
Abstract
Background
The capacity of reef-building corals to tolerate (or adapt to) heat stress is a key factor determining their resilience to future climate change. Changes in coral microbiome composition (particularly for microalgal endosymbionts and bacteria) is a potential mechanism that may assist corals to thrive in warm waters. The northern Red Sea experiences extreme temperatures anomalies, yet corals in this area rarely bleach suggesting possible refugia to climate change. However, the coral microbiome composition, and how it relates to the capacity to thrive in warm waters in this region, is entirely unknown.
Results
We investigated microbiomes for six coral species (Porites nodifera, Favia favus, Pocillopora damicornis, Seriatopora hystrix, Xenia umbellata, and Sarcophyton trocheliophorum) from five sites in the northern Red Sea spanning 4° of latitude and summer mean temperature ranges from 26.6 °C to 29.3 °C. A total of 19 distinct dinoflagellate endosymbionts were identified as belonging to three genera in the family Symbiodiniaceae (Symbiodinium, Cladocopium, and Durusdinium). Of these, 86% belonged to the genus Cladocopium, with notably five novel types (19%). The endosymbiont community showed a high degree of host-specificity despite the latitudinal gradient. In contrast, the diversity and composition of bacterial communities of the surface mucus layer (SML)—a compartment particularly sensitive to environmental change—varied significantly between sites, however for any given coral was species-specific.
Conclusion
The conserved endosymbiotic community suggests high physiological plasticity to support holobiont productivity across the different latitudinal regimes. Further, the presence of five novel algal endosymbionts suggests selection of certain genotypes (or genetic adaptation) within the semi-isolated Red Sea. In contrast, the dynamic composition of bacteria associated with the SML across sites may contribute to holobiont function and broaden the ecological niche. In doing so, SML bacterial communities may aid holobiont local acclimatization (or adaptation) by readily responding to changes in the host environment. Our study provides novel insight about the selective and endemic nature of coral microbiomes along the northern Red Sea refugia.
Similar content being viewed by others
Introduction
Coral reefs have dramatically declined during the last two decades through the mortality of reef-building species driven by frequent and intense heatwaves [1, 2]. Efforts to predict if and how corals will survive into the future has resulted in intensive research to understand coral thermal tolerance across environments [3] and through time [4]. Corals can persist in relatively extreme habitats such as shallow pools [5], reef flats [6], and mangroves [7, 8], or marginally “hot” reef systems such as those within parts of the Persian-Arabian Gulf [9] and the Red Sea [4]. Therefore, coral populations that already exist at high ambient water temperatures have become important model systems to evaluate the different mechanisms with which thermal tolerance may be acquired [10,11,12].
Coral thermal tolerance is ultimately determined by the genetic composition of the holobiont (i.e., the coral host and its associated microbiome: endosymbiotic dinoflagellates, bacteria, virus, fungi, archaea, and endolithic algae—sensu Rohwer et al. [13]). Some corals exhibit a broad capacity to adapt to different thermal histories by frontloading genes that promote heat stress tolerance [3, 14], and/or potentially through shifting their microbial community [15,16,17]. Endosymbiont genotypes or species (family Symbiodiniaceae [18]) associated with corals play an important role in the adaptation of corals living under extreme environments [19, 20]. Endosymbiont response to environmental fluctuations varies greatly between (and within) species/genotypes [21], and the persistence of certain genotypes can influence coral stress tolerance [22]. A new species, Cladocopium thermophilum, resides in extreme warm waters (> 35 °C) of the Persian-Arabian Gulf [19, 20]. Thus, knowledge of the dinoflagellate endosymbiont genetic “identity” is often critical for reconciling ecological patterns of coral species tolerance to environmental stressors.
Bacterial communities associated with the coral host also promote coral acclimatization/adaptation to changing environmental conditions, including transient stress exposure [16, 23, 24]. Bacteria likely play key functional roles in sustaining nutrient cycling [25] or supporting immunity [23], for example, especially in corals that might otherwise be health compromised. Several studies have reported distinct bacterial taxa associated with corals in extreme habitats such as deepwater [26], volcanic vents [27], and warmer back reef pools [16], suggesting the potential for bacteria to play role in enhancing holobiont environmental plasticity. Transplantation experiments have further demonstrated that bacterial communities shift when corals are introduced to new and non-native habitats, suggesting microbiome alteration as an acclimatization strategy to improve holobiont physiology in response to changing environmental conditions such as salinity, nutrients, and water temperature [16, 17, 28].
Indeed, bacterial communities associated with the coral surface mucus layer (SML) are particularly distinct compared to those associated with the tissue and skeleton [29]. The importance of the SML stems from its protective, nutritional, cleansing roles [30, 31], but notably, it acts as a physical barrier against invasion of potential pathogens [32], therefore forming the first line of defense [30]. Removal of the SML (using antibiotics) caused dramatic necrosis and bleaching with symptoms reflecting the invasion by opportunistic and pathogenic bacteria [33]. Further, the microbiome of coral tissue and skeleton are more influenced by intrinsic factors, unlike the SML bacteria that may be more influenced by environmental variables [34]. As such, changes to the SML bacterial community are more closely tied to environmental variance compared to other coral compartments [17, 23, 32. 35].
The Red Sea represents a unique natural laboratory as it covers 15° latitude and coral conspecifics throughout the Red Sea experience a large environmental gradient, particularly temperature ranging from 23.6 ± 0.6 °C in the north to 29 ± 0.4 °C in the south (mean annual ± SD, see [4]). The susceptibility of these conspecifics to thermal anomalies (i.e., temperatures above the long-term summer mean) is highly variable across latitudinal gradients. For instance, corals in the northern Red Sea experience high thermal anomalies of up to 15 Degree Heating Weeks (DHW) without visible bleaching, in comparison to their central and southern counterparts [4]. This is particularly striking when compared to global patterns of coral temperature vulnerability with mass bleaching most often occurring already after 4 DHW and widespread mortality after 8 DHW [36]. Consequently, the northern Red Sea may represent a refuge where corals exist well below their thermal maxima and are thus likely to be among the last to bleach [4, 37]. Notably, investigation of genetic variability of coral hosts (Stylophora pistillata and Pocillopora verrucosa) highlighted low genetic difference and weak isolation between populations across the Red Sea, but strong gene flow [38, 39]. Therefore, the association of corals with different microbiome composition may, at least in part, explain holobiont acclimatization to thermal tolerance within the northern Red Sea.
Here, we examined endosymbiont and SML bacterial communities associated with six coral species collected from two environmental settings: (i) across depths to represent different light regimes and (ii) across 4° of latitude (~ 500 km) detailing sites that varied in mean summer temperatures within the northern Red Sea. We characterized the endosymbiont and bacterial composition using high-throughput metabarcoding to determine how, and therefore if, microbial communities are associated with coral acclimatization under different environmental regimes [4]. We show that while the endosymbiotic dinoflagellate communities for a given host are maintained throughout the investigated region, bacterial diversity and composition were site-specific and varied significantly along the latitudinal gradient. These findings suggest that bacterial communities could aid in holobiont acclimatization or adaptation, while the conserved dinoflagellate community may be able to support productivity throughout the northern Red Sea.
Results
Symbiodiniaceae community structure
Samples were collected from six coral species (two species each of branching and massive scleractinian coral and two species of soft coral) to represent taxonomic and functional diversity, at two depths along five sites (n = 163) with different thermal regimes in the northern Red Sea (Fig. 1, see [4]). DGGE fingerprinting identified a total of 19 endosymbiotic ITS2 types belonging to three genera: Symbiodinium, Cladocopium, and Durusdinium (from previously described clades A, C, and D, respectively [18]). Endosymbionts from Cladocopium were most prevalent (85% of all samples, n = 139) and comprised 14 ITS2 types: C1, C1 variant, C15, C15r, C15q, C170, C170a, C171, C1h, C1h*, C3z*, C41, C65 variant1, and C65 variant2 (Fig. 1). The remaining symbionts were from the genus Symbiodinium (14%, n = 22) and comprised three ITS2 types (A1, A1c, and an A1 variant), and finally Durusdinium trenchii D1a (1%, n = 2, Fig. 1). Of the 19 symbionts, at least five novel types (i.e., not described previously) were recorded (19%, n = 31), namely C15r, C15q in Porites nodifera, C3z* in Favia favus, and C1h* and C171in Xenia umbellata (Additional file 1). Further, four endosymbiont types remained unidentified due to sequencing difficulties (A1 variant, C1 variant, C65 variant1, and C65 variant2).

Endosymbiont distribution for six coral species collected from two depths (2–5 m and 15–18 m) along five different sites at the northern Red Sea (total n = 163). The map shows the long-term mean of sea surface temperature along the Red Sea and the thermal gradient in the northern Red Sea, including sampling sites. Data obtained from Giovanni Ocean color (https://giovanni.gsfc.nasa.gov/giovanni/, MODIS Aqua 4 km satellite, 4 μm night only) for the period between July 2002 and August 2018. The tile plot represents endosymbiont ITS2 types associated with each coral host, depth, and site separately where site represents a latitudinal gradient (sites on y-axis are arranged from the North (top) to South (bottom)). Three distinct patterns are apparent: (i) high degree of host-symbiont specificity, (ii) absence of depth-specific patterns, except for P. damicornis and F. favus, which changed the ratio of dominant clades with depth, and (iii) symbiont community within each host did not change across the latitudinal gradient, except in S.hystrix. White tiles represent missing samples; representative image of coral hosts above tile plot column for each respective species
Overall, the endosymbiont community structure did not vary significantly between the depths (PERMANOVA, F = 1.9, R2 = 0.011, p = 0.059). Nevertheless, few endosymbiont types were observed only in either samples from the shallow (C15r, C15q, C65 variant2, A1c, and D1a) or deep (C170a, C1, C1h*, and A1variant) (Additional file 2: Figure S1). Despite the latitudinal differences and increase of ambient water temperature toward the south by almost 3 °C, the endosymbiont community structure within each coral host generally did not change between sites (PERMANOVA, F = 0.6, R2 = 0.016, p = 0.92). Endosymbiont type shifted only in Seriatopora hystrix from the dominance of C170 (70%) in the northern sites to A1 (20%) and C170a (10%) types in the southernmost (warmer) sites (i.e., Meritte and Wadi El Gemal—Fig. 1).
