
Abstract
Bile acids play key roles in gut metabolism, cell signaling, and microbiome composition. While the liver is responsible for the production of primary bile acids, microbes in the gut modify these compounds into myriad forms that greatly increase their diversity and biological function. Since the early 1960s, microbes have been known to transform human bile acids in four distinct ways: deconjugation of the amino acids glycine or taurine, and dehydroxylation, dehydrogenation, and epimerization of the cholesterol core. Alterations in the chemistry of these secondary bile acids have been linked to several diseases, such as cirrhosis, inflammatory bowel disease, and cancer. In addition to the previously known transformations, a recent study has shown that members of our gut microbiota are also able to conjugate amino acids to bile acids, representing a new set of “microbially conjugated bile acids.” This new finding greatly influences the diversity of bile acids in the mammalian gut, but the effects on host physiology and microbial dynamics are mostly unknown. This review focuses on recent discoveries investigating microbial mechanisms of human bile acids and explores the chemical diversity that may exist in bile acid structures in light of the new discovery of microbial conjugations.
Video Abstract
Similar content being viewed by others
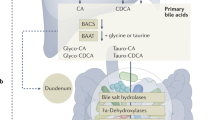

Introduction
The history of bile
Bile has been implicated in human health for millennia. Hippocrates developed the idea of humourism in the third century BC, which describes the body as being composed of four “humors,” two of which involve bile. When these humors are balanced the body is healthy, but illness occurs when any become unbalanced [1]. Even today, we are still trying to understand how the delicate balance between different bile acid (BA) concentrations throughout the body is associated with states of health or disease. Our gut microbiome, the consortium of microorganisms living in our gastrointestinal system, is a major mediator of BA chemistry and, consequently, the development of healthy or diseased states. For example, abnormally high levels of the microbially modified secondary BA deoxycholic acid (3α, 12α-dihydroxy-5β-cholan-24-oic acid, DCA) is associated with gut dysbiosis and disease [2, 3]. There has been increased research in recent years on the connection between our gut microbiome, BA pool composition, and human health, all of which build on our knowledge from the previous two millennia of BA chemistry. This review will describe discoveries from traditional microbial BA modification pathways and provide context to how the newly discovered microbially conjugated BAs affect our understanding of human bile and its transformation by our microbiome.
Bile acid biochemistry and physiology
Primary BAs are those synthesized in the liver from cholesterol [4]. The primary BA pool in humans consists of cholic acid (3α, 7α, 12α-trihydroxy-5β-cholan-24-oic acid, CA), chenodeoxycholic acid (3α, 7α-dihydroxy-5β-cholan-24-oic acid, CDCA), and subsequent C24 taurine- or glycine-bound derivatives (Fig. 1). Glycine and taurine bound BAs are also referred to as bile salts due to decreased pKa and complete ionization resulting in these compounds being present as anions in vivo [8,9,10]. For the purposes of this review, all compounds will be referenced in their protonated form, being named conjugated bile acids in lieu of conjugated bile salts. Primary BAs are heavily modified in the lower gastrointestinal tract to produce a broad range of secondary BAs (Fig. 1). This microbial metabolism is so extensive that instead of primary BAs having the highest prevalence in stool, DCA (a CA derivative) and lithocholic acid (3α-hydroxy-5β-cholan-24-oic acid, LCA, a CDCA derivative), both microbially modified BAs, are the most prevalent [11]. Relevant BAs within humans are not limited to hydroxylation at C3, C7, and C12, but are also found to be hydroxylated at C6 as is the case for α-muricholic acid (3α, 6β, 7α-trihydroxy-5β-cholan-24-oic acid, αMCA) and β-muricholic acid (3α, 6β, 7β-trihydroxy-5β-cholan-24-oic acid, αMCA). Muricholic acids are predominant in mice and scarce in humans, though not absent. MCA forms of bile acids are present in infant urine and feces, though they decrease in concentration to below detectable level in adults [12, 13]. Due to their predominance in mice and rats, MCAs are important in gastrointestinal research using animal models [14].

Diversity of known human bile acids. A All BAs are built off the same sterol backbone with variations in hydroxylated positions, hydroxyl orientation, and the presence of ketones. CA and CDCA, along with GlyCA, GlyCDCA, TaurCA, and TaurCDCA, make up the primary BA pool. Remaining BAs in the list make up secondary and tertiary BA pools as a result of modifications from gut microbes [5,6,7]. Allobile acids, although matching in hydroxyl positions to their standard bile acid counterparts, differ in ring orientation. Standard bile acids have the first ring in the B transorientation, yielding 5β-BAs, while allobile acids have this ring in the C cis-orientation, yielding 5α-BAs
BAs have traditionally been thought to undergo amino acid conjugation solely in the liver. There is a single human enzyme, bile acid-CoA:amino acid N-acyltransferase (hBAAT), that is responsible for acyl-conjugation. These conjugated primary BAs are secreted via the bile canaliculi into the gallbladder where they are stored until consumption of a meal. They are then secreted into the duodenum and travel through the small intestine, only to be subsequently reabsorbed in the terminal ileum and transported to the liver for re-conjugation if necessary, followed by secretion into the gallbladder and recirculation [15]. This enterohepatic circulation is very efficient, recirculating approximately 95% of secreted bile acids, including some of those modified by the microbiota [16]. The remaining 5% undergoes a myriad of transformations throughout the gastrointestinal tract [5, 17]. Although the specific chemistry of BA reabsorption is not completely elucidated, it is generally understood that conjugated BAs are actively transported by ileal transporters and some passive diffusion across the gut epithelium can occur for both conjugated and non-conjugated BAs, specifically those conjugated to glycine [16, 18]. GlyCA and other glycine conjugates may be able to undergo passive diffusion due to the relatively small change in BA biochemistry caused by glycine conjugation.
BAs play an important role in regulating various physiological systems, such as fat digestion, cholesterol metabolism, vitamin absorption, liver function, and enterohepatic circulation through their combined signaling, detergent, and antimicrobial mechanisms [19]. BAs are agonists of the farnesoid X receptor (FXR), with varying degrees of activity depending on the structure of the compound [20]. CDCA is the most potent FXR agonist, followed by DCA, LCA, and lastly, CA. Though their effects on FXR are less clear and more research is needed, conjugated BAs have also been observed to play a role as FXR agonists, notably within the small intestine where concentrations can reach as high as 10 mM [21, 22]. FXR is responsible for regulating several steps in the synthesis of primary BAs CA and CDCA. The loss of FXR activity in mice results in metabolic perturbations and loss of host BA regulation [23]. FXR plays a major role in protecting the small intestine from overgrowth from the large intestine, regulating key antimicrobial pathways including inducible nitric oxide synthase, IL18, angiogenin production, and production of several antimicrobial peptides, such as those within the Defa gene family [21, 24]. TauroBAs, specifically TauroβMCA, have also been shown to act as FXR antagonists, inhibiting BA synthesis via negative regulation [25]. Additionally, BAs are agonists of g-protein coupled receptors such as TGR5 (Takeda G protein-coupled receptor 5) and S1PR2 (sphingosine-1-phosphate receptor 2). S1PR2 is expressed ubiquitously within the liver while TGR5 is expressed primarily in non-parenchymal cells [26]. Expression of both S1PR2 and TGR5 is a balancing act within the liver between homeostasis and damage. S1PR2 is activated by conjugated BAs and results in pro-inflammatory effects that can increase liver damage while TGR5 is activated by all BAs along with several other steroids and results in anti-inflammatory effects in addition to anti-cholestatic and anti-fibrotic effects [26]. These characteristics make S1PR2 inhibitors and TGR5 agonists attractive candidates for drug development.
