
Abstract
Background
Microbiomes play vital roles in plant health and performance, and the development of plant beneficial microbiomes can be steered by organic fertilizer inputs. Especially well-studied are fertilizer-induced changes on bacteria and fungi and how changes in these groups alter plant performance. However, impacts on protist communities, including their trophic interactions within the microbiome and consequences on plant performance remain largely unknown. Here, we tracked the entire microbiome, including bacteria, fungi, and protists, over six growing seasons of cucumber under different fertilization regimes (conventional, organic, and Trichoderma bio-organic fertilization) and linked microbial data to plant yield to identify plant growth-promoting microbes.
Results
Yields were higher in the (bio-)organic fertilization treatments. Soil abiotic conditions were altered by the fertilization regime, with the prominent effects coming from the (bio-)organic fertilization treatments. Those treatments also led to the pronounced shifts in protistan communities, especially microbivorous cercozoan protists. We found positive correlations of these protists with plant yield and the density of potentially plant-beneficial microorganisms. We further explored the mechanistic ramifications of these relationships via greenhouse experiments, showing that cercozoan protists can positively impact plant growth, potentially via interactions with plant-beneficial microorganisms including Trichoderma, the biological agent delivered by the bio-fertilizer.
Conclusions
We show that protists may play central roles in stimulating plant performance through microbiome interactions. Future agricultural practices might aim to specifically enhance plant beneficial protists or apply those protists as novel, sustainable biofertilizers.
Video abstract
Similar content being viewed by others


Background
Soil is the basis for crop production [1, 2] by providing water, nutrients, and the growth matrix for plants [3, 4]. However, space for agricultural use of soils is limited, as other human needs and natural resources compete for this space [5, 6]. Given this limitation for space, intensive agricultural management systems, including continuously growing the same crop, have been developed to help to meet the increasing food demands of a growing human population [7,8,9]. However, such continuous cropping systems commonly suffer from a buildup of soil-borne plant pathogens [10], an imbalance in nutrient availability [8], and a reduction of soil fertility [11,12,13], which together can negatively affect yield [14,15,16]. The application of chemical fertilizers and pesticides used in conventional agricultural practices do help to sustain high crop yields, but the overuse of these agrochemicals can lead to severe environmental problems [7, 17]. Organic soil management might provide a solution to mitigate yield reductions by increasing soil quality and nutrients and reducing soil-borne diseases, with far fewer negative environmental impacts than induced by conventional agricultural practices [18,19,20].
Bacteria and fungi, which represent the most studied soil microbial groups, are known to be impacted by fertilization regimes [11, 18, 21]. Shifts in these communities can be linked to plant productivity and health status through direct mutualistic or pathogenic effects on plants and indirectly by competing with plant-associated microorganisms [22,23,24]. However, bacteria and fungi are embedded in complex soil food webs in which predation can structure their community composition and functioning [25, 26]. The main consumers of soil bacteria and fungi are protists, the most diverse and abundant soil eukaryotes [27]. Protists also display phototrophic, animal parasitic, and plant pathogenic lifestyles [27]. Protistan communities are influenced by numerous environmental factors and soil properties [28, 29], and they have long been proposed as sensitive bio-indicators of soil quality [30]. Logically, differences in fertilization practices affect protistan communities [31], potentially even more than bacterial and fungal communities [32]. Protistan communities might also best predict and potentially control plant health in the presence of soil-borne pathogens [33]. Yet, the functional link between fertilization-induced shifts in protistan communities to their microbial prey and crop yield remains essentially unknown.
To better understand the impact of different fertilization practices on microbiome composition and functioning, we studied the entire soil microbiome, with a particular emphasis on protists, in continuously planted cucumber soils for six growing seasons. Fertilizer treatments consisted of conventional, organic, and bio-organic (organic fertilizer with the addition of Trichoderma fungus) fertilization, in addition to a no-fertilizer control. We then linked microbiome taxonomic and functional shifts to crop yield and performed subsequent greenhouse experiments to validate the functional importance of key protists with and without the presence of other (plant-beneficial) microorganisms on plant performance. We hypothesized that greater yield would be realized in both organic fertilization treatments, with protists representing the microbial group most responsive to different fertilization practices. We further hypothesized that crop yield could be at least partly explained by protist community structure and the relative abundance of specific protistan taxa.
Results
Crop yield and yield contribution of soil microbiome communities
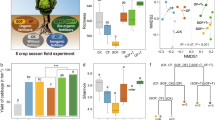
The application of all fertilizers increased crop yield in all six continuous cropping seasons in comparison with the control (P < 0.05; Fig. S1a). Crop yields in OF and BF were higher than that of CF in all seasons combined (P < 0.05; Fig. 1a) and within each season (Fig. S1a). Protistan community structure was the best microbial parameter among the selected microbial indices with respect to explaining crop yield across all treatments (P < 0.05), explaining 11.56 % of the observed variation (Fig. 1b). In contrast, neither the bacterial or fungal community structure, nor the diversity of any microbial group was significantly linked to yield (P > 0.05; Fig. 1b).

Overall effects of different fertilization practices on crop yield (a). The relative importance of bacterial, fungal and protistan diversity and community structure for crop yield (b). The effects of fertilization regime on bacterial, fungal and protistan community structure (c). In panel (a), bars with different letters indicate significant differences as defined by Tukey’s HSD test (P < 0.05). Ctrl: soil without fertilizer amendment; CF: soil amended with chemical fertilizer; OF: soil amended with organic fertilizer; BF: soil amended with bio-organic fertilizer. In panels (b) and (c), ***indicates P < 0.001. Statistical significance for explanatory power was calculated by multiple regression using linear models. Effect value (R2) and statistical significance were calculated by PERMANOVA analysis
Microbial diversity and community structure
Fertilization regimes affected the community structure of all microbial groups, with the strongest impact on protists (protists: R2 = 0.135, P = 0.001; bacteria: R2 = 0.111, P = 0.001; fungi: R2 = 0.123, P = 0.001) (Fig. 1c and Fig. S1b and Table S2). The alpha diversity of protists was marginally affected by fertilization (F = 3.944, P = 0.053), while alpha diversity of both bacteria (F = 0.881, P = 0.491) and fungi (F = 2.166, P = 0.170) was not altered (Table S3). The only significant difference for alpha diversity between treatments was a higher protistan diversity in OF compared with CF (P < 0.05; Fig. S1c). A higher variation of protistan diversity in all fertilization treatments was observed than that in control treatment (Fig. S1d). As protists responded most strongly to fertilization and given their link with yield, we focused subsequent analyses on protists.