The endosymbiont types were strongly linked to coral species identity, indicating a high level of host-specificity (PERMANOVA, F = 48.4, R2 = 0.60, p < 0.001). Each coral species associated predominantly with either single or multiple distinct endosymbiont type(s) that were rarely shared with other coral species (Fig. 1). The only endosymbiont present across multiple host species was A1, which was recorded in Pocillopora damicornis and S. hystrix (Fig. 1). Notably, there was no more than one endosymbiont type detected in any of the sampled colonies along the gradient.
Porites nodifera associated predominantly with C15, but few colonies contained the novel types C15r (n = 1) and C15q (n = 2). F. favus harbored four types, predominantly an unidentified C1 variant (n = 12), the novel C3z* (n = 7), C41 (n = 2), and C1 (n = 1). P. damicornis harbored four types, A1 (n = 11) and A1c (n = 2) in the shallows and mainly C1h (n = 12) in the deep—this C1h type is found commonly in pocilloporids across the Indian Ocean [36, 37]—and a single colony with an unidentified variant of Symbiodinium A1 (Fig. 1). S. hystrix associated with symbionts A1 (n = 6), C170 (n = 21) and C170a (n = 3). The soft coral X. umbellata harbored the novel C171 (n = 20), two colonies with D1a and a single colony had the novel C1h* type. Finally, Sarcophyton trocheliophorum contained two unidentified types closely related to C65 (C65-variant1, n = 24 and C65-variant2, n = 1), which is a common symbiont found in soft corals on the Great Barrier Reef and Indian Ocean [38, 39]. Thus, coral host identity was the main factor determining endosymbiont variability throughout the latitudinal gradient in the northern Red Sea.
Bacterial community structure
Bacterial 16S rRNA gene amplicon sequencing from the SML of six coral species and surrounding seawater samples at two depths (n = 164) yielded 21.3 million sequences, ranging from 38,048 to 1.3 million sequences per sample (median = 117,188 reads; see Additional file 3). Sequence length ranged from 350 bp to 548 bp (median = 427 bp). A total of 6970 OTUs were recorded across all samples, ranging from 159 to 2556 OTUs per sample (median = 656 OTUs—see Additional file 2: Figure S2). OTUs belonged to 40 bacterial phyla, whereby Proteobacteria was the predominant phylum representing 53% of total abundance across all samples (i.e., corals and seawater), followed by Bacteroidetes (16%) and unclassified bacteria (10%). Out of 6970 OTUs, only 14 most dominant OTUs comprised 60.9% of the total bacterial community abundance. The remaining OTUs (n = 6956) were rare (i.e., each contributed < 1% of total abundance), but shaped the remaining microbial community structure (39.1%) without defined dominant taxa (Fig. 2). The most abundant bacterial phylotypes were a single Alteromonas sp. (27.4%) and three Pseudoalteromonas OTUs (16.2%), which together comprised 43.6% of the total bacterial abundance of coral SML and seawater (Fig. 2). Three different Vibrio OTUs cumulatively comprised 6.6% of all sequences, while Endozoicomonas and the photosynthetic Erythrobacter were in low abundance (1.2% each). Soft corals appeared to have similar bacterial composition compared to reef-building corals, with Alteromonas and Pseudoalteromonas comprising the main OTUs; however, X. umbellata had relatively high proportions of Vibrio and Endozoicomonas sp. OTUs (Fig. 2). Similarly, water samples were also dominated by Alteromonas sp. (22.1%), but were markedly comprised of different bacterial phylotypes, such as Roseovarius sp. (4.6%), Rhodobacteraceae (3.8%), and Pelagibacter sp. (2.6%) (Table 1).

Taxonomic profile (genus level) of the abundant bacterial community associated with the surface mucus layer of six coral species and surrounding seawater samples (left) collected from five surveyed sites (right) in the northern Red Sea. Alteromonas and Pseudoalteromonas were the most dominant OTUs and composed combined 43.6% of the total community in both sites and coral species, bacterial community was significantly different between sites and coral hosts. Water samples had markedly distinct bacterial assemblage: over 60% of the bacteria had less than 1% of relative abundance. Unclassified taxa to genus level were denoted by (UC)
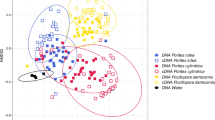
Seawater samples had a distinct bacterial diversity that was significantly richer (i.e., Chao1–F1,162 = 41.4, p < 0.001) and more diverse (i.e., inverse Simpson–F1,162 = 10.7, p < 0.01 and Shannon index–F1,162 = 18.7, p < 0.001) compared to that of coral SML. Seawater bacterial diversity did not significantly vary with either depth or site (all subsequent ANOVA p > 0.05—see Additional file 2: Table S1, Additional file 2: Figure S3). Similarly, the coral SML bacterial richness and diversity did not vary with depth, but in contrast to seawater, differed significantly between sites and coral species (Additional file 2: Table S1). Analysis of bacterial community composition further confirmed this pattern that seawater bacterial communities were significantly different from the coral SML (pairwise PERMANOVA, F = 7.2, R2 = 0.04, p < 0.001—see Fig. 3). Therefore, seawater samples were removed from subsequent analysis. Similar to bacterial diversity, coral SML-associated bacterial community composition did not vary between depths (PERMANOVA, F = 1.4, R2 = 0.01, p = 0.14), but by coral host species (PERMANOVA, F = 5.3, R2 = 0.168, p < 0.01) and site (PERMANOVA, F = 8.4, R2 = 0.174, p < 0.01). PERMANOVA was also performed on each coral species across all sites separately as well as all coral species within each site, confirming that sites and coral host species contribute to the variation in the bacterial community, but not depth (see Additional file 2: Table S2). Principle coordinate analysis (PCoA) confirmed this pattern (after removal of the two most abundant OTUs, only for this visualization but not excluded from statistical analysis, as they obscured the geographic patterns—see also Additional file 2: Figure S4 for PCoA without removal of those OTUs) and bacterial communities were clustered geographically based on site, regardless of depth and coral species (Fig. 3). However, within each site, bacterial communities were distinct between coral species (Additional file 2: Figure S5 and Additional file 2: Table S2). Thus, bacterial community structure varied with host taxa, similar to dinoflagellate endosymbionts association, but it also differed across the latitudinal gradient.

Principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity matrix of bacterial communities associated with six coral species and five sites along the latitudinal gradient in the northern Red Sea. PCoA shows clustering pattern between coral species versus seawater (a) and between different sites (b). Two most abundant OTUs (Alteromonas sp. and Pseudoalteromonas sp.) mask geographic patterns and were therefore excluded for this visualization. Compositional differences in bacterial communities were best explained by site
Bacterial indicator species
We further performed an indicator species analysis to identify bacterial taxa (OTUs) that are representative of specific sites and coral hosts (cutoff level p < 0.05). Due to the similarity between bacterial assemblages at the two depths (PERMANOVA, F = 1.4, R2 = 0.01, p = 0.14), data were pooled across depths. We found a number of OTUs, ranging from 36 OTUs at Abo Galawa to 1111 OTUs at Ras Mohamed (total 2247 out of 6970 OTUs—32%) that were significantly associated with the site (Additional file 2: Figure S6). The abundance of these OTUs relative to the total microbial community varied from 10.7% at Wadi El Gemal to 58.8% at Ras Mohamed (Additional file 2: Figure S6). The main indicator taxa differed between sites, highlighting the heterogeneity of associated taxa, however, Pseudoalteromonas and Alteromonas were reported at all sites as indicator OTUs as well (Additional file 2: Figure S7). For example, Pseudoalteromonas spp. (69.3%) and Endozoicomonas sp. (10%) were the prevalent indicator bacterial OTUs at the northernmost site Abo Ghalloum, but Psychrosphaera sp. (23.6%) and Roseovarius sp. (15.8%) dominated Meritte, and an unclassified Gammaproteobacteria (9.6%), Endozoicomonas sp. (8.3%), and Gplla sp. (i.e., Cyanobacteria—6.3%) dominated Ras Mohamed (Additional file 2: Figure S7). Interestingly, Wadi El Gemal (i.e., the warmest site at the south) was dominated by the photosynthetic Erythrobacter sp. (29%). Notably, its abundance increase southward aligned with the latitudinal gradient (from 0.3 to 3.1%—Additional file 2: Figure S8).
A total of 977 OTUs (14% of total OTUs) were significantly associated with the SML of different coral hosts, ranging from 26 OTUs in P. damicornis to 456 OTUs in P. nodifera (Additional file 2: Figure S6). Abundances varied notably between reef-building corals (i.e., P. nodifera, F. favus, P. damicornis, and S. hystrix; 5.8% to 18.8%), and soft corals (i.e., X. umbellata and S. trocheliophorum; 36.6% and 49%, respectively) relative to the total OTUs (Additional file 2: Figure S6). The main bacterial indicator taxa differed between soft coral hosts, revealing species-specific bacteria, with Pseudoalteromonas, Alteromonas, and Endozoicomonas represented in the SML of all investigated host species (Additional file 2: Figure S7).