Microbial bile acid interactions
Bile acids are potent antimicrobials. As such, they play an important role in the innate immune defense within the intestine. Consequently, modifications of BAs are an essential microbial defense mechanism [27]. BAs have been known to impact susceptible bacteria in both a bacteriostatic and bactericidal fashion since the late 1940s, impacting such genera as Staphylococcus, Balantidium, Pneumococcus, and Enterococcus in addition to members of the phylum Spirochaetes [28]. BAs act as detergents in the gut and support the absorption of fats through the intestinal membrane. These same properties allow for the disruption of bacterial membranes. Primary BAs disrupt membranes in a dose-dependent fashion and non-conjugated BAs exact a greater reduction in viability than their conjugated counterparts when tested against Staphylococcus aureus, several Lactobacillus species, and several Bifidobacterium species [27, 29]. As a result of the conjugation to glycine or taurine, primary BAs are fully ionized at physiological pH. While this is important in the movement of BAs from the liver, complete ionization prevents significant interaction and passive diffusion across bacterial membranes whereas non-conjugated CA and CDCA are able to disrupt membranes, cross them, and cause intracellular damage [30]. Conjugated BAs can have more indirect action on the gut microbiota, however, because at high concentrations in the small intestine they modulate FXR and other ileal receptors which control bile synthesis.
Microbial bile acid transformation pathways
Traditionally, there have been four distinct pathways related to microbial transformations of BAs: deconjugation, dehydroxylation, oxidation, and epimerization. The latter two methods of BA transformations work hand in hand, as formation of oxo-BAs is a key step prior to epimerization. Research into microbial bile salt hydrolases (BSHs) has been the latest boom in health-related BA research since their discovery in the 1970s with over 260 publications listed on PubMed from within the last 10 years (search term ‘bile salt hydrolase’). Additionally, several reviews have been written specifically about the biochemistry, diversity, and implications of microbially transformed BAs on host health [17, 31, 32]. The diversity of BAs has recently been shown to be higher than originally thought as members of the gut microbiota demonstrated the ability to conjugate amino acids to cholic acid independent of the host liver [5].
Deconjugation
Deconjugation of BAs is considered the “gateway reaction” to further modification [33]. There are several hypotheses that could explain the importance of deconjugation. As previously discussed, deconjugated primary BAs can act as signaling molecules which modify the total bile acid pool, and therefore, the microbiota may have evolved the deconjugation mechanism to manipulate bile production further. Deconjugation also results in increased concentrations of antimicrobial BAs, CA and CDCA, that may drive shifts in microbiome composition and act as a possible form of microbial chemical warfare. BSHs (classified as EC 3.5.1.24) are able to deconjugate both glycine- and taurine-bound primary BAs, though differences in activity may indicate BSH substrate specificity [17]. Members of the gut microbiota may also use the liberated glycine and taurine residues as nutrient sources. Regardless, deconjugation is an essential function of the gut microbiome.
Enzymes capable of catalyzing the deconjugation reaction are found across all major bacterial phyla and within major archaeal species, suggesting that the genes encoding them are horizontally transferable [34, 35]. Bacteroides spp. are among one of the phyla suggested to play a major role in deconjugating primary BAs [36]. The diversity of bacteria capable of amino acid hydrolysis includes Gram-positive genera such as Bifidobacterium [37], Lactobacillus [38, 39], Clostridium [40], Enterococcus [41], and Listeria [42]. However, BSH activity is not limited to Gram-positive bacteria. Gram-negatives such as Stenotrophomonas [43], Bacteroides [44], and Brucella [45] also contribute to amino acid hydrolysis within the gut. In the cases of Brucella abortus and Listeria monocytogenes, BSH genes are important for virulence and establishing infection within mouse models. A metagenomic study by Jones et al. found BSH-encoding genes are conserved among all major bacterial and archaeal species within the gut [33]. Bacteria capable of BSH activity comprise 26.03% of identified strains of gut bacteria present in humans, although some of these strains may be in low abundance as only 26.40% of BSH-capable strains are present in human guts throughout the globe [46]. The mere ubiquity of BSHs in the gut exemplifies their importance to our microbiota.
All BSH reactions rely on amide bond hydrolysis in order to free taurine or glycine (Fig. 2A, B). Optimal BSH activity occurs at neutral or slightly acidic pH (5–7) with reported optima around pH 6 [40, 48, 49]. Interestingly, among Bifidobacterium spp. arose three separate classes of BSH [37]. Among the three classes of BSH found within Bifidobacterium spp., two classes had high activity and differed in substrate specificity. Both classes exhibited a preference for glycine-conjugated BAs but varied in activity for taurine-conjugated BAs. Although BSHs may utilize both taurine and glycine conjugates, encoding many BSHs may allow for slight changes in substrate specificity and more specific manipulation of the bile acid pool. BSH enzymes from Ligilactobacillus salivarius (PDB ID: 5HKE) [50, 51], Bifidobacterium longum (PDB ID: 2HF0) [52, 53], Bacteroides thetaiotaomicron (PDB ID: 6UFY) [54, 55], Clostridium perfringens (PDB ID: 2BJF) [56, 57], and Enterococcus faecalis (PDB ID: 4WL3) [58] have been crystalized (Fig. 2C). Comparing structural homology (Fig. 2D), E. faecalis, L. salivarius, B. longum, and B. thetaiotaomicron each maintained the αββα motif indicating that it is essential for activity [46]. The BSH from B. thetaiotaomicron (Fig. 2C, blue) is missing a turn which may be one of the driving factors for the decreased structural homology between the other crystalized BSHs. Analysis of key residues from L. salivarius, B. longum, E. faecalis, and C. perfringens amino acid sequences demonstrated highly conserved residues throughout the BSH active site across each genus [46].

Deconjugation reactions and enzyme homology present between gut bacteria. Regardless of hydroxylation positions, substitution of water for either A glycine or B taurine yields the same products. C Structural homology between subunits from B. thetaiotaomicron (6UFY, blue), L. salivarius (5HKE, red), B. longum (2HF0, yellow), C. perfringens (2BJF, green), and E. faecalis (4WL3, orange) using Visual Molecular Dynamics (VMD) software [47]. D Structural homology (QH) was measured utilizing VMD with a minimum of 0.5804 and a maximum of 0.8533. E. faecalis and L. salivarius BSHs had the greatest similarity while B. thetaiotaomicron was the most dissimilar to all other organisms. These analyses were created de novo for this review
Dehydroxylation at C7
One of the key transformations by gut microbes is BA dehydroxylation at C7. Within Clostridium scindens, the bai operon encodes several proteins needed for the sequential oxidation of CA [59]. The baiG gene encodes for a bile acid transporter, allowing for CA uptake. BaiG is also capable of transporting CDCA and DCA [60]. This is followed by CoA ligation in an ATP-dependent manner by BaiB to form cholyl-CoA. Cholyl-CoA is then oxidized twice, first by BaiA and followed by BaiCD, to yield 3-oxo-Δ4-cholyl-CoA. BaiF is then hypothesized to transfer CoA from 3-oxo-Δ4-cholyl-CoA to CA, yielding 3-oxo-Δ4-CA and cholyl-CoA [6]. BaiF CoA transferase activity has already been observed with DCA-CoA, LCA-CoA, and alloDCA-CoA acting as donors and CA acting as an acceptor [59]. The rate limiting step occurs during the dehydroxylation of C7 via BaiE, a 7α-dehydratase. The genes involved in CA 7α-dehydroxylation are capable of recognizing intermediates in the CDCA dehydroxylation pathway as well. Interestingly, CoA-conjugation at C24 was not necessary for dehydratase activity to occur with CA as the substrate, and in some cases enabled for greater kcat and lower KM [61]. Crystal structures of BaiE have been generated in the ligand-absent conformation from C. scindens (PDB ID: 4LEH) [62], Clostridium hylemonae (PDB ID: 4L8O) [63], and Peptacetobacter hiranonis (formerly Clostridium hiranonis, PDB ID: 4L8P) [64] (Fig. 3). Each unit displayed structural similarity (QH) greater than 85% as calculated in Visual Molecular Dynamics (VMD) [47]. The enzymes responsible for the reductive arm of BA 7α-dehydroxylation within C. scindens are encoded by baiN, which is responsible for the sequential reduction of C6-C7 and C4-C5 after dehydroxylation, and by baiA2, which catalyzes the NADH-dependent 3-oxoreduction of both 3-oxodeoxycholic acid and 3-oxolithocholic acid [65, 66]. BaiO is proposed to carry out a similar function to BaiA2 in the reductive arm of 7α-dehydroxylation though this has not yet been verified experimentally [6]. 7β-dehydroxylation occurs in a similar fashion, the key difference being that BaiH is used in the place of BaiCD for C4 oxidation [67, 68]. 7β-dehydratase activity is likely the rate limiting step in 7β-dehydroxylation similar to BaiE above, though the exact gene has not yet been identified. This indicates that further research is needed to elucidate the impact and prevalence of organisms capable of 7β-dehydroxylation, especially given the relative absence of 7β BAs.

