processed in identical fashion [46, 48, 52] using dada2 [56]. For both datasets, Canberra beta-diversity matrices were calculated, and PERMANOVA tests were performed to quantify significance and effect sizes of ecological distances between cases and controls for each disease. Sample sizes are shown in parentheses encompassing balanced cohorts of cases and controls matched for confounding variables displayed at top left. For HIV cohorts, PERMANOVA statistics were calculated on five total sample groups from two studies [46, 52] including the following: men who have sex with men (n = 76) [46], females (n = 38) [46], men who have sex with women (n = 34) [46], combined females and males (irrespective of sexual behavior (148) [46], and a separate cohort of men who have sex with men (n = 102) [52]
Microbiome in cardiovascular diseases in the general population
It is beyond the scope of this review to give a detailed description of dysbiosis in separate cohorts of CVD in the general population, and for details, we refer to comprehensive reviews by us [57] and others [58]. In brief, most studies published from cohorts of coronary artery disease (CAD) and HF reported depletion of different bacterial genera or species from the Ruminococcaceae and Lachnospiraceae families, which are microbiome patterns that are also observed in PLWH cohorts adjusted for MSM status. Weaknesses of many of the early microbiota studies include limited sample size and lack of essential covariates like diet and clinical data. More recent and comprehensive studies like the MetaCardis cohort have shown a complex interplay between microbiome and metabolomics features of the cardiometabolic disease spectrum from acute coronary syndromes to chronic heart failure [59].
HIV-related or comorbidity-related dysbiosis
Few studies in PLWH have evaluated the relationship of the gut microbiome and cardiovascular disease, and most of the published studies have so far focused on cardiovascular risk factors such as metabolic syndrome, or subclinical atherosclerosis measured in a research setting, including carotid artery plaques and research coronary angiography.
The COCOMO study follows > 1000 PLWH for > 10 years for comorbidities, with available microbiome profiles in > 400 of these participants. Controls were recruited both from the general population and from a pre-exposure prophylaxis (PrEP) cohort of MSM. After separate comparisons of PLWH and controls in MSM and non-MSM strata, depletion of Lachnospiraceae and Ruminococcaceae and increase in Gammaproteobacteria and Desulfovibrionaceae were identified as HIV-related dysbiosis [60]. This HIV-related dysbiosis was associated with a doubled adjusted risk for the metabolic syndrome (MetS), mostly driven by increased risk of diabetes, hypertension, and abdominal obesity among the MetS components. Of note, there was an increasing association between dysbiosis index and MetS in PLWH with nadir CD4 T-cell counts less than 200, whereas in individuals who never developed immunodeficiency, the association was not evident. Furthermore, the HIV-related microbiome was associated with 30 cm2 larger area of visceral adipose tissue on abdominal CT scan, but again, only in those with previous severe immunodeficiency. This could possibly be a result of several factors including long-term viral replication, toxic ART, and a permanently damaged gut mucosa [60].
The AGEhIV study is another well-powered cohort study that has been collecting biological samples for over 10 years with a focus on comorbidities among individuals aged > 45 years and has found concordant results as those above. Namely, Lachnospiraceae and Ruminococcaceae were depleted in PLWH and Gammaproteobacteria and Desulfovibrionaceae were enriched in PLWH [46]. This study found that microbiota diversity was significantly lower in PLWH than controls, and that microbiota diversity was inversely correlated with circulating soluble urokinase plasminogen activator receptor (suPAR) [46]. This marker has been associated with CVD incidence in both PLWH [61, 62] and the general population [63, 64] and may contribute to CVD via activation and recruitment of monocytes [65]. Additionally, work from the AGEhIV cohort found that HIV-associated dysbiosis was significantly greater in PLWH that went on to develop CVD as compared to matched PLWH that did not experience CVD [46]. Associations between nadir CD4 count and HIV-associated dysbiosis were evident in this cohort [46], as it was in the COCOMO cohort and other studies [66, 67].
The largest study of gut microbiota and manifest atherosclerosis in PLWH to date included 361 women in the USA that were assessed by ultrasonography for the presence of carotid artery plaques. The study identified enrichment of Fusobacterium and Proteus and depletion of Odoribacter and Adlercreutzia in women with plaque compared to women without plaque. These bacteria correlated with plasma lipids, which were associated with increased risk of incident carotid artery plaque during 7 years of follow-up. Of note, this was not HIV specific, as the same associations were observed in women without HIV [68]. A smaller study found no significant composition differences between PLWH with and without coronary heart disease (CHD), despite lower alpha diversity in participants with CHD [69].
In a recent work from the COCOMO cohort, we found that PLWH with obstructive CAD assessed by CT angiography had clear shifts in their gut microbiota, with lower alpha diversity, increased beta diversity, compositional shifts including depletion of several bacteria from the Lachnospiraceae and Ruminococcaceae families, and increased relative abundance of Ruminococcus gnavus, a pro-inflammatory microbe associated with inflammatory bowel disease, as well as Veillonella. Of note, we identified no overlapping genera between CAD-related dysbiosis and the previously established HIV-related dysbiosis index, and the HIV-related dysbiosis index was not related to obstructive CAD (Trøseid et al., JID in press).
Whereas much of literature has focused on depletion of Lachnospiraceae and Ruminococcaceae families, enrichment of both Ruminococcus gnavus and Veillonella identified in the COCOMO cohort and Fusobacterium identified in the US cohort [68], have been identified in human carotid plaque studies [70], pointing to a potential causative or contributing role in the atherosclerotic process independent of HIV status. In light of the published studies in the field, it has so far not been possible to identify a clear gut dysbiosis associated with cardiometabolic comorbidities in PLWH across different cohorts. Moreover, the large inter-individual variation in gut microbiota composition has so far made it difficult to apply microbiota signatures as biomarkers for individual risk assessment.
Microbial translocation
Microbial translocation and HIV pathogenesis
A shared feature of the dysbiosis in cohorts of PLWH and cohorts of persons with CVD is the reduced potential for production of short-chain fatty acids (SCFA), including butyrate. Loss of butyrate-producing bacteria may result in a dysfunctional gut mucosal barrier, allowing passive leakage of microbial toxins such as LPS that binds to toll-like receptors and other receptors of the innate immune system, thereby triggering inflammation. This process is called microbial translocation and has been studied in several cohorts of PLWH, since first described by Brenchley et al. in 2006 [71].
Microbial translocation and HIV comorbidities
In the general population, an increased potential for LPS biosynthesis in the microbiome has been reported among patients with CAD [72], and previous studies have linked circulating levels of LPS to insulin resistance [73], glycemic control and abdominal obesity [74], and cardiovascular events (reviewed in [75]). Atherosclerosis is in part an inflammatory process, and several lines of evidence suggest that LPS contributes to this process by fueling a low-grade chronic inflammation and atherothrombosis [75].
In PLWH, we and others have shown that circulating levels of LPS associate with several cardiovascular risk factors, including hypertension [76], insulin resistance, Framingham risk score [77], platelet reactivity [78], metabolic syndrome, central obesity, and hypertriglyceridemia [79]. The latter is possibly due to co-transportation with triglycerides in chylomicrons over the intestinal wall [80]. Despite these associations, circulating levels of LPS have not been linked to incident cardiovascular disease in PLWH. This could partly be due to low sample size in published studies and technical difficulties measuring LPS in bioassays, but it could also reflect that different forms of LPS have different biological properties.
Emerging evidence demonstrates different bioactivity of LPS, where hexa-acylated LPS triggers inflammation, while penta-acylated LPS does not [81]. One report from a CAD cohort showed that genes required for synthesis of the LPS O-antigen were enriched in CAD, whereas the lipid A module was depleted, probably due to depletion of Bacteroides, which produce non-inflammatory penta-acylated lipid A [82].