Linear discriminant analysis (LDA) effect size (LEfSe) analysis supported indicator species analysis and showed that Ras Mohamed was highly enriched by many unclassified bacterial OTUs that drive variation between sites. Overall, 406 OTUs (69 family and 126 genus) were differentially abundant between sites. Ruegeria, Pseudomonas, unclassified Flavobacteriacae, and Oleibacter (LDA > 5, p < 0.001) were the most significant OTUs that were differentially expressed between sites (Additional file 2: Figure S9a). On the other hand, 380 OTUs (53 family and 97 genus) drove most of the variation between the SML of coral species as well as seawater samples, particularly three Endozoicomonas and unclassified Alteromonadaceae OTUs that had the highest LDA score (LDA > 5, p < 0.001). Interestingly, seawater was enrichened by bacteria that drove most variation between coral species compared to seawater, highlighting the distinct bacterial community of seawater (Additional file 2: Figure S9b).
Core microbiome of coral SML
The number of core OTUs (i.e., present in 95% of the samples regardless of their abundance) varied between sites and coral species. The total number of core bacteria associated with seawater was 129 OTUs, while it ranged from 13 in F. favus to 50 OTUs in P. damicornis. Interestingly, only 5 OTUs were common among corals and seawater (Alteromonas, 3 OTUs of Pseudoalteromonas, and Vibrio), but 72 OTUs were exclusively found in the seawater samples highlighting the distinct bacterial community of seawater. Similarly, core bacteria ranged from 56 OTUs at Ras Mohamed to 25 OTUs at Meritte, contributing from 47.3% at Ras Mohamed to 84% at Abo Ghalloum of bacterial abundance (Additional file 2: Table S3). Notably, eight OTUs were shared among all sites (Fig. 4), five of them were the same OTUs shared among all coral species in addition to another 3 OTUs: Vibrio sp., Gplla sp. (i.e., cyanobacteria), and the photosynthetic Erythrobacter sp. There were exclusive OTUs in each site that were consistently observed within SML samples across all coral species and ranged from 23 at Ras Mohamed to 2 OTUs at Wadi El Gemal (Fig. 4 and Additional file 4). Interestingly, two exclusive OTUs at Wadi El Gemal (the warmest site) belong to the chemo/phototroph family Rhodobacteraceae (Additional file 4), but occurred in low abundance and comprised only 0.3% of total bacterial abundance at this site.

Venn diagram illustrating the number of bacterial OTUs that are present in at least 95% of the samples at each site and coral species. The graph shows the number of core OTUs shared among coral species (a). Only five OTUs were common between six corals species and seawater, but seawater samples had 72 exclusive OTUs that were not found in the SML. Similarly, eight OTUs (49.7% of total bacterial abundance) were common between sites (b), five of them were shared between all species in addition to a Vibrio sp., a Gplla sp., and the photosynthetic Erythrobacter sp. Importantly, each site and coral species had a small number of exclusive OTUs (outer region in diagram)
Discussion
Flexibility of coral microbiomes may help enable coral species to tolerate, and adapt to, environmental conditions known to induce stress and mortality [16, 17, 24, 28, 35. 44]. Here, we assessed the composition of coral dinoflagellate endosymbionts and the SML bacterial community for six abundant coral species inhabiting the northern Red Sea, an area recently proposed as a refuge for corals against thermal stress events [4, 45]. Our data provides new insight into whether and how microbiome composition aligns with the tolerance of corals within a region that is characterized by a natural latitudinal gradient of temperature from north to south and also subjected to extreme thermal anomalies [4].
Coral host and dinoflagellates endosymbiont associations
Host species identity contributes to endosymbiotic dinoflagellate distribution and diversity [46, 47]. Similarly, we observed a high level of host specificity for each of the coral species and their endosymbiotic dinoflagellates despite the latitudinal gradient, a trend that is consistent with previous studies in different bioregions [40, 42, 48, 49]. Such specificity may be attributed to one or more factors including (i) compatibility of cellular signals between algal symbiont and coral host during symbiosis establishment [50], (ii) metabolic characteristics of the host [22, 51], (iii) availability of host pigments to facilitate photosynthesis [52], (iv) host-driven factors that influence micro-habitat conditions for the symbiont (e.g., colony morphology and tissue thickness which influence light absorption [53], and (v) host acquisition symbiont strategy (vertical versus horizontal transmission—notably, all coral species investigated here were brooders that use vertical transmission) [54]. Environmental conditions are known to shape distribution patterns of the family Symbiodiniaceae at larger spatial scales [46, 55, 56]. For example, the main reef builder in the Caribbean (Orbicella annularis) exhibits partitioning of the endosymbiont community between the north and south (i.e., 1800 km from the Bahamas to Colombia), which is driven by thermal history rather than host genotype [57]. Thus, flexibility of host-endosymbiont associations (via switching or shuffling) is one strategy for corals to survive across biogeographical regions and under various local environmental stressors [58, 59]. The strong host-endosymbiont associations we observed here likely indicate strong local selection pressure to specific environments of the northern Red Sea [4].
Corals were dominated by Cladocopium spp. (formerly clade C, 86%). Presumably, Cladocopium spp. dominance in the Red Sea reflects the evolutionary origin (and connectivity) of coral taxa from the Indian Ocean where Cladocopium spp. also dominate (see [21, 40, 41, 60,61,62]). This is consistent with Ziegler et al. [55] who noted Cladocopium spp. dominance within the Red Sea (see also Baker et al., [63]) and suggested a strong selection for this genus throughout the entire Arabian region. We found five novel Cladocopium types exclusively within the northern Red Sea highlighting endemism within the region. As such, this “thermal refugia area” may select certain symbiont types that broaden the environmental niche for corals to survive under different (and extreme) conditions [4, 11, 12]. Although the phylogenetic analyses of endosymbionts relative to those in the Indian Ocean are yet to be explored, this may highlight the geographically (semi) isolated nature of the Red Sea that would promote local ecological (and/or genetic) speciation of endosymbionts.
The presence of the genus Symbiodinium with the genus Cladocopium within two branching coral hosts (P. damicornis and S. hystrix) corroborates that many types of Symbiodinium spp. are generalist symbionts within the Red Sea and occupy multiple hosts [64]. This pattern is likely unique for the Red Sea as Symbiodinium is rarely recorded in corals elsewhere and usually found in clams and fire corals in the Indo-Pacific Ocean [40].
Spatial differences of dinoflagellate endosymbiont community
Interestingly, the genetic identity of the algal endosymbionts within a host species did not change from north to south despite the latitudinal gradient. There are two potential explanations for this observation. Firstly, phenotypic plasticity of endosymbionts may enable them to populate different environmental/thermal regimes (e.g., [65]) and therefore influence coral thermal tolerance along the northern Red Sea, possibly via long-term acquisition of endosymbionts locally adapted to high temperature [66]. Howells et al. [67] noted higher photochemical performance and survivorship of Cladocopium spp. C1 isolated from warmer areas than those sampled from cooler regimes. Levin et al. [68], subsequently confirmed that this “warmer-regime” Cladocopium C1 isolate is characterized by enhanced gene expression for heat tolerance. Thus, the history of environmental regimes are likely critical in expanding niche breadth through functional diversity for certain symbiont taxa [22].
Secondly, the resolution of ITS2 as a genetic marker may underestimate the taxonomic diversity of the family Symbiodiniaceae, and as such higher genetic marker resolution is needed to resolve taxonomic diversity [19, 69, 70]. For example, using the bspA gene resolved identical C3 ITS2 fingerprints and demonstrated a distinct monophyletic lineage with a large genetic distance of new species (Cladocopiumthermophilum) compared to other C3 types from the Arabian Gulf ([19], see also [71]). Notably, this novel species C. thermophilum likely consists of different (sub)species [72] that could be resolved using the ITS2 marker alone and the novel analytical framework SymPoral [73]. In our study, we report five novel symbiont types in addition to four unidentified endosymbiont types. This highlights the need to further explore the taxonomy of Symbiodiniaceae within the region using different genetic markers that may provide higher taxonomy resolution. Notably, the recent SymPortal framework that resolve symbiont types from ITS2 next-generation sequencing data overcomes many of the recent limitations.
Bacterial community plasticity across sites
In contrast to the endosymbiont communities, the bacterial communities varied primarily between sites highlighting strong geographical patterns, likely driven by environmental differences, over host-bacterial specificity (Fig. 1). Coral bacterial communities are altered by changing environmental conditions such as pollution [17, 74], nutrient availability [17, 75], environmental anomalies such as temperature [16, 21, 76], salinity [28], pH [77, 78], and eutrophication [79]. Such compositional flexibility indicates a potential capacity for local acclimatization (or adaptation), and thus may be critical for promoting holobiont fitness a. Indeed, our indicator species analysis supports the notion of local acclimatization where a high number of OTUs were significantly associated with sites comprising high relative abundance (Additional file 2: Figure S6). This suggests selection of beneficial bacterial taxa that are presumably important to sustain coral survival under different environmental/thermal regimes. Notably, the stark differences that we observed in bacterial communities of the SML along latitudinal gradients are unlikely to be simply explained by differences in the prevailing environment since the bacterial community differences were also host-specific.
Coral-specific bacteria
Composition of bacterial communities varied between coral species, where 5.8% to 18.8% of the bacterial community associated significantly to specific coral hosts regardless of latitudinal environmental/thermal regimes. Such host-specificity of bacterial communities is consistent with many other studies profiling coral microbiomes [80,81,82,83]. The variation in bacterial community composition between coral taxa could be explained by different factors including (i) species-specific composition of exudates by different coral hosts to the SML (e.g., [84]), (ii) species-specific biochemical composition of coral SML itself [85,86,87,88]; (iii) vertical transmission of bacterial community from parents to offspring [89,90,91,92]. However, which of these factors likely drive differences among bacterial communities of northern Red Sea corals is difficult to discern from the available data.