Dehydroxylation pathway for primary BAs CA (R: -OH) and CDCA (R: -H). A The pathway to complete 7α-dehydroxylation is a multi-stage process that involves progressive substrate oxidation, likely for molecule stability, prior to dehydroxylation, followed by reduction at each previously oxidized position along the sterol backbone [59]. The enzyme capable of dehydroxylation, BaiE, is highly conserved structurally between C. scindens (red), C. hylemonae (blue), and P. hiranonis (yellow), evident in both B side and C top-down views of BaiE
Oxidation and epimerization
Epimerization of BAs is carried out by gut microbes and further diversifies the chemistry of secondary BAs. This occurs in two distinct steps: oxidation of the hydroxyl group by a position-specific hydroxysteroid dehydrogenase, such as a 7α-HSDH, followed by the reduction of another position-specific hydroxysteroid dehydrogenase, 7β-HSDH. Both reactions do not need to be carried out by the same organism and co-cultures of microbes are known to possess epimerization capabilities [69]. CA can be epimerized to form derivatives such as ursocholic acid (3α, 7β, 12α-trihydroxy-5β-cholan-24-oic acid, UCA), 12-epicholic acid (3α, 7α, 12β-trihydroxy-5β-cholan-24-oic acid, 12-ECA), or isocholic acid (3β, 7α,12α-trihydroxy-5β-cholan-24-oic acid, iCA) (Fig. 4A), while CDCA can be epimerized to form either UDCA or isochenodeoxycholic acid (3β,7α-Dihydroxy-5β-cholan-24-oic acid, iCDCA) (Fig. 4B). Both oxidation and subsequent epimerization have been observed at all three CA hydroxyl positions as well as both CDCA hydroxyl positions and are responsible for much of the diversity found in non-conjugated BAs.

Pathways of CA and CDCA epimerization, including corresponding EC identifiers. A CA undergoes three different epimerization pathways leading to the production of iCA (via 3α/β-HSDH), UCA (via 7α/β-HSDH), or 12-ECA (via 12α/β-HSDH) while B CDCA undergoes two distinct epimerization pathways leading to the production of UDCA (via 7α/β-HSDH) or iCDCA (via 3α/β-HSDH). *S. maltophilia transforms CDCA to 7-oxo-CDCA but the enzyme is categorized under EC 1.1.1.159, where the official reaction involves CA 7α-oxidation [70]
Recently, C. scindens, C. hylemonae, C. perfringens, and P. hiranonis have all been observed to produce enzymes capable of hydroxysteroid 3α-dehydrogenation, an important step in the pathway toward 7α-dehydroxylation [35]. However, unlike C. scindens, C. hylemonae, and P. hiranonis, C. perfringens has not been reported to produce LCA or DCA and its growth is inhibited by both secondary BAs [71]. 3α-dehydrogenation also occurs outside of the genus Clostridia and includes other intestinal organisms such as Blautia producta and Eggerthella lenta (formerly Eubacterium lentum) in addition to environmental species such as Acinetobacter lwoffii [65, 72, 73]. Surprisingly, E. lenta 3α-HSDH is capable of utilizing both TaurBAs and GlyBAs as substrates and in the case of CDCA oxidation, 3α-HSDH activity increased when conjugated forms of CDCA were used as substrates [74]. This goes against the notion that bile BA deconjugation is the essential ‘gateway’ reaction and further investigation is required to elucidate if glycine and taurine residues impact molecular mechanisms of catalysis in addition to if conjugated BA oxidation impacts subsequent transformations. 7α-epimerization to UDCA occurs in the gut by members such as Clostridium baratii among other isolates not yet identified [75, 76]. C. baratii has been shown to epimerize CDCA to UDCA but was not capable of epimerizing glyco- and tauro-BAs and instead deconjugated TaurCDCA prior to epimerization [75]. Epimerization of CDCA, independent of conjugation, is important for producing the protective BA UDCA. Ruminococcus gnavus, Clostridium absonum, Stenotrophomonas maltophilia, and Collinsella aerofaciens all contribute to the UDCA pool via conversion of 7-oxo-LCA in an NADH or NADPH-dependent fashion [70, 77,78,79]. Optimum pH varied between species; C. absonum 7β-HSDH functioned optimally at pH 8.5 while R. gnavus and C. aerofaciens functioned optimally at pH 6. 12β-HSDH activity can occur in both acidic and alkaline conditions. R. gnavus, in contrast to C. absonum and C. aerofaciens, displayed a clear preference in catalyzing the conversion of 7-oxo-LCA to UDCA with a specificity constant 55-fold higher than that of the conversion of UDCA to 7-oxo-LCA [77]. This directionality of activity paired with the protective properties of UDCA support R. gnavus as a potential probiotic, and this role should be further investigated.
Several gut bacteria have recently been identified to produce 12α-hydroxysteroid dehydrogenases (12α-HSDH). E. lenta demonstrates 12α-HSDH capabilities in addition to 3α-HSDH. E. lenta 12α-HSDH has an estimated molecular weight of 125 kDa and has a broad pH optimum, between pH 8 and 10.5 [80, 81]. Catalysis requires NAD+ or NADP+ as a cofactor, though there is a preference for NAD+ [65, 81]. E. lenta 12α-HSDH reaction velocity increased when tested with methylated BAs, suggesting a preference for hydrophobic BAs [80]. Similar to its 3α-HSDH, E. lenta 12α-HSDH is capable of utilizing both glycine- and taurine-bound BAs [74]. Enterorhabdus mucosicola is also capable of both 3α and 12α oxidation, although 12α-HSDH activity is limited to when C7 position has already been oxidized [82, 83]. C. scindens, P. hiranonis, and C. hylemonae have since been reported to produce 12α-HSDHs and it is hypothesized that Bacteroides species also encode 12α-HSDHs [35, 65]. Across all three clostridial species, there was a robust preference for 12-oxo-LCA over 12-oxo-CDCA suggesting the C7 hydroxyl group, or lack thereof, plays a large role in determining enzyme activity. Oxidation at C12 occurs for 12β BAs as well and has been observed in strains of Clostridium paraputrificum, Clostridium tertium, and Clostridioides difficile [84]. These 12β-HSDHs are relatively stable at physiological conditions, maintaining activity at 37 °C for approximately 45 minutes at pH 8.5 [85]. Based on the findings by Edenharder and Pfutzner, C. paraputrificum 12β-HSDH behaves in a similar manner to established 12α-HSDHs, as shown by its pH optimum and molecular weight. The gene encoding the 12β-HSDH in C. paraputrificum was recently identified, allowing for investigation into the diversity of potential 12β-HSDH producers [86]. Putative 12β-HSDH genes were found across Firmicutes, Actinobacteria, and Alphaproteobacteria. However, there may be several forms of 12β-HSDH as the authors did not find homologs to the C. paraputrificum 12β-HSDH in C. difficile and C. tertium even though both species are capable of 12β-HSDH activity.