Underlying drivers of protistan taxonomic and functional composition and links with yield
Among protistan functional groups, only microbe-consuming protists positively correlated with yield (P < 0.05; Table S4). Compared with the control, the relative abundance of microbe-consuming protists increased by 4.28 % in BF (P < 0.05), while there were no significant differences across the other treatments (Fig. S1e). The relative abundance of microbe-consuming protists significantly decreased over time only in CF (P < 0.05; Fig. S1f). In addition, we detected lower variations of the relative abundance of microbe-consuming protists in (bio-)organic fertilization treatments (organic and Trichoderma bio-organic fertilization) compared with chemical fertilization treatment (Fig. S1f). We performed SEM to further identify potential links between yield with microbe-consuming protists, bacteria, and fungi. This analysis indicated that microbe-consuming protists were directly linked to yield (P < 0.05; path coefficient = 0.287) through interactions with bacteria (P < 0.01; path coefficient = 0.316) and fungi (P < 0.05; path coefficient = 0.218) (Fig. 2a). Analyses of abiotic factors potentially underlying fertilization-induced changes of microbe-consuming protists revealed soil pH as the major factor among the eight measured soil physicochemical properties (P < 0.05; Fig. S2a, b and c).

Structural equation model (SEM) illustrating the links between bacterial and fungal abundances and the relative abundance of microbe-consuming protists on crop yield (a). Heatmap illustrating the relative abundance of microbe-consuming protistan OTUs associated with crop yield in all treatments (b). In panel (a), *indicates P < 0.05; **indicates P < 0.01. Solid and dashed lines represent significant and non-significant relationships, respectively, with blue arrows depicting positive relationships and red arrows negative relationships. Numbers associated with lines indicate path coefficients, and line width is proportional to the effect size. The low chis-square (χ2 = 0.458), high goodness-of-fit index (GFI = 0.797), low Akaike information criteria (AIC = 18.458), and low root mean-square errors of approximation (RMSEA = 0.000) indicate that our data matches our hypothetical model. In panel (b), the colour key relates the heatmap colours to the standard score (z-score). Plus signs indicate positive, and minus signs negative correlations between OTUs and crop yield. Different letters indicate significant differences as defined by Tukey's HSD test (P < 0.05). Circles are proportional to the average relative abundance of each group across all samples
Fourteen microbe-consuming protistan OTUs positively correlated with crop yield, including nine cercozoan, four ciliophoran, and one ochrophytan taxa (Fig. 2b). Of these, the cercozoan OTU124 (Cercozoa, Cercomonas), OTU4169 (Cercozoa, Euglyphida_XX), and OTU58 (Cercozoa, Rhogostoma−lineage_X) showed significantly higher relative abundance in OF and BF as compared with the Control and CF treatments (P < 0.05; Fig. 2b). Spearman’s correlation analysis, used to further identify potential links between these microbe-consuming protists and bacteria or fungi, indicated that OTU124 was positively correlated with Trichoderma (F_OTU2929) and Aspergillus (F_OTU14), OTU4169 with Pseudomonas (B_OTU1, B_OTU1938) and Aspergillus (F_OTU14, F_OTU23), while OTU58 was negatively correlated with Fusarium (F_OTU3) (Fig. S2d).
Plant growth promotion capability of microbe-consuming protist isolates
Our first greenhouse experiment aimed at mechanistically understanding the observed links between protistan communities and crop yield in the field study. We found that all microbe-consuming protists increased cucumber biomass as compared with the control in natural soils (P < 0.05), but this was not the case in sterilized soils (Fig. 3a). The strongest positive effect on plant performance was induced by the two cercozoan taxa Cercomonas lenta (an increase of 165%) and Cercomonas S24D2 (an increase of 138%) (Fig. 3a). The non-cercozoan microbe-consuming Allovahlkampfia sp. also increased plant biomass compared with the control (64%), but less than Cercomonas lenta and Cercomonas S24D2 (Fig. 3a). The positive effects of the two cercozoan species, Cercomonas lenta and Cercomonas S24D2, on plant performance, were confirmed in a second greenhouse experiment, showing increases of plant biomasses by 101% and 79% compared with control, respectively (Fig. S3).

Fold change of cucumber biomass relative to control in treatments with inoculation of different microbe-consuming protists in a first greenhouse experiment (a) (for the confirmatory second greenhouse experiment see Fig. S3). Cucumber biomass of the third greenhouse experiment testing plant growth promotion capability of Trichoderma stimulated by the inoculation of microbe-consuming protist (b). In panel (a), control indicates that no protists were inoculated; S_ indicates that protists were inoculated into sterilized soil. In panel (b), control indicates that no microbe was inoculated; S_ indicates that protists and fungi were inoculated into sterilized soil. T: Trichoderma; T+C: Trichoderma+Cercomonas lenta. Asterisks indicate significant difference of cucumber biomass in treatments compared with control (Student’s t test, ***indicates P < 0.001; **indicates P < 0.01; NS indicates not significant). The P values indicate significance between T and T+C or S_T and S_T+C under Student’s t test
The third greenhouse experiment was designed to explore potential interactions between microbe-consuming protists and Trichoderma (the biological agent added to organic fertilizers in the BF treatment). In sterilized soil, the strongest positive effects on plant performance were observed when Trichoderma was inoculated without protists (S_T; 193% increase to control) and in Trichoderma+protist inoculations (S_T+C; 197%) (P < 0.05; Fig. 3b). In natural soil, T+C significantly increased the plant biomass when compared with T (P = 0.028), while both T and T+C increased plant biomass by 140% and 196% compared with control, respectively (P < 0.05; Fig. 3b).
