
Abstract
Background
Soil is an important reservoir of antibiotic resistance genes (ARGs), but their potential risk in different ecosystems as well as response to anthropogenic land use change is unknown. We used a metagenomic approach and datasets with well-characterized metadata to investigate ARG types and amounts in soil DNA of three native ecosystems: Alaskan tundra, US Midwestern prairie, and Amazon rainforest, as well as the effect of conversion of the latter two to agriculture and pasture, respectively.
Results
High diversity (242 ARG subtypes) and abundance (0.184–0.242 ARG copies per 16S rRNA gene copy) were observed irrespective of ecosystem, with multidrug resistance and efflux pump the dominant class and mechanism. Ten regulatory genes were identified and they accounted for 13–35% of resistome abundances in soils, among them arlR, cpxR, ompR, vanR, and vanS were dominant and observed in all studied soils. We identified 55 non-regulatory ARGs shared by all 26 soil metagenomes of the three ecosystems, which accounted for more than 81% of non-regulatory resistome abundance. Proteobacteria, Firmicutes, and Actinobacteria were primary ARG hosts, 7 of 10 most abundant ARGs were found in all of them. No significant differences in both ARG diversity and abundance were observed between native prairie soil and adjacent long-term cultivated agriculture soil. We chose 12 clinically important ARGs to evaluate at the sequence level and found them to be distinct from those in human pathogens, and when assembled they were even more dissimilar. Significant correlation was found between bacterial community structure and resistome profile, suggesting that variance in resistome profile was mainly driven by the bacterial community composition.
Conclusions
Our results identify candidate background ARGs (shared in all 26 soils), classify ARG hosts, quantify resistance classes, and provide quantitative and sequence information suggestive of very low risk but also revealing resistance gene variants that might emerge in the future.
Video abstract
Similar content being viewed by others


Background
Antibiotic resistance is a global threat to public health, to which an estimated 700,000 yearly deaths are attributed and is predicted to cause 10 million deaths by 2050 if unchecked [1]. Soils are likely the most significant antibiotic resistance gene (ARG) reservoirs, and large amounts and diversities of ARGs have been found in soils throughout the world [2,3,4], including some in Antarctic surface soils [5]. Many clinically relevant ARGs originated from the soil resistome via horizontal gene transfer [6, 7]. Further studies demonstrate that some anthropogenic activities significantly enrich the abundance of indigenous ARGs in soils [8,9,10]. Understanding soil resistomes at a broad geographic scale and across major ecosystems, especially in native soils which have not been exposed to anthropogenic activities, can help better define the background levels and types of ARGs, which is essential for assessing the potential risk of new human activities.
Different land use practices can significantly alter the soil physicochemical as well as biological properties. Conversion of native soil to crop cultivation has been one of the most common anthropogenic land use changes. For example, 100 years of continuous cultivation significantly changed microbial diversity and structure in a consistent though not major way in North American Midwest prairie soils [11]. Further, the conversion of Amazon rainforest to cattle pasture is another land use change that has expanded in the tropics [12], and has led to homogenization of microbial communities [13]. Since land use change has been and continues to be the most extensive alteration of the terrestrial environment, its impact on the soil resistome is important to understand.
Most of our knowledge of ARGs in soils has come from targeting those genes by real-time quantitative PCR (RT-qPCR) including highly parallel qPCR platforms [14,15,16]. While qPCR is more sensitive, it is limited to the known genes and the specificities imposed by primers. Metagenomics (shotgun sequencing) is now affordable and provides a more comprehensive overview of environmental resistomes [17, 18]. Furthermore, new ARG bioinformatic analysis tools are available to efficiently analyze this large amount of data [19, 20]. Since different ARGs have different levels of risk [21], it is necessary to categorize ARGs by their functional roles, the necessary components for their resistance function, and assess their potential risk separately rather than merely the abundance of total ARGs.
With the increasing availablity of metagenomic data in public databases, a few studies have used that data to provide a global view of the soil resistome [4, 22,23,24,25]. While these studies provide a useful overview, they lose resolution on the effect of different ecologies, which is important to understand soil community assemblies and their relatiohships to its resistome. Here, by using a hierarchical structured (ARGs type-subtype-reference sequence) database and ARGs-OAP pipeline [26, 27], we investigated impacts of land use change on the soil resistome with soils matched for edaphic traits and land form for two major ecosystem types, i.e., U.S. Midwest prairie and Amazon rainforest. We also compared the resistome of these two major ecosystem types with that of an undisturbed Arctic tundra to provide a tropic to polar latitudinal gradient, and all with well-characterized metadata.
We addressed the following objectives: (i) what are the types and quantities of ARGs in soils of three climate regions (tundra, temperate, and tropical), (ii) what are the impacts of major land use changes on the ARG profiles, and (iii) define which ARGs are common, perhaps universal background, and within the ARGs, which are most frequent, do they commonly co-occur and their relevance to risk. The results of this study should improve our understanding of the background level and classes of soil ARGs and allow for better evaluation of the public health risk of ARGs in the environment.
Materials and methods
Experimental design and site description
The metagenomic sequence data used herein were from our previous studies, and used to assess the impact of land use change [13, 28] and global warming [29, 30] on soil microbial communities. The sample site locations of the 26 soil metagenomes used in this study are depicted in Additional file 1: Fig. S1. The Alaskan soils were sampled at 15–25 cm depth (active layer; above permafrost boundary) at a moist acidic tundra area in Interior Alaska near Denali National Park (63° 52′ 59″ N, 149° 13′ 32″ W) in May 2010. The Oklahoma soils were collected by soil core 0–15 cm deep from a tallgrass prairie located at the Great Plain Apiaries in McClain County, Oklahoma, United States (34° 59′ N, 97° 31′ W) in 2011, 2012, and 2013. The site was abandoned from field cropping 40 years ago with light grazing until 2008. Three additional tallgrass prairie ecosystem sites were sampled in the summer of 2009 in the US Midwest from a 750 km transect from Kansas through Iowa to Wisconsin. At each site soil was sampled from a native (never tilled) prairie and an adjacent cultivated (> 100 years) soil matched for soil edaphic traits and landform. The native prairie soils had been grazed by cattle. All cultivated soils had received manure application. The Amazon soils were sampled at the Amazon Rainforest Microbial Observatory site (10° 10′ 05″ S and 62° 49′ 27″ W) in April 2009. Five soil cores of 0–10 cm deep were collected from a primary rainforest and an adjacent 38-year-old converted pasture. The Amazon rainforest soils were never grazed, while the pasture had been continuously used for beef cattle grazing since conversion. Detailed information about the climate, vegetation, soil type, and chemistry at each sampling site were described previously [11, 28, 30, 31].
