
Abstract
Background
Soil bacteria naturally produce antibiotics as a competitive mechanism, with a concomitant evolution, and exchange by horizontal gene transfer, of a range of antibiotic resistance mechanisms. Surveys of bacterial resistance elements in edaphic systems have originated primarily from human-impacted environments, with relatively little information from remote and pristine environments, where the resistome may comprise the ancestral gene diversity.
Methods
We used shotgun metagenomics to assess antibiotic resistance gene (ARG) distribution in 17 pristine and remote Antarctic surface soils within the undisturbed Mackay Glacier region. We also interrogated the phylogenetic placement of ARGs compared to environmental ARG sequences and tested for the presence of horizontal gene transfer elements flanking ARGs.
Results
In total, 177 naturally occurring ARGs were identified, most of which encoded single or multi-drug efflux pumps. Resistance mechanisms for the inactivation of aminoglycosides, chloramphenicol and β-lactam antibiotics were also common. Gram-negative bacteria harboured most ARGs (71%), with fewer genes from Gram-positive Actinobacteria and Bacilli (Firmicutes) (9%), reflecting the taxonomic composition of the soils. Strikingly, the abundance of ARGs per sample had a strong, negative correlation with species richness (r = − 0.49, P < 0.05). This result, coupled with a lack of mobile genetic elements flanking ARGs, suggests that these genes are ancient acquisitions of horizontal transfer events.
Conclusions
ARGs in these remote and uncontaminated soils most likely represent functional efficient historical genes that have since been vertically inherited over generations. The historical ARGs in these pristine environments carry a strong phylogenetic signal and form a monophyletic group relative to ARGs from other similar environments.
Similar content being viewed by others


Background
Antibiotic production and resistance have ancient origins (~ 2 Gyr: [1, 2]). While the spread of antibiotic resistance is one of the preeminent challenges facing global public health in the twenty-first century, antibiotic resistance mechanisms predate anthropogenic antibiotic use. Currently, efforts to restrict the wide-spread dissemination of antibiotic resistance have focused on increasing the repertoire of antibiotics in clinics and by exploring both the origins and mechanisms of antibiotic resistance in soil environments [3, 4]. Understanding the sources of antibiotic resistance genes (ARGs) in remote natural environments which have minimal anthropogenic input may be of benefit in tracking the evolution of resistant pathogens [5].
The three major mechanisms of bacterial antibiotic resistance are broadly categorised as efflux pumps, resistance mutations and antibiotic inactivation strategies [6, 7]. Drug-specific efflux pumps have narrow ranges of antibiotic export mechanism and are specific to a particular antibiotic (e.g. in the removal of tetracycline [8]), whereas multidrug efflux pumps have broad specificity and confer resistance to multiple antibiotics. Resistance to nalidixic acid (a synthetic quinolone antibiotic) is conferred to cells with a mutated gyrA gene, producing a genotype that is recessive to the wild-type [9]. Common drug inactivation mechanisms in soil communities include β-lactamases, which hydrolyse β-lactam antibiotics such as penicillin [10, 11].
Resistance genes are often encoded on the same gene cluster as antibiotic biosynthesis pathway genes [12], conferring resistance in antibiotic-producing species to the products they synthesise [13]. Resistance determinants are acquired through natural selection, such as after exposure to an antibiotic at sub-therapeutic concentrations [14] and through the horizontal transfer of genomic material between species [15]. Some ARGs that are likely to be exchanged between species are typically associated with transposons or other mobile genetic elements (MGE) such as plasmids or integrons [6]. Extracellular DNA harbouring ARGs can persist in soil matrices, by cation binding, for example, facilitating genomic transfer between members of the soil community [16]. Moreover, the persistence of DNA in frigid environments such as permafrost and cold or polar deserts is facilitated by low temperatures that reduce the activity of endoenzymes that degrade cells and their DNA [17].
Soils are environmental reservoirs of ARGs and are sources of resistance in human pathogens [5, 18]. Anthropogenic practices that release antibiotics and/or components of the human microbiome into the environment are significant sources of global ARG proliferation [19, 20]. However, the spectrum of historical (i.e. not of anthropogenic origin) resistance determinants in the soil resistome remains largely unknown, with extreme and remote soil ecosystems particularly underrepresented.
Microbial communities from remote and pristine soils provide a valid genetic resource for exploring the historical evolutionary origins of natural antibiotic resistance from the pre-antibiotic era [6, 21]. Soils with minimum anthropogenic contamination and anthropogenic-induced selection pressures should reflect only the spectrum of natural antibiotics and their cognate resistance mechanisms [1] with little or no genetic impact from selection pressures introduced by the input of the twentieth century synthetic and semi-synthetic antibiotics [22].