Members of the gut microbiota are not only capable of reducing BAs with a single position oxidized, but some also reduce BAs oxidized at two or three positions. Similar trends regarding non-target hydroxyl oxidation have been observed by other Coriobacteriaceae, such as C. aerofaciens, E. lenta, and Lancefieldella parvula (formerly Atopobium parvulum) [82]. Not all members oxidized DCA at both C3 and C12 independent of the other position, but all of the strains observed to modify DCA were shown to oxidize at both positions [82]. C. scindens and P. hiranonis were among the only bacteria capable of completely hydrogenating 3,7,12-trioxolithocholic acid, a fully oxidized derivative of CA, to CA [35]. Oxidation may be a way for microbes to detoxify BAs. By decreasing their amphipathicity, oxidized BAs progressively lose the ability to act as detergents, preventing DNA and membrane damage.
Reconjugation: microbially conjugated bile acids
A novel set of recently discovered BAs were conjugated at the C24 acyl site similarly to the host conjugation mechanism [5]. Instead of the traditional amino acids taurine and glycine, these compounds were conjugated with the amino acids phenylalanine, leucine, and tyrosine on a cholic acid backbone. The initial work associated these molecules with the gut microbiota and follow-up experiments identified the bacterium Enterocloster bolteae, formerly Clostridium bolteae, as a species responsible for their production. In light of their microbial origin and the mechanism mirroring that of host-conjugation, we hereby refer to these compounds as “microbially conjugated bile acids” (MCBAs).
The exact mechanism of this microbially mediated conjugation has yet to be elucidated, although it may rely on a similar mechanism to hBAAT within the liver involving a Cys-Asp-His triad, with cysteine functioning as the catalytic residue for nucleophilic attack [87]. Regardless of their mechanism of production, the addition of unique amino acid chemistry on the BA acyl-site inevitably modifies its chemical properties. Phenylalanine, a large hydrophobic amino acid, will greatly increase the hydrophobicity of the BA itself and possibly induce steric hindrance to any binding mechanisms with ileal receptors or BA transporters. Leucine, too, is a relatively large hydrophobic residue, which may create similar chemical properties to that of phenylalanine. The additional hydroxyl group on the aromatic ring of tyrosine may create some unique properties as this will increase the compound’s hydrophilicity and create a more polar hydrophilic BA, similar to the increase in polarity provided by taurine conjugation to cholic acid. The presence of any of these amino acids at the conjugation site will also alter the BA’s emulsifying properties, as a primary function of these compounds is to solubilize fat from our diet. Although not yet shown in the literature, it is likely that the diversity of MCBAs will increase due to the plethora of amino acid residues available for conjugation and the immense microbial diversity present in the human gut. Until the mechanism of their synthesis is enzymatically elucidated and exhaustive searches into MCBA diversity are performed, our knowledge of the limits on amino acid conjugation of BAs by the human microbiota remains incomplete.
The functions of phenylalanine, leucine, and tyrosine CA conjugates remain mostly unknown, though gavage of mice with these compounds has been shown to result in agonism of FXR. Further investigation into the roles of known and unknown BA conjugates may yield novel drug targets or therapeutic agents for the treatment of numerous enteric diseases. Evidence already points toward BA hydrophilicity playing a major role in activity of several BA modifying enzymes; the three novel conjugates currently reported represent three of the four most lipophilic amino acids based on partition coefficient [88]. Thus, identifying organisms responsible for conjugation of other amino acids to other BAs and amino acid-specific mechanisms are the necessary first steps to determining how microbes are utilizing these compounds to impact the host or competing members of the microbiota.
Molecular diversity of microbially conjugated bile acids
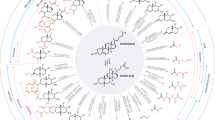
Over 140 amino acids are known to occur in natural proteins [89]. The human BA pool consists of a sterol backbone capable of hydroxylation at four different positions (including C6, observed in MCA), which can be ɑ- or β-hydroxylated, oxidized to form a ketone, or absent. This backbone can also be present as one of two stereoisomers: 5ɑ-sterol or 5β-sterol, significantly broadening potential BA diversity. Limiting the bile acid backbone to only those known to be conjugated by the host (CA and CDCA) in addition to limiting the amino acid conjugated to those naturally occurring in humans, the potential diversity of the human conjugated BA pool increases over 5-fold from what is currently known (Fig. 5). This estimate does not consider non-amino acid conjugates, such as ciliatocholic acid or cholyl-CoA, nor does it include the diversity of potential host hydroxyl modifications, such as sulfonation [90, 91]. Overall, the human bile acid pool is dominated by CA, CDCA, and DCA [92]. Subsequent taurine and glycine conjugation increases this pool to 9 BAs. Limiting the estimate of possible BA-amino acid conjugates to standard amino acids and the three BAs listed above increases the potential human BA pool to 66 unique conjugates. Finally, including all potential oxidized, epimerized, and dehydroxylated states of each hydroxyl group present on CA (C3, C7, C12) in addition to ring orientation expands the number of potential human BA conjugates to over 2800. Although it is unlikely that the number of physiologically relevant MCBAs is this high, one can imagine the potential diversity of MCBAs and the potential for their impact on the gut microbiota and the host.

Potential increased diversity of host BA pool as a result of MCBA production. With the current understanding of BA metabolism, A primary BAs CA and CDCA are known to be conjugated in the liver to taurine and glycine to form B GlyCA, TaurCA, GlyCDCA, and TaurCDCA, completing the pool of primary human BAs. In light of recent research, CA is also known to be conjugated by gut microbes to form C PheCA, LeuCA, and TyrCA [5]. Expanding the potential library of microbially conjugated BAs by including the remaining amino acids conjugates for D CA and E CDCA increases the diversity of human BAs over 5-fold for these backbones alone
Thus far, only relatively hydrophobic amino acids have been reported to be conjugated to cholic acid by microbes, lowering the overall partition coefficient of each molecule. The partition coefficient is the log-ratio of concentrations of a compound in a hydrophobic solvent, such as 1-octanol, compared to a hydrophilic solvent, such as water. This is to say that a higher value indicates that the compound is more present in the hydrophobic phase rather than the hydrophilic phase. As expected, hydrophobicity increases with the reduction of BAs. CA has a partition coefficient of 2.02, which increases to 3.28 when CA is reduced to CDCA and further increases to 3.5 when reduced to DCA [93]. The conjugation of both glycine and taurine to any sterol significantly increases the hydrophilicity of the compound, thus decreasing the partition coefficient for each BA. Therefore, the acyl-conjugation of BAs undoubtedly affects their function. Similarly, microbial conjugation with hydrophobic amino acids would also affect their detergent, signaling, and antimicrobial properties, as well as BA transport. One may wonder then, why do gut microbiota conjugate our bile acids? There are a multitude of possible explanations ranging from enzyme promiscuity to antimicrobial metabolite production, to targeted manipulation of the host BA signaling and regulatory system. Only further research on the genetic, biochemical, and microbiological characterization of the conjugation mechanism and its microbial and host effects will provide the answers. Nevertheless, the MCBA chemical diversity already detected in the mammalian gut, and the potential described above, will invariably diversify the chemical properties of the bile acid pool.