Shotgun sequencing
Sequencing of Alaska, Oklahoma, and Amazon soils was performed on the Illumina HiSeq 2000 platform with 150-bp paired-end strategy by the Joint Genome Institute (JGI). Illumina GAIIx paired-end sequencing augmented with some 454 GS FLX sequencing for the Iowa, Kansas, and Wisconsin soils. The Alaskan soils were also sequenced by Illumina using paired ends but by Los Alamos National Laboratory.
Identification of ARGs in shotgun data
Adapter reads in the sequence data were removed, the remaining reads were filtered to discard bases with a quality score < 20 and length < 50 base pair (bp) by SolexaQA v.3.1.7.1 [32]. To eliminate the differences caused by variations in the sequencing depth among samples, 200 million reads were randomly picked from each sample. The retrieved sequences were then used to search for ARGs following ARGs-OAP v2.0 pipeline as described by Yin et al. [27]. The SARG database identifies ARG types (antibiotic class) and within that class subtypes (e.g., a subtype having > 80% identical aligned bases based on HMM model). Diversity data are derived from the number of subtypes. The parameters used for ARG identification were alignment length cut-off of 75 nucleotides, alignment e value cut-off of 10−7, and alignment identity of 80%. The abundances of ARGs were normalized by 16S rRNA gene expressed as: Abundance = \( \sum \limits_1^n\frac{\mathrm{N}\mathrm{ARG}-\mathrm{like}\kern0.17em \mathrm{sequence}\times \mathrm{Lreads}/\mathrm{L}\mathrm{ARG}\kern0.17em \mathrm{reference}}{\mathrm{N}16\mathrm{S}\kern0.24em \mathrm{sequence}\times \mathrm{Lreads}/\mathrm{L}16\mathrm{S}\kern0.24em \mathrm{sequence}} \) [26]. Analysis of metagenome data of the Earth Microbiome Project shows that the 16S rRNA gene copy number of all soils is very narrow, with a mean of 2.2 16S rRNA copies per cell [33].
Classification of ARG hosts in de novo assembly
De novo assembly was done with MEGAHIT and default parameters. The statistics of assemblies is in Additional file 1: Table S1. ARG-carrying contigs were identified with SARG database and the cutoffs in ARGs-OAP pipeline. The ARG-carrying reads were then classified taxonomically using a contig classification tool, CAT [34].
Identification of clinical ARGs
We define clinical ARGs as those found in human pathogens. Protein sequences of human disease associated bacterial genomes were collected from the Pathosystems Resource Integration Center (PATRIC) [35]. The collected protein sequences were searched against SARG database, hits with identity ≥ 80%, and alignment coverage (alignment length/reference ARG length) ≥ 80% were kept as clinical ARGs. Reads annotated as ARGs by SARG database were extracted from metagenomes of Amazon rainforest and pasture soils, and they were evaluated by BLAST against the protein sequences of clinical ARGs. The hits with identity ≥ 80%, alignment length ≥ 75 bp nucleotides, and e value ≤ 10−7 were regarded as clinical ARGs. The abundance of a clinical ARG was calculated with the same formula to calculate ARG abundance in ARGs-OAP v2.0 pipeline.
To comprehensively understand the homology between ARGs in soil and ARGs found in clinical settings, 12 ARGs in Amazon data sets were assembled with Xander, a target gene assembler [36, 37]. The 12 ARGs were chosen because (1) they were found in human pathogens in PATRIC database; (2) they were detected in Amazon pasture and rainforest soils at sufficient abundance so that assembly of intact/near-intact genes was possible; and (3) hidden Markov models (HMMs) are available. The seed sequences and HMMs were obtained from SARG database [27]. Assembled protein contigs of ≥ 100 amino acids were kept to evaluate sequence similarity with clinical ARG subtypes by BLASTP.
Soil bacterial community
Bacterial taxonomic classification and abundance quantification were analyzed following the SSUsearch pipeline [38]. Briefly, a 16S rRNA gene HMM was used to search against metagenomic data and the hits were annotated with SILVA database. The sequences aligned to a part of 16S V4 variable region (577–657) and with lengths greater than 70 bp were extracted. The extracted 16S rRNA gene sequences were then clustered to estimate OTU number at 95% identity.
Statistical analyses
Only ARGs detected with more than two reads across all samples were retained for further analyses. Bray-Curtis distance-based principal coordinates analysis (PCoA) was performed to estimate the variance of resistome profiles. Procrustes analysis was used to assess the relationship between resistome profile and bacterial community structure, 9999 permutations were used to test the significance. ARGs detected in at least five samples with a maximum read number > 5 in at least one sample were kept for ANOVA and network analyses. The differences in ARG abundances across three soils were tested by ANOVA analysis (least significant difference, p < 0.05). Spearman’s correlation coefficients were calculated based on the read number among soils. Network analysis was performed in Cytoscape to identify ARG clusters; only ARGs with significant (least significant difference, p < 0.01) and strong Spearman coefficients (> 0.9) were used. The ANOVA and Spearman correlation analyses were conducted using SPSS 23.0. PCoA, Adonis and Procrustes analysis were done with R3.5.1.