Putative roles of the dominant SML-associated bacteria
SML bacterial community functional characterization is required to validate whether and how bacterial taxa contribute to niche broadening throughout the northern Red Sea and is beyond the scope of the curernt study [93]. Even so, using the METAGENassist web portal [94] (Additional file 2: Figure S10), functional profiling showed that about half of the samples exhibited potential enrichment of high sulfate and nitrite reducers and dehalogenating bacteria, while the remaining samples had mixed metabolic sources. High enrichment of sulfate reducers may simply reflect the dominance of Alteromonas and Pseudoalteromonas, many of which are known to play a major role in dimethyl-sulfoniopropionate (DMSP) metabolism [95]. High abundance of these genera may indicate elevated levels of DMSP production in Red Sea corals and surrounding waters [96]. DMSP is produced by the endosymbiotic dinoflagellates as well as the coral [97] and metabolized by associated bacteria to form dimethylsulfide (DMS), dimethyl sulfoxide (DMSO), and acrylate. These molecules have potential roles in osmoregulation [98] and antioxidant capacity [99, 100]. High production of DMSP may therefore convey the capacity to survive under high salinity and thermal anomalies that characterize the northern Red Sea [4, 11, 12]. Similarly, the SML of Porites lobata from the central Red Sea was dominated by Pseudoalteromonas [101] and displayed increased levels of sulfur cycling compared to the Arabian Gulf. Therefore, it would be informative to link levels of DMSP (and associated by-products) with tolerance patterns of corals in this region.
Interestingly, the presence of Alteromonas and Pseudoalteromonas within the region may be linked to coral disease resistance. Pseudoalteromonas spp. associated with the SML of several coral species exhibit extracellular antibacterial metabolites which may aid in the defense against invasive pathogens [102, 103]. Wright et al. [104] similarly reported an increased abundance of Alteromonas and Pseudoalteromonas phylotypes in Acropora millepora resisting infection by Vibrio sp. Further, some strains of Alteromonas spp. are known dinitrogen fixers, which may subsequently translocate fixed nitrogen to the algal endosymbionts associated with P. damicornis larvae [92]. Together, these observations highlight that while the detailed role of abundant bacterial phylotypes is yet to be investigated for Red Sea corals, they may provide several functions important to holobiont fitness.
Furthermore, Erythrobacter sp. constituted the dominant indicator taxon at the warmest site (i.e., Wadi El Gemal) where it increased in abundance southwards (Additional file 2: Figure S8). This bacterial group contains bacterial chlorophyll-a (B-Chla) and large amounts of carotenoids [105,106,107,108]. Carotenoids are well known for their antioxidant activities [109], but the relationship between bacterial assemblage associated with coral SML and antioxidant activities is not well resolved. Diaz et al. [110] experimentally measured extracellular superoxide concentrations associated with corals and revealed that the microbial community could produce and importantly regulate ROS in their immediate surroundings, and hence influence coral physiology and health. Hence, Erythrobacter may play a functional role critical toward improving holobiont resistance to heat stress: however, such functional implications have yet to be fully assessed.
Conclusion
We provide the first details of the microbiome communities associated with coral conspecifics across 500 km in the northern Red Sea that showed high thermal tolerance as evidenced by low bleaching susceptibility over the past three decades. Our data highlight that the endosymbiotic communities were highly host-specific with little variation throughout this region. At the same time, we identified five novel types highlighting endemism and the selection of certain genotypes within the region. In contrast, the SML bacterial communities varied significantly between sites and coral hosts, therefore emphasizing how the holobiont composition changes across the latitudinal gradient. Among the associated bacterial OTUs, we identified taxa across the northern Red Sea that may play a role in elevated thermal tolerance and may fill a regional environmental niche that broadens the capacity of corals to survive under extreme conditions. We therefore conclude that the distinct microbiome associated with corals from the northern Red Sea may contribute to the thermal tolerance of corals, previously denoted as a coral reef refuge. Notably, the highly responsive nature of bacterial communities present in the SML provides further justification to investigate their functional role, which may contribute to the success of corals experiencing an increased frequency of thermal stress in the near future.
Materials and methods
Survey sites and sample collection
Sampling sites
Sample collection was conducted at five sites along the northern Gulf of Aqaba and southern Egyptian coast in February 2013, representing a latitudinal gradient mainly varied in temperature. The selected sites were Abo Ghalloum (28.6147°N, 34.5604°E; Gulf of Aqaba), Ras Mohamed (27.7305°N, 34.2691°E; Sinai Peninsula), (3) Abo Galawa (27.3157°N, 33.8097°E), (4) Meritte (27.2485°N, 33.849°E) at Hurghada, and (5) Wadi El Gemal (24.6988°N, 35.1327°E) at the southern Egyptian coast (Fig. 1). All sampling sites were unurbanized and not directly impacted by anthropogenic activities (except Merritte) and characterized by fringing reefs adjacent to the shoreline, except for Abu Galawa which was a patch reef located about 1.5 km off the coast. Sites were located within the thermal gradient where long-term mean (±SD) of summer SST ranged from 26.6 ± 1 °C for the Gulf of Aqaba to 29.3 ± 1.2 °C for the Wadi El Gemal [4]. To ensure that the influence of seawater parameter on coral microbiome composition is minimum, remote sensing data (2003–2012) of chlorophyll a and water attenuation coefficient were used as proxy of water quality. This data showed that there are no significant differences in environmental variables across sites, but the temperature is systematically changing across the region (Additional file 2: Supplementary material). Further, we collected seawater samples to measure ammonia concentration in each study sites, that did not vary significantly between sites (Additional file 2: Supplementary material). Taken together, this highlights that temperature is likely the main driver of compositional change of microbiome along the latitudinal gradient in the northern Red Sea.
Sample collection
Six coral species were sampled at each site across the latitudinal gradient, with the selection of species representing different coral growth forms: massive (Porites nodifera, Favia favus) and branching (Pocillopora damicornis, Seriatopora hystrix) hard coral, as well as soft corals (Xenia umbellata, Sarcophyton trocheliophorum). Specimens were collected from shallow (2–5 m) and deep (15-18 m) reef slopes representing different light regimes as per Kuguru et al., [111]. At each site, three types of samples were collected: coral fragment, coral mucus and seawater. Overall, three replicates x five sites x six species x two depths samples of coral tissue (total n = 164) and coral SML (total n = 141) were collected for endosymbiont ITS2 and bacterial 16S rRNA gene profiling, respectively. Further, three replicates of water samples × 2 depth × 5 sites (total n = 23) were also collected as reference bacterial samples. Specifically, (i) coral fragments (< 1 cm) were collected from three different visibly healthy colonies (> 5 m apart) for each species and depth (i.e., n = 3 per species and depth). Samples were sealed in separate pre-labeled bags filled with in situ seawater [48]. (ii) At each sampled coral colony, associated SML was sampled using sterile 50 ml syringes (n = 3 per species and depth). (iii) Seawater samples (500 ml) were collected in sterilized polyethylene bottles in each site at each depth (n = 3 per site per depth) as environmental bacterial reference samples [102]. All samples were then kept shaded in a cold box until preservation (within 2 h).
Upon return to the laboratory, all coral fragments were preserved directly in pre-loaded 2 ml vials containing DMSO-20% buffer for DNA preservation for subsequent dinoflagellate endosymbionts identification [112]. Each SML and water sample was filtered through sterilized 0.22 μm Cyclopore filter columns (Whatman, UK), and preserved in 2 ml vials preloaded with DMSO-20% buffer for 16S rRNA gene microbial analysis. Preserved coral fragments and filtered bacterial samples were kept at 4 °C until shipping to the UK for genomic analysis, and then stored at − 20 °C.
Symbiodiniaceae identification
The overall purpose of our analysis was to retrieve the dominant endosymbiont type(s). For this reason, we determined DGGE to be the most cost-effective approach that can detect up to 90–95% of the total community present within a single coral colony [113]. Notably, DGGE is not a method to elucidate fine-scale genetic differentiation, which is rather conducted via next-generation sequencing of the ITS2 marker gene [114] and subsequent analysis in SymPortal [73]. Endosymbiont DNA was extracted from approximatly 100 mg of coral tissue using the modified Promega Wizard DNA prep protocol (Madison, WI, USA) as per LaJeunesse et al. [48]. Amplification of the symbiont Internal Transcribed Spacer (ITS2) was performed against a negative control, through two steps as described by Bongaerts et al. [105]: (i) nested PCR was used (10 μl total reaction) to amplify the region between 18S and 28S rDNA (750 bp) using 1 μl of gDNA mixed with “ZITSUPM13” and “ZITSDNM13” primers for 35 cycles as described in Santos et al. [116] (Additional file 2: Table S1); and subsequently (ii) 1 μl of the nested PCR amplicon served as a template to amplify ITS2 (330–360 bp) mixed with ‘ZITS2for’ and GC clamp ‘ZITS2 clamp’ primers as designed by LaJeunesse and Trench [117], and touchdown PCR protocol for 40 cycles were used as per LaJeunesse et al. [43] (Additional file 2: Table S1). ITS2 amplicons were then separated by denaturation gradient gel electrophoresis (DGGE) (45–80% polyacrylamide gel) and aligned against a reference DNA ladder (containing ITS2 Breviolum B1, Cladocopium C1, and Durusdinium D1 samples) at 60 °C for ~ 15 h as per LaJeunesse [48] using a CBS Scientific System (Del Mar, CA, USA). DGGE gels were stained with SYBR green (Molecular Probes, Eugene, OR, USA) and representative bands (n = 3–5 from different samples from each fingerprint found) for each coral species were excised and eluted in 500 μl RNase free water at 4 °C overnight. Subsequently, bands are directly amplified (without gel extraction step) using ZITS2 forward and reverse primers (without the GC clamp) for 30 cycles and sent for sequencing. After that, the ITS2 amplicon was cleaned using USB-EXO SAP-IT PCR cleanup kit (Affymetrix, USA) and sequenced using Applied Biosystems 310 genetic analyzer, USA.