In a previously published probiotic trial including PLWH, we showed that gut bacteria producing hexa-acylated LPS were outnumbered by bacteria-producing penta-acylated LPS by a factor of 25, and that PLWH with a high ratio of hexa- to penta-acylated LPS-producing bacteria exhibited increased levels of systemic inflammation and tryptophan catabolism. Of note, changes in circulating LPS correlated closely to altered abundance of gram-negative bacteria producing penta-acylated LPS, including Bacteroides [83]. Hence, circulating LPS could partly reflect LPS from commensal microbes with low pro-inflammatory potential.
Indirect ways of measuring microbial translocation include measuring markers of immune cell responses to LPS, including circulating levels of soluble CD14 (sCD14) and LPS-binding protein (LBP) which are shed from toll-like receptor 4 upon LPS activation. Both markers have been associated with future cardiovascular events both in the general population [84] and in PLWH [85, 86]. However, both CD14 and LBP are promiscuous molecules with several triggers beyond LPS, and they should be regarded as markers of monocyte activation (sCD14) and general inflammation (LBP) rather than microbial translocation. Also, intestinal fatty acid-binding protein (IFABP) and zonulin are frequently reported in this context but should be regarded as markers of impaired gut barrier function rather than microbial translocation.
More specific quantification of microbes is possible via amplification and sequencing of microbial nucleic acids via 16S rRNA amplicon sequencing or via amplification-free sequencing of total nucleic acids. However, application of these techniques to low biomass samples such as blood and internal organs has been a challenge [87, 88] due to the risk of low-level environmental contaminants dominating results [87,88,89,90,91] and the difficulty in unambiguous distinguishment of such contaminants from the true signal. Such contaminants can come from tubes, tools, the skin of the study participant (as skin must be broken for a needle to collect blood), the skin from study staff, reagents, sample cross-contamination during DNA extraction and amplification, and index hopping during sequencing. This may explain mixed results among studies examining 16S rRNA in the blood of PLWH [92, 93]. Whereas most microbiota studies have focused on the bacteriome, the much less studied fungiome has also been reported to be altered in a few studies including PLWH [94] and could translocate to circulation and trigger inflammation [95]. Interestingly, a study reported that plasma β-d-glucan, a marker of fungal translocation, was higher in PLWH with carotid artery plaque compared to those without plaques [96].
Overall, it has so far remained challenging to measure microbial translocation, making it difficult to assess its potential role as a biomarker for HIV-associated comorbidities and to assess efficacy of experimental therapies that target this mechanism.
Microbial metabolites and cardiovascular risk
Whereas microbiota traits vary from individual to individual and are affected by several confounding factors, including sexual practice and medicines, circulating metabolites may be less variable and therefore easier to evaluate as biomarkers. The microbiome is a complex bioreactor that produces and catabolizes neurotransmitters, amino acids, short-chain fatty acids, lipids, vitamins, and metabolites [97]. Several of these have been linked to different noncommunicable diseases in the general population, but not always to underlying gut microbiota dysbiosis. This section will focus on potential circulating biomarkers associated with CVD and underlying dysbiosis, with discussion of important aspects to consider when applying such biomarkers in PLWH.
Short-chain fatty acids (SCFA)
SCFA are key gut microbial metabolites derived from fiber fermentation that benefit numerous facets of host biology. They are the primary energy source for the epithelial cells that line the colon, they induce tight junction proteins that bolster integrity of the gut epithelial barrier, and they induce regulatory T cells that dampen exuberant inflammation [98,99,100]. All of these functions may be protective in both HIV and CVD, making microbiome-mediated SCFA potentially important in pathology of both of these two disease states.
While over a third of microbial proteins have unknown function [101], many enzymes involved in the production of SCFAs have been identified. For this reason, abundance of these can be quantified in human stool via high-throughput sequencing of microbial DNA (metagenomics) or microbial RNA (metatranscriptomics). Decreased metagenome-encoded potential for SCFA production has been observed across microbiota studies examining individuals with CVD or PLWH [102, 103], suggesting a depletion of bioavailable SCFA is characteristic of these conditions.
Measuring SCFA directly in humans is stymied by several factors. It is estimated that 95% of SCFA produced by gut microbes are absorbed by the time fecal material reaches the rectum [104, 105]. Epithelial transmembrane transporters that are responsible for SCFA uptake into host tissues are upregulated with increasing exposure to SCFA in a dose-dependent fashion [52, 106], suggesting that high SCFA production can be matched by high uptake. For example, an observation of low SCFA in stool could either be the result of high SCFA production having been matched by high expression of SCFA transporters and high SCFA uptake, or of low SCFA production along with low SCFA transport expression and uptake. Thus, the remaining SCFA in feces may be a poor surrogate for microbiome-mediated SCFA production. Indeed, mixed results have been observed when examining SCFA levels in stool of PLWH and controls [107,108,109,110,111]. Murine studies examining microbiome-mediated SCFA production predominantly quantify SCFA in cecal contents [100, 112, 113], which anatomically precede the uptake that occurs in the colon. However, it is not feasible to collect human luminal material at the ileocecal junction, making SCFA quantification in humans challenging. We and others have found that measuring SCFA in serum, the compartment that may represent the SCFA pool post-uptake from the gut lumen, yields biologically meaningful results that are consistent with metagenome-encoded SCFA production capacity [52, 114]. Indeed, serum levels of the SCFA propionate in a cohort of PLWH correlated more strongly with metagenome-encoded abundance of propionate metabolism enzymes than did levels of propionate in stool [52].
Butyrate production from gut microbes may be particularly challenging to quantify in vivo because it is rapidly taken up by epithelial cells which then rapidly consume it, for butyrate is the preferred energy source for colonic epithelial cells. Propionate, on the other hand, is not the preferred energy source [115] and may thus be exported to the serum more so than butyrate. We found that abundance of butyrate-producing enzymes in the microbiome was not correlated with either stool nor serum levels of butyrate [52], highlighting the difficulty of measuring in vivo butyrate production from the gut microbiome.
SCFA in PLWH
Several HIV microbiome studies have found a depletion of SCFA-producing gut bacteria in PLWH compared to controls [116, 117]. Studies have also found lower relative abundance of genes involved in SCFA production within metagenomes of PLWH [102, 103]. In a cohort of matched PLWH and controls, we have recently found that serum levels of the SCFA propionate were significantly reduced in PLWH, and that the conversion of lactate, one of several precursors for SCFA, was associated with CVD in PLWH [52]. While abundance of butyrate-producing enzymes in the microbiome was dramatically reduced in PLWH, we did not find differences in either circulating or stool butyrate levels, possibly because of the aforementioned biological fate of butyrate produced by the microbiome.
SCFA in CVD
Diets rich in fiber, the primary substrate for microbial SCFA production, are promoted as being among the principal effective interventions [118, 119] to reduce blood pressure, a major contributor to CVD [120, 121]. These dietary recommendations are supported by the observed efficacy of dietary fiber intervention trials performed in the general (HIV-seronegative) population for reducing hyptertension [122, 123]. While other aspects of high-fiber diet may contribute to their protective role in CVD, murine studies demonstrate that SCFA alone can lower hypertension and CVD in animal models [124,125,126]. Mechanisms for these cardioprotective effects include induction of inflammation-dampening regulatory T cells [125], which reduce activation of various immune cells linked with CVD progression including macrophages. Another putative mechanism for the effects of SCFA on hypertension includes direct regulation of blood pressure in the kidneys via renal olfactory receptors [124]. Finally, as discussed above, SCFA strengthen gut barrier integrity and help mitigate microbial translocation, which itself may spur CVD in both PLWH and the HIV-negative population.