Results
Detected ARGs and regulatory genes
A total of 268 ARG subtypes potentially conferring resistance to 21 classes of antibiotics were detected in the soils, with most of them belonging to antibiotic deactivation (106 ARGs) and efflux pump (93 ARGs) mechanisms (Fig. 1). More than 58% of the resistome abundance was contributed by efflux pump genes, while only 16% was from the deactivation and cellular protection classes. Ten regulatory genes (mtrR, gadX, tetR, mexT, cAMP-regulatory protein, arlR, ompR, vanS, cpxR, vanR) were detected and they accounted for 13 to 35% of resistome abundances in the studied soils. Five regulatory genes were dominant totaling 0.014–0.141 copies per 16S rRNA gene copy and were observed in all 26 soils (Additional file 1: Fig. S2). Since regulatory genes do not directly confer resistance and they inflate the quantitation, they are not included in most of the following analyses.

Composition of ARGs and regulator genes in 26 soil metagenomes. a Resistance mechanism. b Antibiotic classes
Multidrug resistance genes were most abundant (57.1% of non-regulatory resistome abundance) with 67 subtypes observed, followed by macrolides-lincosamides-streptogramines (MLS) resistance genes, with 28 subtypes that comprised 11.2% of non-regulatory resistome abundance (Fig. 1b). There were 71 beta-lactam, 24 tetracycline, and 20 aminoglycoside resistance gene subtypes detected in these soils, but they on average only accounted for 2.6%, 3.0%, and 1.6% of non-regulatory resistome abundances, respectively.
Non-regulatory ARGs shared by all soils
Fifty-five non-regulatory ARGs were shared by all 26 soils (Fig. 2a). These commonly shared ARGs accounted for 81.5 to 98.6% of non-regulatory resistome abundance across all soils, regardless of native or anthropogenic. The shared ARGs consist of nine classes of antibiotic resistances, but most of them are multidrug resistance genes (31 subtypes, 1.7 × 10−2–6.7 × 10−1 copies per 16S rRNA gene copy). There were six tetracycline resistance genes and five vancomycin resistance genes shared by all soils, and they comprised 1.0 × 10−3–1.2 × 10−2 of resistome abundance. Only two beta-lactam resistance genes were found in all soils, with abundance of 4.6 × 10−4–3.8 × 10−3 copies per 16S rRNA gene copy. The concentrations of shared aminoglycoside and trimethoprim resistance genes differed considerably across the three ecosystems, ranging from 9.4 × 10−5 to 2.0 × 10−3 copies per 16S rRNA gene copy. Efflux pump was the dominant mechanism of these 55 shared ARGs, contributing 80.6% of the total shared ARG abundance.

Diversity and abundance and of ARGs among soils of three ecosystems. a Heatmap showing abundances of 55 background ARGs in 26 soil metagenomes. b diversity and abundance of ARGs by different mechanisms. c ARG abundance of different antibiotic classes. MLS: macrolides-lincosamides-streptogramines
ARGs in native soils
A total of 242 ARG subtypes were observed in the native soils, covering all detected classes of antibiotic resistance (Fig. 2b). There were 144, 191, and 215 ARG subtypes with resistome abundances of 0.195, 0.201, and 0.243 copies per 16S rRNA gene detected in Alaskan tundra soil, Midwestern US native prairie soils and Amazon rainforest soils, respectively. There were no significant differences (p > 0.05) in ARG diversity nor resistome abundance between native and anthropogenic soils. Much of the resistome abundance was contributed by gene components of efflux pump complexes, such as mdtABC-tolC, acrAB, mexEF-oprN, and rosAB (Fig. 3a). Thirteen subtypes of vancomycin resistance genes were detected in native soils, and no significant difference was observed (p = 0.07) between native and anthropogenic soils (Fig. 3b).

Abundance of selected ARGs among soils of the three ecosystems. a Efflux pump complex. b vancomycin resistance genes
Soil resistome at different geographical locations
PCoA analysis clearly demonstrates that soil resistome profiles grouped by geographic location (Fig. 4a). ARG diversity in the tundra was significantly lower (p < 0.01) than that of temperate and tropical areas (Fig. 2b), while no significant difference (p = 0.91) was found between temperate and tropical areas. All ARGs detected in Alaskan soils were found in Midwest America and Amazon soils (Additional file 1: Fig. S3). One-hundred and forty ARGs were observed in both temperate and tropical soils, but not in tundra. The temperate and tropical soils shared 87.6% of ARGs, while 27 ARGs were only found in one or the other. The tropical soils had the highest resistome abundance, but no statistical difference (p > 0.05) was found among the three areas (Fig. 2c). Procrustes analysis showed that the ARG profile was significantly correlated with the bacterial community structure (sum of squares M2 = 0.183, r = 0.904, p < 0.01) (Fig. 4b).

Soil resistome profiles. a PCoA analysis showing profiles of soil resistomes in three ecosystems. Circles represent native soils, and triangles represent anthropogenic soils. b Procrustes analysis of bacterial community and resistome profile. Data point shows the position of a soil sample in the ordination based on resistome profile, and arrow points to its position in the transformed ordination based on bacterial community structure
Effect of cultivated agriculture on soil resistome
There were no observed significant differences (p > 0.05) in either diversity or total ARG abundance between the US Midwest native and the long-term cultivated soils (Fig. 2b). The resistome profiles were very similar in the cultivated soils from the three sampling sites (Fig. 4a). The vancomycin resistance genes in native soils were higher than in cultivated soils, but the difference was not significant (p = 0.56) (Fig. 3b).
Changes of soil resistome during conversion of the Amazon rainforest to pasture
Conversion of Amazon rainforest to pasture significantly (Adonis test, R = 0.148, p < 0.05) altered the soil resistome profile (Fig. 4). The conversion to pasture led to an increase of 23 ARG subtypes in pasture soils (Additional file 1: Fig. S4). All exclusive ARGs (in rainforest or pasture soils), except dfrB2, had a relatively low abundance, ranging from 1.8 × 10−5–3.1 × 10−4 copies per 16S rRNA gene. The non-regulatory resistome abundance in Amazon pasture soil was 11.8% lower than that in the native rainforest soil, although it was not statistically different (p = 0.06).