Bacterial 16S rRNA gene profiling
Due to the rapidly adaptive nature of the surface mucus layer (SML) to local environments and/or stress, bacterial genomic DNA was extracted from coral SML and seawater using the CTAB (Cetyl-trimethyl-ammonium-bromide) method [119]. To amplify the bacterial 16S rRNA gene from SML and water samples, hypervariable regions V3 and V4 of ribosomal DNA were targeted (~550pb) using 341F and 805R universal bacterial primers with an Illumina overhang adaptor (Additional file 2: Table S1) according to the manufacturer’s protocol (Illumina, San Diego, CA, USA). The PCR amplicon was cleaned by an AMPure XP magnetic bead system (Beckman Coulter, Brea, CA, USA), and 5 μl of cleaned PCR amplicon used for indexing PCR using Nextera XT V2 kit (A&B index kit) (Illumina) according to the manufacturer’s protocol. The indexed PCR amplicon was cleaned again by AMPure XP magnetic beads and then quantified using a FLUOstar Omega microplate reader (BMG Labtech, Germany) using Quant-iT PicoGreen dsDNA assay kit (Invitrogen, USA). All samples were then pooled in equimolar ratios. The quality of the final pooled library was checked on a 1% agarose gel as well as on a Bioanalyzer (Agilent 2100, Santa Clara, CA, USA). Version 3 chemistry kit was used in HiSeq and sequencing was conducted at the TGAC genomic analysis center (Norwich, UK).
Data analysis
Symbiodiniaceae analysis
We followed the commonly accepted and widely published protocols for this technique to interpret this type of data (cf. original methodologies in LaJeunesse et al. 2002 [118]). First, symbiont DGGE gels were assessed visually to identify the fingerprint for each coral sample (Additional file 6), and then DNA sequences for representative bands were obtained, trimmed manually, aligned using Geneious (V10), and then blasted against Genbank ‘nr’ database (http://www.ncbi.nlm.nih. gov/BLAST/) for ITS2 type identification. Each identified ITS2 type was tabulated and transformed into presence/absence data matrix for statistical analysis. To test the significance of similarity of symbiont community between sites, coral species, and depth, we performed Permutation Multifactorial Analysis of Variance (PERMANOVA) [120] with 9999 permutations using Jaccard dissimilarity matrix by “adonis” function in R [121] using vegan package in R. Notably, the nature of symbiont community dataset was “presence/absence” while bacterial community was “abundance-based” dataset and therefore they were analyzed separately.
Bacterial bioinformatic analyses
Raw 16S rRNA gene amplicon sequences were trimmed using Sickle version 1.33 [122] at the default quality threshold (Q20) using the paired-end mode. Sequence trimming was performed at the 3’ end, and to ensure high taxonomic resolution, all sequences shorter than 350 bp or having ambiguous bases (Ns) were discarded. The forward and reverse sequences that passed quality filter were then subjected to error correction using Bayes Hammer implemented in SPAdes v3.7.1 with default settings [123, 124]. Paired-end sequences were aligned and primers removed using the PEAR algorithm implemented in PANDAseq version 1.33 [125, 126]. Chimeric check was performed using RDP 16S rRNA gene database to ensure sequences quality [117], and paired reads were then de-replicated, sorted by abundance, and clustered into operational taxonomic units (OTUs) at 97% similarity threshold using VSEARCH v1.11.1 (Rognes, https://github.com/torognes/vsearch). Low abundance sequences (< 5 occurrences over all samples) and non-bacterial OTUs (i.e., mitochondria, chloroplast, archaea, eukaryote, and unknown sequences) were then removed. Taxonomic divisions were assigned as OTU centroids using the RDP classifier [127] as implemented in QIIME [128], with a minimum confidence level of 0.7, and relative abundances of taxa were computed using QIIME’s “summarize_taxa.py” script.
Bacterial community analysis
The OTU abundance matrix of the microbial community (using non-normalized approach [129]—see Additional file 5) was used to calculate microbial diversity indices (i.e., Chao1 richness estimator, inverse Simpson, and Shannon diversity indices) for each coral sample (total n = 164). Normality of diversity indices outcome was checked using the Shapiro test [130], and log-transformed to assess the influence of site, coral species, and depth on microbial diversity using multifactorial ANOVA. The bacterial communities associated with soft coral species (X. umbellata and S. trocheliophorum) appeared similar in diversity and composition to those associated with reef-building corals (see Additional file 2: Figure S3), and therefore soft corals were included in the remaining analysis.
Multivariate analysis was further used to test the statistical difference of microbial community structure. Permutation Multifactorial Analysis of Variance (PERMANOVA) [120] with 9999 permutations using Bray-Curtis dissimilarity matrix by “adonis” function in R was performed on (i) all coral samples to assess the influence of site, coral species, and depth and their interactions on microbial community structure, (ii) on each coral species across sites to investigate the effect of site on each coral species separately, and (iii) on each site to include all coral species (i.e., all corals within each site) to assess the influence of coral species on microbial composition at each site separately. Principal coordinate analysis (PCoA) ordination based on Bray-Curtis dissimilarity was used to visualize the dispersion of microbial community among sites, coral species, and depth.
Indicator species analysis was performed to test the association between bacterial community and between both, sites and coral host, using indicspecies package in R [131]. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was also performed to obtain the most differentially abundant bacteria between sites and coral species using the Microbiome Analysis web portal (https://www.microbiomeanalyst.ca/) with default settings [132]. To investigate OTUs that were consistently associated with coral SML and whether/how they changed with the increase of the ambient temperature across sites, data were transformed into a presence/absence data matrix, and the core mucus microbiome calculated as the occurrence of each OTU in 95% of the samples (i.e., 95% occurrence threshold) across sites. All plots and statistical analysis were performed in R version 3.2.3 [133].
Change history
21 February 2020
Following publication of the original article [1], the authors reported an error on the legend of of P.damicornis in Fig. 1.
21 February 2020
Following publication of the original article [1], the authors reported an error on the legend of of P.damicornis in Fig. 1.
References
Hughes TP, Anderson KD, Connolly SR, Heron SF, Kerry JT, Lough JM, et al. Spatial and temporal patterns of mass bleaching of corals in the Anthropocene. Science. 2018;359:80–3.
Hughes TP, Kerry JT, Álvarez-Noriega M, Álvarez-romero JG, Anderson KD, Baird A, et al. Global warming and recurrent mass bleaching of corals. Nature. 2017;543:373–7 https://doi.org/10.1038/nature21707.
Dixon GB, Davies SW, Aglyamova GA, Meyer E, Bay LK, Matz MV. Coral reefs. Genomic determinants of coral heat tolerance across latitudes. Science. 2015;348:1460–2 https://doi.org/10.1126/science.1261224.
Osman EO, Smith DJ, Ziegler M, Kürten B, Conrad C, El-Haddad KM, et al. Thermal refugia against coral bleaching throughout the northern Red Sea. Glob Chang Biol. 2018;24:474–84 https://doi.org/10.1111/gcb.13895.
Oliver TA, Palumbi SR. Do fluctuating temperature environments elevate coral thermal tolerance? Coral Reefs. 2011;30:429–40 https://doi.org/10.1007/s00338-011-0721-y.
Schoepf V, Stat M, Falter JL, McCulloch MT. Limits to the thermal tolerance of corals adapted to a highly fluctuating, naturally extreme temperature environment. Sci Rep. 2015;5:17639 https://doi.org/10.1038/srep17639.
Camp EF, Smith DJ, Evenhuis C, Enochs I, Manzello D, Woodcock S, et al. Acclimatization to high-variance habitats does not enhance physiological tolerance of two key Caribbean corals to future temperature and pH. Proc R Soc Lond B Biol Sci. 2016;283:20160442 https://doi.org/10.1098/rspb.2016.0442.
Camp EF, Nitschke MR, Rodolfo-Metalpa R, Houlbreque F, Gardner SG, Smith DJ, et al. Reef-building corals thrive within hot-acidified and deoxygenated waters. Sci Rep. 2017;7:2434 https://doi.org/10.1038/s41598-017-02383-y.
D’Angelo C, Hume BCC, Burt J, Smith EG, Achterberg EP, Wiedenmann J. Local adaptation constrains the distribution potential of heat-tolerant Symbiodinium from the Persian/Arabian Gulf. ISME J. 2015;9:1–10 https://doi.org/10.1038/ismej.2015.80.
Camp EF, Schoepf V, Mumby PJ, Hardtke LA, Rodolfo-Metalpa R, Smith DJ, et al. The future of coral reefs subject to rapid climate change: lessons from natural extreme environments. Front Mar Sci. 2018;5:4 https://doi.org/10.3389/fmars.2018.00004.
Gegner HM, Ziegler M, Rä Decker N, Buitrago-Loez C, Aranda M, Voolstra CR. High salinity conveys thermotolerance in the coral model Aiptasia. Biol Open. 2017;6:1943–8 https://doi.org/10.1242/bio.028878.
Gegner HM, Rädecker N, Ochsenkühn M, Barreto MM, Ziegler M, Reichert J, Schubert P, Wilke T, Voolstra CR. High levels of floridoside at high salinity link osmoadaptation with bleaching susceptibility in the cnidarian-algal endosymbiosis. Biology Open. 2019;8(12):bio045591.
Rohwer F, Seguritan V, Azam F, Knowlton N. Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser. 2002;243:1–10.
Palumbi SR, Barshis DJ, Traylor-Knowles N, Bay RA, Hoegh-Guldberg O, Hughes TP, et al. Mechanisms of reef coral resistance to future climate change. Science. 2014;344:895–8 https://doi.org/10.1126/science.1251336.