Carnitine metabolites
The most compelling evidence of a link between the gut microbiome and CVD has been related to microbial metabolism of the dietary factors phosphatidylcholine and L-carnitine to trimethylamine-N-oxide (TMAO). The source of TMAO is TMA which is produced by the gut microbiota from nutrients containing L-carnitine or phosphatidylcholine and subsequently oxidized in the liver by flavin-containing monooxygenases to TMAO [127]. In particular, carnitine is abundant in red meat; hence, TMAO and other carnitine metabolites are potential links between dietary factors, gut microbiota, and CVD. In a landmark paper from the Hazen group [128], TMAO was identified as a strong predictor of CAD, and subsequent studies have linked TMAO to other types of CVD including acute coronary syndrome and chronic HF [129,130,131,132]. TMAO has been mechanistically linked to thromboembolic events as it enhances thrombus formation [133]. Furthermore, precursors of TMAO promote foam cell formation and atherosclerosis in animal models, but not when adding antibiotics to the drinking water, suggesting a microbiota dependent mechanism [134]. However, a firm link to disease-specific dysbiosis has not been convincingly demonstrated [57].
TMAO has been assessed in several cohorts of PLWH with conflicting results. Some studies have found an association with CVD [135], others did not [136], and one study showed a U-shaped association between TMAO and CVD in PLWH [137]. In a prospective cohort of 520 PLWH in the USA, plasma TMAO was associated with increased risk of incident carotid artery plaque, independent of traditional and HIV-related risk factors, during a median follow-up of 7 years, although the association was attenuated after further adjustment for markers of monocyte activation [135]. In contrast, in a longitudinal nested case–control study of first-time MI in PLWH from Denmark, we found no evidence for increased TMAO levels across several time points before onset of MI. However, TMAO levels increased significantly after initiation of ART, in particular in those starting a protease inhibitor-containing regimen [136]. Hence, we speculate that certain drugs including ART could interfere with microbial generation of TMA or with hepatic oxidation from TMA to TMAO, making TMAO a less suitable biomarker in PLWH.
Interestingly, a separate work reported that TMA was associated with carotid atherosclerosis in PLWH [138]. However, TMA is more volatile than TMAO, making it difficult to measure. Furthermore, another study showed that one of the TMAO precursors choline, but not TMAO, was associated with progression of carotid atherosclerosis in PLWH [135]. Other TMAO precursors such as trimethyl lysine (TML) have been associated with atherosclerosis in the general population [139,140,141], but to the best of our knowledge, not in PLWH. However, TMAO precursors such as carnitine, choline, and TML are probably more diet-related than microbiota-related metabolites and will in most likely have a limited role in advancing our understanding the contribution of the gut microbiota on comorbidities in PLWH.
Uremic toxins
The role of microbiota-derived uremic toxins could be of particular relevance for cardiovascular risk in relation to chronic kidney disease (reviewed in [142]). Emerging evidence suggests that one such uremic toxin, phenylacetylglutamine (PAGln), which accumulates in children with urea cycle disorders, provides prognostic information on cardiovascular risk in association with chronic kidney disease [143] and even in populations without renal failure [144]. Similar to TMAO, PAGln is mainly a bacterial degradation product, which is derived from phenylalanine-rich food and undergoes subsequent conjugation with glutamine in the liver [144]. In the general population, PAGln has been associated with the risk of ischemic stroke and atrial fibrillation [145] and recently also with coronary CAD [146] and HF [147].
PAGln signals within host cells via G protein-coupled receptors, including adrenergic receptors [148].The link between PAGln and CVD was first established by an untargeted metabolomics approach, demonstrating that the gut microbiome contributes to circulating levels of PAGln, and that PAGIn could enhance platelet adhesion and thrombus formation [148].
So far, there are limited data on PAGln in PLWH. Similar to TMAO, levels of PAGln were reported to increase in PLWH treated with ART, with higher levels in PLWH with hyperglycemia and/or hyperlipidemia [149]. Studies of PAGln in relation to cardiovascular comorbidities in PLWH are yet to be performed but should take into account potential impact of ART and renal dysfunction in the study design.
Secondary bile acids
Whereas bile acids are traditionally regarded as emulsifiers to facilitate the absorption of dietary fat and fat-soluble vitamins, bile acids are also recognized as signaling molecules that interact with plasma membranes as well as nuclear receptors, exerting regulatory effects on energy homeostasis [150], lipid and glucose metabolism [151], and other physiological processes [152]. In the gut, primary bile acids undergo metabolism to secondary bile acids, before reabsorption as a part of the enterohepatic cycle (reviewed in [153]). These microbial bile acid modifications have major impact on the agonist activity on the bile acid receptors such as the farnesoid X receptor which has several pleiotropic effects [154] and could represent a link between the gut microbiome and CVD.
We have previously analyzed the circulating bile acid pool in patients with HF and healthy controls and found an increased ratio of secondary to primary bile acids in HF which was associated with reduced overall survival in unadjusted, but not in adjusted analyses [155]. Bile acids are technically difficult to measure. With the exception of a study reporting higher levels of primary and secondary bile acids, as well as microbiome alterations in PLWH with chronic HCV infection and a history of major depression [156], data on circulating bile acid pool is so far limited in PLWH.
Tryptophan metabolites of the kynurenine pathway
Kynurenine pathway metabolites can be produced via the catabolism of tryptophan by the host enzyme indoleamine 2,3-dioxygenase 1 (IDO1), which is induced in the setting of inflammation. This enzymatic pathway serves to limit T-cell proliferation via tryptophan starvation and by the direct action of kynurenine compounds (e.g., kynurenine, 3-hydroxyanthranilic acid) on T cells [157] including the induction of regulatory T cells. Kynurenine compounds also diminish differentiation of Th17 cells, which are critical mediators of gut barrier integrity and are characteristically depleted in the gut of PLWH that initiated treatment during the chronic phase [158]. This gut Th17 cell depletion is associated with elevated markers of inflammation and possibly microbial translocation [158]. Serum kynurenine/tryptophan (KT) ratio, a surrogate marker for activity of the kynurenine metabolic pathway, is in turn associated with mortality and Th17 cell depletion in PLWH [159, 160]. While IDO1 is induced by inflammatory cytokines and is expressed highly in the gut of PLWH with progressive infection [160], its expression is diminished in the treated PLWH despite persistently elevated KT ratios in this subject group [44]. We previously found that gut-resident microbes encode enzymes with analogous functions to that of IDO1, and that the abundance of gut bacteria that encoded such enzymes correlated with KT ratios in treated PLWH, while gut IDO1 expression itself did not [44]. Fecal metabolomics have concordantly found kynurenine metabolites elevated in PLWH [161], further suggesting that microbes may contribute to the immunomodulatory kynurenine pathway of tryptophan catabolism in PLWH.
In the general population, several studies have linked increased KT ratio to increased risk of diabetes and CAD [162, 163]. In PLWH, several studies have reported the kynurenine pathway to associate with mortality [159, 164, 165], non-AIDS comorbidities, aging, and inflammation, and the kynurenine pathway has been suggested to be of particular importance in connecting gut inflammation with age-related comorbidities [30]. Studies in PLWH have linked tryptophan metabolism to gut microbiota alterations and different aspects of atherosclerosis, including endothelial dysfunction [166] and carotid atherosclerosis [167,168,169], although the links between dysbiosis, tryptophan catabolism, and cardiovascular disease have been incompletely defined. In the COCOMO cohort, we found that increased KT ratio mediates around 10% of the association between gut microbiota alterations and visceral adipose tissue accumulation [170], suggesting this metabolic pathway may also be linked with adiposity. Some of the strongest associations between mortality and KT ratios are evident in sub-Saharan African populations [159, 164, 165]. Although tryptophan levels have been consistently lower in several cohorts of PLWH [30], tryptophan levels were in general markedly lower in a sub-Saharan cohort than those reported for developed countries, suggesting that lower tryptophan intake related to malnutrition could be of importance in addition to the inflammatory-induced tryptophan depletion [171]. Given the primary source of bioavailable tryptophan is dietary, the interplay between diet and microbiota in influencing kynurenine pathway activity and its links to these important adverse biological phenomena merits further exploration.