Identification of ARG hosts in de novo assemblies
We identified 77–208 ARG-containing contigs in de novo assemblies of studied soils, with N50 lengths from 414 to 14,414 bp (Additional file 1: Table S1). Approximately 81.5% of contigs had ARG coverage less than 30%, only four contigs contained intact ARGs (one catB, one dfrB2 and three cAMP-regulatory proteins) (Additional file 1: Fig. S5). Hosts of 59 ARG subtypes were classified to phylum, among them 20 were multidrug resistance genes, 7 were vancomycin resistance genes, 6 were beta-lactam resistance genes, and 5 were tetracycline resistance genes (Fig. 5). Proteobacteria, Actinobacteria, Firmicutes, and Acidobacteria were primary ARG hosts, they carried 28, 21, 11, and 9 ARGs respectively. bacA, mdtB, mdtC, and multidrug_transporter gene had most wide host phyla (≥ 4). Regulatory genes vanR and vanS were classified to Proteobacteria, Firmicutes and Actinobacteria.

Network showing identified hosts of ARGs at phylum level. Different colors represent different classes of ARGs
Detection of clinical ARGs in soil resistome
Since most of the above assembled ARGs are multidrug and regulatory genes and are not recognized in the PATRIC database, we targeted the 12 that were detected and clinically important to evaluate at the sequence level. These include two beta-lactam (ampC and FEZ-1), one quinolone (mfpA), three aminoglycoside (aac(2’)-I, aac(6’)-I, and aph(6’)-I), and six tetracycline resistance genes in Amazon rainforest and pasture soils (Fig. 6). Clinical-similar reads were detected in all 12 ARGs except mfpA at amino acid identity cut-off of 80%. Among them, more than 74% of FEZ-1, aac(2’)-I, tetC, tetO, and tetV reads had this level of amino acid identity to their clinical types. Only six ARGs were recovered with 90% sequence similarity. Highly similar (97% identity) clinical ARG reads were detected for FEZ-1, tetC, and tetX, but they only account for 0.9–4.4% of their environmental (clinical and non-clinical) abundance and none had sequences identical to those ARGs in clinical pathogens. tetV was the most abundant clinical ARG, with abundance of 4.1 × 10−4 and 2.4 × 10−4 copies per 16S rRNA gene in Amazon rainforest and pasture soils, respectively. No statistically significant difference (p > 0.05) was found in abundance of clinical ARGs at all chosen identity levels between Amazon rainforest and pasture soils. Clinical-similar ampC was only detected in Amazon pasture soil at identity level ≥ 90% but its abundance was as low as 5.3 × 10−7 copies per 16S rRNA gene.

Abundance of 12 clinical ARGs in Amazon rainforest and pasture soils. The blue column represents the ARG abundance quantified with SARG database. The yellow and red columns are abundances of clinical ARGs at 80%, 90%, 95%, and 97% amino acid identities. Bars are standard errors
Only three of the 12 selected ARGs could be assembled using the more sensitive target gene assembler, namely ampC, tetM, and tetO. Assembled ampC was only found in Amazon forest soils, and it shared 46–56% of amino acid similarity with clinical ampC. Assembled tetM and tetO shared 62–71% and 69–74% of similarity to their clinical types found in human pathogens. Consistent to the results by SARG pipeline, no significant differences were found in assembled tetM and tetO abundances between Amazon forest and pasture soils.
Identification of ARG clusters in soils
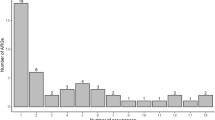
Network analysis was performed to identify ARG clusters present in all soils (Additional file 1: Fig. S6). We found wide co-occurrence of multiple genes despite their distant geographic locations, various ecosystems, and with or without anthropogenic activities. A total of eight types of ARG clusters were observed, comprised of 67 ARG subtypes for 12 classes of antibiotics. Most of the co-occurred ARGs are multidrug resistance genes (27 subtypes), followed by tetracycline resistance genes (9 subtypes) and beta-lactamase resistance genes (8 subtypes). Four aminoglycoside resistance genes were found in ARG clusters and they had the highest average linkage with 4.8 to other ARGs. The largest ARG cluster was comprised of 25 ARGs, where ThinB and aac(3)-IV were the hub genes connecting 10 and 9 other ARGs, respectively.
Discussion
The high diversity and abundance of the soil resistome in Alaskan tundra, temperate prairie, and tropical ecosystems support the view that ARGs are naturally ubiquitous, and in widely different terrestrial ecosystems. The ARGs detected in native soils include those that can potentially confer resistance to all major antibiotics used to treat humans and animals, such as beta-lactams (LRA, PER, TEM, and OXA genes), macrolides and lincosamides (erm genes), quinolones (qepA), aminoglycosides (aac and aph genes), and tetracyclines (tet genes). It is not surprising that ARGs naturally exist in native soils [3] because many antibiotics are produced by soil microorganisms, and indeed were the original source of pharmaceutical products [39]. In accord with our observation, previous studies also identified divergent beta-lactamase resistance genes and a novel chloramphenicol resistance gene from undisturbed Alaskan soil [40, 41]. Vancomycin is regarded as the last line of defense against MRSA strains, but thirteen subtypes of vancomycin resistance genes were detected in these native soils, including vanH, vanA, and vanX which are found in clinical pathogens Staphylococcus aureus and vancomycin-resistant enterococci [42]. Similarly, D’Costa et al. detected the three vancomycin genes in 30,000-year-old permafrost sediments, and further analyses confirmed the similarity in structure and function between the ancient vanA and their modern variants [43]. Our ARG host analysis showed that vanH, vanA, and vanX were harbored by Actinobacteria which are vancomycin producing bacteria [44].