Very few studies have explored antibiotic resistance in so-called pristine soils. Soils from a remote Alaskan environment showed abundant β-lactamases [10], while genes encoding resistance to tetracycline and glycopeptide antibiotics were found in ancient permafrost DNA and isolated cave samples contained multiple antibiotic resistance genes for macrolide glycosylation [1, 23]. However, very little is known about the abundance or diversity of ARGs in pristine Antarctic soils, which represent some of the few remaining environmental niches which are essentially undisturbed by human activity. Antibiotic resistance elements have been found in Antarctic seawater [24, 25] and in McMurdo Dry Valley soils [26], although these sites cannot be considered ‘pristine’ due to the on-going and long-term research programs carried out in these regions. Here we used a metagenomic-based approach to identify the natural diversity of antibiotic resistance genes in silico from remote and pristine Antarctic soils of the Mackay Glacier region. These sites have no known exposure to anthropogenic antibiotics, are accessible to a few researchers, and can be validly considered to be ‘antibiotic naïve’.
Methods
Sites, sampling and physicochemical analysis
Surface soil samples were collected in January 2015 from 17 sites in ice-free areas in the vicinity and to the north of Mackay Glacier, South Victoria Land, Antarctica, which spans ~ 100 km (Fig. 1). Five aliquots of 50 g soil (0–5 cm depth; sieved to 2 mm mesh size) were collected from approximately 1 m2 area at each sampling site using sterile methods. Samples were stored in sterile 50 ml polypropylene Falcon tubes (Grenier, Bio-One) at below 0 °C in the field and during transport to the University of Pretoria (South Africa). Soils were analysed for soil pH, total nitrogen, carbon, phosphorus and major cations (K+, Na+, Ca2+ and Mg2+) at the Stellenbosch Central Analytical Facilities, Stellenbosch University, South Africa, using standard quality control procedures [27]. Elemental analysis was performed using a LECO TruSpec® Elemental Determinator by combustion analysis. X-ray fluorescence spectrometry for major cations was performed on a Philips PW1404 XRF. Soil pH was measured using 2.5:1 (mass:volume) soil suspensions in deionised water.

Satellite image of the Mackay Glacier ecotone with the 17 sampling sites indicated. Source: Landsat Image Mosaic of Antarctica (LIMA) Digital Database and Google Earth
Sample preparation and DNA sequencing
Metagenomic DNA was extracted from each soil sample aliquot in duplicate using an established buffer-chloroform/phenol protocol [28]. Samples with the highest DNA concentration and purity from each site (n = 17) were submitted for sequencing at a commercial supplier (MR DNA Lab, Shallowater, TX, USA). Sequencing was performed on a HiSeq 2500 Ultra-High-Throughput Sequencing system (Illumina) using paired-ends (2 × 250 bp) for 500 cycles as per the manufacturers’ instructions.
Metagenomic assembly and ARG taxonomic identification
Raw reads were quality-filtered, trimmed and screened using Prinseq-lite v0.20.4 [29] in combination with in-house scripts. We used FLASH v1.2.11 (fast length adjustment of short reads) to align high-quality paired-end reads [30]. Sequences were de novo assembled using metaSPAdes v3.9.0 [31], as recommended [32]. The quality of each assembled metagenome (n = 17) was assessed using MetaQUAST v4.3 [33]. To provide taxonomic assignments, all contigs were compared to the entire NCBI protein non-redundant database using DIAMOND v0.7.9.58 at an E-value cutoff of 1 × 10− 5 [34]. The proportion of species-assigned ARGs at each site was calculated by dividing the number of ARG-containing species by the total number of species present in each community.
ARG database construction
A local non-redundant ARG database was created by concatenating the Antibiotic Resistance Genes Database (ARDB) [35] and the Comprehensive Antibiotic Resistance Database (CARD) [36]. The concatenated database, noradab (non-redundant antibiotic resistance database, is available online (noradab.bi.up.ac.za).
Resistome identification
Gene prediction for the 17 metagenomes was performed using Prodigal v2.6.3 [37] with the meta option specified. Genes predicted by Prodigal were compared against the local noradab database by means of BLASTp with an E value threshold of 1 × 10− 6. Results were filtered for hits with a minimum percentage identity of 75% and alignment length of at least 25 amino acids. For each predicted gene adhering to these parameters, only the hit with the highest score was annotated as an ARG. We used the Markov Cluster (MCL) algorithm to resolve ARG family redundancy [38]. All ARGs were compared against each other in an all-against-all BLASTp approach [39] with a cutoff E value of 1 × 10− 10. The resulting E values were used in the MCL algorithm to cluster ARGs into families with an inflation parameter set to 1.1 and log-transformed E values with a limit set at 100. ARG families were annotated according to the corresponding ARDB or CARD database descriptions for all members of the cluster [35, 36].
The relative abundances of ARGs were calculated as the total number of ARGs per sample divided by the total number of Prodigal predicted genes. The relative ARG family abundance was inferred by calculating the number of different ARG families represented by the number of different ARGs in the sample. The local non-redundant ARG database contained 4485 unique AR protein sequences.