Microbial bile acid products and host health
Though BAs themselves function as important antimicrobial agents, microbial modification of BAs is equally important in disease prevention and maintenance of a healthy gut microbiome. Though C. difficile infections are devastating, fecal microbiota transplant can be a successful treatment in some cases. Successful transplants correlate with an increase in BSH copy number compared to levels prior to transplant, suggesting that microbial modifications of primary BAs play a role in protecting the host against microbial infection [94]. The host microbiota plays an important role in protection against colonization by pathogenic organisms, and the involvement of BA modification in this protective effect is only beginning to be understood [95]. Decreased bile acid deconjugation correlates with several irritable bowel diseases, such as ulcerative colitis, Crohn’s disease, and irritable bowel syndrome [34]. Supplementing diets with microbially transformed BAs can have profound beneficial effects on host pathology. LCA production, a result of CDCA dehydroxylation, is one of the more interesting transformations by gut microbes with a known impact on host health. LCA has been observed to act as an anti-inflammatory agent and protect against colitis in a mouse model [96]. However, LCA and DCA, another the secondary BA, are known carcinogens. While primary human bile acids are known to induce DNA damage within bacteria, LCA and DCA have been observed to damage DNA within mammalian cells [97]. DCA exposure has also been correlated with increased apoptosis and increased production of reactive oxygen and nitrogen species. Subsequently, LCA and DCA are the most prevalent bile acids in human colorectal cancer [98].
Epimerized BAs also influence host health. UDCA, the 7β epimer of CDCA, exhibits protective effects in the gut, specifically through inhibition of TNFα, IL-1β, and IL-6 release [96]. UDCA use has been shown to counteract the apoptotic effects of DCA [99]. UDCA has also been approved for use in gallstone dissolution and in treating primary biliary cholangitis, the later indication as a result of the ability of UDCA to increase bile acid biosynthesis [96, 99]. One caveat of UDCA use is that, at high doses (28–30 mg/kg/day), long-term use leads to increased risk of colorectal cancer in patients with ulcerative colitis and primary sclerosing cholangitis [100].
It is possible that MCBAs may also play a role in disease mechanisms, as PheCA, TyrCA, and LeuCA were more prevalent in patients with inflammatory bowel disease and cystic fibrosis (and though not disease related, were also found in infants) [5]. However, one cannot know, simply by detection in a diseased population, whether MCBAs, or any BA for that matter, are cause or consequence of a particular diseased state; a conundrum that is well known in the microbiome field. There is evidence that at least one microbe that produces MCBAs, E. bolteae (referred to as C. bolteae in the referenced manuscript), may be involved in severe IBD and Crohn’s disease, as it was identified as one of the most transcriptionally active microbes in the dysbiotic and diseased gut and MCBAs were elevated in these same samples [5, 101]. This association indicates that MCBAs may be involved in severe IBD, but future research is required. Regardless, BAs can serve as markers for various disease states [97, 98] and can themselves be used as therapeutics, such as in the case of UDCA, making them an important group of compounds for identification and treatment of human disease.
Conclusions
Although BAs have been studied for centuries, recent discoveries show that we still have much to learn. The host BA pool controls microbial diversity, but so too does microbial metabolism of these BAs drive host physiology. In this sense, BAs act as the language of an intricate molecular cross-talk between humans and their gut microbiota. Mechanisms of microbial modification of host BAs continue to be elucidated as do the roles BA metabolism plays in host health. The presence of MCBAs in the human BA pool demonstrates the need for further study of microbial BA modification and further expands the chemical language our gut microbiota uses to communicate with its host.
Availability of data and materials
Not applicable
Abbreviations
- BA:
-
Bile acid
- CA:
-
Cholic acid
- 12-ECA:
-
12-epicholic acid
- iCA:
-
Isocholic acid
- UCA:
-
Ursocholic acid
- HCA:
-
Hyocholic acid
- αMCA:
-
α-muricholic acid
- βMCA:
-
β-muricholic acid
- CDCA:
-
Chenodeoxycholic acid
- UDCA:
-
Ursodeoxycholic acid
- iCDCA:
-
Isochenodeoxycholic acid
- DCA:
-
Deoxycholic acid
- HDCA:
-
Hyodeoxycholic acid
- LCA:
-
Lithocholic acid
- GlyCA:
-
Glycocholic acid
- TaurCA:
-
Taurocholic acid
- GlyCDCA:
-
Glycochenodeoxycholic acid
- TaurCDCA:
-
Taurochenodeoxycholic acid
- PheCA:
-
Phenylalanocholic acid
- TyrCA:
-
Tyrosocholic acid
- LeuCA:
-
Leucholic acid
- HSDH:
-
Hydroxysteroid dehydrogenase
- BSH:
-
Bile salt hydrolase
- hBAAT:
-
Human bile acid-CoA:amino acid N-acyltransferase
- FXR:
-
Farnesoid X receptor
References
Goodacre CJ, Naylor WP. Evolution of the temperament theory and mental attitude in complete denture prosthodontics: from hippocrates to M.M. House. J Prosthodontics. 2020;29:594–8 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/jopr.13215.
Liu L, Dong W, Wang S, Zhang Y, Liu T, Xie R, et al. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. 2018;9:5588–97 Available from: https://pubs.rsc.org/en/content/articlelanding/2018/FO/C8FO01143E#!divAbstract.
Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int J Cancer. 2017;140:2545–56 Available from: https://pubmed.ncbi.nlm.nih.gov/28187526/.
Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–58 Available from: https://jamanetwork.com/journals/jamainternalmedicine/fullarticle/1105662.
Quinn RA, Melnik AV, Vrbanac A, Fu T, Patras KA, Christy MP, et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature. 2020;579:123–9. https://doi.org/10.1038/s41586-020-2047-9. Accessed 27 May 2020.
Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome. 2019;7:75 Available from: https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-019-0689-3.
Noronha A, Modamio J, Jarosz Y, Guerard E, Sompairac N, Preciat G, et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Re. 2019;47:D614–24 Available from: https://academic.oup.com/nar/article/47/D1/D614/5146204.
Bortolini O, Medici A, Poli S. Biotransformations on steroid nucleus of bile acids. Steroids. 1997:564–77 Available from: https://www.sciencedirect.com/science/article/pii/S0039128X97000433?via%3Dihub.
Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74 Available from: http://www.annualreviews.org/doi/10.1146/annurev.biochem.72.121801.161712.
Moini J. Epidemiology of diet and diabetes mellitus. Epidemiol Diab. 2019:57–73. https://doi.org/10.1016/B978-0-12-816864-6.00005-5.
Kakiyama G, Muto A, Takei H, Nittono H, Murai T, Kurosawa T, et al. A simple and accurate HPLC method for fecal bile acid profile in healthy and cirrhotic subjects: Validation by GC-MS and LC-MS. J Lipid Res. 2014;55:978–90 Available from: https://www.jlr.org/article/S0022-2275(20)37516-7/fulltext.