We identified 55 structural ARGs and 5 regulatory ARGs that were shared by all soils regardless of ecosystem type and geography, and hence are candidate common or “background” soil ARGs. These ARGs were also found across various terrestrial ecosystems in previous studies. Thirty of our background ARGs were detected in paddy soils [4]; ten were found in dryland (peanut) soils [9]; at least sixteen were observed in greenhouse soils [45] and ten were found in Antarctic soils [5]. Most of the shared ARGs are multidrug resistance genes with efflux pump as the dominant mechanism. For example, 11 of the background ARGs are involved in Mex-Opr efflux pump systems, and they are known to play a prominent role in the multidrug resistance of gram-negative bacteria [46, 47]. The AcrAB efflux pump plays a physiologic role of pumping out bile acids, fatty acids, and various toxic compounds [48, 49]. Thus, we argue that these commonly shared ARGs should be considered as a separate category, generally of low risk, when evaluating ARG risk in soil environments. However, this does not mean that background ARGs are risk free, since some of them have been found in plasmids and can be enriched with anthropogenic activity. For example, macB can be easily acquired by mobile elements, and thus spread macrolide resistance [50]. Background ARGs acrA, vanC, and mexF were found significantly enriched by the application of sewage sludge and chicken manure to soil [51]. These results imply that compared to the abundance of ARGs, assessment of ARG mobility may be more important for ARG risk evaluation since ARG transfer into pathogens is a primary risk factor.
The soil resistome profile had a significant geographic pattern, which was greater than land use change. The significant correlation (p < 0.01, R2 = 0.795) between ARG diversity and bacterial diversity (Additional file 1: Fig. S7A) suggests that the lower bacterial diversity may explain the lower ARG diversity in tundra. Similar to our findings, Wang et al. [5] detected a positive correlation (R2 = 0.39, p = 0.0001) between bacterial Shannon index and Shannon index of ARGs in Antarctic soils. In contrast, no significant correlation (R2 = 0.152) was observed between ARG abundance and bacterial diversity (Additional file 1: Fig. S7b), which is inconsistent with Bahram et al. [23] who investigated the microbiome of global topsoil samples (189 sites, 7560 subsamples, 12 ecosystems) and found significant negative correlation between ARG abundance and bacterial diversity. The inconsistency could be due to the difference in soil edaphic traits, vegetation, land use history, and which genes are included. For example, Bahram et al.’s study used ARDB for ARG annotation and included regulatory genes such as vanR and vanS in analyses while all regulatory genes were excluded from our analyses. Significant correlation between the resistome profile and microbial community structure was observed, indicating that the differences in ARG profile is primarily driven by bacterial composition. This is consistent with previous studies which also found strong correlation between ARG profile and bacterial community structure in various environments [52,53,54]. Thus, the variation of soil resistomes at different geographical locations was probably related to the differing vegetation, climate, and edaphic factors such as pH and soil organic matter [55,56,57] which will select different populations (and hence the ARGs they carry) or some ARGs by their alternative function(s).
It is well known that the introduction of selective or co-selective pressure by human activities is primarily responsible for the enrichment of ARGs in soils. For example, irrigation with reclaimed water led to enrichment of 60 ARGs [8]. Long-term application of pig manure significantly enhanced the abundance of tetL, tetB(P), tetO, tetW, sul1, ermB, and ermF as compared with inorganic fertilizers [58]. However, it is not clear whether the normal agricultural activities such as crop production affect the soil resistome. In this study, no significant change was observed in either ARG diversity or resistome abundance after long-term continuous cultivation. However, the cultivated soils from the three Midwest sites tend to have similar resistome profiles which may be due to selection for similar adaptations of the bacterial community to agronomic production. Cropping system type, fertilization, and other soil management practices are thought to be factors that can influence the soil resistome [9]. In these study sites, antibiotics and heavy metals were not used so external factors would not have provided for selection. Overall, our results suggest that standard cultivation and fertilization practices of US Midwest (primarily moldboard plow, inorganic N.P.K, and low levels of manure, e.g., cattle grazing) did not increase the public health risk of ARGs in soil. Twenty-three new ARGs emerged and both enrichment and attenuation of ARGs were observed after conversion of Amazon forest to pasture. We speculate that the grass vegetation (Urochloa brizantha, Urochloa decumbens, Panicum maximum) and/or cattle grazing, which includes their manures, may be responsible for the changes in ARGs by selecting different microbial populations and/or increasing their diversity [13].
Growing evidence has shown that some ARGs in pathogens are acquired from environmental bacteria through horizontal gene transfer. For example, the CTX-M extended spectrum beta-lactamase originated from chromosomal genes of an environmental genus, Kluyvera [59], and the clinical vanA has been found in environmental Bacilli [60]. Thus, we selected 12 ARGs which are clinically important and could transfer between bacteria [61] and assessed their sequence similarity to clinical ARGs in human pathogens. The ARGs we detected in Amazon soils are distinct from those found in human pathogens, implying that most ARGs in the natural soil resistome are not demonstrated as problematic or at least not yet entered the clinical realm. Only a few clinical-similar reads (> 90% amino acid identity) of our tested ARGs were observed, but further evaluation of target gene assemblies confirmed that most of them were aligned to conserved regions of these genes. For example, ampC codes clinically important cephalosporinases which confers resistance to cephalothin, cefazolin, cefoxitin, and most penicillins, but the assembled ampC in Amazon soils shared less than 54% of similarity with those found in human pathogens. It is noteworthy that most researchers used short-read based BLAST for ARG search, which provides a sensitive detection but will also recover non-functional pseudo genes or conserved domains. By contrast, ARG evaluation with assembled genes will miss some low abundance ARGs but should better reflect the presence, abundance and sequence similarity of potentially functional ARGs.
There are approximately one ARG in 10 cells in our soils (assuming 2.2 copies of 16S rRNA genes per cell for soil bacteria [33]). Despite of the high resistome abundance, most of ARGs cannot be well assembled by de novo assembly. This is because 80% of soil resistome abundance was contributed by 9–17 ARGs (Additional file 1: Fig. S8). About 50–69% of ARG subtypes were less abundant than 1 in 10,000 cells. We estimated the average coverage of our soil sequence data with NonPareil [62] which showed that approximately 1.6∼11.4 terabytes of sequence data are required for 95% abundance-weighted average coverage of the temperate and tropical soil communities (Additional file 1: Fig. S9). These results show the genetic complexity of the soil microbiome and hence the difficulty of assembling more than the dominant ARGs using Illumina short reads with today’s resources. As one example, we checked the coverage of vanS regulatory genes and found an uneven distribution by position (Additional file 1: Fig. S10). A further BLASTX of reads annotated as vanS against nr database demonstrated that most of the reads are HAMP domain (present in Histidine kinases, Adenylate cyclases, Methyl accepting proteins, and Phosphatases). HAMP domain is approximately 50 amino acids long and is commonly found in integral membrane proteins and two-component regulatory systems [63]. It indicates that at least part of resistome abundance from the short-read alignments may include conserved protein domains, which would lead to overestimates of ARGs.