Redundancy analysis
Redundancy analysis was performed in R v3.2.3 (R Foundation for Statistical Computing; https://www.r-project.org) with the vegan package (v2.4.0) [40]. ARG presence/absence transformed values and a nine-factor environmental dataset (soil pH, percentage C and N, P (ppm), K+ (mg/kg), Na+ (cmol(+)/kg), Ca2+ (cmol(+)/kg), Mg2+ (cmol(+)/kg) and site altitude) were used to evaluate the effect of soil abiotic features on ARG distribution across the environment.
Mobile genomic elements
To find evidence of mobile genetic elements (MGE) associated with ARGs, we extracted the amino acid sequences from all contigs that were predicted to harbour ARGs. These contigs, together with the peptides identified in all other contigs in the metagenomes, were then compared to the NCBI Conserved Domain Database (CDD)-COG database using reverse PSI-BLAST (RPS-BLAST). For all comparisons, an E value of 1 × 10− 3 was used to screen for the presence of COGs related to mobile genetic elements [41].
Network analysis
Co-occurrence networks were produced by obtaining Spearman correlation coefficients (ρ) from the bacterial and archaeal relative abundance data for genera present in all 17 communities against all ARGs found in each metagenome. Correlations with rho coefficients greater than ρ = 0.6, or below ρ = − 0.6, and with significant P values (P < 0.05) were included in the analysis. Cytoscape v.3.5.1 was used for network visualisation [42].
Phylogenetic analysis
Phylogenetic analyses were performed using three highly abundant ARG types from three distinct ARG families that were present in at least seven sites. For each site, all predicted proteins identified as the specific ARG and the noradab protein with which sequence similarity was ascertained were aligned, together with similarly annotated bacterial protein sequences from a diverse set of soil habitats obtained from the NCBI database online. Multiple sequence alignments were performed with MAFFT [43], and the resulting sequences were trimmed using trimAl [44]. Phylogenetic trees were constructed by RAxML [45] with 1000 bootstraps and automated substitution model selection. A description of the selected ARGs chosen from noradab is available in Additional file 1: Table S2.
Results and discussion
We compiled noradab by combining the existing ARDB and CARD databases to remove redundant sequences present in both repositories. ARDB is a widely used and informative collection of ARGs containing 7828 sequences. However, ARDB contains multiple redundant sequences [46, 47], in addition to three sequences in nucleic acid instead of amino acid format [47]. These nucleic acid sequences were removed, and the resulting 7825 protein sequences were inspected for redundancy. We found that 4826 of these sequences shared 100% similarity, as determined previously [47]. The resulting non-redundant ARDB database consisted of 2999 unique protein sequences.
The CARD protein homology sequences (December 2017 release) contain antimicrobial resistance genes but do not include mutations as resistance mechanisms. CARD contains 2169 protein sequences, of which 11 were redundant sequences. Overall, there were 664 shared sequences among the non-redundant ARDB and non-redundant CARD databases. As such, we constructed a non-redundant antibiotic resistance gene database—noradab—containing 4493 unique protein sequences with a description or header inclusive of all the descriptions found across all redundant sequences within ARDB or CARD. Clustering resulted in 140 ARG family clusters, which included 47 singletons.
Assembled shotgun metagenomic sequences (contigs) can be used effectively to access full-length antibiotic resistance genes (ARGs) in environmental resistomes [48]. In the 17 assembled Antarctic soil metagenomes, we identified an average of 265,000 open reading frames (ORFs) per metagenome, of which only 177 were annotated as potentially encoding antibiotic resistance. The low levels of ARGs in the soil metagenomes may be due to the stringent selection parameters implemented here (see the “Methods” section) but are more likely to reflect the very low level of anthropogenic impact, and the effective absence of an antibiotic burden, on these soils. The low proportion of ARGs (c.f., total ORFs) is comparable to previous estimates of resistance genes in paddy field soils [49], temperate soils [50] and glacial cores and surface snow samples [21]. Globally, hot and cold desert soil metagenomes are characterised by a lower proportion of ARGs (1.5% of annotated reads) compared to temperate metagenomes (4.8%), which is thought to indicate reduced competition between members of desert soil communities [51].
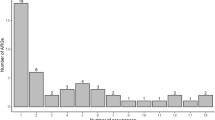
In total, the identified ARGs spanned 23 ARG families (Table 1) and represent all known generic mechanisms of antibiotic resistance (resistance mutations, antibiotic efflux and antibiotic inactivation [7]). The distribution of ARGs was highly variable across the remote, pristine sites, ranging from 2 (sites MS2-2 and MS3-5) to 16 (sites MGM-3 and MS5-1) ARGs per site (Fig. 2). This variable dispersal extended to the frequency of unique ARGs per site, with MGM-3 (n = 7) and MS5-1 (n = 6) containing the most unique ARGs (Table 2). The relative abundance of ARGs was highest in sample CN-4 and the lowest in samples BG12-3 and MS2-2.