Torda G, Donelson JM, Aranda M, Barshis DJ, Bay L, Berumen ML, et al. Rapid adaptive responses to climate change in corals. Nat Clim Chang. 2017;7:627–36 https://doi.org/10.1038/nclimate3374.
Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat Commun. 2017;8:14213 https://doi.org/10.1038/ncomms14213.
Ziegler M, Grupstra CGB, Barreto MM, Eaton M, BaOmar J, Zubier K, Al-Sofyani A, Turki AJ, Ormond R, Voolstra CR. Coral bacterial community structure responds to environmental change in a host-specific manner. Nat Commun. 2019;10(1).
LaJeunesse TC, Parkinson JE, Gabrielson PW, Jeong HJ, Reimer JD, Voolstra CR, et al. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral Endosymbionts. Curr Biol. 2018;28(16):2570–2580.e6.
Hume BCC, Angelo CD, Smith EG, Stevens JR, Burt J, Wiedenmann J. Symbiodinium thermophilum sp. nov., a thermotolerant symbiotic alga prevalent in corals of the world’s hottest sea, the Persian/Arabian Gulf. Sci Rep. 2015;5:8562.
Hume BCC, Voolstra CR, Arif C, D’Angelo C, Burt JA, Eyal G, et al. Ancestral genetic diversity associated with the rapid spread of stress-tolerant coral symbionts in response to Holocene climate change. Proc Natl Acad Sci U S A. 2016;113:4416–21 https://doi.org/10.1073/pnas.1601910113.
Gardner SG, Camp EF, Smith DJ, Voolstra CR, Kahlke T, Osman EO, et al. Coral microbiome diversity reflects mass coral bleaching susceptibility during the 2016 El Niño heat wave. Ecol Evol. 2019;9:938–56 https://onlinelibrary.wiley.com/doi/full/10.1002/ece3.4662.
Suggett DJ, Warner ME, Leggat W. Symbiotic dinoflagellate functional diversity mediates coral survival under ecological crisis. Trends Ecol Evol. 2017;32(10):735–45 https://www.sciencedirect.com/science/article/abs/pii/S0169534717301908.
Reshef L, Koren O, Loya Y, Zilber-Rosenberg I, Rosenberg E. The coral probiotic hypothesis. Environ Microbiol. 2006;8:2068–73 https://doi.org/10.1111/j.1462-2920.2006.01148.x.
Bang C, Dagan T, Deines P, Dubilier N, Duschl WJ, Fraune S, Hentschel U, Hirt H, Hülter N, Lachnit T, Picazo D, Pita L, Pogoreutz C, Rädecker N, Saad MM, Schmitz RA, Schulenburg H, Voolstra CR, Weiland-Bräuer N, Ziegler M, Bosch TCG. Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology. 2018;127:1–19.
Gates RD, Ainsworth TD. The nature and taxonomic composition of coral symbiomes as drivers of performance limits in scleractinian corals. J Exp Mar Biol Ecol. 2011;408:94–101.
Röthig T, Yum LK, Kremb SG, Roik A, Voolstra CR. Microbial community composition of deep-sea corals from the Red Sea provides insight into functional adaption to a unique environment. Sci Rep. 2017:44714 https://doi.org/10.1038/srep44714.
Morrow KM, Bourne DG, Humphrey C, Botté ES, Laffy P, Zaneveld J, et al. Natural volcanic CO2 seeps reveal future trajectories for host–microbial associations in corals and sponges. ISME J. 2015;9:894–908 https://doi.org/10.1038/ismej.2014.188.
Röthig T, Ochsenkühn MA, Roik A, van der Merwe R, Voolstra CR. Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol Ecol. 2016;25:1308–23 https://doi.org/10.1111/mec.13567.
Sweet MJ, Croquer A, Bythell JC. Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs. 2011;30:39–52.
Shnit-Orland M, Kushmaro A. Coral mucus-associated bacteria: a possible first line of defense. FEMS Microbiol Ecol. 2009;67:371–80 https://doi.org/10.1111/j.1574-6941.2008.00644.x.
Brown BE, Bythell JC. Perspectives on mucus secretion in reef corals. Mar Ecol Prog Ser. 2005;296:291–309.
Krediet CJ, Ritchie KB, Alagely A, Teplitski M. Members of native coral microbiota inhibit glycosidases and thwart colonization of coral mucus by an opportunistic pathogen. ISME J. 2013;7:980–90 https://doi.org/10.1038/ismej.2012.164.
Glasl B, Herndl GJ, Frade PR. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 2016;8:1–13 https://doi.org/10.1038/ismej.2016.9.
Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, et al. Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat Commun. 2018;9:4921 https://doi.org/10.1038/s41467-018-07275-x.
Rosado PM, DCA L, GAS D, Chaloub RM, Jospin G, Nunes da Rocha U, et al. Marine probiotics: increasing coral resistance to bleaching through microbiome manipulation. ISME J. 2018;1 https://doi.org/10.1038/s41396-018-0323-6.
Eakin CM, Morgan JA, Heron SF, Smith TB, Liu G, Alvarez-Filip L, et al. Caribbean corals in crisis: record thermal stress, bleaching, and mortality in 2005. PLoS One. 2010;5:1–9.
Fine M, Gildor H, Genin A. A coral reef refuge in the Red Sea. Glob Chang Biol. 2013;19:3640–7 https://doi.org/10.1111/gcb.12356.
Arrigoni R, Benzoni F, Terraneo TI, Caragnano A, Berumen ML. Recent origin and semi-permeable species boundaries in the scleractinian coral genus Stylophora from the Red Sea. Sci Rep. 2016;6 https://www.nature.com/articles/srep34612.
Robitzch V, Banguera-Hinestroza E, Sawall Y, Al-Sofyani A, Voolstra CR. Absence of genetic differentiation in the coral Pocillopora verrucosa along environmental gradients of the Saudi Arabian Red Sea. Front Mar Sci. 2015;2:5 https://doi.org/10.3389/fmars.2015.00005.
LaJeunesse TC, Pettay DT, Sampayo EM, Phongsuwan N, Brown B, Obura DO, et al. Long-standing environmental conditions, geographic isolation and host-symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium. J Biogeogr. 2010;37:785–800 https://doi.org/10.1111/j.1365-2699.2010.02273.x.
Chauka LJ. Diversity of symbiotic algae in the genus Symbiodinium in Scleractinian corals of Tanzania. West Indian Ocean J Mar Sci. 2013;11:67–76 http://www.ajol.info/index.php/wiojms/article/view/78566%0Apapers3://publication/uuid/9CEF6E2C-AF3A-4880-B32A-FC476DE647C2.
LaJeunesse TC, Bhagooli R, Hidaka M, DeVantier L, Done T, Schmidt GW, et al. Closely related Symbiodinium spp. differ in relative dominance in coral reef host communities across environmental, latitudinal and biogeographic gradients. Mar Ecol Prog Ser. 2004;284:147–61.
Tonk L, Sampayo EM, LaJeunesse TC, Schrameyer V, Hoegh-Guldberg O. Symbiodinium (Dinophyceae) diversity in reef-invertebrates along an offshore to inshore reef gradient near Lizard Island. Great Barrier Reef J Phycol. 2014;50:552–63 https://doi.org/10.1111/jpy.12185.
Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M, et al. ISME J. 2015:1–14. https://doi.org/10.1038/ismej.2015.39.
Fine M, Gildor H, Genin A. A coral reef refuge in the Red Sea. Glob Chang Biol. 2013;19(12):3640–7.
Tonk L, Sampayo EME, Weeks SSS, Magno-Canto M, Hoegh-Guldberg O, Hoegh-Guldberg O, et al. Host-specific interactions with environmental factors shape the distribution of Symbiodinium across the great barrier reef. PLoS One. 2013;8:e68533 https://doi.org/10.1371/journal.pone.0068533.
Tonk L, Sampayo EM, Chai A, Schrameyer V, Hoegh-Guldberg O. Symbiodinium (Dinophyceae) community patterns in invertebrate hosts from inshore marginal reefs of the southern great barrier reef. Australia J Phycol. 2017;53:589–600.
LaJeunesse TC, Loh WKW, van Woesik R, Hoegh-Guldberg O, Schmidt GW, Fitt WK. Low symbiont diversity in southern great barrier reef corals relative to those of the Caribbean. Limnol Oceanogr. 2003;48:2046–54.
van Oppen MJ, Palstra FP, Piquet AM, Miller DJ. Patterns of coral-dinoflagellate associations in Acropora: significance of local availability and physiology of Symbiodinium strains and host-symbiont selectivity. Proc Biol Sci / R Soc. 2001;268:1759–67 https://doi.org/10.1098/rspb.2001.1733.
Davy SK, Allemand D, Weis VM. Cell Biology of Cnidarian-Dinoflagellate Symbiosis. Microbiol Mol Biol Rev. 2012;76(2):229–61 https://mmbr.asm.org/content/76/2/229.
Rädecker N, Raina J-B, Pernice M, Perna G, Guagliardo P, Kilburn MR, Aranda M, Voolstra CR. Using Aiptasia as a Model to Study Metabolic Interactions in Cnidarian-Symbiodinium Symbioses. Front Physiol. 2018;9.
Roth MS. The engine of the reef: photobiology of the coral-algal symbiosis. Front Microbiol. 2014;422 https://doi.org/10.3389/fmicb.2014.00422.
Enríquez S, Méndez ER, Iglesias-Prieto R. Multiple scattering on coral skeletons enhances light absorption by symbiotic algae. Limnol Oceanogr. 2005;50:1025–32.
Stat M, Loh WKW, Hoegh-Guldberg O, Carter DA. Symbiont acquisition strategy drives host-symbiont associations in the southern great barrier reef. Coral Reefs. 2008;27:763–72.