Microbiota-derived indoles
In addition to kynurenines, tryptophan can also be metabolized into serotonin (5-hydroxytryptamine) as well as into indole and its derivatives, the latter through the gut microbiota-dependent indole pathway [172]. Indole and its derivatives have been linked to protective (e.g., indole-3-propionic acid; IPA) and detrimental (e.g., indoxyl sulfate) effects on inflammation and vascular disease. Whereas indole and IPA are important for the gut mucosal barrier function in part by exerting anti-inflammatory activities through activation of aryl hydrocarbon receptor and pregnane X receptors, indoxyl sulfate has cardiotoxic and nephrotoxic properties [173, 174]. Indoxyl sulfate is associated with cardiovascular risk related to chronic kidney disease [174], and as it accumulates with decreased renal clearance, it is also considered a microbiota-derived uremic toxin, as discussed above.
A recent study of women with and without HIV evaluating a broad range of tryptophan metabolites along the kynurenine and indole pathway found that plasma levels of IPA and IPA/kynurenic acid ratio were inversely associated with carotid artery plaque, regardless of HIV serostatus [175]. Of note, five gut bacterial genera and many affiliated species were positively associated with IPA, including Roseburia sp., Eubacterium sp., Lachnospira sp., and Coprobacter sp., whereas no bacterial genera were found to be associated with kynurenic acid, suggesting a beneficial role of IPA and its bacterial sources in atherosclerosis and CVD [175].
The histidine metabolite imidazole propionate
Imidazole propionate (ImP) is a microbially produced histidine metabolite. ImP has been linked to insulin resistance and type 2 diabetes through the mammalian target of rapamycin complex (mTORC) pathway [176, 177] and was recently reported to provide prognostic information and to be related to dysbiosis in patients with HF [178, 179].
ImP production has been linked to certain bacteria, including Ruminococcus gnavus and Veillonella [176]. As we found in the COCOMO cohort these bacteria to be related to obstructive CAD in PLWH, we hypothesized that circulating ImP levels could be a potential biomarker of obstructive CAD. We found elevated ImP levels to be associated with both obstructive CAD and the underlying dysbiosis [180]. However, whereas dysbiosis index was independently associated with obstructive CAD, the association with ImP was attenuated and no longer significant in multivariable analyses.
Our findings are in line with a recent report of ImP being associated with carotid atherosclerosis and underlying dysbiosis in women living with HIV [181]. Further analysis identified additional bacterial species and bacterial hutH gene (encoding enzyme histidine ammonia-lyase in ImP production) associated with plasma ImP levels, and that a gut microbiota score based on these ImP-associated species was positively associated with plaque and several pro-inflammatory markers [181]. Hence, ImP seems to capture cardiovascular comorbidities and underlying dysbiosis in PLWH irrespective of gender or mode of transmission. So far, no studies have reported ImP in relation to CAD in the general population, but, in light of our findings, such studies are warranted.
Integrating dysbiosis and microbial metabolites
Unbiased versus targeted omics approach
With the combined genes of the microbiome approaching that of the total human genome, and each microbe having the potential to turn on and off the production of hundreds of metabolites, several undiscovered microbiota-related metabolites are likely to be relevant for HIV-associated comorbidities. Whereas most studies to date have been based on sequencing the 16S rRNA gene, metagenomic sequencing presents an opportunity to better define functional changes in the gut microbiome. Ultimately, combined analyses of the actual byproducts of microbial activity (unbiased metabolomics and/or proteomics analyses of parallel plasma samples) and microbiota (16S rRNA or metagenomics) analyses controlling for relevant confounders may augment discovery [57, 181]. Furthermore, the gut virome and mycobiome are underexplored areas in PLWH, and both may impact immune function [182, 183].
A recent study applying unbiased multi-omics approach on the COCOMO cohort identified separate clusters of PLWH with different metabolic risk profiles. Although analyses were adjusted for confounders including sexual preference, the high-risk cluster was partly driven by a Prevotella-enriched gut microbiota with a high proportion of MSM, and there was little overlap between microbiota profiles and plasma metabolomics and lipidomics, respecitively [184]. In contrast, a recent study of PLWH discovered ImP as the most promising of several soluble markers through an unbiased multi-omics approach and also found a clear correlation between ImP and dysbiosis as well as carotid atheroslcerosis [181]. Of note, the latter study only included women living with HIV; hence, the confounding effect of MSM was not relevant.
There is a risk that unsupervised, unbiased multi-omics approach in data set with a large proportion of MSM will not be able to filter out the MSM signal without a clear strategy, preferably by including seronegative control groups of MSM and non-MSM, either as part of the study cohort or by applying data from data repositories. An alternative is to establish an HIV-associated dysbiosis index in a data set with relevant control groups or to establish a comorbidity-related dysbiosis index if the MSM proportion is equal in those with and without comorbidities.
A more targeted approach is needed to confirm or reject promising findings from unbiased discovery studies. The choice of candidate biomarkers can be made by different strategies, either based on specific hypotheses or by certain traits in gut dysbiosis pointing to specific biomarkers to be tested, i.e., depletion of Ruminococcaceae and Lachnospiraceae in relation to circulating SCFAs [52], alteration of tryptophan metabolizing bacteria in relation to KT-ratio [44] or IPA levels [175], or increase in Ruminococcus gnavus in relation to ImP levels [180].
From biomarkers to clinical application
Risk stratification beyond traditional risk factors
For translation to a clinical setting, biomarkers that are easily measurable in a reproducible way in plasma, urine or other body fluids, will probably be easier to implement than individual microbiota signatures, given the complexity and variability of the latter. A clinically relevant microbiota-related biomarker should preferably be associated with the disease-related dysbiosis or other microbiota traits, as well as with the comorbidity in question, independent of relevant covariates. For HIV-related comorbidities, this should include traditional and HIV-related risk factors, as well as potential confounders, such as mode of transmission, antibiotics, other relevant drugs, and dietary data.
However, for use in a clinical setting, a novel biomarker should also provide additional information beyond established biomarkers or at least independent of established biomarkers. In non-HIV cohorts, TMAO has been shown to provide information on risk of major cardiovascular events after myocardial infarction independent of troponin levels and in independent cohorts [129] but is yet to be established as a risk marker in an acute clinical setting [130].
In PLWH, a major advance was recently published on screening for precursors of anal cancer by measuring microbial proteins from anal swabs [185]. Of note, microbiota differences were limited and driven by outliers, whereas microbiota-derived proteomics separated clearly and converged on common pathways related to energy metabolism. Of note, measurements of two downstream substances, cobalamin and succinyl-CoA, in two independent cohort from Madrid and Milano, were able to increase sensitivity and specificity dramatically compared to anal cytology [185]. These data need to be reproduced in independent studies before affecting screening algorithms for anal cancer in PLWH, and similar requirements should be made for emerging biomarkers for other comorbidities.
Therapeutic target
Another potential clinical application of the gut microbiota is as a therapeutic target. With respect to comorbidities, a microbiota-directed intervention should preferably demonstrate improvement of the comorbidity in question or its risk factors. Several attempts have been made to target the microbiota with probiotics (live beneficial bacteria), prebiotics (food for beneficial bacteria), or synbiotics (probiotics combined with prebiotics) in PLWH. In a comprehensive review summarizing these trials, there is no evidence that any of these interventions are clinically helpful in PLWH [186]. Although several trials have reported effect on one or two biomarkers, typically a cytokine or a subset of immune cells, these biomarkers differ between trials, the primary end point is often not clearly defined, and most trials have been underpowered [186].