García-Cañaveras JC, Donato MT, Castell JV, Lahoz A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res. 2012;53:2231–41 Available from: https://www.jlr.org/article/S0022-2275(20)43200-6/fulltext.
Goto J, Hasegawa K, Nambara T, Iida T. Studies on steroids. CCLIV. Gas chromatographic-mass spectrometric determination of 4- and 6-hydroxylated bile acids in human urine with negative ion chemical ionization detection. Chromatogr. 1992;574:1–7 Available from: https://pubmed.ncbi.nlm.nih.gov/1629271/. Accessed 15 Mar 2021.
Li J, Dawson PA. Animal models to study bile acid metabolism. Biochim Biophys Acta. 2019:895–911. https://doi.org/10.1016/j.bbadis.2018.05.011.
Hofmann AF. The enterohepatic circulation of bile acids in mammals: Form and functions. Front Biosci. 2009;14:2584–98 Available from: https://pubmed.ncbi.nlm.nih.gov/19273221/.
Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015:1085–99 Available from: https://www.jlr.org/article/S0022-2275(20)31721-1/fulltext.
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–59 Available from: https://www.jlr.org/article/S0022-2275(20)33626-9/.
Aldini R, Roda A, Lenzi PL, Ussia G, Vaccari MC, Mazzella G, et al. Bile acid active and passive ileal transport in the rabbit: effect of luminal stirring. Eur J Clin Invest. 1992;22:744–50. https://doi.org/10.1111/j.1365-2362.1992.tb01439.x. Accessed 15 Mar 2021.
de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metabolism. 2013:657–69. https://doi.org/10.1016/j.cmet.2013.03.013.
Shin DJ, Wang L. Bile acid-activated receptors: a review on FXR and other nuclear receptors. Handbook Exp Pharmacol. 2019:51–72 Available from: https://link.springer.com/chapter/10.1007/164_2019_236. Accessed 23 Oct 2020.
Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–5. Available from: https://www.pnas.org/content/103/10/3920. Accessed 23 Oct 2020.
Hofmann AF, Eckmann L. How bile acids confer gut mucosal protection against bacteria. Proc Natl Acad Sci USA. 2006:4333–4. Available from: https://www.pnas.org/content/103/12/4333. Accessed 23 Oct 2020.
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44 Available from: http://www.cell.com/article/S0092867400000623/.
Tremblay S, Romain G, Roux M, Chen XL, Brown K, Gibson DL, et al. Bile acid administration elicits an intestinal antimicrobial program and reduces the bacterial burden in two mouse models of enteric infection. Infect Immun. 2017;85. Available from: https://doi.org/10.1128/IAI.00942-16. Accessed 15 Mar 2021.
Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metabolism. 2013;17:225–35. Available from: https://www.cell.com/action/showPdf?pii=S1550-4131. Accessed 15 Mar 2021.
Keitel V, Stindt J, Häussinger D. Bile acid-activated receptors: GPBAR1 (TGR5) and other G protein-coupled receptors. Handb Exp Pharmacol. 2019:19–49 Available from: https://link.springer.com/chapter/10.1007/164_2019_230.
Sannasiddappa TH, Lund PA, Clarke SR. In vitro antibacterial activity of unconjugated and conjugated bile salts on Staphylococcus aureus. Front Microbiol. 2017;8 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5572772/.
Stacey M, Webb M. Studies on the antibacterial properties of the bile acids and some compounds derived from cholanic acid. Proc R Soc Med. 1947;134:523–37. Available from: https://royalsocietypublishing.org/doi/10.1098/rspb.1947.0029. Accessed 23 Oct 2020.
Kurdi P, Kawanishi K, Mizutani K, Yokota A. Mechanism of growth inhibition by free bile acids in Lactobacilli and Bifidobacteria. J Bacteriol. 2006;188:1979–86. Available from: https://jb.asm.org/content/188/5/1979. Accessed 8 Dec 2020.
Urdaneta V, Casadesús J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med. 2017:163 Available from: https://www.frontiersin.org/articles/10.3389/fmed.2017.00163/.
Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016:22–39 Available from: http://www.tandfonline.com/doi/full/10.1080/19490976.2015.1127483.
Gustafsson BE, Midtvedt T, Norman A. Isolated fecal microorganisms capable of 7-alpha-dehydroxylating bile acids. J Exp Med. 1966;123:413–32 Available from: http://rupress.org/jem/article-pdf/123/2/413/1082316/413.pdf.
Jones BV, Begley M, Hill C, CGM G, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–5 Available from: https://www.pnas.org/content/105/36/13580.
Joyce SA, Gahan CGM. Disease-associated changes in bile acid profiles and links to altered gut microbiota. Dig Dis. 2017;35:169–77 Available from: https://www.karger.com/Article/FullText/450907.
Doden H, Sallam LA, Devendran S, Ly L, Doden G, Daniel SL, et al. Metabolism of oxo-bile acids and characterization of recombinant 12α- hydroxysteroid dehydrogenases from bile acid 7α-dehydroxylating human gut bacteria. Appl Environ Microbiol. 2018;84:235–53 Available from: https://aem.asm.org/content/84/10/e00235-18.
Ovadia C, Perdones-Montero A, Spagou K, Smith A, Sarafian MH, Gomez-Romero M, et al. Enhanced Microbial Bile Acid Deconjugation and Impaired Ileal Uptake in Pregnancy Repress Intestinal Regulation of Bile Acid Synthesis. Hepatology. 2019;70:276–93 Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.30661.
Kim GB, Yi SH, Lee BH. Purification and characterization of three different types of bile salt hydrolases from Bifidobacterium strains. J Dairy Sci. 2004;87:258–66. Available from: https://doi.org/10.3168/jds.S0022-0302(04)73164-1.
Elkins CA, Moser SA, Savage DC. Genes encoding bile salt hydrolases and conjugated bile salt transporters in Lactobacillus johnsonii 100-100 and other Lactobacillus species. Microbiol. 2001;147:3403–12 Available from: https://www.microbiologyresearch.org/content/journal/micro/10.1099/00221287-147-12-3403.
Corzo G, Gilliland SE. Bile salt hydrolase activity of three strains of Lactobacillus acidophilus. J Dairy Sci. 1999;82:472–80 Available from: https://www.journalofdairyscience.org/article/S0022-0302(99)75256-2/.
Coleman JP, Hudson LL. Cloning and characterization of a conjugated bile acid hydrolase gene from Clostridium perfringens. Appl Environ Microbiol. 1995;61:2514–20 Available from: https://aem.asm.org/content/61/7/2514.
Wijaya A, Hermann A, Abriouel H, Specht I, Yousif NMK, Holzapfel WH, et al. Cloning of the bile salt hydrolase (bsh) gene from Enterococcus faecium FAIR-E 345 and chromosomal location of bsh genes in food Enterococci. J Food Protect. 2004;67:2772–8 Available from: http://meridian.allenpress.com/jfp/article-pdf/67/12/2772/1676188/0362-028x-67_12_2772.pdf.
Dussurget O, Cabanes D, Dehoux P, Lecuit M, Buchrieser C, Glaser P, et al. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol Microbiol. 2002;45:1095–106 Available from: http://doi.wiley.com/10.1046/j.1365-2958.2002.03080.x.
Dean M, Cervellati C, Casanova E, Squerzanti M, Lanzara V, Medici A, et al. Characterization of cholylglycine hydrolase from a bile-adapted strain of Xanthomonas maltophilia and its application for quantitative hydrolysis of conjugated bile salts. Appl Environ Microbiol. 2002;68:3126–8 Available from: https://aem.asm.org/content/68/6/3126.