ARG frequencies across sampled sites. Number of different ARGs indicated in green, with the number of unique ARGs displayed in red, axis on the left. The relative ARG abundances are shown as a black line, axis on the right
We found a strong linear correlation between the number of unique ARGs and the number of unique taxa in each community (Pearson’s correlation, r = 0.89, P < 1.62e−06; Additional file 1: Figure S1). This trend supports a common feature in ecology, whereby higher levels of biodiversity are generally reflected by greater functional heterogeneity [52]. In contrast to other Antarctic regions, such as McMurdo Dry Valley soils [26] and glacial cores [21], we found a very diverse set of ARGs. This may be due to the sensitivity of our approach compared to functional screening, for example [10, 53]. An alternative reason for this difference could be due to antibiotic consumption by heterotrophic bacteria in these hyperoligotrophic soils (C and N concentrations were near accurate detection limits; Additional file 1: Table S1). Bacteria are known to be capable of surviving on a limited number of antibiotics, even using these inhibitory molecules as sole carbon sources [54], and a history of heterotrophic antibiotic degradation might minimise the pressure for ARG evolution.
We predicted the source phylum of each ARG using comparisons to the entire NCBI protein non-redundant database (Fig. 3). Most ARGs (126 of 177; 71%) belonged to Gram-negative bacteria, while 35 ARGs (20%) could not be confidently classified beyond the kingdom level. A further 16 ARGs (9%) were assigned to Gram-positive bacteria, exclusively the Bacilli (Firmicutes) and Actinobacteria. The Gram-negative Bacteroidetes (41 ARGs) and Acidobacteria (37 ARGs) were the primary sources of ARGs across all sites. Together, these two phyla accounted for just under half of the ARGs found (78 out of 177) and both harboured all major antibiotic resistance strategies (Fig. 3). Bacterial phyla encoding multiple resistance mechanisms included Proteobacteria (30 ARGs), Firmicutes (16 ARGs), Cyanobacteria (8 ARGs) and Actinobacteria (5 ARGs). By contrast, Nitrospira (2 ARGs), Chlorobi (1 ARG), Gemmatimonadetes (1 ARG) and Marinimicrobia (1 ARG) contributed very few ARGs, which mirrored their low abundance within this environment [55].

Co-occurrence network of ARG mechanisms showing resistance mechanisms encoded by diverse soil bacterial phyla. Phyla from all 17 soils that were assigned an ARG are presented here (diamond-shaped nodes), with significant co-occurrences with a specific ARG (circles) indicated (edges)
Many clinical pathogens are members of the Proteobacteria [56]. Proteobacteria have been shown to harbour the greatest number of ARGs in different soil niches, reflecting their taxonomic dominance in those samples [26, 50, 54]. Similarly, the most abundant prokaryotic taxa in the Mackay Glacier soils, Bacteroidetes and Acidobacteria, contributed the most ARGs overall. This contrasts with the phylogenetic affiliations of ARGs in the human gut, for example, which were primarily assigned to abundant Firmicutes [57]. Overall, we support the contention that ARG distribution broadly reflects community membership [51]. In addition, our results serve to highlight the prevalence of ARGs in bacteria with large genomes (larger than 6 Mb) [54].
The two most abundant ARG families identified were undecaprenyl pyrophosphate phosphatases (UppP) and genes for efflux/transporter systems. We found that UppP genes, which confer resistance to Bacitracin, were assigned to the three most abundant bacterial phyla in these communities, Bacteroidetes (9 ARGs), Acidobacteria (7 ARGs) and Proteobacteria (3 ARGs), while a large proportion could not be assigned to a known taxonomic group (7 ARGs). These genes were present in 12 of the 17 metagenomes, suggesting that this is a relatively common mechanism of antibiotic resistance in soil communities, which extend to globally distributed soil biomes including forests, tundra and grassland ecosystems [50, 58].
Efflux transporters, which mediate the export of antibiotics across the cell surface and reduce intracellular antibiotic load [8], are some of the most common mechanisms of resistance in microorganisms [6]. ORFs annotated as efflux mechanisms were found in 13 of the 17 metagenomes, confirming that this is a common resistance strategy in soil bacterial populations. The most common transporter genes in these samples were the ancient super-family, adenosine triphosphate (ATP)-binding cassette (ABC) efflux/transporters [59]. We found a total of 50 ARGs assigned as ABC transporters (Table 3). ABC transporters couple ATP hydrolysis to solute efflux to actively transport compounds across the cell membrane [60]. We also found the rosA gene in many samples (19 ARGs), the product of which is an antiporter efflux pump. ARGs encoding macrolide transporter ATP-binding/permease proteins were some of the most common exporters and were present in 9 of the 17 soil resistomes. Tetracycline efflux pumps were uncommon (8 ARGs), and we found no other efflux pumps specific to a single class of antibiotics. Some communities possessed MDS (membrane-spanning domain) efflux pumps (2 ARGs) and multidrug efflux pumps (11 ARGs), which have the potential to remove a broad spectrum of antibiotics from the cell.