Other attempts have been made to use fecal microbiota transplant (FMT) with different target population and donor selection strategies. Although FMT appears safe, there is limited evidence for better immunological recovery or efficacy in modulating other established, clinically relevant outcomes in PLWH [187, 188]. Of note, engraftment was transient and limited after one-time transplant [189] and even with repeated weekly inoculations regardless of antibiotic pre-treatment [188], underscoring the challenges of inducing lasting changes in gut microbiota composition via FMT.
Alternative approaches for therapeutic strategies include attempts to target the enzymatic pathways of bacterial metabolites, such as production of TMAO or ImP, in a “drug the bug” strategy [190]. So far, identifying suitable biomarkers will be required before choosing more targeted approaches in PLWH.
Tool for precision medicine
It was recently shown in the REPRIEVE trial that pitavastatin, a cholesterol-lowering drug, reduced the relative risk of a major cardiovascular event by around one third in PLWH at low to moderate risk of cardiovascular disease [191]. As the risk reduction occurred irrespective of baseline LDL cholesterol levels, other mechanisms such as anti-inflammatory effects of statins could be relevant. Of note, there is at present no single biomarker to guide selection of candidates for statin therapy, beyond global risk assessment.
The tremendous inter-individual variation in gut microbiota composition is clearly a limitation in biomarker studies, but this variation could potentially be used as a tool for precision medicine. An elegant study showing substantial inter-individual variation of glycemic responses after different meals also showed that integration of microbiota profiles and metadata in a machine learning model made it possible to precisely predict individual glycemic responses in order to personalize nutritional advice [192]. Whether such an approach could be used to tailor individualized prophylaxis, including statin therapy to prevent HIV-related comorbidities is promising but yet unknown. Indeed, the gut microbiome can metabolize therapeutic drugs [193], and the gut microbiomes across individuals exhibit substantial heterogeneity in their capacity to metabolize different drugs [194]. Studies to test the prediction of optimal individualized therapies would require real-time integration of microbiota profiles (or related metabolites), drugs, traditional risk factors, and HIV-specific factors to assess, i.e., 10-year risk of myocardial infarction or stroke. Such a prediction tool should be able to outperform established risk score algorithms, which generally underestimate CVD risk in PLWH, and be fairly easy to use in a clinical setting. Nevertheless, with rapid advances in artificial intelligence, such an approach could be feasible in the near future.
Future research
As shown in Table 1, several metabolites demonstrated to be of relevance in the general population are understudied in PLWH. This includes among others circulating bile acids, uremic toxins and partly histidine metabolites, and carnitine metabolites beyond TMAO. On the other hand, studies performed in PLWH should also inspire studies in the general population, such as circulating propionate levels, which should be investigated as a potential dysbiosis-related cardiovascular biomarker in HIV and non-HIV cohorts.
Furthermore, gut microbiota analysis beyond the bacteriome should be applied to precisely define the HIV-related mycobiome and virome and further assess the cross-kingdom microbiome in relation to comorbidities. Moreover, with the rapid advances in artificial intelligence, it is of importance to make clear strategies on how to overcome specific confounding factors, such as sexual preference, when planning unbiased multi-omics analyses.
Finally, once promising gut microbiota-related candidate markers have been identified in various studies, such markers should be independently validated in adequately powered multicenter prospective cohorts designed to assess biomarkers in relation to incident comorbidities, such as the MISTRAL study which is nested in the EuroSIDA cohort. Such studies should ultimately lay the foundation for future precision medicine, including novel strategies for personalized risk assessment and intervention studies targeting the gut microbiota to reduce the risk of HIV-related comorbidities.
Availability of data and materials
No datasets were generated or analysed during the current study.
References
Rodger AJ, et al. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS. 2013;27:973–9.
Achhra AC, et al. Immunodeficiency and the risk of serious clinical endpoints in a well studied cohort of treated HIV-infected patients. AIDS. 2010;24:1877–86.
Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382:1525–33.
Trickey A, et al. Life expectancy after 2015 of adults with HIV on long-term antiretroviral therapy in Europe and North America: a collaborative analysis of cohort studies. Lancet HIV. 2023;10:e295–307.
Guaraldi G, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis. 2011;53:1120–6.
Schouten J, et al. Cross-sectional comparison of the prevalence of age-associated comorbidities and their risk factors between HIV-infected and uninfected individuals: the AGEhIV cohort study. Clin Infect Dis. 2014;59:1787–97.
Verheij E, et al. Long-term evolution of comorbidities and their disease burden in individuals with and without HIV as they age: analysis of the prospective AGE(h)IV cohort study. Lancet HIV. 2023;10:e164–74.
Obel N, et al. Impact of non-HIV and HIV risk factors on survival in HIV-infected patients on HAART: a population-based nationwide cohort study. PLoS One. 2011;6:e22698.
Marcus JL, et al. Comparison of overall and comorbidity-free life expectancy between insured adults with and without HIV infection, 2000–2016. JAMA Netw Open. 2020;3:e207954.
Althoff KN, et al. The shifting age distribution of people with HIV using antiretroviral therapy in the United States. AIDS. 2022;36:459–71.
GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204–22.
Alonso A, et al. HIV infection and incidence of cardiovascular diseases: an analysis of a large healthcare database. J Am Heart Assoc. 2019;8:e012241.
Freiberg MS, et al. Association between HIV infection and the risk of heart failure with reduced ejection fraction and preserved ejection fraction in the antiretroviral therapy era: results from the veterans aging cohort study. JAMA Cardiol. 2017;2:536–46.
Lam JO, et al. Variation in heart failure risk by HIV severity and sex in people with HIV infection. J Acquir Immune Defic Syndr. 2022;91:175–81.
Feinstein MJ, et al. Adjudicated heart failure in HIV-infected and uninfected men and women. J Am Heart Assoc. 2018;7:e009985.
Knudsen AD, et al. Prevalence and risk factors of prolonged QT interval and electrocardiographic abnormalities in persons living with HIV. AIDS. 2019;33:2205–10.
Bloomfield GS, et al. Prevalence and correlates of electrocardiographic abnormalities in adults with HIV: insights from the randomized trial to prevent vascular events in HIV (REPRIEVE). J Acquir Immune Defic Syndr. 2022;89:349–59.
Freiberg MS, et al. HIV Infection and the risk of World Health Organization-defined sudden cardiac death. J Am Heart Assoc. 2021;10:e021268.
Tseng ZH, et al. Sudden cardiac death and myocardial fibrosis, determined by autopsy, in persons with HIV. N Engl J Med. 2021;384:2306–16.
Knudsen AD, et al. Brief report: prevalence of peripheral artery disease is higher in persons living with HIV compared with uninfected controls. J Acquir Immune Defic Syndr. 2018;79:381–5.
Beckman JA, et al. Association of human immunodeficiency virus infection and risk of peripheral artery disease. Circulation. 2018;138:255–65.
Cedarbaum E, et al. Contributions of HIV, hepatitis C virus, and traditional vascular risk factors to peripheral artery disease in women. AIDS. 2019;33:2025–33.
Høgh J, et al. HIV infection is associated with thoracic and abdominal aortic aneurysms: a prospective matched cohort study. Eur Heart J. 2021;42:2924–31.
Filipkowski AM, et al. Association of HIV infection and incident abdominal aortic aneurysm among 143 001 veterans. Circulation. 2023;148:135–43.
Mdodo R, et al. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: cross-sectional surveys. Ann Intern Med. 2015;162:335–44.
Rasmussen LD, et al. Myocardial infarction among Danish HIV-infected individuals: population-attributable fractions associated with smoking. Clin Infect Dis. 2015;60:1415–23.