Kawamoto K, Horibe I, Uchida K. Purification and characterization of a new hydrolase for conjugated bile acids, chenodeoxycholyltaurine hydrolase, from Bacteroides vulgatus. J Biochem. 1989;106:1049–53 Available from: https://academic.oup.com/jb/article-lookup/doi/10.1093/oxfordjournals.jbchem.a122962.
Delpino MV, Marchesini MI, Estein SM, Comerci DJ, Cassataro J, Fossati CA, et al. A bile salt hydrolase of Brucella abortus contributes to the establishment of a successful infection through the oral route in mice. Infect Immun. 2007;75:299–305 Available from: https://iai.asm.org/content/75/1/299. Accessed 8 Dec 2020.
Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome. 2019;7:9 Available from: https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-019-0628-3.
Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graphics. 1996; Available from: https://www.sciencedirect.com/science/article/abs/pii/0263785596000185?via%3Dihub.
Percy-Robb IW. Bile Acids: A pH Dependent Antibacterial System in the Gut? Br Med J. 1972;3:813–5 Available from: https://www.bmj.com/content/3/5830/813.
Stellwag EJ, Hylemon PB. Purification and characterization of bile salt hydrolase from Bacteroides fragilis subsp. fragilis. Biochim Biophys Acta. 1976;452:165–76 Available from: https://www.sciencedirect.com/science/article/abs/pii/0005274476900681?via%3Dihub.
Xu F, Guo F, Hu XJ, Lin J. Crystal structure of bile salt hydrolase from Lactobacillus salivarius. Acta Crystallogr F Struct Biol Commun. 2016;72:376–81 Available from: http://scripts.iucr.org/cgi-bin/paper?S2053230X16005707.
Hu X-J. bile salt hydrolase from Lactobacillus salivarius; 2016. https://doi.org/10.2210/pdb5HKE/pdb.
Kumar RS, Brannigan JA, Prabhune AA, Pundle AV, Dodson GG, Dodson EJ, et al. Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum reveals an evolutionary relationship with penicillin V acylase. J Biol Chem. 2006;281:32516–25 Available from: https://www.jbc.org/article/S0021-9258(20)86826-4/.
Suresh CG, Kumar RS, Brannigan JA. Bifidobacterium longum bile salt hydrolase; 2006. https://doi.org/10.2210/pdb2HF0/pdb.
Seegar TCM. B. theta Bile Salt Hydrolase; 2019. https://doi.org/10.2210/pdb6UFY/pdb.
Adhikari AA, Seegar TCM, Ficarro SB, McCurry MD, Ramachandran D, Yao L, et al. Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nat Chem Biol. 2020;16:318–26 Available from: https://www.nature.com/articles/s41589-020-0467-3.
Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP, Saenger W. Crystal structure of conjugated bile acid hydrolase from Clostridium perfringens in complex with reaction products taurine and deoxycholate; 2005. https://doi.org/10.2210/pdb2BJF/pdb.
Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP, Saenger W. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product. Biochemistry. 2005;44:5739–48 Available from: https://pubs.acs.org/doi/abs/10.1021/bi0473206.
Ramasamy S, Chand D, Suresh C. Crystal structure determination of Bile Salt Hydrolase from Enterococcus feacalis; 2015. https://doi.org/10.2210/pdb4wl3/pdb.
Ridlon JM, Hylemon PB. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J Lipid Res. 2012;53:66–76 Available from: https://www.jlr.org/article/S0022-2275(20)40800-4/.
Mallonee DH, Hylemon PB. Sequencing and expression of a gene encoding a bile acid transporter from Eubacterium sp. strain VPI 12708. J Bacteriol. 1996;178:7053–8 Available from: https://jb.asm.org/content/178/24/7053.
Bhowmik S, Chiu HP, Jones DH, Chiu HJ, Miller MD, Xu Q, et al. Structure and functional characterization of a bile acid 7α dehydratase BaiE in secondary bile acid synthesis. Proteins. 2016;84:316–31. https://doi.org/10.1002/prot.24971.
Joint Center for Structural Genomics. RCSB PDB - 4LEH: Crystal structure of a bile-acid 7-alpha dehydratase (CLOSCI_03134) from Clostridium scindens ATCC 35704 at 2.90 A resolution; 2013. https://doi.org/10.2210/pdb4LEH/pdb.
Joint Center for Structural Genomics. RCSB PDB - 4L8O: Crystal structure of a bile-acid 7-alpha dehydratase (CLOHYLEM_06634) from Clostridium hylemonae DSM 15053 at 2.20 A resolution; 2013. https://doi.org/10.2210/pdb4L8O/pdb.
Joint Center for Structural Genomics. RCSB PDB - 4L8P: Crystal structure of a bile-acid 7-alpha dehydratase (CLOHIR_00079) from Clostridium hiranonis DSM 13275 at 1.60 A resolution; 2013. https://doi.org/10.2210/pdb4L8P/pdb.
Harris SC, Devendran S, Méndez- García C, Mythen SM, Wright CL, Fields CJ, et al. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243 T. Gut Microbes. 2018;9:523–39. Available from: https://www.tandfonline.com/doi/full/10.1080/19490976.2018.1458180. [cited 2021 Apr 18]
Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature. 2020;582:566–70 Available from: https://www.nature.com/articles/s41586-020-2396-4.
Bhowmik S, Jones DH, Chiu HP, Park IH, Chiu HJ, Axelrod HL, et al. Structural and functional characterization of BaiA, an enzyme involved in secondary bile acid synthesis in human gut microbe. Proteins. 2014;82:216–29 Available from: https://onlinelibrary.wiley.com/doi/10.1002/prot.24353.
Kang DJ, Ridlon JM, Moore DR, Barnes S, Hylemon PB. Clostridium scindens baiCD and baiH genes encode stereo-specific 7α/7β-hydroxy-3-oxo-Δ4-cholenoic acid oxidoreductases. Biochim Biophys Acta. 2008;1781:16–25 Available from: https://www.sciencedirect.com/science/article/abs/pii/S1388198107002156?via%3Dihub.
Hirano S, Masuda N. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J Lipid Res. 1981;22:1060–8 Available from: https://www.jlr.org/article/S0022-2275(20)40663-7/.
Pedrini P, Andreotti E, Guerrini A, Dean M, Fantin G, Giovannini PP. Xanthomonas maltophilia CBS 897.97 as a source of new 7β- and 7α-hydroxysteroid dehydrogenases and cholylglycine hydrolase: Improved biotransformations of bile acids. Steroids. 2006;71:189–98 Available from: https://www.sciencedirect.com/science/article/abs/pii/S0039128X05002308?via%3Dihub.
Wang S, Martins R, Sullivan MC, Friedman ES, Misic AM, El-Fahmawi A, et al. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome. 2019;7:1–20 Available from: https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-019-0740-4.
Eggert T, Bakonyi D, Hummel W. Enzymatic routes for the synthesis of ursodeoxycholic acid. J Biotechnol. 2014;191:11–21 Available from: https://www.sciencedirect.com/science/article/abs/pii/S0168165614008050?via%3Dihub.
Giovannini PP, Grandini A, Perrone D, Pedrini P, Fantin G, Fogagnolo M. 7α- and 12α-Hydroxysteroid dehydrogenases from Acinetobacter calcoaceticus lwoffii: a new integrated chemo-enzymatic route to ursodeoxycholic acid. Steroids. 2008;73:1385–90 Available from: https://www.sciencedirect.com/science/article/pii/S0039128X08002031. Accessed 24 Apr 2021.