Discussion
We found that protists, especially microbe-consumers, were the microorganisms most strongly affected by fertilization practices. Protistan communities as well as specific protistan taxa were mostly strongly explaining crop yield variation via interactions with bacterial and fungal communities. Our findings suggest that the protistan community plays a previously neglected role in determining plant performance, which can be at least as important as bacterial and fungal communities that are typically targeted in soil and rhizosphere microbiome studies. This extends previous studies that show protists to be the most sensitive microbial group to fertilization [32] and the best microbial community determinant of plant health [33]. Indeed, we here show evidence for a potential positive link between protistan communities and plant yield, a finding that we support with findings from greenhouse studies that show protist-induced plant growth increases. The importance of different protistan communities and species might be explained by species-specific protistan feeding differences [34, 35] that might specifically shape a plant growth-promoting microbiome [26].
Modification in abiotic soil parameters through fertilization, particularly the associated soil acidification, was a driver of changes in protistan diversity, community structure, and functional composition, which confirms the importance of abiotic parameters as global and local determinants of protistan communities [28]. Indeed, our study suggests that pH might be of similar importance in structuring protists at the local scale as soil moisture at both local and global scales [29, 36]. Especially phagotrophic microbe-consumers, as the dominant soil protistan functional group [36], responded strongly to changes in soil physicochemical properties, potentially partly due to co-occurring changes in bacterial and fungal prey communities. The addition of Trichoderma to organic fertilizer (BF treatment) also affected protistan communities, confirming that microbial introductions can change protistan communities [37]. Our results also show the tight link between communities of bacteria and protists as repeatedly shown before [38], highlighting protists as key predators of soil bacteria [27, 39]. We also show links between fungi and protists that might be linked to their emerging role as important fungivores [40]. This ancient co-evolutionary predator-prey relationship likely resulted in the abovementioned species-specific feeding differences as well as highly sophisticated antagonism mechanisms, in which bacteria and fungi defend against predators [26, 41].
The strong link between organic fertilization enriched microbe-consuming protists, particularly cercozoan taxa, and plant yield in the field might be explained by a combination of effects including enhanced nutrient turnover, a promotion of plant-beneficial microorganisms and a manipulation of the plant hormonal balance [27, 42, 43]. Indeed, microbe-consuming protists are suggested to enhance plant performance by preying on plant-detrimental microbes or increasing the performance of plant growth-promoting microorganisms through predation on their competitors [26, 42]. Our greenhouse experiments confirmed the positive plant growth-promoting effect of microbe-consuming protists (Cercomonas lenta; Cercomonas S24D2; Allovahlkampfia sp.), particularly of Cercozoa, globally one of the most abundant and functionally important soil protist group [36, 44]. These cercozoan taxa observed here might increase plant yield through negative links with plant pathogens (Fusarium) and positive links with plant-beneficial microorganisms (Pseudomonas, Trichoderma, and Aspergillus), a link that we highlight in our greenhouse experiments (Fig. 3b).
Our third greenhouse experiment provided direct evidence for the functional importance of Trichoderma in enhancing plant performance [45]. The stronger positive effect on plant performance of Trichoderma in sterilized than in natural soils suggests that competitive interactions with other soil organisms reduce the positive effect of Trichoderma [46, 47], while other indigenous soil-borne pathogens might also directly inhibit plant performance [33]. No significant difference was observed between “S_T” and “S_T+C,” indicating that Cercomonas lenta might not directly interact with Trichoderma. The stronger positive effect on plant performance was induced in “T+C” compared with the “T,” suggesting that Cercomonas lenta may increase the competitive ability of Trichoderma in natural soil that increases its positive effects on plant performance. Those results provide a novel viewpoint to manipulate the soil microbiome by combining protists with plant-beneficial fungi, which might help increase agro-ecosystem functions in a sustainable way.
Conclusions
We show that protistan communities, and in particular microbe-consuming taxa, are important microbial determinants of crop yield via their specific enhancement of plant-beneficial microorganisms. Microbe-consuming protists represented the microbial group most strongly affected by fertilization. Therefore, these protists may represent a powerful tool for targeted and environmentally friendly microbiome manipulation to increase crop yield. We highlight the need to include protists in studies seeking to evaluate the impacts and mechanisms involved in bio-organic fertilizer applications, and future research should evaluate the use of protists as bioadditives for the development of new strategies for sustainable protist-supported plant growth promotion.
Methods
Field description, experimental design, and soil sample collection
The long-term continuous cropping field was initiated in March 2014 and is located at the Nanjing Institute of Vegetable Science, Jiangsu province, China (31° 43′ N, 118° 46′ E). It is situated in a subtropical monsoon climate area with an annual average temperature of 15.4 °C and an average precipitation of 1106 mm.
Cucumber was the crop that was continuously planted from March 2014 through October 2016, and treatments were established in a randomized complete block design with three replicates for each fertilizer treatment. Briefly, the four fertilization treatments were: Ctrl, soil amended with no fertilizer; CF, soil amended with chemical fertilizer; OF, soil amended with organic fertilizer and BF, soil amended with bio-organic fertilizer. A detailed fertilization scheme (average amount of six seasons) is shown in Table S1. In brief, the organic fertilizer is a mixture of compounds including liquid amino acids from animal carcasses and chicken manure compost at a ratio of 1:5 (v/w). The bio-organic fertilizer is composed of organic fertilizer with approximately 5.0×107 CFU dry weight of Trichoderma guizhouense NJAU 4742 g−1 dry fertilizer. Here, the strain Trichoderma guizhouense NJAU 4742 has a strong growth-promoting ability and is a widely used commercial biological agent in China [48, 49]. A rotary tiller was used to apply different fertilizers before cucumber planting, and all treatments were managed in the same way throughout the experiment. Soil samples were collected and crop yield was detected during full bearing period in June 2014 (subsequently termed “Crop 1”), October 2014 (“Crop 2”), June 2015 (“Crop 3”), October 2015 (“Crop 4”), June 2016 (“Crop 5”) and October 2016 (“Crop 6”). For each replicate, a composite soil sample was generated from 8 random soil cores taken to a depth of 5 cm using a 25-mm soil auger. Samples were thoroughly homogenized and sieved through a 2-mm mesh-size sieve. One portion of each sample was air-dried for chemical analyses, and the other portion was stored at −80 °C for subsequent DNA extraction.