Genes involved in antibiotic inactivation strategies were less common than either UppP genes or efflux pumps in the Antarctic soil metagenomes. Genes encoding β-lactamases, which confer resistance to β-lactam antibiotics by intracellular enzymatic degradation, are the most common antibiotic-inactivating ARGs in most microbial communities [2]. Surprisingly, we found a very low abundance of β-lactamases, despite identifying class A, B and C β-lactamase genes in a single sample (site MGM-3). This is in contrast with a recent study, based on GeoChip analysis, that reported a high diversity of β-lactamase genes in Antarctic McMurdo Dry Valley soil and lithic niches [26]. β-Lactamase genes are common components of the temperate soil resistome and have also recently been found at low levels in undisturbed cold soil ecosystems such as Arctic permafrost [11], glacial ice cores [21] and Alaskan soils [10, 61]. We note that the Mackay Glacier region soil samples used in our study come from much more remote sites than those of the McMurdo Dry Valley studies and are much less likely to have been exposed to anthropogenic input.
Interestingly, we found that rifampicin phosphorylation (8 ARGs) and erythromycin inactivation (1 ARG) were unique resistance mechanisms in Gram-positive bacteria, including members of the Bacillus spp. which are known to be resistant to aminoglycosides in the natural environment [11]. Gram-negative bacteria also harboured exclusive antibiotic inactivation mechanisms, such as genes for modifying naturally occurring antibiotics such as novobiocin (8 ARGs) and chloramphenicol (6 ARGs). Unexpectedly, we found a single ARG encoding resistance to trimethoprim, a synthetic antibiotic that inhibits DNA synthesis (Fig. 3). This finding supports the contention that aerosol transport within and to the Antarctic continent can result in the introduction of non-indigenous microorganisms [62, 63], serving as a mechanism for the introduction of antibiotic resistance genes into remote and pristine soil communities [5]. An alternative explanation for this finding could be the presence of thymidylate synthetase (thyE), which converts deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), at the 5′ end of the drfA sequence that encodes resistance to trimethoprim.
While we identified ARGs belonging to all the generic mechanisms of antibiotic resistance, we identified only 23 families of the 140 ARG families present in the concatenated noradab database, in all 17 metagenomes. Many of the ‘missing’ ARG families were those involved in the inactivation of synthetic antibiotics, such as Florfenicol and semi-synthetic derivatives such as Ciprofloxacin, although resistance genes for some natural antibiotics, such as Gentamycin, were not identified. We argue that the absence of ARGs targeting synthetic antibiotics reflects the pristine nature of the soil communities. This is entirely consistent with the observation that agricultural soil communities, that have been impacted by human and animal activities and high concentrations of antibiotics over relatively long periods, contain resistance genes for numerous synthetic antibiotics [48].
Our data showed substantial differences in AR families for different sample sites (Table 1, Fig. 4). The number of different ARG families in each community also varied considerably. For example, site MGM-3 contained a total of 11 different AR families, whereas MS2-2 and MS3-5 were the most ARG naïve, with only two ARG families in each community (Additional file 1: Figure S2). The number of unique ARG families per site did not exceed two, indicating shared resistance strategies across all communities. Overall, nine ARG families were present in five or more of the communities (Table 3). Only five of the sites contained a unique ARG family (Table 4).

ARG family frequencies across sampled sites. The number of different ARG families are indicated in green, with the number of unique ARG families displayed in red, axis on the left. The black line represents the relative ARG family abundance, axis on the right
To explain the differences in ARG distribution between soil communities across the range of sample sites, we used a redundancy analysis (RDA) based on environmental physicochemical parameters (Additional file 1: Table S1). The diversity of ARGs was significantly driven by the percentage soil nitrogen (N) (P < 0.03; Additional file 1: Figure S3). No significant trends related to abiotic features were found for ARG families (data not shown), which may reflect the loss of discriminatory power at broader gene classification levels. Although most of the environmental factors measured here did not significantly influence the resistome family portfolio of individual sites, our observation that soil N influenced ARG composition in these hyperoligotrophic soils is consistent with studies showing significant differences in soil diversity and ARG composition resulting from N fertilisation [64, 65].
Strikingly, the number of ARGs per sample showed a significant negative correlation with the number of species per site (Pearson’s correlation; r = − 0.49, P < 0.05, Fig. 5). This is an interesting and novel result and might be attributed to the competitive exclusion of species by antibiotic exposure, potentially via the inhibition of the growth and activity of competing species [66]. This could favour the selection of pre-existing genotypes rather than leading to the development and acquisition of novel resistance mechanisms, as previously proposed [65]. However, if antibiotic concentrations do not reach levels of inhibition, as indicated in many soil environments [6], antibiotic production may instead serve to disrupt cellular signalling by acting as signal quenchers [67].