Ziegler M, Arif C, Burt JA, Dobretsov S, Roder C, LaJeunesse TC, et al. Biogeography and molecular diversity of coral symbionts in the genus Symbiodinium around the Arabian peninsula. J Biogeogr. 2017;44:674–86 https://doi.org/10.1111/jbi.12913.
Ziegler M, Arif C, Voolstra CR. Symbiodiniaceae Diversity in Red Sea Coral Reefs & Coral Bleaching. In: Voolstra C, Berumen M, editors. Coral Reefs of the Red Sea. Coral Reefs of the World, vol. 11. Cham: Springer; 2019.
Kennedy EV, Tonk L, Foster NL, Chollett I, Ortiz J-C, Dove S, et al. Symbiodinium biogeography tracks environmental patterns rather than host genetics in a key Caribbean reef-builder, Orbicella annularis. https://doi.org/10.1098/rspb.2016.1938.
Silverstein RN, Cunning R, Baker AC. Change in algal symbiont communities after bleaching, not prior heat exposure, increases heat tolerance of reef corals. Glob Chang Biol. 2015;21:236–49.
Boulotte NM, Dalton SJ, Carroll AG, Harrison PL, Putnam HM, Peplow LM, et al. Exploring the Symbiodinium rare biosphere provides evidence for symbiont switching in reef-building corals. ISME J. 2016:1–9 https://doi.org/10.1038/ismej.2016.54.
Yang SY, Keshavmurthy S, Obura D, Sheppard CRC, Visram S, Chen CA. Diversity and distribution of symbiodinium associated with seven common coral species in the Chagos Archipelago, Central Indian ocean. PLoS One. 2012;7:1–9 https://doi.org/10.1371/journal.pone.0035836.
McClanahan T, Baker A, Ateweberhan M. Preparing for climate change in the western Indian Ocean Identifying climate refugia, biodiversity responses and preferred management. WIOMSA Book Series No. 12. viii; 2011.
Baker A, McClanahan T, Starger C, Boonstra R. Long-term monitoring of algal symbiont communities in corals reveals stability is taxon dependent and driven by site-specific thermal regime. Mar Ecol Prog Ser. 2013;479:85–97 https://doi.org/10.3354/meps10102.
Baker AC, Jones SHIV, Lee TS, Abuzinada AH, Joubert E, Krupp F. Symbiont diversity in Arabian corals and its relation to patterns of contemporary and historical environmental stress. In: Proceedings of an international symposium on: the extent and impact of coral bleaching in the Arabian region: Riyadh (5–9 February 2000) Kingdom of Saudi Arabia; 2005. p. 24–36.
Sawall Y, Al-sofyani A, Banguera-hinestroza E, Voolstra CR. Spatio-temporal analyses of Symbiodinium physiology of the coral Pocillopora verrucosa along large-scale nutrient and temperature gradients in the Red Sea. Photobiology. 2014;9:1–12 https://doi.org/10.1371/journal.pone.0103179.
Hoadley KD, Pettay DT, Dodge D, Warner ME. Contrasting physiological plasticity in response to environmental stress within different cnidarians and their respective symbionts. Coral Reefs. 2016;35:529–42 https://doi.org/10.1007/s00338-016-1404-5.
Chakravarti LJ, Beltran VH, van Oppen MJH. Rapid thermal adaptation in photosymbionts of reef-building corals. Glob Chang Biol. 2017;23:4675–88 https://doi.org/10.1111/gcb.13702.
Howells EJ, Beltran VH, Larsen NW, Bay LK, Willis BL, van Oppen MJH. Coral thermal tolerance shaped by local adaptation of photosymbionts. Nat Clim Chang. 2012;2:116–20 https://doi.org/10.1038/nclimate1330.
Levin RA, Beltran VH, Hill R, Kjelleberg S, McDougald D, Steinberg PD, et al. Sex, scavengers, and chaperones: transcriptome secrets of divergent symbiodinium thermal tolerances. Mol Biol Evol. 2016;33:3032 https://doi.org/10.1093/molbev/msw119.
Thornhill DJ, Howells EJ, Wham DC, Steury TD, Santos SR. Population genetics of reef coral endosymbionts ( Symbiodinium , Dinophyceae). Mol Ecol. 2017;26:2640–59 https://doi.org/10.1111/mec.14055.
Thornhill DJ, Lewis AM, Wham DC, LaJeunesse TC. Host-specialist lineages dominate the adaptive radiation of reef coral endosymbionts. Evolution. 2014;68:352–67 https://doi.org/10.1111/evo.12270.
LaJeunesse TC, Thornhill DJ. Improved resolution of reef-coral Endosymbiont (Symbiodinium) species diversity, ecology, and evolution through psbA non-coding region genotyping. PLoS One. 2011;6:e29013 https://doi.org/10.1371/journal.pone.0029013.
Smith EG, Ketchum RN, Burt JA. Host specificity of Symbiodinium variants revealed by an ITS2 metahaplotype approach. ISME J. 2017;11(6):1500–3.
Hume BCC, Smith EG, Ziegler M, Warrington HJM, Burt JA, LaJeunesse TC, et al. SymPortal: A novel analytical framework and platform for coral algal symbiont next-generation sequencing ITS2 profiling. Mol Ecol Resour. 2019:1755–0998.13004 https://doi.org/10.1111/1755-0998.13004.
Ziegler M, Roik A, Porter A, Zubier K, Mudarris MS, Ormond R, et al. Coral microbial community dynamics in response to anthropogenic impacts near a major city in the Central Red Sea. Mar Pollut Bull. 2015; https://doi.org/10.1016/j.marpolbul.2015.12.045.
Zaneveld JR, Burkepile DE, Shantz AA, Pritchard CE, McMinds R, Payet JP, et al. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat Commun. 2016;7:1–12.
Wang L, Shantz AA, Payet JP, Sharpton TJ, Foster A, Burkepile DE, et al. Corals and their microbiomes are differentially affected by exposure to elevated nutrients and a natural thermal anomaly. Front Mar Sci. 2018;5:1–16 https://doi.org/10.3389/fmars.2018.00101.
Webster NS, Negri AP, Botté ES, Laffy PW, Flores F, Noonan S, et al. Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci Rep. 2016;6:19324 https://doi.org/10.1038/srep19324.
Grottoli AG, Dalcin Martins P, Wilkins MJ, Johnston MD, Warner ME, Cai W-J, et al. Coral physiology and microbiome dynamics under combined warming and ocean acidification. PLoS One. 2018;13:e0191156 https://doi.org/10.1371/journal.pone.0191156.
Jessen C, Villa Lizcano JF, Bayer T, Roder C, Aranda M, Wild C, et al. In-situ effects of eutrophication and overfishing on physiology and bacterial diversity of the Red Sea coral Acropora hemprichii. PLoS One. 2013;8:e62091 https://doi.org/10.1371/journal.pone.0062091.
Carlos C, Torres TT, Ottoboni LMM. Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci Rep. 2013;3:1–7 https://doi.org/10.1038/srep01624.
Lee OO, Yang J, Bougouffa S, Wang Y, Batang Z, Tian R, et al. Spatial and species variations in bacterial communities associated with corals from the Red Sea as revealed by pyrosequencing. Appl Environ Microbiol. 2012;78:7173–84 https://doi.org/10.1128/AEM.01111-12.
Morrow KM, Moss AG, Chadwick NE, Liles MR. Bacterial associates of two caribbean coral species reveal species-specific distribution and geographic variability. Appl Environ Microbiol. 2012;78:6438–49.
Neave MJ, Rachmawati R, Xun L, Michell CT, Bourne DG, Apprill A, Voolstra CR. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2017;11(1):186–200.
Nelson CE, Goldberg SJ, Wegley Kelly L, Haas AF, Smith JE, Rohwer F, et al. Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J. 2013;7:962–79 https://doi.org/10.1038/ismej.2012.161.
Meikle P, Richards GN, Yellowlees D. Structural investigations on the mucus from six species of coral. Mar Biol. 1988;99:187–93 https://doi.org/10.1007/BF00391980.
Ducklow HW, Mitchell R. Composition of mucus released by coral reef coelenteratesl. Limnol Oceanogr. 1979;24:706–14.
Lee STM, Davy SK, Tang S-L, Kench PS. Mucus sugar content shapes the bacterial community structure in thermally stressed Acropora muricata. Front Microbiol. 2016;7:371 https://doi.org/10.3389/fmicb.2016.00371.
Hadaidi G, Gegner HM, Ziegler M, Voolstra CR. Carbohydrate composition of mucus from scleractinian corals from the central Red Sea. Coral Reefs. 2019;38(1):21–7.
Leite DCA, Leão P, Garrido AG, Lins U, Santos HF, Pires DO, et al. Broadcast spawning coral Mussismilia hispida can vertically transfer its associated bacterial Core. Front Microbiol. 2017;8:176 https://doi.org/10.3389/fmicb.2017.00176.
Thompson JR, Rivera HE, Closek CJ, Medina M. Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front Cell Infect Microbiol. 2015;4:176 https://doi.org/10.3389/fcimb.2014.00176.
Sharp KH, Distel D, Paul VJ. Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J. 2012;6:790–801 https://doi.org/10.1038/ismej.2011.144.
Ceh J, Kilburn MR, Cliff JB, Raina J-BB, Van Keulen M, Bourne DG. Nutrient cycling in early coral life stages: Pocillopora damicornis larvae provide their algal symbiont (Symbiodinium) with nitrogen acquired from bacterial associates. Ecol Evol. 2013;3(8):2393–400 https://onlinelibrary.wiley.com/doi/full/10.1002/ece3.642.
Neave MJ, Apprill A, Ferrier-Pagès C, Voolstra CR. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol. 2016;100:8315–24 https://doi.org/10.1007/s00253-016-7777-0.