Borges ÁH, et al. Interleukin 6 is a stronger predictor of clinical events than high-sensitivity C-reactive protein or D-dimer during HIV infection. J Infect Dis. 2016;214:408–16.
Hoel H, et al. Soluble markers of interleukin 1 activation as predictors of first-time myocardial infarction in HIV-infected individuals. J Infect Dis. 2020;221:506–9.
Ridker PM, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31.
MacCann R, Landay AL, Mallon PWG. HIV and comorbidities - the importance of gut inflammation and the kynurenine pathway. Curr Opin HIV AIDS. 2023;18:102–10.
Sukumaran L, et al. Association between inflammatory biomarker profiles and cardiovascular risk in individuals with and without HIV. AIDS. 2023;37:595–603.
Vos AG, Idris NS, Barth RE, Klipstein-Grobusch K, Grobbee DE. Pro-inflammatory markers in relation to cardiovascular disease in HIV infection. A systematic review. PLoS One. 2016;11:e0147484.
De Francesco D, Sabin CA, Reiss P, Kootstra NA. Monocyte and T cell immune phenotypic profiles associated with age advancement differ between people with HIV, lifestyle-comparable controls and blood donors. Front Immunol. 2020;11:581616.
Savinelli S, et al. Obesity in HIV infection: host-pathogen interaction. AIDS. 2022;36:1477–91.
Gelpi M, et al. Higher risk of abdominal obesity, elevated low-density lipoprotein cholesterol, and hypertriglyceridemia, but not of hypertension, in people living with human immunodeficiency virus (HIV): results from the Copenhagen comorbidity in HIV infection study. Clin Infect Dis. 2018;67:579–86.
Gelpi M, et al. Abdominal adipose tissue is associated with alterations in tryptophan-kynurenine metabolism and markers of systemic inflammation in people with human immunodeficiency virus. J Infect Dis. 2020;221:419–27.
Nguyen KA, et al. Metabolic syndrome in people living with human immunodeficiency virus: an assessment of the prevalence and the agreement between diagnostic criteria. Int J Endocrinol. 2017;2017:1613657.
Hernandez-Romieu AC, Garg S, Rosenberg ES, Thompson-Paul AM, Skarbinski J. Is diabetes prevalence higher among HIV-infected individuals compared with the general population? Evidence from MMP and NHANES 2009–2010. BMJ Open Diabetes Res Care. 2017;5:e000304.
Noguera-Julian M, et al. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine. 2016;5:135–46.
Kelley CF, et al. The rectal mucosa and condomless receptive anal intercourse in HIV-negative MSM: implications for HIV transmission and prevention. Mucosal Immunol. 2017;10:996–1007.
Vujkovic-Cvijin I, Somsouk M. HIV and the gut microbiota: composition, consequences, and avenues for amelioration. Curr HIV/AIDS Rep. 2019;16:204–13.
Monaco CL, et al. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe. 2016;19:311–22.
Lee SC, et al. Enrichment of gut-derived Fusobacterium is associated with suboptimal immune recovery in HIV-infected individuals. Sci Rep. 2018;8:14277.
Vujkovic-Cvijin I, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra191.
Yu G, Fadrosh D, Ma B, Ravel J, Goedert JJ. Anal microbiota profiles in HIV-positive and HIV-negative MSM. AIDS. 2014;28:753–60.
Vujkovic-Cvijin I, et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat Commun. 2020;11:2448.
Armstrong AJS, et al. An exploration of Prevotella-rich microbiomes in HIV and men who have sex with men. Microbiome. 2018;6:198.
Vujkovic-Cvijin I, et al. Host variables confound gut microbiota studies of human disease. Nature. 2020;587:448–54.
Justice A, Sullivan L, Fiellin D, Veterans Aging Cohort Study Project, T. HIV/AIDS, comorbidity, and alcohol: can we make a difference? Alcohol Res Health. 2010;33:258–66.
Dillon SM, et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7:983–94.
Pinto-Cardoso S, et al. Fecal bacterial communities in treated HIV infected individuals on two antiretroviral regimens. Sci Rep. 2017;7:43741.
Sereti I, et al. Impaired gut microbiota-mediated short-chain fatty acid production precedes morbidity and mortality in people with HIV. Cell Rep. 2023;42:113336.
McDonald D, et al. American gut: an open platform for citizen science microbiome research. mSystems. 2018;3:1–28.
Duko B, Ayalew M, Ayano G. The prevalence of alcohol use disorders among people living with HIV/AIDS: a systematic review and meta-analysis. Subst Abuse Treat Prev Policy. 2019;14:52.
Mayer KH, et al. The persistent and evolving HIV epidemic in American men who have sex with men. Lancet. 2021;397:1116–26.
Callahan BJ, et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Trøseid M, Andersen G, Broch K, Hov JR. The gut microbiome in coronary artery disease and heart failure: current knowledge and future directions. EBioMedicine. 2020;52:102649.
Tang WHW, Bäckhed F, Landmesser U, Hazen SL. Intestinal microbiota in cardiovascular health and disease: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2089–105.
Fromentin S, et al. Microbiome and metabolome features of the cardiometabolic disease spectrum. Nat Med. 2022;28:303–14.
Gelpi M, et al. Impact of human immunodeficiency virus-related gut microbiota alterations on metabolic comorbid conditions. Clin Infect Dis. 2020;71:e359–67.
Hoenigl M, et al. Soluble urokinase plasminogen activator receptor is predictive of non-AIDS events during antiretroviral therapy-mediated viral suppression. Clin Infect Dis. 2019;69:676–86.
Rasmussen LJ, et al. Soluble urokinase plasminogen activator receptor (suPAR) is a novel, independent predictive marker of myocardial infarction in HIV-1-infected patients: a nested case-control study. HIV Med. 2016;17:350–7.
Eapen DJ, et al. Soluble urokinase plasminogen activator receptor level is an independent predictor of the presence and severity of coronary artery disease and of future adverse events. J Am Heart Assoc. 2014;3:e001118.
Eugen-Olsen J, et al. Circulating soluble urokinase plasminogen activator receptor predicts cancer, cardiovascular disease, diabetes and mortality in the general population. J Intern Med. 2010;268:296–308.
Hindy G, et al. Increased soluble urokinase plasminogen activator levels modulate monocyte function to promote atherosclerosis. J Clin Invest. 2022;132:e1587884.
Guillen Y, et al. Low nadir CD4+ T-cell counts predict gut dysbiosis in HIV-1 infection. Mucosal Immunol. 2019;12:232–46.
Lu D, et al. Association between CD4(+) T cell counts and gut microbiota and serum cytokines levels in HIV-infected immunological non-responders. BMC Infect Dis. 2021;21:742.
Wang Z, et al. Gut microbiota, plasma metabolomic profiles, and carotid artery atherosclerosis in HIV infection. Arterioscler Thromb Vasc Biol. 2022;42:1081–93.
Kehrmann J, et al. Gut microbiota in human immunodeficiency virus-infected individuals linked to coronary heart disease. J Infect Dis. 2019;219:497–508.
Figuero E, et al. Detection of periodontal bacteria in atheromatous plaque by nested polymerase chain reaction. J Periodontol. 2011;82:1469–77.
Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71.
Zhu Q, et al. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol Genomics. 2018;50:893–903.
Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72.
Trøseid M, et al. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: evidence from bariatric surgery. Diabetes Care. 2013;36:3627–32.
Violi F, et al. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat Rev Cardiol. 2023;20:24–37.
Manner IW, et al. Markers of microbial translocation predict hypertension in HIV-infected individuals. HIV Med. 2013;14:354–61.
Pedersen KK, et al. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance, and risk of myocardial infarction. J Acquir Immune Defic Syndr. 2013;64:425–33.