Analysis of soil physicochemical properties
Soil pH was measured in a 1:5 soil/water suspension with a glass electrode. Soil organic matter (SOM) was measured using the K2Cr2O7-H2SO4 oxidation-reduction colorimetric method [32]. Total nitrogen (TN) was analyzed by an elemental analyzer (multi EA® 5000, Analytik Jena, Germany). Total phosphorus (TP) was determined by the molybdenum blue method after wet digestion with H2SO4+HClO4 [50]. Total potassium (TK) was determined after digesting the sample with HNO3+HClO4 by flame photometry [51]. Available nitrogen (AN) was measured with the alkaline-hydrolysable diffusion method [52]. Available phosphorus (AP) in the soil was extracted with sodium bicarbonate and determined using the molybdenum blue method. Available potassium (AK) in the soil was extracted with ammonium acetate and determined by flame photometry [53].
DNA extraction, real-time PCR assay, and Illumina MiSeq sequencing
For each composite soil sample, total DNA was extracted from 10 g of soil using the DNeasy® Power max® Soil Kit (Qiagen, Germany), according to the manufacturer’s instructions. Genomic DNA concentration and purity were measured using a NanoDrop ND-2000 (NanoDrop Technologies, Wilmington, DE) spectrophotometer. Abundances of bacteria and fungi as estimated by 16S rRNA gene and ITS region, respectively. Copy numbers were determined with the primer combinations Eub338F/Eub518R and ITS1f/5.8s, respectively, following established protocols [18] on a qTOWER Real-Time PCR System (Analytik Jena, Germany). We set up 20-μl reaction mixtures containing 10 μl of the SYBR®Premix Ex Taq™ (TaKaRa, Japan), 0.4 μl of each primer (10 μM), 2 μl of template DNA, and 7.2 μl of ddH2O. Standard curves were generated by using 10-fold serial dilutions of a plasmid containing a full-length copy of the 16S rRNA gene from Escherichia coli or the 18S rRNA gene from Saccharomyces cerevisiae. Thermal-cycling conditions for each sample were run as follows: 30 s at 95 °C for initial denaturation, 40 cycles of 5 s at 95 °C, and 34 s at 60 °C, and the results were expressed as log copy numbers g−1 dry soil.
Prokaryote-, fungal-, and eukaryote-wide primers sets were used for high-throughput Illumina MiSeq sequencing: 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) to amplify prokaryotic 16S rRNA gene V4 regions [54], ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′) to amplify fungal ITS1 regions [55, 56], and V4_1f (5′-CCAGCASCYGCGGTAATWCC-3′) and TAReukREV3 (5′-ACTTTCGTTCTTGATYRA-3′) to amplify eukaryotic 18S rRNA gene V4 regions [57]. These primer pairs were modified for sequencing by adding the forward Illumina Nextera adapter, a two base pair “linker” sequence, and a unique 7-bp barcode sequence at the 5′ end of the forward primer, and the appropriate reverse Illumina Nextera adapter and linker sequence to the 5′ end of the reverse primer. PCR amplification was performed in a 25 μl volume: 5 μl of 5 × reaction buffer, 5 μl of 5 × GC buffer, 2 μl dNTPs (2.5 mM), 1 μl of each primer (10 uM), 0.25 μl of high-fidelity DNA polymerase, 2 μl of DNA template and 8.75 μl of ddH2O. The PCR thermal cycling conditions were performed with the following temperature regime: initial denaturation (98 °C for 2 min), followed by 30 cycles of denaturation (98 °C for 15 s), annealing (55 °C for 30 s), extension (72 °C for 30 s), and a final extension (72 °C for 5 min). Each sample was amplified in triplicate, pooled in equimolar concentrations of 20 ng μl−1, and then purified with a PCR Purification Kit (Axygen Bio, USA). Paired-end sequencing was performed on an Illumina MiSeq sequencer at Personal Biotechnology Co., Ltd (Shanghai, China). All raw sequence data are deposited in the NCBI Sequence Read Archive (SRA) under the BioProject PRJNA599073.
Bioinformatic analyses
The bacterial, fungal, and protistan raw sequences were processed according to previously established protocols [58, 59]. Briefly, low-quality sequences and singletons were removed. After that, the remaining sequences were assigned to OTUs at a 97% similarity threshold, and chimeras were filtered using UCHIME [60]. Finally, representative sequences for bacterial and fungal OTUs were classified using the RDP classifier against the RDP Bacterial 16S rRNA gene database and the UNITE Fungal ITS database, respectively [61]. 18S rRNA gene sequences were matched against the Protist Ribosomal Reference database (PR2) [62]. We discarded OTUs assigned as Rhodophyta, Streptophyta, Metazoa, Fungi, and unclassified Opisthokonta sequences to generate a protistan-focused OTU table for subsequent analyses. We further assigned taxonomic protistan OTUs into different functional groups (microbe-consuming, phototrophic, plant pathogenic, animal parasitic, and saprophytic), according to their feeding mode based [37, 44, 63]. Relative abundances of each protistan taxonomic and functional group in relation to total protistan reads were used for later analyses.