The number of ARGs and number of species per site. Linear model indicated in red and lowess in blue (Pearson’s correlation r = − 0.49, P < 0.05)
Interestingly, the contigs containing ARGs did not possess any flanking regions that shared similarities with the COG mobilomes categories. However, many contigs which did not possess an ARG were enriched with genes that shared similarities with a variety of transposons, phage, integrase and plasmid elements (n = 91,408). This result indicates that although contigs with ARGs lacked traces of mobile genetic elements, there is evidence of the capacity for substantial horizontal gene transfer in all 17 pristine Antarctic soil metagenomes, due to the presence of multiple mobilome-associated genes.
In order to shed light on the evolutionary history of the Antarctic soil resistome, we interrogated the phylogenetic placement of the three most abundant ARG classes, with respect to other environmental ARG sequences (Fig. 5), including cold and hot desert soil communities and many temperate soil biomes. Our analysis showed distinct clustering of our sequences from all other environmentally derived ARGs for all three resistance mechanisms, i.e. dihydrofolate reductase (drfE) genes (Fig. 6a), macrolide transporter ATP-binding permease proteins (Fig. 6b), and major facilitator superfamily transporter sequences (rosA) (Fig. 6c). Together, these results provide strong evidence that ARG homologues found in these remote and uncontaminated Antarctic soils represent legacy genes that were acquired, or evolved, in the distant past that over time formed part of the essential gene pools and have undergone niche-specific selective pressure. We argue that the low similarity to modern ARG variants could reflect either parallel evolutionary processes or the outward transport of historical ARGs as templates for subsequent evolution in more temperate environments. Phylogenetic analyses have placed the origins of some β-lactamase genes at over 2 billion years ago [2].

Unrooted Bayesian phylogeny of microbial antibiotic resistance gene sequences identified across the 17 Antarctic soil metagenomes. a Dihydrofolate reductase (drfE) genes, b Macrolide transporter ATP-binding permease proteins, and c major facilitator superfamily transporter sequences (rosA) are shown; blue denotes ORFs identified in the Antarctic metagenomes. Reference sequences are provided with accession numbers, and the protein sequence present in noradab is shown in orange
Conclusions
Many studies have explored the distribution of antibiotic resistance in natural environments; however the spectrum of resistance determinants in remote pristine soil communities had not yet been elucidated. This is the first detailed metagenomic study of antibiotic resistance genes in remote, pristine soils that are naïve to anthropogenic antibiotic use. We have found 177 naturally occurring, historical genes conferring resistance to natural antibiotics. Our theory that most antibiotic resistance genes found here originate primarily from antibiotic-producing species was supported by the presence of antibiotic biosynthesis genes in many phyla encoding resistance and is further highlighted by the complete absence of mobile genetic elements (transposons, integrons and recombinases) flanking all putative ARGs found here. Antibiotic resistance appears to be transferred vertically over generations, with limited to no horizontal movement of ARGs between species. Thus, community members with antibiotic resistance may proliferate at the expense of susceptible counterparts [66]. This evidence supports the concept [65] that phylogeny, rather than HGT, drives differences in soil resistome content in the environment. This is entirely consistent with our finding that ARG abundance is negatively correlated with inferred species richness, indicating that communities with a high proportion of resistance elements exclude susceptible species, subsequently reducing soil diversity. Future challenges include understanding how both direct and indirect human-induced modulations influence the composition of these distinct soil communities, likely resulting in alterations to soil resistome properties.
Abbreviations
- ABC:
-
Adenosine triphosphate (ATP)-binding cassette
- ARBD:
-
Antibiotic Resistance Genes Database
- ARG:
-
Antibiotic resistance gene
- BG:
-
Benson Glacier
- CARD:
-
Comprehensive Antibiotic Resistance Database
- CN:
-
Cliff Nunatak
- HGT:
-
Horizontal gene transfer
- MG:
-
Mackay Glacier
- MGE:
-
Mobile genetic elements
- MGM:
-
Mount George Murray
- MS1–MS7:
-
Mount Seuss
- MtG:
-
Mount Gran
- Myr:
-
Million years
- noradab :
-
Non-redundant antibiotic resistance database
- PT:
-
Pegtop Mountain
- RDA:
-
Redundancy analysis
- TG:
-
Towle Glacier
References
D'costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R. Antibiotic resistance is ancient. Nature. 2011;477(7365):457.
Hall BG, Barlow M. Evolution of the serine β-lactamases: past, present and future. Drug Resist Updat. 2004;7(2):111–23.
Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8(4):260–71.
WHO. Antimicrobial resistance: global report on surveillance 2014. Geneva: WHO; 2014.
Cytryn E. The soil resistome: the anthropogenic, the native, and the unknown. Soil Biol Biochem. 2013;63:18–23.
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol. 2010;8(4):251–9.
Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406(6797):775–81.
Li X-Z, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs. 2004;64(2):159–204.