It is noteworthy that some regulatory genes are included in the SARG database as well as the widely used CARD database [64]. It is problematic as to whether regulatory genes should be counted as ARGs since they only control expression, and not only of ARGs. For example, vanR and vanS cannot confer resistance to vancomycin, but vanR can promote co-transcription of vanA, vanH, and vanX when activated by vanS [65]. A high abundance of regulatory genes was detected and they differed in soils from the several ecosystems (Fig. 2b, c). We removed regulatory genes from our further analyses since the potential risk of ARGs is largely from the horizontal transfer of structural genes which code for functional proteins. In addition to the regulatory genes, some ARGs are components of a functional complex, for which an individual ARG cannot code antibiotic resistance without others. For example, many ARGs detected in our study are components of mdtABC-tolC, acrAB-tolC, and mexEF-oprN efflux complexes. Thus, the addition of ARGs belonging to a complex can inflate the total resistome abundance.
Granted, soil is an important reservoir of ARGs; it harbors background ARGs that may or may not become problematic, probably harbors ARGs not yet emerged, and can harbor clinical ARGs, most likely to have entered soil from human or animal waste disposal. We recommend that more attention be paid to ARG genes or gene sets necessary for resistance function, for their status relative to common ARG backgrounds, for linkage to mobile genetic elements, and their correspondence or linkage to host populations. Sequence similarity may or may not be indicative of potential ARG function but it is a strong indicator of whether the ARG source was from a known clinical resistance and detectable by methods targeting the clinical gene variant.
Conclusions
Soil harbors ARGs that may or may not become problematic, and some that are yet to emerge. We show that the ARG reservoir in soil is global, huge, and exhibits significant geographic patterns. We identified 55 structural and 5 regulatory ARGs as common in all samples of these diverse ecosystems and suggest that these candidate background ARGs be considered as a separate category for health risk evaluation. Further, soil ARGs shared low sequence similarities with those commonly found in human pathogens. We recommend that more attention be paid to ARG genes or gene sets necessary for resistance function, for their status relative to common ARG backgrounds, for linkage to mobile genetic elements and their correspondence or linkage to host populations to evaluate risk.
Availability of data and materials
All sequence data used in the study is available at European Nucleotide Archive (no. PRJEB10725) and JGI (Project Ids 1077701–1077706 and 1080879–1080888).
References
O’Neill J. The review on antimicrobial resistance. In: Tackling drug-resistant infections globally: final report and recommendations. London: HM Government and the Wellcome Trust; 2016.
Braga LPP, Alves RF, Dellias MTF, Navarrete AA, Basso TO, Tsai SM. Vinasse fertirrigation alters soil resistome dynamics: an analysis based on metagenomic profiles. Biodata Min. 2017;10(1):17. https://doi.org/10.1186/s13040-017-0138-4.
Cytryn E. The soil resistome: The anthropogenic, the native, and the unknown. Soil Biol Biochem. 2013;63:18–23. https://doi.org/10.1016/j.soilbio.2013.03.017.
Xiao KQ, Li B, Ma LP, Bao P, Zhou X, Zhang T, et al. Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMS Microbiol Ecol. 2016;92(3):fiw023. https://doi.org/10.1093/femsec/fiw023.
Wang F, Stedtfeld RD, Kim OS, Chai B, Yang L, Stedtfeld TM, et al. Influence of soil characteristics and proximity to Antarctic research stations on abundance of antibiotic resistance genes in soils. Environ Sci Technol. 2016;50(23):12621–9. https://doi.org/10.1021/acs.est.6b02863.
Dantas G, Sommer MOA, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science. 2008;320(5872):100–3. https://doi.org/10.1126/science.1155157.
Wellington EMH, Boxall ABA, Cross P, Feil EJ, Gaze WH, Hawkey PM, et al. The role of the natural environment in the emergence of antibiotic resistance in gram-negative bacteria. Lancet Infect Dis. 2013;13(2):155–65. https://doi.org/10.1016/S1473-3099(12)70317-1.
Wang FH, Qiao M, Su JQ, Chen Z, Zhou X, Zhu YG. High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ Sci Technol. 2014;48(16):9079–85. https://doi.org/10.1021/es502615e.
Wang F, Xu M, Stedtfeld RD, Sheng H, Fan J, Liu M, et al. Long-term effect of different fertilization and cropping systems on the soil antibiotic resistome. Environ Sci Technol. 2018;52(22):13037–46. https://doi.org/10.1021/acs.est.8b04330.
Wu D, Huang XH, Sun JZ, Graham DW, Xie B. Antibiotic resistance genes and associated microbial community conditions in aging landfill systems. Environ Sci Technol. 2017;51(21):12859–67. https://doi.org/10.1021/acs.est.7b03797.
Mackelprang R, Grube AM, Lamendella R, Jesus EDC, Copeland A, Liang C, et al. Response of the soil microbiome to cultivation in native tallgrass prairie soils of the Midwestern United States. Front Microbiol. 2018;9:1775. https://doi.org/10.3389/fmicb.2018.01775.
Soares BS, Nepstad DC, Curran LM, Cerqueira GC, Garcia RA, Ramos CA, et al. Modelling conservation in the Amazon basin. Nature. 2006;440(7083):520–3. https://doi.org/10.1038/nature04389.
Rodrigues JLM, Pellizari VH, Mueller R, Baek K, Jesus ED, Paula FS, et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc Natl Acad Sci U S A. 2013;110(3):988–93. https://doi.org/10.1073/pnas.1220608110.
Sandberg KD, LaPara TM. The fate of antibiotic resistance genes and class 1 integrons following the application of swine and dairy manure to soils. FEMS Microbiol Ecol. 2016;92(2). https://doi.org/10.1093/femsec/fiw001.