Pot experiments to test the effects of microbe-consuming protistan isolates on cucumber growth
Firstly, we tested the effects of microbe-consuming protists on cucumber growth in the first and second greenhouse experiments. We used two Cercomonas cultures (Cercomonas lenta strain ECO-P-01 and Cercomonas sp. strain S24D2) as these were indicative for high plant growth. Furthermore, we used another protist, Allovahlkampfia sp. strain NL10 as a control to test how protists not linked to high yield affect plant growth. Secondly, microbe-consuming protists were tested for their stimulatory effect on the plant growth promotion capacity of Trichoderma in the third greenhouse experiment. Experimental soil was collected from the control treatment in the above-mentioned long-term field experiment site. Soils were mixed and passed through a 2-mm sieve. One portion of the soil was stored at room temperature, and the other sterilized by gamma irradiation (60 KGy). Cucumber seeds were sterilized by immersion in sodium hypochlorite (3%) for 30 seconds and ethanol (75%) for 2 minutes, followed by 20-fold immersion in sterile water. Pot experiments were constructed using polypropylene pots filled with 100 g dry soil and hydrated with sterile water and cultivated in a Plant Illuminated Incubator (daytime: 12 hours and 28 °C, night: 12 hours and 25 °C, all-day average humidity of 50 %) with periodic randomization throughout the experiment.
-
1)
Greenhouse experiments 1 and 2: Effects of microbe-consuming protists on cucumber growth
Seven inoculation treatments were designed in the first greenhouse experiment as follows: (1) Cercomonas lenta: Cercomonas lenta strain ECO-P-01 in 5 ml PAS buffer (Page’s Amoeba Saline: 120 mg NaCl, 4 mg MgSO4·7H2O, 4 mg CaCl2·2H2O, 142 mg Na2HPO4, and 136 mg KH2PO4 in 1 liter of distilled water) [64] was inoculated into natural soil, (2) Cercomonas S24D2: Cercomonas sp. strain S24D2 with 5 ml PAS buffer was inoculated into natural soil, (3) Allovahlkampfia sp.: Allovahlkampfia sp. strain NL10 in 5 ml PAS buffer was inoculated into natural soil, (4) S_ Cercomonas lenta: Cercomonas lenta strain ECO-P-01 in 5 ml PAS buffer was inoculated into sterilized natural soil, (5) S_ Cercomonas S24D2: Cercomonas sp. strain S24D2 in 5 ml PAS buffer was inoculated into sterilized natural soil, (6) S_ Allovahlkampfia sp.: Allovahlkampfia sp. strain NL10 in 5 ml PAS buffer was inoculated into sterilized natural soil, and (7) Control, 5 ml PAS buffer was added into natural soil. A follow-up experiment was conducted in the same way as described above, but focusing on Cercomonas lenta and Cercomonas S24D2 and a non-protist Control to validate the effects observed in the first experiment. Protists were inoculated by adding 1.0 × 104 protistan cells g−1 dry soil in 5 ml PAS buffer in sterilized and natural soils and added sterile water to 40% soil moisture. The three protist strains used for experimentation, Cercomonas sp. strain S24D2, Cercomonas lenta ECO-P-01, and Allovahlkampfia sp. strain NL10, were isolated and identified by [65].
-
2)
Greenhouse experiment 3: Testing interactions between protists and plant beneficial Trichoderma on the resident microbiome and plant growth
Trichoderma sp. isolates were randomly selected after recovery from soil collected from the BF treatment in the field experiment. In brief, 10 g of soil was suspended in a 250-ml Erlenmeyer flask containing 90 ml of sterile distilled water. After stirring at 180 rpm for 40 min, serial dilutions were spread onto plates containing Martin’s semi-selective medium (per liter: 18 g agar, 10 g dextrose, 0.5 g MgSO4, 0.5 g peptone, 0.5 g beef extract, 0.05 g bengal pink and 0.3 g chloramphenicol), and plates were incubated at 28 ± 1°C for 7 days. Colonies with typical Trichoderma morphology were transferred to potato-dextrose agar (PDA), incubate at 28 ± 1 °C, and identified based on the ITS region sequence analysis as described previously [66]. Six treatments in sterilized and non-sterilized soils were implemented with controls consisting of 5 ml PAS buffer added to sterilized (S_Control) and alive (Control) soils. Trichoderma treatments were created by adding 1.0 × 104 Trichoderma spores g−1 dry soil in 5 ml PAS buffer in sterilized (S_T) and non-sterilized (T) soils. Trichoderma+Cercomonas lenta treatments consisted of 1.0×104 Trichoderma spores g−1 dry soil and 1.0 × 104 Cercomonas lenta strain ECO-P-01 cells g−1 dry soil in 5 ml of PAS buffer in sterilized (S_T+C) and non-sterilized (T+C) soils. All solutions were evenly inoculated into soils, and sterile water to 40% soil moisture was added.
Plant samples were collected after two weeks for experiments 1 and 3 and after 1 week for experiment 2. Shoots were oven-dried at 65 °C for 5 days before measuring dry biomass.