Arndt D, Xia J, Liu Y, Zhou Y, Guo AC, Cruz JA, et al. METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012;40.
Raina J-B, Tapiolas D, Willis BL, Bourne DG. Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol. 2009;75:3492–501 https://doi.org/10.1128/AEM.02567-08.
Bourne DG, Dennis PG, Uthicke S, Soo RM, Tyson GW, Webster N. Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J. 2013;7:1452–8 https://doi.org/10.1038/ismej.2012.172.
Raina JB, Tapiolas DM, Forêt S, Lutz A, Abrego D, Ceh J, et al. DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature. 2013;502:677–80.
Gardner SG, Nielsen DA, Laczka O, Shimmon R, Beltran VH, Ralph PJ, et al. Dimethylsulfoniopropionate, superoxide dismutase and glutathione as stress response indicators in three corals under short-term hyposalinity stress. Proc R Soc B Biol Sci. 2016;283:20152418 https://doi.org/10.1098/rspb.2015.2418.
Hopkins FE, Bell TG, Yang M, Suggett DJ, Steinke M. Air exposure of coral is a significant source of dimethylsulfide (DMS) to the atmosphere. Sci Rep. 2016;6:36031 https://doi.org/10.1038/srep36031.
Sunda W, Kieber DJ, Kiene RP, Huntsman S. An antioxidant function for DMSP and DMS in marine algae. Nature. 2002;418:317–20.
Hadaidi G, Röthig T, Yum LK, Ziegler M, Arif C, Roder C, et al. Stable mucus-associated microbial communities in bleached and healthy corals of Porites lobata from the Arabian seas. Sci Rep. 2017;7:45362 https://doi.org/10.1038/srep45362.
Shnit-Orland M, Sivan A, Kushmaro A. Antibacterial activity of Pseudoalteromonas in the coral Holobiont. Microb Ecol. 2012;64:851–9 https://doi.org/10.1007/s00248-012-0086-y.
Holmström C, Kjelleberg S, Baumann L, Baumann P, Mandel M, Allen RD, et al. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol Ecol. 1999;30:285–93 https://doi.org/10.1111/j.1574-6941.1999.tb00656.x.
Wright RM, Kenkel CD, Dunn CE, Shilling EN, Bay LK, Matz MV. Intraspecific differences in molecular stress responses and coral pathobiome contribute to mortality under bacterial challenge in Acropora millepora. Sci Rep. 2017;7:2609.
Denner EBM, Vybiral D, Koblízek M, Kämpfer P, Busse H-J, Velimirov B. Erythrobacter citreus sp. nov., a yellow-pigmented bacterium that lacks bacteriochlorophyll a, isolated from the western Mediterranean Sea. Int J Syst Evol Microbiol. 2002;52(Pt 5):1655–61 https://doi.org/10.1099/00207713-52-5-1655.
Koblízek M, Béjà O, Bidigare RR, Christensen S, Benitez-Nelson B, Vetriani C, et al. Isolation and characterization of Erythrobacter sp. strains from the upper ocean. Arch Microbiol. 2003;180:327–38 https://doi.org/10.1007/s00203-003-0596-6.
Yurkov V, Stackebrandt E, Holmes A, Fuerst JA, Hugenholtz P, Golecki J, et al. Phylogenetic positions of novel aerobic, bacteriochlorophyll a-containing bacteria and description of Roseococcus thiosulfatophilus gen. nov., sp. nov., Erythromicrobium ramosum gen. nov., sp. nov., and Erythrobacter litoralis sp. nov. Int J Syst Bacteriol. 1994;44:427–34.
Yuki ST, Hamasaki K, Suzuki K. Photosynthetic characteristics of marine aerobic anoxygenic phototrophic bacteria Roseobacter and Erythrobacter strains. Arch Microbiol. 2012;194:331–41.
Fiedor J, Burda K. Potential role of carotenoids as antioxidants in human health and disease. Nutrients. 2014;6:466–88.
Diaz JM, Hansel CM, Apprill A, Brighi C, Zhang T, Weber L, et al. Species-specific control of external superoxide levels by the coral holobiont during a natural bleaching event. Nat Commun. 2016;7:13801 https://doi.org/10.1038/ncomms13801.
Kuguru B, Winters G, Beer S, Santos SR, Chadwick NE. Adaptation strategies of the corallimorpharian Rhodactis rhodostoma to irradiance and temperature. Mar Biol. 2007;151:1287–98 https://doi.org/10.1007/s00227-006-0589-5.
Gaither MR, Szabó Z, Crepeau MW, Bird CE, Toonen RJ. Preservation of corals in salt-saturated DMSO buffer is superior to ethanol for PCR experiments. Coral Reefs. 2011;30:329–33.
Mieog JC, van Oppen MJH, Berkelmans R, Stam WT, Olsen JL. Quantification of algal endosymbionts ( Symbiodinium ) in coral tissue using real-time PCR. Mol Ecol Resour. 2009;9:74–82 https://doi.org/10.1111/j.1755-0998.2008.02222.x.
Arif C, Daniels C, Bayer T, Banguera-Hinestroza E, Barbrook A, Howe CJ, et al. Assessing Symbiodinium diversity in scleractinian corals via next-generation sequencing-based genotyping of the ITS2 rDNA region. Mol Ecol. 2014;23:4418–33 https://doi.org/10.1111/mec.12869.
Bongaerts P, Sampayo EM, Bridge TCL, Ridgway T, Vermeulen F, Englebert N, et al. Symbiodinium diversity in mesophotic coral communities on the great barrier reef: a first assessment. Mar Ecol Prog Ser. 2011;439:117–26 https://doi.org/10.3354/meps09315.
Santos SR, Taylor DJ, Coffroth MA. Genetic comparisons of freshly isolated versus cultured symbiotic dinoflagellates: implications for extrapolating to the intact symbiosis. J Phycol. 2001;37:900–12 https://doi.org/10.1046/j.1529-8817.2001.00194.x.
LaJeunesse T, Trench R. Biogeography of two species of Symbiodinium (Freudenthal) inhabiting the intertidal sea anemone Anthopleura elegantissima (Brandt). Biol Bull. 2000;199:126–34 https://doi.org/10.2307/1542872.
LaJeunesse TC. Diversity and community structure of symbiotic dinoflagellates from Caribbean coral reefs. Mar Biol. 2002;141:387–400 https://doi.org/10.1007/s00227-002-0829-2.
Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol. 2000;66:5488–91.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46 https://doi.org/10.1111/j.1442-9993.2001.tb00081.x.
Clarke KR. Non-parametric multivariate analyses of changes in community structure: Blackwell Publishing Ltd; 1993. https://doi.org/10.1111/j.1442-9993.1993.tb00438.x.
Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at https://github.com/najoshi/sickle, vol. 2011; 2011.
Nikolenko SI, Korobeynikov AI, Alekseyev MA. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14(Suppl 1):S7 https://doi.org/10.1186/1471-2164-14-S1-S7.
Nurk S, Bankevich A, Antipov D, Gurevich A, Korobeynikov A, Lapidus A, et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); 2013. p. 158–170.
Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina paired-end reAd mergeR. Bioinformatics. 2014;30:614–20.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31 https://doi.org/10.1186/1471-2105-13-31.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6 https://doi.org/10.1038/nmeth.f.303.
Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(27) https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-017-0237-y.
Shapiro SS, Francia RS. An approximate analysis of variance test for normality. J Am Stat Assoc. 1972;67:215–6 https://doi.org/10.2307/2284728.
De Cáceres M, Legendre P. Associations between species and groups of sites: indices and statistical inference. Ecology. 2009;90:3566–74 https://doi.org/10.1890/08-1823.1.
Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017.
R Developement Core Team. R: A Language and Environment for Statistical Computing. R Found Stat Comput. 2015;1:409 https://doi.org/10.1007/978-3-540-74686-7.
Acknowledgements
The authors would like to thank Dr. Rasha Sharaf, the Egyptian Culture Bureau in London, who facilitated travel and paperwork. Samples were collected under the Red Sea National Park authority permits, and authors would specially thank Ahmad Ghallab, Tamer Kamal, Yasser Saied, Ayman Gharabawy, Ahmed El-Sadek, Ismail Abdelazeem, Khaled El-Haddad, Emad El-Aydi, and Dr. Mahmoud Maaty for logistics support and sampling help. Also, thanks to Prof. Mohamed Abdel Al-Wahab, National Institute of Oceanography and Fishers (NOIF), Hurghada, for facilitating logistics.
Funding
This work is primarily funded by Egyptian Ministry of Higher Education, Cultural Affairs and Missions Sector, PhD grant to E.O. Osman. Also, the contribution of D.J. Suggett was funded through an Australian Research Council Discovery Project (DP160100271). Research was further supported by baseline funds to C.R. Voolstra from the King Abdullah University of Science and Technology (KAUST) and funds to M.E. Warner by the National Science Foundation (grant no.1258065 and 1635695).
Author information
Authors and Affiliations
Contributions
EOO, DJSmith, and DJSuggett designed the experiment. EOO conducted fieldwork and sampling. EOO, MEW, EMS and DTP contributed to symbiont identification lab work and analysis, while EOO, CRV, CP, and DRC worked on bacterial community, bioinformatics, and analysis. EOO wrote the first draft with all other authors editing and approving the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Novel ITS2 Sequances.
Additional file 2:
Supplementary methods, figures and tables.
Additional file 3.
Bacterial OTU table.
Additional file 4.
Core microbiome.
Additional file 5.
Normalization and-beta-diversity.
Additional file 6.
Endoymbiont DGGE fingerprints.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Osman, E.O., Suggett, D.J., Voolstra, C.R. et al. Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome 8, 8 (2020). https://doi.org/10.1186/s40168-019-0776-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-019-0776-5