Hane MW, Wood TH. Escherichia coli K-12 mutants resistant to nalidixic acid: genetic mapping and dominance studies. J Bacteriol. 1969;99(1):238–41.
Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil. ISME J. 2009;3(2):243–51.
Perron GG, Whyte L, Turnbaugh PJ, Goordial J, Hanage WP, Dantas G, Desai MM. Functional characterization of bacteria isolated from ancient arctic soil exposes diverse resistance mechanisms to modern antibiotics. PLoS One. 2015;10(3):e0069533.
Martin JF, Liras P. Organization and expression of genes involved in the biosynthesis of antibiotics and other secondary metabolites. Annu Rev Microbiol. 1989;43(1):173–206.
Hopwood DA. How do antibiotic-producing bacteria ensure their self-resistance before antibiotic biosynthesis incapacitates them? Mol Microbiol. 2007;63(4):937–40.
Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. Antibiotic tolerance facilitates the evolution of resistance. Science. 2017; https://doi.org/10.1126/science.aaj2191.
Pärnänen K, Karkman A, Tamminen M, Lyra C, Hultman J, Paulin L, Virta M. Evaluating the mobility potential of antibiotic resistance genes in environmental resistomes without metagenomics. Sci Rep. 2016;6:35790.
Levy-Booth DJ, Campbell RG, Gulden RH, Hart MM, Powell JR, Klironomos JN, Pauls KP, Swanton CJ, Trevors JT, Dunfield KE. Cycling of extracellular DNA in the soil environment. Soil Biol Biochem. 2007;39(12):2977–91.
Pietramellara G, Ascher J, Borgogni F, Ceccherini M, Guerri G, Nannipieri P. Extracellular DNA in soil and sediment: fate and ecological relevance. Biol Fertil Soils. 2009;45(3):219–35.
Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337(6098):1107–11.
Zhu Y, Zhao Y, Li B, Huang C, Zhang S, Yu S, Chen Y, Zhang T, Gillings M, Su J. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat Microbiol. 2017;2:16270.
Wang H, Sangwan N, Li H-Y, Su J-Q, Oyang W-Y, Zhang Z-J, Gilbert JA, Zhu Y-G, Ping F, Zhang H-L. The antibiotic resistome of swine manure is significantly altered by association with the Musca domestica larvae gut microbiome. ISME J. 2017;11(1):100–11.
Segawa T, Takeuchi N, Rivera A, Yamada A, Yoshimura Y, Barcaza G, Shinbori K, Motoyama H, Kohshima S, Ushida K. Distribution of antibiotic resistance genes in glacier environments. Environ Microbiol Rep. 2013;5(1):127–34.
Martinez JL. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ Pollut. 2009;157(11):2893–902.
Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, Johnston MD, Barton HA, Wright GD. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One. 2012;7(4):e34953.
De Souza M-J, Nair S, Bharathi PL, Chandramohan D. Metal and antibiotic-resistance in psychrotrophic bacteria from Antarctic Marine waters. Ecotoxicology. 2006;15(4):379–84.
Miller RV, Gammon K, Day MJ: Antibiotic resistance among bacteria isolated from seawater and penguin fecal samples collected near Palmer Station, Antarctica This article is one of a selection of papers in the Special Issue on Polar and Alpine Microbiology. Can J Microbiol 2009, 55(1):37-45.
Wei ST, Higgins CM, Adriaenssens EM, Cowan DA, Pointing SB. Genetic signatures indicate widespread antibiotic resistance and phage infection in microbial communities of the McMurdo Dry Valleys, East Antarctica. Polar Biol. 2015;38(6):919–25.
Sparks DL, Page A, Helmke P, Loeppert R, Soltanpour P, Tabatabai M, Johnston C, Sumner M: Methods of soil analysis. Part 3-chemical methods: Soil Science Society of America Inc.; 1996.
Miller D, Bryant J, Madsen E, Ghiorse W. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol. 1999;65(11):4715–24.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–4.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017; https://doi.org/10.1101/gr.213959.116.
van der Walt AJ, van Goethem MW, Ramond J-B, Makhalanyane TP, Reva O, Cowan DA. Assembling metagenomes, one community at a time. BMC Genomics. 2017;18(1):521.
Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2015; https://doi.org/10.1093/bioinformatics/btv697.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60.
Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 2009;37(suppl 1):D443–7.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–73.
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;11(1):1.
Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–84.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Oksanen J, Blanchet FG, Kindt R, Oksanen MJ, Suggests M. Package ‘vegan’. Community Ecol. 2013;2:0.
Galperin MY, Makarova KS, Wolf YI, Koonin EV. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2014;43(D1):D261–9.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Katoh K, Misawa K, Kuma Ki MT. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Fitzpatrick D, Walsh F. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol Ecol. 2016;92(2):fiv168.
Yang Y, Jiang X, Chai B, Ma L, Li B, Zhang A, Cole JR, Tiedje JM, Zhang T. ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics. 2016;32(15):2346–51.