Statistical analyses
We estimated bacterial, fungal, and protistan α-diversity using non-parametric Shannon indexes. For β-diversity, principal coordinate analysis (PCoA) based on the unweighted UniFrac distance was used to explore differences of bacterial, fungal, and protistan community structures across all soil samples. Linear mixed models (LMM) were performed to assess the effects of fertilization and crop season on the diversity of the soil microbiome, with the plot position serial number (i _ j, are the row and column number of the plot, respectively) as a random effect in “lme4” and “lmerTest” packages [67] in R (version 3.4.3). The permutational multivariate analysis of variance (PERMANOVA) [68] was performed to assess the effects of fertilization and crop season on soil microbiome community structures through the adonis function with 999 permutations in the “vegan” package in R (version 3.4.3). The diversity (Shannon index) and community structure (PCoA1) of bacteria, fungi, and protists were selected as the six main microbial predictors, and the significance of effects of microbial predictors on crop yield was calculated using multiple regression by linear models in R (version 3.4.4). The analysis of the relative importance of the microbial predictors was run using the “relaimpo” package [69] in R (version 3.4.3). Analysis of similarity (ANOSIM) was performed to evaluate significant differences in microbial community structures across the four fertilizer treatments through the “anosim” command in Mothur [70]. As the community composition of protists was strongly affected by fertilization and was the only microbial group significantly linked with crop yield, we focused subsequent analyses on the protistan community. For that, One-way ANOVA with Tukey’s HSD test was performed to determine significant differences between treatments, and regression models were fitted in SPSS v20.0 (SPSS Inc. USA). Redundancy analysis (RDA) was performed to examine the relationships between abiotic factors (environmental variables) and the functional composition of protistan communities in the “vegan” package in R (version 3.4.3). Observed differences were tested for significance using the “envfit” function with 999 permutations. Further analyses focused on microbe-consuming protists as the only functional group showing significant correlations with crop yield. We used Spearman’s correlation coefficient to evaluate the correlation between abundant microbe-consuming protistan OTUs (top 50) and crop yield, abundant bacterial OTUs (average relative abundance > 0.5%), and abundant fungal OTUs (average relative abundance > 0.5%), respectively. Heat map analysis of the microbe-consuming protistan OTUs linked to crop yield across all treatments was carried out with the “pheatmap” package in R (version 3.4.3). All Spearman’s correlation coefficients were calculated with the “corr.test” function in the “psych” package in R (version 3.4.3). The P values were adjusted using the false discovery rate method [71]. Normal distribution was tested by the Shapiro-Wilk test, and non-normal data was log transformed [72].
Structural equation models were constructed to quantify links between crop yield and abundances of microbe-consuming protists, bacteria, and fungi. All variables were standardized by Z transformation (mean = 0, standard deviation = 1) to improve normality using the scale function in R [32]. The SEM construction and analysis were run using AMOS 17.0 (SPSS, Chicago, IL, USA). The covariance matrix was fit into the model using maximum likelihood estimation. The following metrices were used to ensure model fitting to the data: Chi-square (a model fits a given dataset well when χ2 is low), goodness-of-fit-index (GFI > 0.90), and root mean square error of approximation (RMSEA < 0.05) [73]. The fold change of plant biomass in different microbe-consuming protists inoculation relative to the control in pot experiment was calculated using the following formula: (X − Control)/Control, in which X is the plant biomass in different treatments with inoculation of different microbe-consuming protists, and Control represents the plant biomass in treatment without inoculation of microbe-consuming protists.
Availability of data and materials
All raw sequence data have been made available in the NCBI Sequence Read Archive (SRA) database under the BioProject PRJNA599073.
References
Lal R. Soil carbon sequestration impacts on global climate change and food security. Science. 2004;304:1623–7.
Li X, Lewis EE, Liu Q, Li H, Bai C, Wang Y. Effects of long-term continuous cropping on soil nematode community and soil condition associated with replant problem in strawberry habitat. Sci Rep. 2016;6:30466.
Kibblewhite MG, Ritz K, Swift MJ. Soil health in agricultural systems. Phil Trans R Soc B. 2008;363:685–701.
Lal R. Managing world soils for food security and environmental quality. In: Sparks DL, editor. Advances in agronomy. San Diego: Academic; 2001. p. 155–92.
Blum WEH. Soil and land resources for agricultural production: general trends and future scenarios-a worldwide perspective. Int Soil Water Conserv Res. 2013;1:1–14.
Borrelli P, Robinson DA, Fleischer LR, Lugato E, Ballabio C, Alewell C, et al. An assessment of the global impact of 21st century land use change on soil erosion. Nat Commun. 2017;8:2013.
Tilman D, Fargione J, Wolff B, D’Antonio C, Dobson A, Howarth R, et al. Forecasting agriculturally driven global environmental change. Science. 2001;292:281–4.
Zhou X, Wu F. Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol Ecol. 2012;80:469–78.
Diaz-Ambrona CGH, Maletta E. Achieving global food security through sustainable development of agriculture and food systems with regard to nutrients, soil, land, and waste management. Curr Sustain Renewable Energy Rep. 2014;1:57–65.
Dong L, Xu J, Feng G, Li X, Chen S. Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system. Sci Rep. 2016;6:31802.
Acosta-Martínez V, Burow G, Zobeck TM, Allen VG. Soil microbial communities and function in alternative systems to continuous cotton. Soil Sci Soc Am J. 2010;74:1181–92.
Balota EL, Colozzi-Filho A, Andrade DS, Dick RP. Microbial biomass in soils under different tillage and crop rotation systems. Biol Fertil Soils. 2003;38:15–20.
Garbeva P, van Veen JA, van Elsas JD. Microbial diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu Rev Phytopathol. 2004;42:243–70.
Liu X, Zhang J, Gu T, Zhang W, Shen Q, Yin S, et al. Microbial community diversities and taxa abundances in soils along a seven-year gradient of potato monoculture using high throughput pyrosequencing approach. PLoS One. 2014;9:e86610.
Yang J, Ruegger PM, McKenry MV, Becker JO, Borneman J. Correlations between root-associated microorganisms and peach replant disease symptoms in a California soil. PLoS One. 2012;7:e46420.
Bonner J, Galston AW. Toxic substances from the culture media of guayule which may inhibit growth. Bot Gaz. 1944;106:185–98.
Bhardwaj D, Ansari MW, Sahoo RK, Tuteja N. Biofertilizers function as key player in sustainable agriculture by improving soil fertility, plant tolerance and crop productivity. Microb Cell Fact. 2014;13:66.
Liu H, Xiong W, Zhang R, Hang X, Wang D, Li R, et al. Continuous application of different organic additives can suppress tomato disease by inducing the healthy rhizospheric microbiota through alterations to the bulk soil microflora. Plant Soil. 2018;423:229–40.