Mythen SM, Devendran S, Méndez-García C, Cann I, Ridlon JM. Targeted synthesis and characterization of a gene cluster encoding NAD(P)H-dependent 3α-, 3β-, and 12α-hydroxysteroid dehydrogenases from Eggerthella CAG:298, a gut metagenomic sequence. Appl Environ Microbiol. 2018:84 Available from: https://aem.asm.org/content/84/7/e02475-17.
Lepercq P, Gérard P, Béguet F, Raibaud P, Grill J-P, Relano P, et al. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by Clostridium baratii isolated from human feces. FEMS Microbiol Lett. 2004;235:65–72 Available from: https://academic.oup.com/femsle/article-lookup/doi/10.1111/j.1574-6968.2004.tb09568.x.
Edenharder R, Knaflic T. Epimerization of chenodeoxycholic acid to ursodeoxycholic acid by human intestinal lecithinase-lipase-negative Clostridia. J Lipid Res. 1981;22:652–8 Available from: https://www.jlr.org/article/S0022-2275(20)37375-2/. [cited 2021 Apr 25].
Lee JY, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54:3062–9 Available from: https://www.jlr.org/article/S0022-2275(20)35057-4/.
Ferrandi EE, Bertolesi GM, Polentini F, Negri A, Riva S, Monti D. In search of sustainable chemical processes: Cloning, recombinant expression, and functional characterization of the 7α- and 7β-hydroxysteroid dehydrogenases from Clostridium absonum. Appl Microbiol Biotechnol. 2012;95:1221–33 Available from: https://link.springer.com/article/10.1007/s00253-011-3798-x.
Liu L, Aigner A, Schmid RD. Identification, cloning, heterologous expression, and characterization of a NADPH-dependent 7β-hydroxysteroid dehydrogenase from Collinsella aerofaciens. Appl Microbiol Biotechnol. 2011;90:127–35 Available from: https://link.springer.com/article/10.1007/s00253-010-3052-y.
MacDonald IA, Jellett JF, Mahony DE, Holdeman LV. Bile salt 3α- and 12α-hydroxysteroid dehydrogenases from Eubacterium lentum and related organisms. Appl Environ Microbiol. 1979;37:992–1000 Available from: https://aem.asm.org/content/37/5/992.long.
MacDonald IA, Mahony DE, Jellet JF, Meier CE. Nad-dependent 3α- and 12α-hydroxysteroid dehydrogenase activities from Eubacterium lentum ATCC no. 25559. Biochimica et Biophysica Acta (BBA)/Lipids and Lipid. Metabolism. 1977;489:466–76. Available from: https://www.sciencedirect.com/science/article/pii/0005276077901679.
Wegner K, Just S, Gau L, Mueller H, Gérard P, Lepage P, et al. Rapid analysis of bile acids in different biological matrices using LC-ESI-MS/MS for the investigation of bile acid transformation by mammalian gut bacteria. Anal Bioanal Chem. 2017;409:1231–45 Available from: https://link.springer.com/article/10.1007/s00216-016-0048-1.
Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, et al. Genome-based taxonomic classification of the phylum actinobacteria. Front Microbiol. 2018;9:2007 Available from: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02007.
Edenharder R, Schneider J. 12β-Dehydrogenation of bile acids by Clostridium paraputrificum, C. tertium, and C. difficle and epimerization at carbon-12 of deoxycholic acid by cocultivation with 12α-dehydrogenating Eubacterium lentum. Appl Environ Microbiol. 1985;49:964–8 Available from: https://aem.asm.org/content/49/4/964.
Edenharder R, Pfützner A. Characterization of NADP-dependent 12β-hydroxysteroid dehydrogenase from Clostridium paraputrificum. Biochim Biophys Acta. 1988;962:362–70 Available from: https://www.sciencedirect.com/science/article/abs/pii/0005276088902664?via%3Dihub.
Doden HL, Wolf PG, Gaskins HR, Anantharaman K, Alves JMP, Ridlon JM. Completion of the gut microbial epi-bile acid pathway. Gut Microbes. 2021;13:1–20 Available from: https://www.tandfonline.com/doi/full/10.1080/19490976.2021.1907271.
Sfakianos MK, Wilson L, Sakalian M, Falany CN, Barnes S. Conserved residues in the putative catalytic triad of human bile acid coenzyme A:Amino acid N-acyltransferase. J Biol Chem. 2002;277:47270–5. Available from: https://www.jbc.org/article/S0021-9258(19)71454-9/. Accessed 19 Oct 2020.
van de Waterbeemd H, Karajiannis H, el Tayar N. Lipophilicity of amino acids. Amino Acids. 1994:129–45 Available from: https://link.springer.com/article/10.1007/BF00814156.
Ambrogelly A, Palioura S, Söll D. Natural expansion of the genetic code. Nat Chem Biol. 2007;3:29–35 Available from: https://www.nature.com/articles/nchembio847.
Tamari M, Ogawa M, Kametaka M. A new bile acid conjugate, ciliatocholic acid, from bovine gall bladder bile. J Biochem. 1976;80:371–7.
Chiang JYL. Bile acids: Regulation of synthesis. J Lipid Res. 2009:1955–66. Available from: https://www.jlr.org/article/S0022-2275(20)30703-3/.
Chiang JYL. Recent advances in understanding bile acid homeostasis [version 1; peer review: 2 approved]. F1000Res. 2017;6(F1000 Faculty Rev):2029. https://doi.org/10.12688/f1000research.12449.1.
Roda A, Minutello A, Angellotti MA, Fini A. Bile acid structure-activity relationship: Evaluation of bile acid lipophilicity using 1-octanol/water partition coefficient and reverse phase HPLC. J Lipid Res. 1990;31:1433–43 Available from: https://www.jlr.org/article/S0022-2275(20)42614-8/.
Mullish BH, McDonald JAK, Pechlivanis A, Allegretti JR, Kao D, Barker GF, et al. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut. 2019;68:1791–800. https://doi.org/10.1136/gutjnl-2018-317842.
Pickard JM, Zeng MY, Caruso R. Núñez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017:70–89. https://doi.org/10.1111/imr.12567.
Ward JBJ, Lajczak NK, Kelly OB, O’Dwyer AM, Giddam AK, Ní Gabhann J, et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol. 2017;312:G550–8 Available from: https://www.physiology.org/doi/10.1152/ajpgi.00256.2016. Accessed 23 Nov 2020.
Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005:47–65 Available from: https://www.sciencedirect.com/science/article/abs/pii/S1383574204000560?via%3Dihub. Accessed 24 Apr 2021.
Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011:863–71 Available from: https://link.springer.com/article/10.1007/s00204-011-0648-7.
Goossens JF, Bailly C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol Ther. 2019;203:107396 Available from: https://www.sciencedirect.com/science/article/abs/pii/S0163725819301391?via%3Dihub. Accessed 15 Mar 2021.
Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, VAC L, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. 2011:1638–45 Available from: https://journals.lww.com/00000434-201109000-00014.
Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–62. https://doi.org/10.1038/s41586-019-1237-9.
Acknowledgements
The authors would like to acknowledge and thank Kerri A. Neugebauer for her help editing this manuscript.
Funding
No relevant funding.
Author information
Authors and Affiliations
Contributions
DVG and RAQ wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication of this article.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guzior, D.V., Quinn, R.A. Review: microbial transformations of human bile acids. Microbiome 9, 140 (2021). https://doi.org/10.1186/s40168-021-01101-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01101-1