D'costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311(5759):374–7.
Xiao K-Q, Li B, Ma L, Bao P, Zhou X, Zhang T, Zhu Y-G. Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMS Microbiol Ecol. 2016;92(3):fiw023.
Noronha MF, Júnior GVL, Gilbert JA, de Oliveira VM. Taxonomic and functional patterns across soil microbial communities of global biomes. Sci Total Environ. 2017;609:1064–74.
Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci. 2012;109(52):21390–5.
Lefcheck JS, Byrnes JE, Isbell F, Gamfeldt L, Griffin JN, Eisenhauer N, Hensel MJ, Hector A, Cardinale BJ, Duffy JE. Biodiversity enhances ecosystem multifunctionality across trophic levels and habitats. Nat Commun. 2015;6:6936.
Walsh F, Duffy B. The culturable soil antibiotic resistome: a community of multi-drug resistant bacteria. PLoS One. 2013;8(6):e65567.
Dantas G, Sommer MO, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science. 2008;320(5872):100–3.
Adriaenssens EM, Kramer R, Van Goethem MW, Makhalanyane TP, Hogg I, Cowan DA. Environmental drivers of viral community composition in Antarctic soils identified by viromics. Microbiome. 2017;5(1):83.
Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12.
Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013;4:2151.
Nesme J, Cécillon S, Delmont TO, Monier J-M, Vogel TM, Simonet P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr Biol. 2014;24(10):1096–100.
Higgins CF. ABC transporters: physiology, structure and mechanism–an overview. Res Microbiol. 2001;152(3):205–10.
Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73(1):241–68.
Schloss PD, Allen HK, Klimowicz AK, Mlot C, Gross JA, Savengsuksa S, McEllin J, Clardy J, Ruess RW, Handelsman J. Psychrotrophic strain of Janthinobacterium lividum from a cold Alaskan soil produces prodigiosin. DNA Cell Biol. 2010;29(9):533–41.
Cowan DA, Chown SL, Convey P, Tuffin M, Hughes K, Pointing S, Vincent WF. Non-indigenous microorganisms in the Antarctic: assessing the risks. Trends Microbiol. 2011;19(11):540–8.
Bottos EM, Woo AC, Zawar-Reza P, Pointing SB, Cary SC. Airborne bacterial populations above desert soils of the McMurdo Dry Valleys, Antarctica. Microb Ecol. 2014;67(1):120–8.
Fierer N, Lauber CL, Ramirez KS, Zaneveld J, Bradford MA, Knight R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012;6(5):1007–17.
Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, Dantas G. Bacterial phylogeny structures soil resistomes across habitats. Nature. 2014;509(7502):612–6.
Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8(1):15–25.
Romero D, Traxler MF, López D, Kolter R. Antibiotics as signal molecules. Chem Rev. 2011;111(9):5492–505.
Acknowledgements
The authors thank New Zealand Antarctic Research Institute (NZARI) and Antarctica New Zealand for logistics and financial support.
Funding
We gratefully acknowledge our sources of funding: National Research Foundation of South Africa (NRF) under the following grant numbers (Grant IDs. 93074 (DAC), 97891 (MWVG) and 99320 (TPM)) and the University of Pretoria Genomics Research Institute (GRI). The funders had no input on the design of the study nor the analyses and interpretation of the data.
Availability of data and materials
The quality-filtered, unassembled metagenomic sequences are available on the MG-RAST server [42] under the accession numbers 4667018.3 through 4667036. All contigs longer than 200 bp from the assembled metagenomes are deposited on the NCBI under the BioProject PRJNA376086. The concatenated antibiotic resistance database, noradab (non-redundant antibiotic resistance database), is available online (noradab.bi.up.ac.za).
Author information
Authors and Affiliations
Contributions
DAC, TPM, OKIB and RP made substantial contributions to the design of the study. DAC collected the samples. MWVG and RP analysed and interpreted the data with substantial contributions from OKIB and TPM. MWVG, RP, OKIB and TPM drafted the manuscripts, and all authors revised it. All authors approve the final version to be published and agree to be accountable for the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Environmental factors of the 17 sampled sites. Table S2. The selected ARGs chosen from noradab, including the names, gene and ARG description, and ARG families. Figure S1. The number of unique ARGs and number of unique AR hosts per site. Linear model indicated in red and lowess in blue (Pearson’s correlation r = 0.89, P = 1.62e-06). Figure S2. ARG host frequencies across sampled sites. The number of different ARG hosts is indicated in green with the number of unique ARG hosts displayed in red, axis on the left. The black line represents the relative abundance, axis on the right. Figure S3. ARG redundancy analysis. The only environmental factor to display a significant impact was percentage N (P =0.024). (DOCX 129 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Van Goethem, M.W., Pierneef, R., Bezuidt, O.K.I. et al. A reservoir of ‘historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 6, 40 (2018). https://doi.org/10.1186/s40168-018-0424-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-018-0424-5