Maeder P, Fliessbach A, Dubois D, Gunst L, Fried P, Niggli U. Soil fertility and biodiversity in organic farming. Science. 2002;296:1694–7.
Reeves DW. The role of soil organic matter in maintaining soil quality in continuous cropping systems. Soil Tillage Res. 1997;43:131–67.
Bonanomi G, De Filippis F, Cesarano G, La Storia A, Ercolini D, Scala F. Organic farming induces changes in soil microbiota that affect agro-ecosystem functions. Soil Biol Biochem. 2016;103:327–36.
Beneduzi A, Ambrosini A, Passaglia LMP. Plant growth-promoting rhizobacteria (PGPR): their potential as antagonists and biocontrol agents. Genet Mol Biol. 2012;35:1044–51.
Berendsen RL, Pieterse CMJ, Bakker PAHM. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012;17:478–86.
van der Heijden MGA, Bardgett RD, van Straalen NM. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett. 2008;11:296–310.
Thakur MP, Geisen S. Trophic regulations of the soil microbiome. Trends Microbiol. 2019;27:771–80.
Gao Z, Karlsson I, Geisen S, Kowalchuk G, Jousset A. Protists: puppet masters of the rhizosphere microbiome. Trends Plant Sci. 2019;24:165–76.
Geisen S, Mitchell EAD, Adl S, Bonkowski M, Dunthorn M, Ekelund F, et al. Soil protists: a fertile frontier in soil biology research. FEMS Microbiol Rev. 2018;42:293–323.
Dupont AÖC, Griffiths RI, Bell T, Bass D. Differences in soil micro-eukaryotic communities over soil pH gradients are strongly driven by parasites and saprotrophs. Environ Microbiol. 2016;18:2010–24.
Geisen S, Bandow C, Römbke J, Bonkowski M. Erratum to “Soil water availability strongly alters the community composition of soil protists” [Pedobiologia – J. Soil Ecol. 57 (4–6) (2014) 205–213]. Pedobiologia. 2015;58:55.
Foissner W. Protozoa as bioindicators in agroecosystems, with emphasis on farming practices, biocides, and biodiversity. Agric Ecosyst Environ. 1997;62:93–103.
Lentendu G, Wubet T, Chatzinotas A, Wilhelm C, Buscot F, Schlegel M. Effects of long-term differential fertilization on eukaryotic microbial communities in an arable soil: a multiple barcoding approach. Mol Ecol. 2014;23:3341–55.
Zhao Z, He J, Geisen S, Han L, Wang J, Shen J, et al. Protist communities are more sensitive to nitrogen fertilization than other microorganisms in diverse agricultural soils. Microbiome. 2019;7:33.
Xiong W, Song Y, Yang K, Gu Y, Wei Z, Kowalchuk GA, et al. Rhizosphere protists are key determinants of plant health. Microbiome. 2020;8:27.
Pedersen AL, Winding A, Altenburger A, Ekelund F. Protozoan growth rates on secondary-metabolite-producing Pseudomonas spp. correlate with high-level protozoan taxonomy. FEMS Microbiol Lett. 2011;316:16–22.
Schulz-Bohm K, Geisen S, Wubs ERJ, Song C, de Boer W, Garbeva P. The prey’s scent – volatile organic compound mediated interactions between soil bacteria and their protist predators. ISME J. 2017;11:817–20.
Oliverio AM, Geisen S, Delgado-Baquerizo M, Maestre FT, Turner BL, Fierer N. The global-scale distributions of soil protists and their contributions to belowground systems. Sci Adv. 2020;6:eaax8787.
Xiong W, Li R, Guo S, Karlsson I, Jiao Z, Xun W, et al. Microbial amendments alter protist communities within the soil microbiome. Soil Biol Biochem. 2019;135:379–82.
Wilschut RA, van der Putten WH, Garbeva P, Harkes P, Konings W, Kulkarni P, et al. Root traits and belowground herbivores relate to plant–soil feedback variation among congeners. Nat Commun. 2019;10:1564.
Bonkowski M. Protozoa and plant growth: the microbial loop in soil revisited. New Phytol. 2004;162:617–31.
Geisen S, Koller R, Hünninghaus M, Dumack K, Urich T, Bonkowski M. The soil food web revisited: diverse and widespread mycophagous soil protists. Soil Biol Biochem. 2016;94:10–8.
Jousset A. Ecological and evolutive implications of bacterial defences against predators. Environ Microbiol. 2012;14:1830–43.
Jousset A. Application of protists to improve plant growth in sustainable agriculture. In: Mehnaz S, editor. Rhizotrophs: plant growth promotion to bioremediation. Singapore: Springer; 2017. p. 263–73.
Bonkowski M, Clarholm M. Stimulation of plant growth through interactions of bacteria and protozoa: testing the auxiliary microbial loop hypothesis. Acta Protozool. 2012;51:237–47.
Dumack K, Fiore-Donno AM, Bass D, Bonkowski M. Making sense of environmental sequencing data: ecologically important functional traits of the protistan groups Cercozoa and Endomyxa (Rhizaria). Mol Ecol Resour. 2020;20:398–403.
Stewart A, Hill R. Applications of Trichoderma in plant growth promotion. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy MG, editors. Biotechnology and biology of Trichoderma. Amsterdam: Elsevier; 2014. p. 415–28.
Thakur MP, van der Putten WH, Cobben MMP, van Kleunen M, Geisen S. Microbial invasions in terrestrial ecosystems. Nat Rev Microbiol. 2019;17:621–31.
Mallon CA, van Elsas JD, Salles JF. Microbial invasions: the process, patterns, and mechanisms. Trends Microbiol. 2015;23:719–29.
Zhang Y, Wang X, Pang G, Cai F, Zhang J, Shen Z, et al. Two-step genomic sequence comparison strategy to design Trichoderma strain-specific primers for quantitative PCR. AMB Express. 2019;9:179.