Haugaard AK, et al. Discrepant coagulation profile in HIV infection: elevated D-dimer but impaired platelet aggregation and clot initiation. AIDS. 2013;27:2749–58.
Manner IW, et al. Plasma lipopolysaccharide and triglycerides are independently associated and both markers correlate with the development of metabolic syndrome in HIV infection. J Acquir Immune Defic Syndr. 2014;65:e158-161.
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–7.
Vatanen T, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–53.
Jie Z, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:845.
Storm-Larsen C, et al. Microbial translocation revisited: targeting the endotoxic potential of gut microbes in HIV-infected individuals. AIDS. 2019;33:645–53.
Awoyemi A, Trøseid M, Arnesen H, Solheim S, Seljeflot I. Effects of dietary intervention and n-3 PUFA supplementation on markers of gut-related inflammation and their association with cardiovascular events in a high-risk population. Atherosclerosis. 2019;286:53–9.
Sandler NG, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203:780–90.
Trevillyan JM, Moser C, Currier JS, Sallam T. Immune biomarkers in the prediction of future myocardial infarctions in people with human immunodeficiency virus. Clin Infect Dis. 2020;70:1764–7.
de Goffau MC, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. 2019;572:329–34.
Kennedy KM, et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature. 2023;613:639–49.
Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016;8:24.
Nearing JT, Comeau AM, Langille MGI. Identifying biases and their potential solutions in human microbiome studies. Microbiome. 2021;9:113.
Salter SJ, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87.
Ferri E, et al. Plasma levels of bacterial DNA in HIV infection: the limits of quantitative polymerase chain reaction. J Infect Dis. 2010;202:176–7. author reply 178.
Svard J, Sonnerborg A, Vondracek M, Molling P, Nowak P. On the usefulness of circulating bacterial 16S rDNA as a marker of microbial translocation in HIV-1-infected patients. J Acquir Immune Defic Syndr. 2014;66:e87-89.
Li S, et al. Neglected mycobiome in HIV infection: alterations, common fungal diseases and antifungal immunity. Front Immunol. 2022;13:1015775.
Weiner LD, et al. Fungal translocation is associated with immune activation and systemic inflammation in treated HIV. AIDS Res Hum Retroviruses. 2019;35:461–72.
Isnard S, et al. Circulating β-d-glucan as a marker of subclinical coronary plaque in antiretroviral therapy-treated people with human immunodeficiency virus. Open Forum Infect Dis. 2021;8:ofab109.
Sonnenburg JL, Bäckhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64.
Duscha A, et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell. 2020;180:1067-1080 e1016.
Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–5.
Smith PM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–73.
Zhang Y, et al. Discovery of bioactive microbial gene products in inflammatory bowel disease. Nature. 2022;606:754–60.
Rocafort M, et al. Evolution of the gut microbiome following acute HIV-1 infection. Microbiome. 2019;7:73.
Vazquez-Castellanos JF, et al. Interplay between gut microbiota metabolism and inflammation in HIV infection. ISME J. 2018;12:1964–76.
den Besten G, et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325–40.
Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81:1031–64.
Borthakur A, et al. Regulation of monocarboxylate transporter 1 (MCT1) promoter by butyrate in human intestinal epithelial cells: involvement of NF-kappaB pathway. J Cell Biochem. 2008;103:1452–63.
El-Far M, et al. Upregulated IL-32 expression and reduced gut short chain fatty acid caproic acid in people living with HIV with subclinical atherosclerosis. Front Immunol. 2021;12:664371.
Liu J, et al. Among older adults, age-related changes in the stool microbiome differ by HIV-1 serostatus. EBioMedicine. 2019;40:583–94.
Chen Y, et al. Signature changes in gut microbiome are associated with increased susceptibility to HIV-1 infection in MSM. Microbiome. 2021;9:237.
Serrano-Villar S, et al. The effects of prebiotics on microbial dysbiosis, butyrate production and immunity in HIV-infected subjects. Mucosal Immunol. 2017;10:1279–93.
Gonzalez-Hernandez LA, et al. Alterations in bacterial communities, SCFA and biomarkers in an elderly HIV-positive and HIV-negative population in western Mexico. BMC Infect Dis. 2019;19:234.
Rivera-Chavez F, et al. Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe. 2016;19:443–54.
Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50.
Xiong R, et al. Multi-’omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe. 2023;31:273-287 e275.
Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83:424–9.
Dillon SM, et al. Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS. 2017;31:511–21.
Ishizaka A, et al. Unique gut microbiome in HIV patients on antiretroviral therapy (ART) suggests association with chronic inflammation. Microbiol Spectr. 2021;9:e0070821.
Lichtenstein AH, et al. 2021 Dietary guidance to improve cardiovascular health: a scientific statement from the American Heart Association. Circulation. 2021;144:e472–87.
National Heart, Lung and Blood Institute. 2019. Why the DASH Eating Plan Works. April 16, 2024. {https://www.nhlbi.nih.gov/resources/why-dash-eating-plan-works}
Group, S.R., et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–16.
Fuchs FD, Whelton PK. High blood pressure and cardiovascular disease. Hypertension. 2020;75:285–92.
Streppel MT, Arends LR, van ’t Veer P, Grobbee DE, Geleijnse JM. Dietary fiber and blood pressure: a meta-analysis of randomized placebo-controlled trials. Arch Intern Med. 2005;165:150–6.
Evans CE, et al. Effects of dietary fibre type on blood pressure: a systematic review and meta-analysis of randomized controlled trials of healthy individuals. J Hypertens. 2015;33:897–911.
Pluznick JL, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110:4410–5.
Bartolomaeus H, et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. 2019;139:1407–21.
Marques FZ, et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–77.
Koeth RA, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–85.
Tang WH, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–84.
Li XS, et al. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: a prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur Heart J. 2017;38:814–24.
Trøseid M. Gut microbiota and acute coronary syndromes: ready for use in the emergency room? Eur Heart J. 2017;38:825–7.
Tang WH, et al. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–14.
Trøseid M, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–26.
Zhu W, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–24.
Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63.
Shan Z, et al. Gut microbial-related choline metabolite trimethylamine-N-oxide is associated with progression of carotid artery atherosclerosis in HIV infection. J Infect Dis. 2018;218:1474–9.
Haissman JM, et al. Microbiota-dependent metabolite and cardiovascular disease marker trimethylamine-N-oxide (TMAO) is associated with monocyte activation but not platelet function in untreated HIV infection. BMC Infect Dis. 2017;17:445.
Miller PE, et al. Brief report: intestinal microbiota-produced trimethylamine-N-oxide and its association with coronary stenosis and HIV serostatus. J Acquir Immune Defic Syndr. 2016;72:114–8.
Srinivasa S, et al. Plaque burden in HIV-infected patients is associated with serum intestinal microbiota-generated trimethylamine. AIDS. 2015;29:443–52.
Skagen K, et al. The carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis. 2016;247:64–9.
Bjørnestad E, et al. Circulating trimethyllysine and risk of acute myocardial infarction in patients with suspected stable coronary heart disease. J Intern Med. 2020;288:446–56.
Li XS, et al. Untargeted metabolomics identifies trimethyllysine, a TMAO-producing nutrient precursor, as a predictor of incident cardiovascular disease risk. JCI Insight. 2018;3:e99096.
Velasquez MT, Centron P, Barrows I, Dwivedi R, Raj DS. Gut microbiota and cardiovascular uremic toxicities. Toxins (Basel). 2018;10:287.
Poesen R, et al. Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J Am Soc Nephrol. 2016;27:3479–87.
Awoyemi A, Hov JR, Trøseid M. Phenylacetylglutamine from the gut microbiota: a future therapeutic target in heart failure? Circ Heart Fail. 2023;16:e010222.