Johnson TA, Stedtfeld RD, Wang Q, Cole JR, Hashsham SA, Looft T, et al. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. Mbio. 2016;7(2):e02214–5. https://doi.org/10.1128/mBio.02214-15.
Zhu YG, Johnson TA, Su JQ, Qiao M, Guo GX, Stedtfeld RD, et al. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc Natl Acad Sci U S A. 2013;110(9):3435–40. https://doi.org/10.1073/pnas.1222743110.
Li B, Yang Y, Ma LP, Ju F, Guo F, Tiedje JM, et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015;9(11):2490–502. https://doi.org/10.1038/ismej.2015.59.
Ma L, Xia Y, Li B, Yang Y, Li LG, Tiedje JM, et al. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken, and human feces. Environ Sci Technol. 2016;50(1):420–7. https://doi.org/10.1021/acs.est.5b03522.
Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 2018;6:1–15.
Li L, Yin X, Zhang T. Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome. 2018;6(1):93. https://doi.org/10.1186/s40168-018-0480-x.
Martínez JL, Coque TM, Baquero F. What is a resistance gene? Ranking risk in resistomes. Nat Rev Microbiol. 2015;13(2):116–23. https://doi.org/10.1038/nrmicro3399.
Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DGJ. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics. 2015;16(1):964. https://doi.org/10.1186/s12864-015-2153-5.
Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, et al. Structure and function of the global topsoil microbiome. Nature. 2018;560(7717):233–7. https://doi.org/10.1038/s41586-018-0386-6.
Van Goethem MW, Pierneef R, Bezuidt OKI, Van De Peer Y, Cowan DA, Makhalanyane TP. A reservoir of ‘historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome. 2018;6(1):40. https://doi.org/10.1186/s40168-018-0424-5.
Yuan K, Yu K, Yang R, Zhang Q, Yang Y, Chen E, et al. Metagenomic characterization of antibiotic resistance genes in Antarctic soils. Ecotox Environ Safe. 2019;176:300–8. https://doi.org/10.1016/j.ecoenv.2019.03.099.
Yang Y, Jiang XT, Chai BL, Ma LP, Li B, Zhang AN, et al. ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinform. 2016;32(15):2346–51. https://doi.org/10.1093/bioinformatics/btw136.
Yin XL, Jiang XT, Chai BL, Li LG, Yang Y, Cole JR, et al. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinform. 2018;34:2263–70.
Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT. Tackling soil diversity with the assembly of large, complex metagenomes. P Natl Acad Sci U S A. 2014;111(13):4904–9. https://doi.org/10.1073/pnas.1402564111.
Cheng L, Zhang NF, Yuan MT, Xiao J, Qin YJ, Deng Y, et al. Warming enhances old organic carbon decomposition through altering functional microbial communities. ISME J. 2017;11(8):1825–35. https://doi.org/10.1038/ismej.2017.48.
Johnston ER, Rodriguez-R LM, Luo C, Yuan MM, Wu L, He Z, et al. Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska tundra ecosystem. Front Microbiol. 2016;7:579.
Natali SM, Schuur EAG, Trucco C, Pries CEH, Crummer KG, Lopez AFB. Effects of experimental warming of air, soil and permafrost on carbon balance in Alaskan tundra. Glob Chang Biol. 2011;17(3):1394–407. https://doi.org/10.1111/j.1365-2486.2010.02303.x.
Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010;11(1):485. https://doi.org/10.1186/1471-2105-11-485.
Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457–63. https://doi.org/10.1038/nature24621.
von Meijenfeldt FAB, Arkhipova K, Cambuy DD, Coutinho FH, Dutilh BE. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biology. 2019;20(1):217. https://doi.org/10.1186/s13059-019-1817-x.
Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017;45:535–42.
Wang Q, Fish JA, Gilman M, Sun Y, Brown CT, Tiedje JM, et al. Xander: employing a novel method for efficient gene-targeted metagenomic assembly. Microbiome. 2015;3(1):32. https://doi.org/10.1186/s40168-015-0093-6.
Guo J, Quensen J, Sun Y, Wang Q, Brown CT, Cole JR, et al. Review, evaluation and directions for gene-targeted assembly for ecologic analyses of metagenomes. Front. Genet. 2019;10:957. https://doi.org/10.3389/fgene.2019.00957.
Guo JR, Cole JR, Zhang QP, Brown CT, Tiedje JM. Microbial community analysis with ribosomal gene fragments from shotgun metagenomes. Appl Environ Microbiol. 2016;82(1):157–66. https://doi.org/10.1128/AEM.02772-15.
Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol R. 2010;74(3):417–33. https://doi.org/10.1128/MMBR.00016-10.
Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J. 2009;3(2):243–51. https://doi.org/10.1038/ismej.2008.86.
Lang KS, Anderson JM, Schwarz S, Williamson L, Handelsman J, Singer RS. Novel florfenicol and chloramphenicol resistance gene discovered in Alaskan soil by using functional metagenomics. Appl Environ Microbiol. 2010;76(15):5321–6. https://doi.org/10.1128/AEM.00323-10.
Wright GD. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5(3):175–86. https://doi.org/10.1038/nrmicro1614.
D'Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, et al. Antibiotic resistance is ancient. Nature. 2011;477(7365):457–61. https://doi.org/10.1038/nature10388.
Yushchuk O, Binda E, Marinelli F. Glycopeptide antibiotic resistance genes: distribution and function in the producer actinomycetes. Front Microbiol. 2020;11. https://doi.org/10.3389/fmicb.2020.01173.
Fang H, Wang H, Cai L, Yu Y. Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ Sci Technol. 2015;49(2):1095–104. https://doi.org/10.1021/es504157v
Li Y, Mima T, Komori Y, Morita Y, Kuroda T, Mizushima T, et al. A new member of the tripartite multidrug efflux pumps, MexVW-OprM, in Pseudomonas aeruginosa. J Antimicrob Chemoth. 2003;52(4):572–5. https://doi.org/10.1093/jac/dkg390.
Aendekerk S, Diggle SP, Song Z, Høiby N, Cornelis P, Williams P, et al. The MexGHI-OpmD multidrug efflux pump controls growth, antibiotic susceptibility and virulence in Pseudomonas aeruginosa via 4-quinolone-dependent cell-to-cell communication. Microbiology. 2005;151(4):1113–25. https://doi.org/10.1099/mic.0.27631-0.
Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol. 1996;178(1):306–8. https://doi.org/10.1128/JB.178.1.306-308.1996.
Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DEJ. Structural basis of multiple drugbinding capacity of the AcrB multidrug efflux pump. Science. 2003;300(5621):976–80. https://doi.org/10.1126/science.1083137.
Roberts MC. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett. 2008;282(2):147–59. https://doi.org/10.1111/j.1574-6968.2008.01145.x.
Chen QL, An XL, Li H, Su JQ, Ma YB, Zhu YG. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ Int. 2016;92-93:1–10.
Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015;9(1):207–16. https://doi.org/10.1038/ismej.2014.106.
Guo N, Wang YK, Tong TZ, Wang SG. The fate of antibiotic resistance genes and their potential hosts during bio-electrochemical treatment of high-salinity. Water Res. 2018;133:79–86. https://doi.org/10.1016/j.watres.2018.01.020.
Qian X, Sun W, Gu J, Wang XJ, Sun JJ, Yin YN, et al. Variable effects of oxytetracycline on antibiotic resistance gene abundance and the bacterial community during aerobic composting of cow manure. J Hazard Mater. 2016;315:61–9. https://doi.org/10.1016/j.jhazmat.2016.05.002.
Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. P Natl Acad Sci U S A. 2006;103(3):626–31. https://doi.org/10.1073/pnas.0507535103.
Nottingham AT, Fierer N, Turner BL, Whitaker J, Ostle NJ, McNamara NP, et al. Microbes follow Humboldt: temperature drives plant and soil microbial diversity patterns from the Amazon to the Andes. Ecology. 2018;99(11):2455–66. https://doi.org/10.1002/ecy.2482.
Prober SM, Leff JW, Bates ST, Borer ET, Firn J, Harpole WS, et al. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol Lett. 2015;18(1):85–95. https://doi.org/10.1111/ele.12381.
Peng S, Feng YZ, Wang YM, Guo XS, Chu HY, Lin XG. Prevalence of antibiotic resistance genes in soils after continually applied with different manure for 30 years. J Hazard Mater. 2017;340:16–25. https://doi.org/10.1016/j.jhazmat.2017.06.059.
Poirel L, Kampfer P, Nordmann P. Chromosome-encoded Ambler class A beta-lactamase of Kluyvera georgiana, a probable progenitor of a subgroup of CTX-M extendedspectrum beta-lactamases. Antimicrob Agents Chemother. 2002;46(12):4038–40. https://doi.org/10.1128/AAC.46.12.4038-4040.2002.
Patel R, Piper K, Cockerill FR, Steckelberg JM, Yousten AA. The biopesticide Paenibacillus popilliae has a vancomycin resistance gene cluster homologous to the Enterococcal VanA vancomycin resistance gene cluster. Antimicrob Agents Chemother. 2000;44(3):705–9. https://doi.org/10.1128/AAC.44.3.705-709.2000.
van Hoek AH, Mevius D, Guerra B, Mullany P, Roberts AP, Aarts HJ. Acquired antibiotic resistance genes: an overview. Front Microbiol. 2011;2:203.
Rodriguez-R LM, Gunturu S, Tiedje JM, Cole JR, Konstantinidis KT. Nonpareil 3: Fast estimation of metagenomic coverage and sequence diversity. mSystems. 2018;3:e00039–18.
Hulko M, Berndt F, Gruber M, Linder JU, Truffault V, Schultz A, et al. The HAMP domain structure implies helix rotation in transmembrane signaling. Cell. 2006;126(5):929–40. https://doi.org/10.1016/j.cell.2006.06.058.
Jia BF, Raphenya AR, Alcock B, Waglechner N, Guo PY, Tsang KK, et al. Card 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:566–73.
Arthur M, Molinas C, Courvalin P. The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriolo. 1992;174(8):2582–91. https://doi.org/10.1128/JB.174.8.2582-2591.1992.
Acknowledgements
We thank Professor Tong Zhang and his students Xiaole Yin and Xiaotao Jiang at The University of Hong Kong for providing help on using ARGs-OAP pipeline. We thank Dr. Lisa Boughner for assistance with manuscript editing.
Funding
This study was supported by Interdisciplinary Research Center for Soil Microbial Ecology and Land Sustainable Productivity in Dry Areas at Northwest A&F University and by Center for Health Impacts in Agriculture at Michigan State University.
Author information
Authors and Affiliations
Contributions
JMT designed the experiment. XQ analyzed the data and wrote the manuscript. SG, GJR, CBL, and JRC guided the bioinformatic analyses. JMT and JG provided advice and revised this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Statistics of de novo assemblies and ARG-carrying contigs. Fig. S1. Geographical distribution of sampling sites. Fig. S2. Antibiotic resistance regulatory genes in soils with arlR, cpxR, ompR, vanR and vanS found in all 26 soils and part of shared background. Fig. S3. Venn diagram showing shared ARGs among Alaska, Midwest USA, and Amazon soils. Fig. S4. (A) The shared and exclusive ARGs between Amazon rainforest soils and pasture soils. (B) The composition of the exclusive ARGs. Fig. S5. ARG coverage (length of an assembled contig divided by length of the intact ARG) on de novo assemblies. Fig. S6. Network analysis assessing the ARG cluster across soils from tundra, temperate prairie and tropical ecosystems. Fig. S7. Pearson correlation between (A) ARG diversity and bacterial diversity; (B) resistome abundance and bacterial diversity. Fig. S8. Rank percentage of soil resistome abundance of top 50 ARGs. Fig. S9. Nonpareil curves showing estimated average coverage in soil datasets. Fig. S10. Per base coverage of vanS regulatory gene in 26 soils.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, X., Gunturu, S., Guo, J. et al. Metagenomic analysis reveals the shared and distinct features of the soil resistome across tundra, temperate prairie, and tropical ecosystems. Microbiome 9, 108 (2021). https://doi.org/10.1186/s40168-021-01047-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01047-4