Yang X, Chen L, Yong X, Shen Q. Formulations can affect rhizosphere colonization and biocontrol efficiency of Trichoderma harzianum SQR-T037 against Fusarium wilt of cucumbers. Biol Fertil Soils. 2011;47:239–48.
Zheng H, Ouyang Z, Wang X, Fang Z, Zhao T, Miao H. Effects of regenerating forest cover on soil microbial communities: a case study in hilly red soil region, Southern China. For Ecol Manage. 2005;217:244–54.
Kaushik P, Garg VK. Vermicomposting of mixed solid textile mill sludge and cow dung with the epigeic earthworm Eisenia foetida. Bioresour Technol. 2003;90:311–6.
Quan M, Liang J. The influences of four types of soil on the growth, physiological and biochemical characteristics of Lycoris aurea (L’ Her.) Herb. Sci Rep. 2017;7:43284.
Shen Z, Zhong S, Wang Y, Wang B, Mei X, Li R, et al. Induced soil microbial suppression of banana fusarium wilt disease using compost and biofertilizers to improve yield and quality. Eur J Soil Biol. 2013;57:1–8.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS. 2011;108:4516–22.
Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol. 1993;2:113–8.
White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR protocols. San Diego: Academic; 1990. p. 315–22.
Bass D, Silberman JD, Brown MW, Pearce RA, Tice AK, Jousset A, et al. Coprophilic amoebae and flagellates, including Guttulinopsis, Rosculus and Helkesimastix, characterise a divergent and diverse rhizarian radiation and contribute to a large diversity of faecal-associated protists. Environ Microbiol. 2016;18:1604–19.
Shen Z, Xue C, Penton CR, Thomashow LS, Zhang N, Wang B, et al. Suppression of banana Panama disease induced by soil microbiome reconstruction through an integrated agricultural strategy. Soil Biol Biochem. 2019;128:164–74.
Xiong W, Jousset A, Guo S, Karlsson I, Zhao Q, Wu H, et al. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2018;12:634–8.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013;41:D597–604.
Adl SM, Simpson AGB, Lane CE, Lukesˇ J, Bass D, Bowser SS, et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012;59:429–93.
Thomas V, Herrera-Rimann K, Blanc DS, Greub G. Biodiversity of amoebae and amoeba-resisting bacteria in a hospital water network. Appl Environ Microbiol. 2006;72:2428–38.
Gao Z. Soil protists: from traits to ecological functions. Utrecht: Utrecht University; 2020.
Kullnig-Gradinger CM, Szakacs G, Kubicek CP. Phylogeny and evolution of the genus Trichoderma: a multigene approach. Mycol Res. 2002;106:757–67.
Wagner MR, Lundberg DS, del Rio TG, Tringe SG, Dangl JL, Mitchell-Olds T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat Commun. 2016;7:12151.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
Grömping U. Relative importance for linear regression in R: the package relaimpo. J Stat Softw. 2006;17(1):1–27.
Clarke KR. Non-parametric multivariate analyses of changes in community structure. Austral Ecol. 1993;18:117–43.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
Mcdonald JH. Handbook of biological statistics. Baltimore: Sparky House Publishing; 2009. p. 317.
Grace JB, Keeley JE. A structural equation model analysis of postfire plant diversity in California shrublands. Ecol Appl. 2006;16:503–14.
Funding
This study was funded by the National key research and development program (2017YFD0200805), the Fundamental Research Funds for the Central Universities (KYXK202009 and KYZ201871), the Innovative Research Team Development Plan of the Ministry of Education of China (IRT_17R56), the 111 Project of China (B12009), and the Priority Academic Program Development of the Jiangsu Higher Education Institutions (PAPD). Stefan Geisen was supported by an NWO-VENI grant from the Netherlands Organisation for Scientific Research (016.Veni.181.078).
Author information
Authors and Affiliations
Contributions
Sai Guo, Rong Li, Wu Xiong, Qirong Shen, and Stefan Geisen wrote the manuscript. Sai Guo, Rong Li, Hongjun Liu, Zhilei Gao, Nan Zhang, George A. Kowalchuk, Stefan Geisen, and Qirong Shen developed the ideas and designed the experimental plans. Sai Guo, Rong Li, Xinnan Hang, Zixuan Jiao, and Yani Mo performed the experiments. Sai Guo, Rong Li, Wu Xiong, and Stefan Geisen analyzed the data. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Effects of different fertilization managements on cucumber yield across six cropping seasons (a). Protistan community based on unweighted unifrac distance of different fertilization managements across six cropping seasons (b). Effects of different fertilization managements (c) and crop seasons (d) on protistan diversity. Effects of different fertilization managements (e) and crop seasons (f) on microbe-consuming protists. Fig. S2. Redundancy analysis of the relationship between environmental variables and protistan functional groups (a). The relative importance of soil physicochemical properties for protistan diversity (b) and community structure (c). Heatmap illustrating the relationship between microbe-consuming protistan OTUs that are positively associated with crop yield and bacterial OTUs or fungal OTUs (relative abundance > 0.5%) in all treatments (d). Fig. S3. Fold change increase of cucumber biomass relative to the control in treatments with inoculation of two cercozoan species in the confirmatory second greenhouse experiment. Table S1. Fertilization scheme for chemical fertilizer (CF), organic fertilizer (OF) and bio-organic fertilizer (BF) in this study. Table S2. The effects of fertilization and crop season on distinct microbial groups based on PERMANOVA analysis. Table S3. The effects of fertilization and crop season on distinct microbial groups based on linear mixed model (LMM). Table S4. Spearman’ s rank correlation coefficient between cucumber yield and functional groups. Table S5. Soil physicochemical properties of different treatments in continuous cropping. Table S6. Detailed information of indicator protistan taxa (OTUs) based on PR2 database and GenBank.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, S., Xiong, W., Hang, X. et al. Protists as main indicators and determinants of plant performance. Microbiome 9, 64 (2021). https://doi.org/10.1186/s40168-021-01025-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-021-01025-w