Fang C, et al. PAGln, an atrial fibrillation-linked gut microbial metabolite, acts as a promoter of atrial myocyte injury. Biomolecules. 2022;12:1120.
Ottosson F, et al. The gut microbiota-related metabolite phenylacetylglutamine associates with increased risk of incident coronary artery disease. J Hypertens. 2020;38:2427–34.
Romano KA, et al. Gut microbiota-generated phenylacetylglutamine and heart failure. Circ Heart Fail. 2023;16:e009972.
Nemet I, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180:862-877.e822.
Yuan ZW, et al. Evaluation of characteristic metabolites of aromatic amino acids in patients with HIV infection at different stages of disease. J Clin Lab Anal. 2023;37:e24795.
Watanabe M, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–9.
Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32 Suppl 2:S237-245.
Vallim TQ, Edwards PA. Bile acids have the gall to function as hormones. Cell Metab. 2009;10:162–4.
Guzior DV, Quinn RA. Review: microbial transformations of human bile acids. Microbiome. 2021;9:140.
Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–66.
Mayerhofer CCK, et al. Increased secondary/primary bile acid ratio in chronic heart failure. J Card Fail. 2017;23:666–71.
Taylor BC, et al. Depression in individuals coinfected with HIV and HCV is associated with systematic differences in the gut microbiome and metabolome. mSystems. 2020;5:e00465-20.
Stone TW, Williams RO. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol Sci. 2023;44:442–56.
Wacleche VS, Landay A, Routy JP, Ancuta P. The Th17 lineage: from barrier surfaces homeostasis to autoimmunity, cancer, and HIV-1 pathogenesis. Viruses. 2017;9:303.
Byakwaga H, et al. The kynurenine pathway of tryptophan catabolism, CD4+ T-cell recovery, and mortality among HIV-infected Ugandans initiating antiretroviral therapy. J Infect Dis. 2014;210:383–91.
Favre D, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2:32ra36.
Serrano-Villar S, et al. HIV infection results in metabolic alterations in the gut microbiota different from those induced by other diseases. Sci Rep. 2016;6:26192.
Pedersen ER, et al. Associations of plasma kynurenines with risk of acute myocardial infarction in patients with stable angina pectoris. Arterioscler Thromb Vasc Biol. 2015;35:455–62.
Rebnord EW, et al. The kynurenine:tryptophan ratio as a predictor of incident type 2 diabetes mellitus in individuals with coronary artery disease. Diabetologia. 2017;60:1712–21.
Martinez P, et al. Reversal of the kynurenine pathway of tryptophan catabolism may improve depression in ART-treated HIV-infected Ugandans. J Acquir Immune Defic Syndr. 2014;65:456–62.
Lee S, et al. Immunologic pathways that predict mortality in HIV-infected Ugandans initiating antiretroviral therapy. J Infect Dis. 2017;215:1270–4.
Hoel H, et al. Impact of HIV and type 2 diabetes on gut microbiota diversity, tryptophan catabolism and endothelial dysfunction. Sci Rep. 2018;8:6725.
Luo K, et al. Tryptophan metabolism, gut microbiota, and carotid artery plaque in women with and without HIV infection. AIDS. 2024;38:223–33.
Qi Q, et al. Plasma tryptophan-kynurenine metabolites are altered in human immunodeficiency virus infection and associated with progression of carotid artery atherosclerosis. Clin Infect Dis. 2018;67:235–42.
Siedner MJ, et al. Persistent immune activation and carotid atherosclerosis in HIV-infected Ugandans receiving antiretroviral therapy. J Infect Dis. 2016;213:370–8.
Gelpi M, et al. Association of the kynurenine pathway of tryptophan metabolism with human immunodeficiency virus-related gut microbiota alterations and visceral adipose tissue accumulation. J Infect Dis. 2022;225:1948–54.
Bipath P, Levay PF, Viljoen M. Tryptophan depletion in context of the inflammatory and general nutritional status of a low-income South African HIV-infected population. J Health Popul Nutr. 2016;35:5.
Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe. 2018;23:716–24.
Ye X, et al. Dual role of indoles derived from intestinal microbiota on human health. Front Immunol. 2022;13:903526.
Hung SC, Kuo KL, Wu CC, Tarng DC. Indoxyl sulfate: a novel cardiovascular risk factor in chronic kidney disease. J Am Heart Assoc. 2017;6:e005022.
Luo K, et al. Tryptophan metabolism, gut microbiota, and carotid artery plaque in women with and without HIV infection. AIDS. 2024;38:223–33.
Molinaro A, et al. Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat Commun. 2020;11:5881.
Koh A, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175:947-961.e917.
Molinaro A, et al. Microbially produced imidazole propionate is associated with heart failure and mortality. JACC Heart Fail. 2023;11:810–21.
Raju SC, et al. Microbial-derived imidazole propionate links the heart failure-associated microbiome alterations to disease severity. Genome Med. 2024;16:27.
Trøseid M, et al. Gut microbiota alterations and circulating imidazole propionate levels are associated with obstructive coronary artery disease in people with HIV. J Infect Dis. 2024;229:898–907.
Wang Z, et al. Gut microbiota, circulating inflammatory markers and metabolites, and carotid artery atherosclerosis in HIV infection. Microbiome. 2023;11:119.
Dallari S, et al. Enteric viruses evoke broad host immune responses resembling those elicited by the bacterial microbiome. Cell Host Microbe. 2021;29:1014-1029 e1018.
Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–7.
Mikaeloff F, et al. Network-based multi-omics integration reveals metabolic at-risk profile within treated HIV-infection. Elife. 2023;12:e82785.
Serrano-Villar S, et al. Microbiome-derived cobalamin and succinyl-CoA as biomarkers for improved screening of anal cancer. Nat Med. 2023;29:1738–49.
Meyer-Myklestad MH, et al. Probiotics to HIV-infected immunological nonresponders: altered mucosal immunity and microbial diversity restricted to ileum. J Acquir Immune Defic Syndr. 2022;89:77–86.
Caira-Chuquineyra B, et al. Fecal microbiota transplantation for people living with human immunodeficiency virus: a scoping review. AIDS Res Hum Retroviruses. 2022;38:700–8.
Serrano-Villar S, et al. Fecal microbiota transplantation in HIV: a pilot placebo-controlled study. Nat Commun. 2021;12:1139.
Vujkovic-Cvijin I, et al. Limited engraftment of donor microbiome via one-time fecal microbial transplantation in treated HIV-infected individuals. Gut Microbes. 2017;8:440–50.
Jonsson AL, Bäckhed F. Drug the Bug! Cell. 2015;163:1565–6.
Grinspoon SK, et al. Pitavastatin to prevent cardiovascular disease in HIV infection. N Engl J Med. 2023;389:687–99.
Zeevi D, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163:1079–94.
Wallace BD, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–5.
Javdan B, et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell. 2020;181:1661-1679 e1622.
Sandler NG, et al. Sevelamer does not decrease lipopolysaccharide or soluble CD14 levels but decreases soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol levels in individuals with untreated HIV infection. J Infect Dis. 2014;210:1549–54.
Acknowledgements
Not applicable.
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital) I.V.-C. was supported by DP1HL174182.
Author information
Authors and Affiliations
Contributions
MT made the first draft of the manuscript. SN drafted the part of cardiovascular comorbidities. ICV drafted the part of disease-related dysbiosis and prepared Fig. 1. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
SDN reports grants from the Novo Nordic Foundation, advisory boards for Gilead, MSD and Takeda, and travelling grants from Gilead, and MT reports advisory board for Lilly, not related to the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Trøseid, M., Nielsen, S.D. & Vujkovic-Cvijin, I. Gut microbiome and cardiometabolic comorbidities in people living with HIV. Microbiome 12, 106 (2024). https://doi.org/10.1186/s40168-024-01815-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-024-01815-y